

Abstract
A hallmark of Alzheimer's disease (AD) pathology is the appearance of senile plaques, which are composed of β-amyloid (Aβ) peptides. Aβ is produced by sequential cleavages of amyloid precursor protein (APP) by β- and γ-secretases. These cleavages take place in endosomes during intracellular trafficking of APP through the endocytic and recycling pathways. Genome-wide association studies have identified several risk factors for late-onset AD, one of which is CD2-associated protein (CD2AP), an adaptor molecule that regulates membrane trafficking. Although CD2AP's involvement in APP trafficking has recently been reported, how APP trafficking is regulated remains unclear. We sought to address this question by investigating the effect of CD2AP overexpression or knockdown on the intracellular APP distribution and degradation of APP in cultured COS-7 and HEK293 cells. We found that overexpression of CD2AP increases the localization of APP to Rab7-positive late endosomes, and decreases its localization to Rab5-positive early endosomes. CD2AP overexpression accelerated the onset of APP degradation without affecting its degradation rate. Furthermore, nutrient starvation increased the localization of APP to Rab7-positive late endosomes, and CD2AP overexpression stimulated starvation-induced lysosomal APP degradation. Moreover, the effect of CD2AP on the degradation of APP was confirmed by CD2AP overexpression and knockdown in primary cortical neurons from mice. We conclude that CD2AP accelerates the transfer of APP from early to late endosomes. This transfer in localization stimulates APP degradation by reducing the amount of time before degradation initiation. Taken together, these results may explain why impaired CD2AP function is a risk factor for AD.
Keywords: amyloid precursor protein (APP), Rab, endosome, membrane trafficking, protein degradation, Alzheimer disease, CD2AP, CIN85, Rab7
Introduction
Alzheimer's disease (AD)3 is the most common and progressive neurodegenerative cause of dementia. A pathological hallmark of AD is the presence of senile plaques in the brain tissue of affected patients, which are mainly composed of β-amyloid (Aβ) peptides (1, 2). Aβ is generated by the sequential cleavages of amyloid precursor protein (APP) by two membrane proteases. The first cleavage is carried out by β-secretase (BACE1) to produce a β-cleaved C-terminal fragment (β-CTF), which is further cleaved by γ-secretase (presenilin complex), resulting in the generation of the Aβ peptides (3–5). It is thought that extracellular Aβ accumulation in senile plaques is a major cause of AD development (1, 2).
APP is a type I transmembrane protein whose physiological function has yet to be fully elucidated; however, many functions such as neurite outgrowth, cell adhesion, migration of neuronal precursor cells, synaptogenesis, and axon guidance have been suggested (6–8). APP is present in both plasma membrane and endosomes, being recycled between the two (9, 10). During this intracellular trafficking, a part of APP is subjected to an additional cleavage by α-secretase (ADAM10) to generate N-terminal secreted APP (sAPPα) and the C-terminal fragment (α-CTF) (11). APP is degraded in lysosomes when it is delivered to the late endosome from the early endosome (3–5, 9, 10). In healthy neurons, β-cleavage rarely occurs, thus trafficking and degradation of APP is thought to be precisely regulated. However, the molecular mechanism of APP physiological trafficking and its disruption in AD are not fully understood.
Genome-wide association studies have identified many risk factors for late-onset AD (12–16). A group of these risk factors affect membrane trafficking proteins, one which is CD2-associated protein (CD2AP). Interestingly, CD2AP was found to be an AD susceptibility allele associated with neuritic plaque pathology (17), and its homologue, CIN85 (Cbl-interacting protein of 85 kDa)/SH3KBP1, has been identified as a suppressor of Aβ toxicity in yeast (18), lending further evidence to its involvement in AD.
CD2AP and CIN85 are adaptor proteins with three SH3 domains in their N-terminal region, as well as a proline-rich, actin-binding motif and a coiled-coil in their C-terminal half (19, 20). The SH3 domains of CD2AP and CIN85 have a unique binding preference to a PXP(A)XPR sequence (21, 22), rather than to the canonical PXXP SH3-binding sequence (23). A number of proteins that bind to this SH3 domain have been reported, including ALG2-interacting protein X (ALIX), RIN3, and disabled 1 (Dab1), all of which influence different steps of endocytosis and endosomal trafficking (21, 24–26). In addition, CD2AP can modulate protein trafficking by interacting with actin-capping protein or cortactin to facilitate the formation of branched actin filaments (27).
Recent reports suggest CD2AP plays a role in APP trafficking and Aβ production (28, 29). Knockdown of CD2AP decreases the extracellular Aβ levels in Neuro-2a cells (28) and increases intracellular Aβ in the cell body and dendrites of primary hippocampal neurons (29). Therefore, it seems evident that CD2AP regulates the endocytic transport and degradation of APP. However, which process of APP transport and/or degradation of CD2AP regulates has not yet been determined. Recent studies on CD2AP have also produced conflicting results; one study indicated that depletion of CD2AP leads to decreased APP in the plasma membrane (28), but another described less degradation of internalized APP with CD2AP loss (29). Therefore, how CD2AP regulates APP metabolism is still an open question.
Here, we have examined the role of CD2AP in the intracellular transport of APP and its degradation using cultured cell lines and primary neurons. We found that CD2AP accelerates the transit of APP from early endosomes to the lysosomal pathway for degradation.
Results
Overexpression of CD2AP alters intracellular distribution of APP
CD2AP is localized to endosomal vesicles and regulates membrane trafficking (21, 30). We first examined the role of CD2AP on the intracellular distribution of APP. APP-Myc was cotransfected with FLAG-CD2AP in HEK293 cells. Their localization was observed with anti-tag antibodies, the specificity of which were confirmed by immunostaining of untransfected control cells (for example, CD2AP-negative cells indicated by arrowheads in Fig. S1a). APP showed localization to the perinuclear region and plasma membrane with weak staining throughout the cytoplasm (Fig. 1a), consistent with previous observations (31, 32). The coexpression of CD2AP did not appear to affect the localization of APP (Fig. 1a, right). Because endogenous APP was mainly found at the perinuclear region in either Neuro-2a cells or primary neurons (see Figs. 5h and 11), we focused on APP in the cytoplasmic endosomes hereafter.
Figure 1.

Overexpression of CD2AP alters the intracellular localization of APP. a, effect of CD2AP on the intracellular distribution of APP. Myc-APP was transfected into HEK293 cells in the absence (left, −) or presence (right, +) of CD2AP and was observed after staining with anti-Myc antibody. A higher magnification view of the white box is shown on the right. The nucleus (N) is indicated by dotted lines. Bars represent 20 and 5 μm for low and high magnification, respectively. b, effect of CD2AP overexpression on the endosomal localization of APP. Myc-APP was cotransfected into HEK293 cells with EGFP-Rab4, -Rab5, -Rab7, -Rab8 or -Rab11, markers for fast recycling endosomes, early endosomes, late endosomes, Golgi-derived secretory vesicles, or recycling endosomes, in the absence (left, −) or presence (right, +) of CD2AP. Rabs were visualized by EGFP fluorescence and APP was immunostained with anti-Myc antibody. APP is shown in the 1st column, Rabs in the 2nd column, and merge in the 3rd column. The right panels are a higher magnification view of the white box in the merges. Bar for the left three panels, 20 μm; right panels, 5 μm. c, percentage of APP colocalized with each Rab. Data are expressed as the mean ± S.E. (n = 10 for each Rab). d, relative level of APP colocalization with each Rab in the presence (+) and absence (−) of CD2AP (mean ± S.E. n = 10 for Rab4, Rab5, Rab8, and Rab11, and n = 20 for Rab7. ns, not significant. *, p < 0.05, Student's t test).
Figure 5.
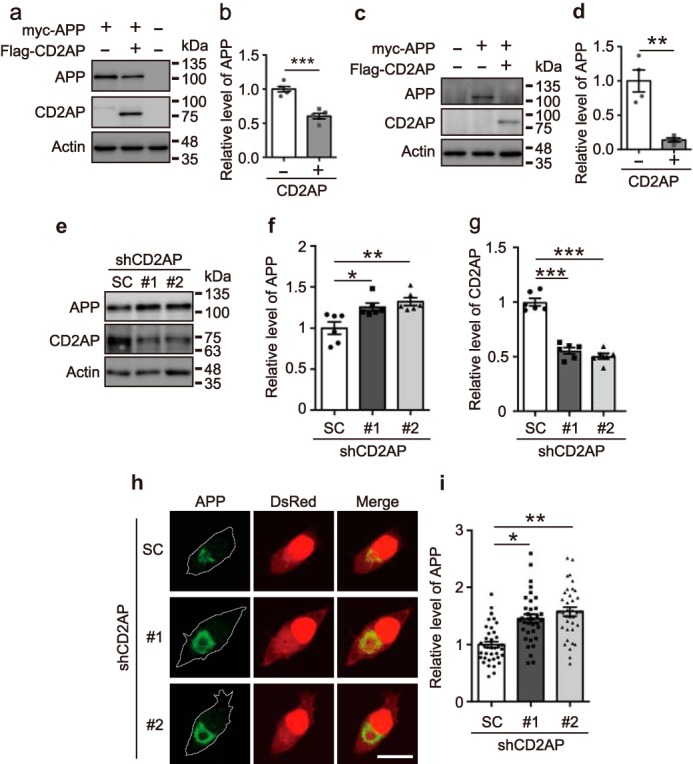
Overexpression or knockdown of CD2AP changes expression levels of APP. a, effect of CD2AP overexpression on protein levels of APP. Myc-APP was cotransfected with FLAG-CD2AP into COS-7 cells. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting with anti-Myc for APP and anti-FLAG for CD2AP. Actin was the loading control. b, level of APP. Data are expressed as the mean ± S.E. (n = 5. ***, p < 0.001, Student's t test). c, effect of CD2AP overexpression on APP levels in neurons. Myc-APP was cotransfected with FLAG-CD2AP into cultured primary neurons. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting with anti-Myc for APP and anti-FLAG for CD2AP. Actin was the loading control. d, level of APP. Data are expressed as the mean ± S.E. (n = 4. **, p < 0.01, Student's t test). e, knockdown of CD2AP by shRNAs. Neuro-2a cells were transfected with shCD2AP #1, #2, or SC and the cell lysates were immunoblotted with anti-APP and anti-CD2AP. f and g, levels of APP and CD2AP, respectively. Data are expressed as the mean ± S.E. (n = 5. *, p < 0.05; **, p < 0.01; ***, p < 0.001, one-way ANOVA analysis, followed by Tukey's multiple comparisons test). h, effect of CD2AP knockdown on levels of APP. APP was cotransfected in Neuro-2a cells with shRNA #1 or #2 of CD2AP and DsRed and then visualized by immunostaining with anti-APP and DsRed fluorescence. APP is shown in the left, DsRed in the middle, and the merge in the right panels. Scale bar, 20 μm. i, relative level of fluorescence intensity of APP. Data are expressed as the mean ± S.E. (n = 37 for SC, n = 35 for #1, n = 36 for #2. *, p < 0.05; **, p < 0.01, one-way ANOVA analysis, followed by Tukey's multiple comparisons test).
Figure 11.

Overexpression or knockdown of CD2AP changes APP levels in neurons. a, effect of CD2AP overexpression on APP levels in neurons. Cultured cortical neurons were transfected with DsRed in the presence (+) or absence (−) of FLAG-CD2AP at DIV6. CD2AP and endogenous APP were detected by immunostaining with anti-FLAG and anti-APP, respectively, at DIV7. DsRed was used as a cell fill. Bar, 20 μm. b, fluorescence intensity of APP in neurons expressing DsRed. Data are expressed as the mean ± S.E. (n = 46 for CD2AP−, n = 59 for CD2AP+. *, p < 0.05, Student's t test). c, effect of the knockdown of CD2AP on APP levels in neurons. Cultured cortical neurons were transfected with shRNA of CD2AP at DIV6 and then visualized by immunostaining with anti-APP and fluorescence of DsRed at DIV8. APP is shown in the left, DsRed in the middle, and the merge in the right panels. Bar, 20 μm. d, relative fluorescence intensity of APP. Data are expressed as the mean ± S.E. (n = 20 for each of SC, shCD2AP #1 and #2. *, p < 0.05; **, p < 0.01, one-way ANOVA analysis, followed by Tukey's multiple comparisons test). e, effect of bafilomycin A1 (BafA1) on the starvation-induced degradation of APP. Cultured cortical neurons at DIV4 were treated with 0.1 μm BafA1 for 2 h, and then starved in the absence (Cont) or presence (BafA1) of BafA1 for an additional 1 or 2 h. The cell lysates were immunoblotted with anti-APP. Actin was the loading control. f, effect of knockdown of CD2AP on the starvation-induced degradation of APP. Cultured cortical neurons were transfected with shRNA of CD2AP at DIV6 and starved for 1 or 2 h at DIV8. APP was visualized by immunostaining with anti-APP. Bar, 20 μm. g, relative fluorescence intensity of APP in neurons expressing DsRed. Data are expressed as the mean ± S.E. (n = 15 for each of SC, shCD2AP #1 and #2. *, p < 0.05; ***, p < 0.001, two-way ANOVA analysis, followed by Sidak's multiple comparisons test).
Previous studies have reported that APP is localized to several different endosomes (33, 34), but it remains unclear if APP preferentially localizes to a specific type of endosome. To clarify APP's vesicular distribution, APP-Myc was cotransfected with EGFP-Rab4, -Rab5, -Rab7, -Rab8, or -Rab11, which are markers for fast recycling endosomes, early endosomes, late endosomes, Golgi-derived secretory vesicles, and recycling endosomes, respectively, in the presence or absence of FLAG-CD2AP (Fig. 1b). APP and CD2AP were visualized by immunostaining with anti-Myc and anti-FLAG antibodies, respectively. Most HEK293 cells expressing both APP and CD2AP were also labeled with EGFP-Rabs (Fig. S1b); 98, 100, 91, 96, and 100% positive for Rab4, Rab5, Rab7, Rab8, and Rab11, respectively. Although expression of these Rab constructs should increase the relative levels of their respective endosomes, it is still possible to analyze the preferential localization of APP in each type of endosome. APP was found at a similar level in all endosomal vesicles, from early to recycling vesicles, with the exception of late endosomes, in which only a small amount of APP was present (Fig. 1b). Although APP was observed as small puncta in the presence of Rab5 and Rab11, it accumulated strongly in the perinuclear region in the presence of Rab8 (Fig. 1b). The colocalization indexes of APP are 41, 46, 6, 63, and 68% with Rab4, Rab5, Rab7, Rab8, and Rab11, respectively (Fig. 1c). The expression of CD2AP increased the localization to Rab4 slightly and to Rab7 significantly, with a concomitant decrease of ∼0.8-fold to Rab5 (Fig. 1d and higher magnification images of Fig. 1b). To verify that the effect of CD2AP was not due to its overexpression, we overexpressed an unrelated protein, DsRed, with APP and examined the colocalization of APP with Rab5 and Rab7. DsRed did not change the relative level of APP colocalized with Rab5 or Rab7 (Fig. S2), indicating that the shift of APP localization to Rab7+ late endosomes is a CD2AP-specific effect. We also confirmed that APP is in fact present on Rab5- and Rab7-positve endosomes by reconstruction of confocal Z-stack images. These images revealed colocalization of APP with Rab5 and Rab7 (Fig. 2). Considering that Rab7-positive late endosomes are one of the immediate recipients of cargo from Rab5-positive early endosomes, these results suggest that CD2AP promotes the transition of APP from early to late endosomes.
Figure 2.
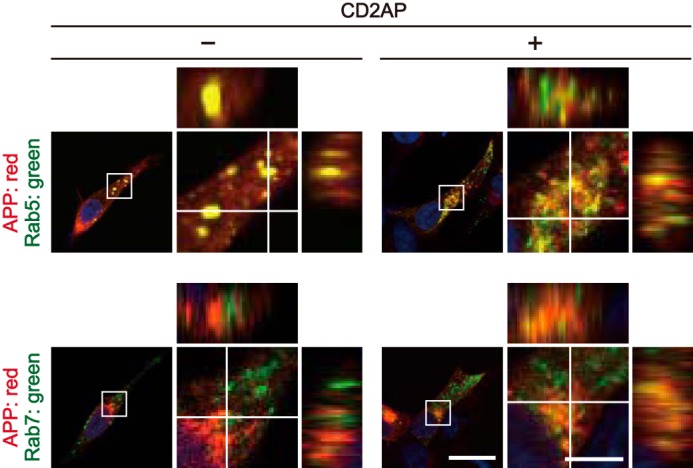
CD2AP-dependent colocalization of APP with Rab5 and Rab7 visualized by orthogonal projection. X-Y confocal images of HEK293 cells transfected with APP and Rab5 (upper row) or Rab7 (bottom row) in the absence (−) or presence (+) of CD2AP. Rabs were visualized by EGFP fluorescence and APP was immunostained with anti-Myc antibody. The right panels are a higher magnification view of the white box in the merges. The X-Z and Y-Z reconstructions of the white line in the X-Y image are shown at the upper and right side of each panel, respectively. Bars for the low magnification panels, 20 μm; high magnification panels, 5 μm.
As previously mentioned, overexpression of Rabs exaggerates the vesicle compartments where they reside. Therefore, we also wanted to observe the localization of APP in early endosomes and late endosomes using their endogenous markers, EEA1 and Rab7, respectively, without overexpression of Rab5 and Rab7. APP was transfected in HEK293 cells in the presence and absence of CD2AP, and its localization was compared with those of EEA1 and Rab7. In the absence of exogenous CD2AP, a portion of APP in the perinuclear region colocalized with EEA1 (Fig. 3a, top row). Rab7 was also found in the perinuclear region, but mostly in the region where APP staining was absent (Fig. 3a, third row). Their orthogonal colocalizations are shown in the right column of Fig. 3a. The colocalization indexes of X-Y images are 23.4 and 8.0% for EEA1 and Rab7, respectively (Fig. 3b). Thus, APP was present to a greater extent in early endosomes than in late endosomes. Expression of CD2AP appeared to make APP distribution slightly more dispersed (Fig. 3a). Furthermore, CD2AP decreased the colocalization of APP with EEA1 and increased the colocalization with Rab7 (Fig. 3a, second and fourth rows). The colocalization index of APP with Rab7 increased from 8 to 13% with CD2AP overexpression. To exclude the possibility of an artificial effect due to protein overexpression, we expressed DsRed instead of CD2AP and quantified the colocalization of APP with EEA1 or Rab7. DsRed did not influence the localization of APP (Fig. S3).
Figure 3.

Overexpression of CD2AP reduces the localization of APP in early endosomes and increases localization in late endosomes. a, effect of CD2AP on the colocalization of APP with EEA1 or Rab7, markers for early endosomes or late endosomes. Myc-APP was transfected into HEK293 cells in the absence (−) or presence (+) of CD2AP. APP, EEA1, and Rab7 were immunostained with anti-Myc, anti-EEA1, and anti-Rab7 antibodies, respectively. APP is shown in the left, EEA1 or Rab7 in the 2nd, and the merge in the 3rd panels. The right panels are a higher magnification view of the white box in the merges. The X-Z and Y-Z reconstruction along the white lines of the X-Y image is shown at the upper and right side of each panel, respectively. Bars for the left three panels, 20 μm; right panels, 5 μm. b, percentage of APP colocalization with EEA1 or Rab7. Data are expressed as the mean ± S.E. (n = 20 for EEA1 and Rab7). c, relative level of APP colocalized with EEA1 or Rab7 in the presence or absence of CD2AP (mean ± S.E. n = 20 for EEA1 and Rab7. *, p < 0.05, **, p < 0.01, Student's t test).
All of the above results are from experiments using overexpression of CD2AP. Consequently, we next examined the effect of CD2AP knockdown on the colocalization of APP with Rab5 or Rab7 (Fig. 4). We used Neuro-2a cells, which are derived from mouse brain, and two different shRNA sequences to knockdown mouse CD2AP, shCD2AP #1 and #2 (see Fig. 5, e and g). Both shRNAs increased the colocalization of APP with Rab5, and decreased its colocalization with Rab7 (Fig. 4), consistent with the results of the overexpression experiments. These results suggest that CD2AP promotes the transition of APP from early endosomes to late endosomes.
Figure 4.

The effect of CD2AP knockdown on the colocalization of APP with Rab5 or Rab7. a, the effect of knockdown vectors of CD2AP on the colocalization of APP with Rab5 or Rab7, markers for early endosomes or late endosomes. Myc-APP and EGFP-Rab5 or -Rab7 were cotransfected in Neuro-2a cells with CD2AP shRNA #1, #2 or scramble control (SC). Rabs were visualized by EGFP fluorescence and APP was immunostained with anti-Myc antibody. APP is shown in the 1st panels, Rabs in the 2nd panels, and merge in the 3rd panels. The right panels are a higher magnification view of the white box in the merges. Bars for the left three panels, 20 μm; right panels, 5 μm. b, relative level of APP colocalization with each Rab. Data are expressed as the mean ± S.E. (n = 20 for SC, #1 and #2, **, p < 0.01, one-way ANOVA analysis, followed by Tukey's multiple comparisons test).
Overexpression or knockdown of CD2AP changes protein levels of APP
Endocytosed APP is thought to be transported to the plasma membrane through recycling endosomes or degraded in lysosomes through late endosomes (9, 10). If this is indeed the case, the aforementioned results suggest that CD2AP directs APP for degradation in lysosomes. We then asked whether overexpression of CD2AP affects the protein level of APP. The expression levels of APP were compared between cells cotransfected with CD2AP or not. The APP levels were decreased to 61% in COS-7 cells by coexpressing CD2AP (Fig. 5, a and b). Similar results were obtained with the other cell lines, Neuro-2a and HEK293 (Fig. S4), where APP separated into two bands. This is due to a difference in glycosylation of APP depending on cell types (35). Of note, coexpression of CD2AP did not affect the maturation of APP (Fig. S4). Reduction of APP was also observed in cultured primary neurons (Fig. 5, c and d). In contrast, CD2AP overexpression did not affect the endogenous CD2AP expression level (Fig. S5a). To control for possible effects of protein overexpression, we compared the expression levels of APP in the presence and absence of EGFP in COS-7 cells (Fig. S6, a and b) and cultured primary neurons (Fig. S6, c and d). EGFP did not affect APP expression. These results indicate that CD2AP reduces APP levels in neurons, reflecting its effect in cultured cell lines.
Next, we examined the effect of CD2AP knockdown on APP protein expression. Both shRNAs, shCD2AP #1 and #2, decreased CD2AP expression as shown by immunoblotting (Fig. 5, e and g) and immunofluorescent staining (Fig. S5, b and c), and these shRNAs increased the expression level of APP by ∼1.25- and 1.32-fold, respectively, in Neuro-2a cells (Fig. 5f). APP was cotransfected with shRNA for CD2AP in Neuro-2a cells and was detected by immunostaining with anti-APP antibodies (Fig. 5h). DsRed was used as a marker of cell area of the transfected cells. The expression of APP was measured in DsRed-positive cells. The knockdown of CD2AP by both of the shRNAs increased the immunofluorescent staining of APP. The APP levels, which were estimated by dividing the APP fluorescence intensity by the cell area, indeed increased by ∼150% in the CD2AP knockdown cells (Fig. 5i), indicating that CD2AP regulates APP levels.
Overexpression of CD2AP enhances the degradation of APP
The rate of APP degradation was measured by chase experiments using cycloheximide (CHX), a protein synthesis inhibitor, in the presence or absence of CD2AP. CHX treatment decreased expression levels of APP, but not CD2AP or actin at a 3.5-h time point (Fig. 6a), indicating a faster turnover rate for APP than for CD2AP. To precisely compare the degradation rates of APP in the presence and absence of CD2AP, the amount of APP protein at time 0 before CHX treatment was normalized between conditions (Fig. 6b), and the subsequent decrease was quantified. APP displayed a gradual decrease in the presence of CHX (Fig. 6c), but demonstrated an accelerated initiation of degradation in the presence of CD2AP. Although APP was decreased by only 7% during the initial 30 min of CHX treatment in the absence of CD2AP, this decrease was enhanced by 35% with the addition of CD2AP (Fig. 6c). The rates of APP decrease were similar thereafter, in the presence or absence of CD2AP (Fig. 6c). We repeated this CHX-chase experiment and overexpressed EGFP as a control. The expression levels of APP were not affected by EGFP (Fig. S7), suggesting that CD2AP specifically promotes the commitment of APP to a degradation pathway.
Figure 6.
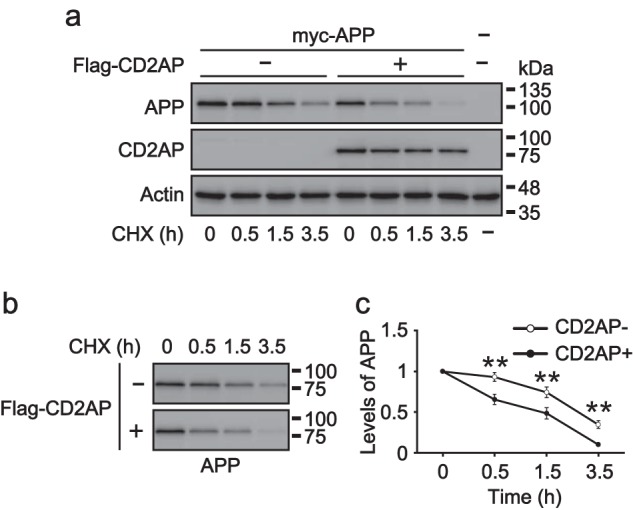
Effect of CD2AP on the degradation of APP. a, effect of CD2AP overexpression on the levels of APP in cells treated with CHX. Myc-APP was transfected with (+) or without (−) FLAG-CD2AP into COS-7 cells. After 18 h, the cells were treated with CHX (50 μg/ml) for the indicated times. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting with anti-Myc for APP and anti-FLAG for CD2AP. Actin was the loading control. b, the immunoblot of APP in a is redisplayed here after adjustment of the APP signal at time 0 between the presence (+) and absence (−) of CD2AP. c, levels of APP in the presence (+) and absence (−) of CD2AP. Data are expressed as the mean ± S.E. (n = 5; **, p < 0.01, two-way ANOVA analysis, followed by Sidak's multiple comparisons test).
Nutrient starvation alters APP distribution
Proteins transported in late endosomes are delivered to lysosomes for degradation through two transport pathways: either direct delivery to the lysosome under steady-state conditions, or indirect transport through the autophagy pathway under nutrient-poor conditions. The autophagy pathway is induced by nutrient starvation. It is reported that the nutrient starvation stimulates degradation of APP (36). Therefore, we speculated that the nutrient-dependent degradation of APP is also stimulated with CD2AP. First, we examined whether APP is degraded in the lysosome after starvation by treating HEK293 cells with NH4Cl, which inhibits lysosomal proteases by neutralizing the internal pH of lysosomes (37). NH4Cl suppressed the decrease of APP (Fig. S8), confirming that APP is degraded in lysosomes under starvation conditions (36). Next, APP was cotransfected in HEK293 cells with EGFP-Rab4, -Rab5, -Rab7, -Rab8, or -Rab11, and the distribution of APP was examined after starvation (Fig. 7a). APP was found in all of the endosomal Rabs, similar to the results in Fig. 1c. Starvation increased the colocalization of APP with Rab7 by ∼4.44-fold, with a 0.36-fold decrease in localization to Rab5 (Fig. 7b and higher magnification view of Fig. 7a).
Figure 7.

Cellular distribution of APP in starved cells. a, effect of starvation on the intracellular distribution of APP. HEK293 cells were transfected with Myc-APP and EGFP-Rab4, -Rab5, -Rab7, -Rab8, or -Rab11. After 18 h, Rabs were visualized by EGFP fluorescence, and APP was immunostained with anti-myc antibody before (−, left side) or after (+, right side) 1 h starvation. APP is shown in the 1st panels, Rabs in the 2nd, merge in the 3rd, and the right panels are a higher magnification of the white box in the merges. Bars for the left three panels, 20 μm; right panels, 5 μm. b, relative level of APP colocalized with each Rab under normal (−) or starvation (+) conditions. Data are expressed as the mean ± S.E. (n = 10; ns, not significant. **, p < 0.01; ***, p < 0.001, Student's t test).
Again, we confirmed the colocalization of APP with Rab5 and Rab7 by Z-stack reconstruction. The Z-X and Z-Y images obtained by confocal microscopy revealed not only their colocalization, but also higher localization of APP with Rab7 in starved cells (Fig. 8), consistent with the results of Fig. 7.
Figure 8.
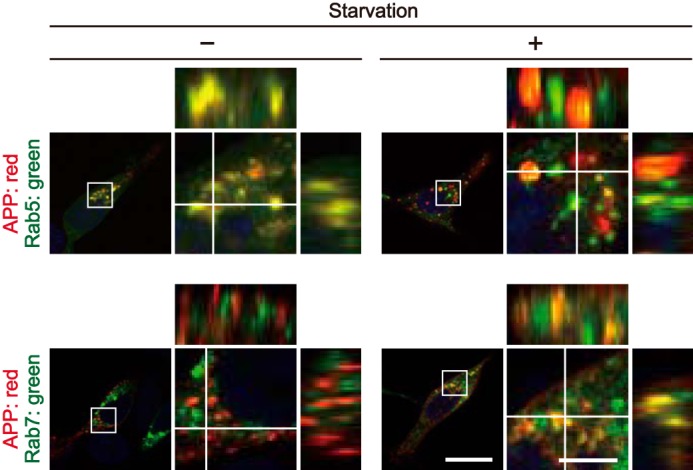
Starvation-dependent colocalization of APP with Rab5 and Rab7 visualized by orthogonal projections. The X-Y confocal images of HEK293 cells expressing APP and Rab5 (upper row) or Rab7 (lower row) in the absence (−) or presence (+) of starvation. Rabs were visualized by EGFP fluorescence and APP was immunostained with anti-Myc antibody. The right panels are a higher magnification view of the white box in the merges. The X-Z and Y-Z reconstructions along the white lines in the X-Y image are shown at the upper and right side of each panel, respectively. Bars for the low magnification panels, 20 μm; high magnification panels, 5 μm.
Next, we examined the localization of APP in early endosomes and late endosomes using their endogenous markers EEA1 and Rab7, respectively, rather than overexpression. APP was transfected into HEK293 cells, which were then nutrient-starved and immunostained for APP and endogenous EEA1 and Rab7 (Fig. 9a). Starvation increased the colocalization of APP with Rab7 by ∼1.60-fold, with a 0.63-fold decrease in localization to EEA1 (Fig. 9b). These results corroborate our previous findings, which suggest that cell starvation stimulates the transport of APP from early to late endosomes.
Figure 9.

Starvation reduces the localization of APP in early endosomes and increases the localization in late endosomes. a, the X-Y confocal images of APP and EEA1 (upper two rows) or Rab7 (lower two rows) in HEK293 cells starved (+) or not (−). APP, EEA1, and Rab7 were immunostained with anti-Myc, anti-EEA1, and anti-Rab7 antibodies, respectively. APP is shown in the 1st panel, EEA1 or Rab7 in the 2nd, and the merge in the 3rd panel. The right panels are a higher magnification view of the white box in the merges. The X-Z and Y-Z reconstruction along the white lines in the X-Y image is shown at the upper and right side of each panel, respectively. Bar for the left three panels, 20 μm; right panels, 5 μm. b, relative level of APP colocalization with EEA1 or Rab7 under normal (−) or starvation (+) conditions. Data are expressed as the mean ± S.E. (n = 20 for EEA1 and Rab7. *, p < 0.05, **, p < 0.01, Student's t test).
Overexpression of CD2AP accelerates the start of the starvation-induced APP degradation
The above observation prompted us to examine how CD2AP influences APP degradation under starvation conditions. COS-7 cells transfected with APP were starved, and the expression levels of APP were followed over time. Starvation abolished APP almost completely after treatment for 6 h (Fig. 10a). Overexpression of CD2AP accelerated APP's disappearance, with very little expression observed after 4 h of starvation. Under this condition, CD2AP and actin retained consistent levels of protein, which illustrates their expression stability. To estimate the degradation rate of APP, the APP protein level at time 0, before starvation, was normalized between conditions with and without CD2AP (Fig. 10b), then quantified for 6 h after starvation. The degradation of APP started 1 h after starvation in the presence of CD2AP, and proceeded gradually in both conditions (Fig. 10c). The expression of CD2AP reduced the lag time before the start of APP degradation. APP was decreased by 19% after 1 h of starvation in the presence of CD2AP, whereas no decrease was detected in its absence (Fig. 10c). The rate of the decrease was linear and appeared to be the same between both conditions (Fig. 10c), suggesting that CD2AP does not affect the degradation itself. To eliminate the possible effect of protein overexpression, similar experiments were performed with overexpression of EGFP. Levels of APP were not affected by EGFP (Fig. S9). These results suggest that CD2AP regulates the commitment of APP to the lysosomal degradation pathway, even when its degradation is induced by starvation.
Figure 10.

Overexpression of CD2AP reduces the lag time before APP degradation induced by starvation. a, effect of CD2AP on the starvation-induced degradation of APP. COS-7 cells were cotransfected with Myc-APP and FLAG-CD2AP and were treated with starvation for the indicated times. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting with anti-Myc for APP and anti-Flag for CD2AP. Actin was the loading control. B, the immunoblot of APP in panel a is redisplayed here after adjustment of the APP signal at time 0 between the presence (+) and absence (−) of CD2AP. c, levels of APP in the presence (+) and absence (−) of CD2AP. Data are expressed as the mean ± S.E. (n = 6; *, p < 0.05, two-way ANOVA analysis, followed by Sidak's multiple comparisons test).
Overexpression of CD2AP decreases levels of APP in neurons
Given that CD2AP appeared to play a role in APP degradation in cell lines, we next examined its role in neuronal APP metabolism. Endogenous APP was observed in cultured primary neurons by immunostaining with anti-APP antibodies after transfection with CD2AP. Cotransfected DsRed was used as a marker of transfected neurons and cell area. APP displayed a vesicular staining pattern, particularly in the perinuclear region, whereas CD2AP exhibited a punctate staining pattern, distributed throughout the cytoplasm (Fig. 11a). The expression of CD2AP slightly decreased the immunofluorescence intensity of APP (Fig. 11a). Quantification of APP immunostaining indicated a significant decrease, 13% lower in CD2AP-expressing neurons than in nontransfected control neurons (Fig. 11b). Although the decrease was much less compared with that of exogenous APP coexpressed with CD2AP (Fig. 5, c and d), we think this difference may be due to the differences between two experiments, the detection of overexpressed APP with anti-myc (Fig. 5, c and d) versus the detection of endogenous APP with anti-APP (Fig. 11b). APP levels in primary neurons were not affected by overexpression of a control EGFP construct (Fig. S10). In contrast, the knockdown of CD2AP by shRNA increased the fluorescence intensity of APP staining by ∼130–140% (Fig. 11, c and d). These results suggest that CD2AP regulates the expression of APP in neurons.
Next, we confirmed whether APP is degraded through the lysosomal pathway after starvation by using bafilomycin A1, a lysosome inhibitor, whose target is vacuolar H+-ATPase. Bafilomycin A1 suppressed the degradation of APP induced by starvation (Fig. 11e), indicating that APP is degraded in lysosomes under starvation conditions. We then tested the effect of CD2AP on starvation-induced APP degradation. APP immunofluorescence intensity was decreased by 32% at 1 h after starvation in the control. When CD2AP shRNA was introduced, this decrement lessened to 24 and 22%, with shCD2AP #1 and #2, respectively. Interestingly, they also suppressed further degradation of APP over time (Fig. 11, f and g), suggesting that shCD2AP effectively inhibits APP degradation after 1 h of neuronal starvation (Fig. 11g). These results suggest that the depletion of CD2AP abrogates the degradation of APP in primary neurons that is normally induced by starvation.
Overexpression of CD2AP decreases level of soluble APP-β (sAPPβ) in the culture medium
The identification of CD2AP as a risk factor for late-onset AD, along with its known influence on APP trafficking, suggest its involvement in Aβ generation. Consequently, we evaluated the effect of CD2AP on β-cleavage of APP by measuring the amount of sAPPβ in the culture medium. We validated our sAPPβ ELISA by coexpression of BACE1 β-secretase and APP in COS-7 cells. Although the expression of APP modestly increased sAPPβ levels, coexpression of BACE1 increased sAPPβ by ∼4.43-fold (Fig. 12a), indicating that the ELISA worked as anticipated. We then examined the effect of CD2AP on the production of sAPPβ in cultured cells treated with CHX (Fig. 12b). The level of sAPPβ was lower in cells overexpressing CD2AP compared with those without, displaying a 58% reduction at 0.5 h after CHX addition (Fig. 12b). Longer CHX treatments produced similar sAPPβ trends, with lower levels in CD2AP-treated conditions. These results suggest that CD2AP decreases the occurrence of APP β-cleavage.
Figure 12.
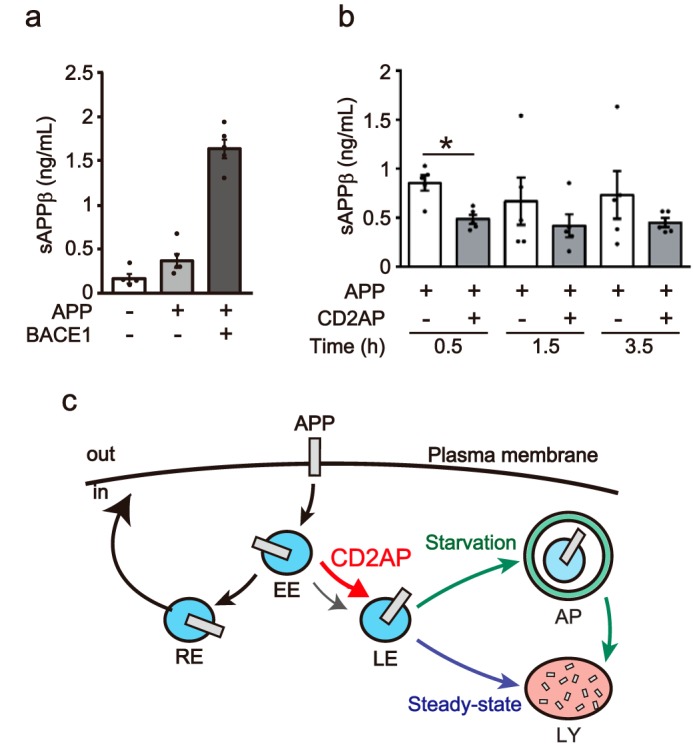
Overexpression of CD2AP decreases the level of sAPPβ in the medium of CHX-treated cells. a, measurement of sAPPβ by ELISA. Myc-APP was cotransfected with FLAG-CD2AP or EGFP-BACE1 into COS-7 cells. The culture media was collected 18 h after transfection and sAPPβ was measured by ELISA. Data are expressed as the mean ± S.E. (n = 5). b, the effect of CD2AP on level of sAPPβ in the culture medium of cells treated with CHX. Myc-APP was transfected with (+) or without (−) FLAG-CD2AP into COS-7 cells. The cells were treated with CHX (50 μg/ml) 18 h after transfection for the indicated times. sAPPβ in the culture medium was measured by ELISA. Data are expressed as the mean ± S.E. (n = 5. *, p < 0.05, Student's t test). c, schematic representation of the role of CD2AP in APP trafficking. Endocytosed APP is mostly transferred to the recycling endosomes (RE) after the early endosomes (EE). CD2AP promotes the transit of APP to late endosomes (LE) for degradation in lysosomes (LY), either through a direct route under steady-state conditions, or through an autophagosome (AP) under starvation conditions, thus maintaining the proper level of cellular APP.
Discussion
Healthy neurons require strict regulation of APP intracellular trafficking and degradation to prevent unwarranted β-cleavage and Aβ plaque generation. Dysregulation of these pathways increase the risk of AD. As a result, it is critical to understand the underlying mechanisms governing the intercellular trafficking and metabolism of APP.
CD2AP has been recently identified as a genetic risk factor for late-onset AD (14–16). Although two subsequent publications indicate its involvement in APP trafficking (28, 29), their findings suggest conflicting mechanisms. As a result, how CD2AP participates in APP trafficking remains unclear. In this study, we start to unravel this question, by showing that CD2AP controls the transition of APP from early to late endosomes, thereby regulating the lysosomal degradation of APP.
These data are critical, because APP transits through multiple subcellular compartments, and could therefore be regulated in many ways. APP is synthesized in endoplasmic reticulum and transported to the plasma membrane by an exocytic pathway (3–5, 9, 10). A subset of APP is cleaved on the plasma membrane by α-secretase (ADAM10) (11), leading to shedding of the extracellular N-terminal region as soluble APP (sAPPα), and retention of the CTF in plasma membrane. The other population of APP is internalized and recycled back to the cell surface, or is degraded in lysosomes through late endosomes. Thus, APP is shuttled between the cell surface and internal vesicles by the endocytic and recycling pathways. During its intracellular trafficking, APP could be subjected to β-cleavage by BACE1, producing Aβ after subsequent cleavage with γ-secretase. In contrast, the total protein amount of APP is mainly determined by its degradation in lysosomes, following its delivery by Rab7-positive late endosomes.
Our data revealed, however, that APP colocalization with Rab7 in late endosomes was considerably less than with other endosomal markers (Figs. 1 and 7), suggesting that only a minor fraction of APP is directed to late endosomes for degradation under basal conditions in cultured cells. Once APP is transported to late endosomes, it is degraded immediately, as if this step is rate-limiting. Understanding how APP is sorted to either the recycling or degradation pathway is crucial, because its degradation rate is strongly affected by the amount of time APP resides in each endosomal compartment. Here, we found that the expression of CD2AP increased the ratio of APP present in Rab7-labeled late endosomes and its degradation. These data demonstrate that CD2AP is a sorting factor directing APP from early endosomes to late endosomes (Fig. 12c), potentially acting as a mechanistic switch between the two pathways.
Endosomal trafficking appears to influence neurodegeneration, as multiple human genome-wide association studies studies have identified endosomal regulators as genetic risk factors for late-onset AD, including phosphatidylinositol-binding clathrin assembly protein (PICALM), bridging integrator 1 (BIN1), sortilin-related receptor 1 (SORL1), and CD2AP (12–16). Recent studies have confirmed their role in APP trafficking and metabolism. For example, PICALM modulates Aβ production via internalization of APP and γ-secretase (38, 39); BIN1 affects Aβ generation through endosomal BACE1 trafficking (29); and SORL1 regulates APP transport between the trans-Golgi network and the endosome and acts as a negative regulator of Aβ generation (34). CD2AP has also been indicated to regulate the endocytic transport and the production of Aβ (28, 29). However, the effect of CD2AP on Aβ production was different between two studies; one showed a decrease in Aβ production (28) and the other indicated an increase (29) when CD2AP levels were reduced. Furthermore, both studies did not determine where in the endosomal network CD2AP works, but one suggested that APP accumulates in early endosomes when the expression of CD2AP is reduced (29). These results are consistent with our data, as knockdown of CD2AP with shRNA increased the localization of APP in the early endosomes. We also found that only a minor population of APP resided in late endosomes, but the expression of CD2AP increased this localization and accelerated the degradation of APP by reducing the lag time of degradation initiation. These results suggest that down-regulation of CD2AP increases Aβ production, and we confirmed this hypothesis by measuring sAPPβ released into the culture medium. These results also agreed with data from Ubelmann et al. (29), but not with data obtained from another study using CD2AP heterozygous mice. Although Aβ levels are influenced by many factors, including γ-cleavage, clearance, and aggregation, sAPPβ measurements provide an output for the overall effect of CD2AP on β-cleavage. Thus, taken together, our data show that CD2AP regulates the protein levels of APP by sorting it to the late endosomes for eventual degradation.
Impairments in autophagy are also well-demonstrated in AD (40). Accumulation of autophagosomes or late autophagic vacuoles is observed in AD brains (41, 42). Additionally, cell starvation induces autophagy (43) and stimulates the degradation of APP (36). Furthermore, endosomal Rab7 regulates autophagy (44–46). Based on these previous reports, we examined the role of CD2AP in APP degradation under starvation conditions.
Previous data suggest that CD2AP may influence autophagy. For instance, CD2AP is reported to interact with Alix, which functions at the multivesicular body formation by interacting with ESCRT components (24). Because ESCRT participates in autophagy pathway (48), we cannot exclude the possibility that CD2AP plays a role promoting APP degradation in a autophagy process. In our experiments, however, CD2AP decreased the lag time before the beginning of degradation in starved cells (Figs. 7 and 10). These effects of CD2AP were similar to those observed with CHX-treated cells, where the autophagic activity is not so high, suggesting that CD2AP functions mainly at the transition from early to late endosomes, which corresponds to the entrance site to the starvation-induced autophagic-lysosomal degradation. Thus, CD2AP may stimulate APP transport to late endosomes in both autophagy-dependent and autophagy-independent AD.
The exact mechanism of how CD2AP regulates the transition of APP from early to late endosomes remains unclear. CD2AP plays a role in multiple steps of endosomal trafficking by interacting with different proteins, as described previously. In addition, APP has a highly conservative YENPTY motif in its cytoplasmic domain, which can bind to a number of adaptor proteins, such as Fe65, X11/Mint, Numb, SNX17, and Dab1 to modulate its movement (22, 49–53). Intriguingly, one of these proteins, Dab1, is capable of binding both APP and CD2AP/CIN85. Dab1 has a PTB domain in its N-terminal region that could bind to the YENPTY sequence of APP, and a PKPAPR sequence in its C-terminal region that could bind the SH3 domain of CD2AP/CIN85. It is possible that CD2AP interacts with APP via Dab1 or a Dab1-like linker protein. Moreover, these interactions could be regulated by phosphorylation, as previous studies have showed that Cdk5-p35 regulates Dab1's interaction with CIN85/CD2AP (22) and Fyn mediates its binding with APP (54). Future work should focus on determining which factors mediate CD2AP-APP interactions in neurons, and how these mechanisms influence neurodegeneration.
In conclusion, the overexpression of CD2AP increases localization of APP to late endosomes and accelerates the lysosome-dependent or starvation-dependent degradation of APP. CD2AP regulates the sorting of APP from early endosomes to late endosomes. Down-regulation of CD2AP lengthens the time APP resides in the endosomes, which should increase Aβ production by enhancing the frequency interactions between APP and BACE1. Thus, the present results help to explain how CD2AP acts as a risk factor for AD.
Experimental procedures
Antibodies, chemicals, and plasmids
Anti-APP (number 18961) was obtained from IBL, anti-APP 22C11 (number MAB348) and anti-Myc 4A6 (number 05-724) were purchased from Millipore, anti-EEA1 (number 2411) and anti-Rab7 (number 9367) were obtained from Cell Signaling Technology, anti-CD2AP (HPA003326), anti-FLAG M2 (number F1804), and anti-actin (number A2066) were purchased from Sigma, anti-CD2AP (number sc-9137) was purchased from Santa Cruz Biotechnology, and anti-GFP (number 11814460001) was obtained from Roche. Secondary antibodies conjugated with Alexa Fluor 488, Alex Fluor 546, or Alexa Fluor 647 were purchased from Invitrogen. CHX was purchased from Sigma and bafilomycin A1 was from Cayman Chemical. The pEBOS-Myc-APP plasmid was constructed from pEBOS-APP, which was provided by Dr. F. Kametani at the Tokyo Metropolitan Institute of Medical Science. The 3× FLAG-CD2AP was provided by Dr. M. Taoka at Tokyo Metropolitan University. EGFP-Rab4A, -Rab5A, -Rab7A, -Rab8A, and -Rab11A were previously described (55), as was pCAGGS-DsRed (56). EGFP-BACE1 was provided by Dr. S. Roy at the University of California San Diego (57). The CD2AP knockdown shRNA sequences were 5′-GGAACCAATGAAGATGAACTTACA-3′ for shCD2AP #1 and 5′-AGGGTGAACTGAACGGTAAAG-3′ for shCD2AP #2. The SC sequence was 5′-ACTTGACGATAACGAGACAAGATA-3′ for the control. Each knockdown sequence was individually inserted into a pSUPER.puro vector (Oligoengine).
Cell cultures and transfection
COS-7 and HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin at 37 °C in 5% CO2. Neuro-2a cells were maintained in minimum essential medium (Gibco) supplemented with 10% (v/v) fetal bovine serum, nonessential amino acids (Gibco), 100 units/ml of penicillin, and 0.1 mg/ml of streptomycin at 37 °C in 5% CO2. COS-7, HEK293, and Neuro-2a cells were plated at 0.75–1 × 105, 0.5–1.0 × 105, and 0.5–1.0 × 105 cells/ml, respectively, and transfected with the indicated plasmids using Lipofectamine 2000 or Lipofectamine 3000 (Invitrogen), according to the manufacturer's instruction.
Mouse primary neuron culture and transfection
ICR mice were obtained from Sankyo Laboratory Service (Tokyo, Japan). All mouse experiments were performed according to the guidelines for animal experimentation of Tokyo Metropolitan University. The studies were approved by the Research Ethics Committee of Tokyo Metropolitan University (approval numbers, 28-7 and G29-8). All efforts were made to reduce the suffering of the animals. Primary cortical neurons were prepared from mouse brains at embryonic day 16, as previously described (47). Neuronal cells were plated at 0.3–7.5 × 105 cells/ml. They were maintained in neurobasal medium (Gibco) supplemented with 2% (v/v) B27 (Gibco) and 0.5 μm l-glutamine at 37 °C in 5% CO2 and were individually transfected with the indicated plasmids using Lipofectamine 2000 or Lipofectamine 3000 at 3 or 6 days in vitro (DIV). Neurons accounted for about 75% of cells in the cultures at 7 DIV when they were assessed by immunostaining for Tuj1, a neuronal marker.
Starvation
Nutrient starvation was induced according to the method by Jaeger et al. (36). Briefly, the cells were washed twice with phosphate-buffered saline (PBS) and incubated in Hank's balanced salt solution (Gibco) containing 1.8 mm CaCl2 and 0.81 mm MgSO4 for the indicated times.
Immunofluorescence staining
Cells plated on a coverslip were fixed with 4% (w/v) paraformaldehyde in PBS for 20 min and then treated with 0.1% (v/v) Triton X-100 and 5% (w/v) skim milk in PBS for permeabilization and blocking. The cells were probed with the primary antibodies in PBS, 0.1% (v/v) Triton X-100, 5% (w/v) skim milk for 1 h at room temperature and were then incubated with a secondary antibody and DAPI (4′,6-diamidino-2-phenylindole; Dojindo, Kumamoto, Japan) in PBS, 0.1% (v/v) Triton X-100, 5% (w/v) skim milk for 1 h at room temperature, followed by observation under a confocal laser scanning microscope LSM710 (Carl Zeiss). The specificity of anti-Myc, anti-FLAG, anti-APP, or anti-CD2AP was validated by the staining of cells overexpressing the antigen protein. The specificity of anti-EEA1 or anti-Rab7 was confirmed by control experiments where secondary antibodies were used alone (no primary antibodies).
SDS-PAGE and immunoblotting
SDS-PAGE and immunoblotting were carried out as previously described (47).
ELISA of sAPPβ
COS-7 cells were transfected with the indicated plasmids. The culture medium was directly collected from the plates 24 h after transfection and then centrifuged at 200 × g at 4 °C for 10 min. The supernatant was stored at −80 °C until the quantitation. The secreted sAPPβ was measured by the human sAPPβ-w (highly sensitive) assay kit (number 27732, IBL) according to the manufacturer's protocol.
Quantification and statistical analysis
Immunoreactions were visualized by acquiring a digital image with a Fusion SL4 imager (Vilber Lourmat). The band intensities were quantified using ImageJ. The fluorescent intensity of APP was measured using ZEN imaging software (Carl Zeiss). The colocalization indices were defined as the number of pixels that were fluorescent for both APP and Rab or APP and EEA1, over the total APP fluorescence. Data were collected and analyzed by a blinded co-author using Zen imaging software. Quantitative data are expressed as the mean ± S.E. and were subjected to Student's t test for single comparisons, one-way ANOVA, followed by Tukey's multiple comparisons test for multiple comparisons, or two-way ANOVA, followed by Sidak's multiple comparisons test.
Author contributions
K. F. and S.-i. H. conceptualization; K. F., T. Tomita, and M. F. formal analysis; K. F., T. Takasugi, Y.-W. C., and Y. H. investigation; K. F., T. Takasugi, Y.-W. C., Y. H., T. Tomita, and M. F. methodology; T. Takasugi and Y. H. validation; T. Tomita data curation; M. F. resources; S.-i. H. funding acquisition; S.-i. H. writing-original draft; S.-i. H. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Fuyuki Kametani, Tokyo Metropolitan Institute of Medical Sciences, for providing APP expression constructs, Masato Taoka, Tokyo Metropolitan University, for providing an expression plasmid of CD2AP, and Dr. S. Roy, University of California San Diego, for providing EGFP-BACE1. We also thank Dr. J. Courtland, Department of Cell Biology, Duke University Medical School, for reading the manuscript.
This work was supported in part by MEXT Grant-in-aid project, Scientific Research on Innovation Area (Brain Protein Aging and Dementia Control) of Japan Grant 26117004 (to S. H.) and MEXT Grant-in-aid numbers 16K07060 (to S. H.) and 18J01378 (to K. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S10.
- AD
- Alzheimer's disease
- APP
- amyloid precursor protein
- Aβ
- amyloid β
- α-CTF
- α-cleaved C-terminal fragment
- β-CTF
- β-cleaved C-terminal fragment
- CD2AP
- CD2-associated protein
- CIN85
- Cbl-interacting protein of 85kDa
- CHX
- cycloheximide
- DIV
- days in vitro
- sAPPα
- soluble APP-α
- sAPPβ
- soluble APP-β
- ALIX
- ALG2-interacting protein X
- Dab1
- disabled 1
- PICALM
- phosphatidylinositol-binding clathrin assembly protein
- BIN1
- bridging integrator 1
- SORL1
- sortilin-related receptor 1
- SH3
- Src homology 3
- EGFP
- enhanced growth factor protein
- ANOVA
- analysis of variance
- shRNA
- short hairpin RNA.
References
- 1. Nelson P. T., Alafuzoff I., Bigio E. H., Bouras C., Braak H., Cairns N. J., Castellani R. J., Crain B. J., Davies P., Del Tredici K., Duyckaerts C., Frosch M. P., Haroutunian V., Hof P. R., Hulette C. M., et al. (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Selkoe D. J., and Hardy J. (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol.Med. 8, 595–608 10.15252/emmm.201606210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Small S. A., and Gandy S. (2006) Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 10.1016/j.neuron.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thinakaran G., and Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rajendran L., and Annaert W. (2012) Membrane trafficking pathways in Alzheimer's disease. Traffic 13, 759–770 10.1111/j.1600-0854.2012.01332.x [DOI] [PubMed] [Google Scholar]
- 6. Naumann N., Alpár A., Ueberham U., Arendt T., and Gärtner U. (2010) Transgenic expression of human wild-type amyloid precursor protein decreases neurogenesis in the adult hippocampus. Hippocampus 20, 971–979 [DOI] [PubMed] [Google Scholar]
- 7. Siemes C., Quast T., Kummer C., Wehner S., Kirfel G., Muller U., and Herzog V. (2006) Keratinocytes from APP/APLP2-deficient mice are impaired in proliferation, adhesion and migration in vitro. Exp. Celll Res. 312, 1939–1949 10.1016/j.yexcr.2006.02.025 [DOI] [PubMed] [Google Scholar]
- 8. Young-Pearse T. L., Bai J., Chang R., Zheng J. B., LoTurco J. J., and Selkoe D. J. (2007) A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 27, 14459–14469 10.1523/JNEUROSCI.4701-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Musardo S., Saraceno C., Pelucchi S., and Marcello E. (2013) Trafficking in neurons: searching for new targets for Alzheimer's disease future therapies. Eur. J. Pharmacol. 719, 84–106 10.1016/j.ejphar.2013.07.019 [DOI] [PubMed] [Google Scholar]
- 10. Wang X., Zhou X., Li G., Zhang Y., Wu Y., and Song W. (2017) Modifications and trafficking of APP in the pathogenesis of Alzheimer's disease. Front. Mol. Neurosci. 10, 294 10.3389/fnmol.2017.00294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuhn P. H., Wang H., Dislich B., Colombo A., Zeitschel U., Ellwart J. W., Kremmer E., Rossner S., and Lichtenthaler S. F. (2010) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032 10.1038/emboj.2010.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bertram L., McQueen M. B., Mullin K., Blacker D., and Tanzi R. E. (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23 10.1038/ng1934 [DOI] [PubMed] [Google Scholar]
- 13. Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Williams A., Jones N., Thomas C., Stretton A., Morgan A. R., Lovestone S., et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hollingworth P., Harold D., Sims R., Gerrish A., Lambert J. C., Carrasquillo M. M., Abraham R., Hamshere M. L., Pahwa J. S., Moskvina V., Dowzell K., Jones N., Stretton A., Thomas C., Richards A., et al. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naj A. C., Jun G., Beecham G. W., Wang L. S., Vardarajan B. N., Buros J., Gallins P. J., Buxbaum J. D., Jarvik G. P., Crane P. K., Larson E. B., Bird T. D., Boeve B. F., Graff-Radford N. R., De Jager P. L., et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43, 436–441 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lambert J. C., Grenier-Boley B., Harold D., Zelenika D., Chouraki V., Kamatani Y., Sleegers K., Ikram M. A., Hiltunen M., Reitz C., Mateo I., Feulner T., Bullido M., Galimberti D., Concari L., et al. (2013) Genome-wide haplotype association study identifies the FRMD4A gene as a risk locus for Alzheimer's disease. Mol. Psychiatry 18, 461–470 10.1038/mp.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shulman J. M., Chen K., Keenan B. T., Chibnik L. B., Fleisher A., Thiyyagura P., Roontiva A., McCabe C., Patsopoulos N. A., Corneveaux J. J., Yu L., Huentelman M. J., Evans D. A., Schneider J. A., Reiman E. M., De Jager P. L., and Bennett D. A. (2013) Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol. 70, 1150–1157 10.1001/jamaneurol.2013.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Treusch S., Hamamichi S., Goodman J. L., Matlack K. E., Chung C. Y., Baru V., Shulman J. M., Parrado A., Bevis B. J., Valastyan J. S., Han H., Lindhagen-Persson M., Reiman E. M., Evans D. A., Bennett D. A., et al. (2011) Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science 334, 1241–1245 10.1126/science.1213210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dikic I. (2002) CIN85/CMS family of adaptor molecules. FEBS Lett. 529, 110–115 10.1016/S0014-5793(02)03188-5 [DOI] [PubMed] [Google Scholar]
- 20. Hernandez-Valladares M., Kim T., Kannan B., Tung A., Aguda A. H., Larsson M., Cooper J. A., and Robinson R. C. (2010) Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat. Struct. Mol. Biol. 17, 497–503 10.1038/nsmb.1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rouka E., Simister P. C., Janning M., Kumbrink J., Konstantinou T., Muniz J. R., Joshi D., O'Reilly N., Volkmer R., Ritter B., Knapp S., von Delft F., Kirsch K. H., and Feller S. M. (2015) Differential recognition preferences of the three Src homology 3 (SH3) domains from the adaptor CD2-associated protein (CD2AP) and direct association with Ras and Rab interactor 3 (RIN3). J. Biol. Chem. 290, 25275–25292 10.1074/jbc.M115.637207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato Y., Taoka M., Sugiyama N., Kubo K., Fuchigami T., Asada A., Saito T., Nakajima K., Isobe T., and Hisanaga S. (2007) Regulation of the interaction of Disabled-1 with CIN85 by phosphorylation with Cyclin-dependent kinase 5. Genes Cells 12, 1315–1327 10.1111/j.1365-2443.2007.01139.x [DOI] [PubMed] [Google Scholar]
- 23. Saksela K., and Permi P. (2012) SH3 domain ligand binding: what's the consensus and where's the specificity? FEBS Lett. 586, 2609–2614 10.1016/j.febslet.2012.04.042 [DOI] [PubMed] [Google Scholar]
- 24. Morita E., Sandrin V., Chung H. Y., Morham S. G., Gygi S. P., Rodesch C. K., and Sundquist W. I. (2007) Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 26, 4215–4227 10.1038/sj.emboj.7601850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fuchigami T., Sato Y., Tomita Y., Takano T., Miyauchi S. Y., Tsuchiya Y., Saito T., Kubo K., Nakajima K., Fukuda M., Hattori M., and Hisanaga S. (2013) Dab1-mediated colocalization of multi-adaptor protein CIN85 with Reelin receptors, ApoER2 and VLDLR, in neurons. Genes Cells 18, 410–424 10.1111/gtc.12045 [DOI] [PubMed] [Google Scholar]
- 26. Homayouni R., Rice D. S., Sheldon M., and Curran T. (1999) Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J. Neurosci. 19, 7507–7515 10.1523/JNEUROSCI.19-17-07507.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao J., Bruck S., Cemerski S., Zhang L., Butler B., Dani A., Cooper J. A., and Shaw A. S. (2013) CD2AP links cortactin and capping protein at the cell periphery to facilitate formation of lamellipodia. Mol. Cell. Biol. 33, 38–47 10.1128/MCB.00734-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao F., Jiang H., Srivatsan S., Xiao Q., Lefton K. B., Yamada K., Mahan T. E., Lee J. M., Shaw A. S., and Holtzman D. M. (2015) Effects of CD2-associated protein deficiency on amyloid-β in neuroblastoma cells and in an APP transgenic mouse model. Mol. Neurodegener. 10, 12 10.1186/s13024-015-0006-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ubelmann F., Burrinha T., Salavessa L., Gomes R., Ferreira C., Moreno N., and Guimas Almeida C. (2017) Bin1 and CD2AP polarise the endocytic generation of β-amyloid. EMBO Rep. 18, 102–122 10.15252/embr.201642738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cormont M., Metón I., Mari M., Monzo P., Keslair F., Gaskin C., McGraw T. E., and Le Marchand-Brustel Y. (2003) CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 4, 97–112 10.1034/j.1600-0854.2003.40205.x [DOI] [PubMed] [Google Scholar]
- 31. Quast T., Wehner S., Kirfel G., Jaeger K., De Luca M., and Herzog V. (2003) sAPP as a regulator of dendrite motility and melanin release in epidermal melanocytes and melanoma cells. FASEB J. 17, 1739–1741 10.1096/fj.02-1059fje [DOI] [PubMed] [Google Scholar]
- 32. Nizzari M., Venezia V., Repetto E., Caorsi V., Magrassi R., Gagliani M. C., Carlo P., Florio T., Schettini G., Tacchetti C., Russo T., Diaspro A., and Russo C. (2007) Amyloid precursor protein and Presenilin 1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J. Biol. Chem. 282, 13833–13844 10.1074/jbc.M610146200 [DOI] [PubMed] [Google Scholar]
- 33. Toh W. H., Tan J. Z., Zulkefli K. L., Houghton F. J., and Gleeson P. A. (2017) Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic 18, 159–175 10.1111/tra.12465 [DOI] [PubMed] [Google Scholar]
- 34. Eggert S., Gonzalez A. C., Thomas C., Schilling S., Schwarz S. M., Tischer C., Adam V., Strecker P., Schmidt V., Willnow T. E., Hermey G., Pietrzik C. U., Koo E. H., and Kins S. (2018) Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cell. Mol. Life Sci. 75, 301–322 10.1007/s00018-017-2625-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakagawa K., Kitazume S., Oka R., Maruyama K., Saido T. C., Sato Y., Endo T., and Hashimoto Y. (2006) Sialylation enhances the secretion of neurotoxic amyloid-β peptides. J. Neurochem. 96, 924–933 10.1111/j.1471-4159.2005.03595.x [DOI] [PubMed] [Google Scholar]
- 36. Jaeger P. A., and Wyss-Coray T. (2010) Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 67, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 37. Orhon I., and Reggiori F. (2017) Assays to monitor autophagy progression in cell cultures. Cells 6, e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiao Q., Gil S. C., Yan P., Wang Y., Han S., Gonzales E., Perez R., Cirrito J. R., and Lee J. M. (2012) Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem. 287, 21279–21289 10.1074/jbc.M111.338376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanatsu K., Morohashi Y., Suzuki M., Kuroda H., Watanabe T., Tomita T., and Iwatsubo T. (2014) Decreased CALM expression reduces Abeta42 to total Aβ ratio through clathrin-mediated endocytosis of γ-secretase. Nat. Commun. 5, 3386 10.1038/ncomms4386 [DOI] [PubMed] [Google Scholar]
- 40. Colacurcio D. J., Pensalfini A., Jiang Y., and Nixon R. A. (2018) Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's disease. Free Rad. Biol. Med. 114, 40–51 10.1016/j.freeradbiomed.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu W. H., Kumar A., Peterhoff C., Shapiro Kulnane L., Uchiyama Y., Lamb B. T., Cuervo A. M., and Nixon R. A. (2004) Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Inter. J. Biochem. Cell Biol. 36, 2531–2540 10.1016/j.biocel.2004.05.010 [DOI] [PubMed] [Google Scholar]
- 42. Yoon S. Y., Choi J. E., Kweon H. S., Choe H., Kim S. W., Hwang O., Lee H., Lee J. Y., and Kim D. H. (2008) Okadaic acid increases autophagosomes in rat neurons: implications for Alzheimer's disease. J. Neurosci. Res. 86, 3230–3239 10.1002/jnr.21760 [DOI] [PubMed] [Google Scholar]
- 43. Deter R. L., and De Duve C. (1967) Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 33, 437–449 10.1083/jcb.33.2.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gutierrez M. G., Munafo D. B., Beron W., and Colombo M. I. (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117, 2687–2697 10.1242/jcs.01114 [DOI] [PubMed] [Google Scholar]
- 45. Jäger S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., and Eskelinen E. L. (2004) Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117, 4837–4848 10.1242/jcs.01370 [DOI] [PubMed] [Google Scholar]
- 46. Kuchitsu Y., Homma Y., Fujita N., and Fukuda M. (2018) Rab7 knockout unveils regulated autolysosome maturation induced by glutamine starvation. J. Cell Sci. 131, jcs215442 10.1242/jcs.215442 [DOI] [PubMed] [Google Scholar]
- 47. Furusawa K., Asada A., Urrutia P., Gonzalez-Billault C., Fukuda M., and Hisanaga S. I. (2017) Cdk5 Regulation of the GRAB-mediated Rab8-Rab11 cascade in axon outgrowth. J. Neurosci. 37, 790–806 10.1523/JNEUROSCI.2197-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lefebvre C., Legouis R., and Culetto E. (2018) ESCRT and autophagies: endosomal functions and beyond. Semin. Cell Dev. Biol. 74, 21–28 10.1016/j.semcdb.2017.08.014 [DOI] [PubMed] [Google Scholar]
- 49. King G. D., and Scott Turner R. (2004) Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp. Neurol. 185, 208–219 10.1016/j.expneurol.2003.10.011 [DOI] [PubMed] [Google Scholar]
- 50. Minami S. S., Hoe H. S., and Rebeck G. W. (2011) Fyn kinase regulates the association between amyloid precursor protein and Dab1 by promoting their localization to detergent-resistant membranes. J. Neurochem. 118, 879–890 10.1111/j.1471-4159.2011.07296.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee J., Retamal C., Cuitino L., Caruaño-Yzermans A., Shin J. E., van Kerkhof P., Marzolo M. P., and Bu G. (2008) Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 283, 11501–11508 10.1074/jbc.M800642200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sullivan S. E., Dillon G. M., Sullivan J. M., and Ho A. (2014) Mint proteins are required for synaptic activity-dependent amyloid precursor protein (APP) trafficking and amyloid beta generation. J. Biol. Chem. 289, 15374–15383 10.1074/jbc.M113.541003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kyriazis G. A., Wei Z., Vandermey M., Jo D. G., Xin O., Mattson M. P., and Chan S. L. (2008) Numb endocytic adapter proteins regulate the transport and processing of the amyloid precursor protein in an isoform-dependent manner: implications for Alzheimer disease pathogenesis. J. Biol. Chem. 283, 25492–25502 10.1074/jbc.M802072200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoe H. S., Minami S. S., Makarova A., Lee J., Hyman B. T., Matsuoka Y., and Rebeck G. W. (2008) Fyn modulation of Dab1 effects on amyloid precursor protein and ApoE receptor 2 processing. J. Biol. Chem. 283, 6288–6299 10.1074/jbc.M704140200 [DOI] [PubMed] [Google Scholar]
- 55. Tsuboi T., and Fukuda M. (2006) Rab3A and Rab27A cooperatively regulate the docking step of dense-core vesicle exocytosis in PC12 cells. J. Cell Sci. 119, 2196–2203 10.1242/jcs.02962 [DOI] [PubMed] [Google Scholar]
- 56. Tanabe K., Yamazaki H., Inaguma Y., Asada A., Kimura T., Takahashi J., Taoka M., Ohshima T., Furuichi T., Isobe T., Nagata K., Shirao T., and Hisanaga S. (2014) Phosphorylation of drebrin by cyclin-dependent kinase 5 and its role in neuronal migration. PloS One 9, e92291 10.1371/journal.pone.0092291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Das U., Scott D. A., Ganguly A., Koo E. H., Tang Y., and Roy S. (2013) Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79, 447–460 10.1016/j.neuron.2013.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.