

Abstract
Exocytosis mediates the release of neurotransmitters and hormones from neurons and neuroendocrine cells. Tandem C2 domain proteins in the synaptotagmin (syt) and double C2 domain (Doc2) families regulate exocytotic membrane fusion via direct interactions with Ca2+ and phospholipid bilayers. Syt1 is a fast-acting, low-affinity Ca2+ sensor that penetrates membranes upon binding Ca2+ to trigger synchronous vesicle fusion. The closely related Doc2β is a slow-acting, high-affinity Ca2+ sensor that triggers spontaneous and asynchronous vesicle fusion, but whether it also penetrates membranes is unknown. Both syt1 and Doc2β bind the dynamically regulated plasma membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2), but it is unclear whether PIP2 serves only as a membrane contact or enables specialized membrane-binding modes by these Ca2+ sensors. Furthermore, it has been shown that PIP2 uncaging can trigger rapid, syt1-dependent exocytosis in the absence of Ca2+ influx, suggesting that current models for the action of these Ca2+ sensors are incomplete. Here, using a series of steady-state and time-resolved fluorescence measurements, we show that Doc2β, like syt1, penetrates membranes in a Ca2+-dependent manner. Unexpectedly, we observed that PIP2 can drive membrane penetration by both syt1 and Doc2β in the absence of Ca2+, providing a plausible mechanism for Ca2+-independent, PIP2-dependent exocytosis. Quantitative measurements of penetration depth revealed that, in the presence of Ca2+, PIP2 drives Doc2β, but not syt1, substantially deeper into the membrane, defining a biophysical regulatory mechanism specific to this high-affinity Ca2+ sensor. Our results provide evidence of a novel role for PIP2 in regulating, and under some circumstances triggering, exocytosis.
Keywords: synaptotagmin; synapse; exocytosis; calcium-binding protein; membrane biophysics; membrane protein; calcium sensor; phosphatidylinositol (4,5)-bisphosphate (PIP2); poly-anionic phospholipid; tandem C2 domain protein
Introduction
Exocytosis, a fundamental physiologic process, relies on the fusion of cellular membranes. In many cases, membrane fusion is mediated by soluble N-ethylmaleimide–sensitive factor attachment protein receptors (SNAREs)2 along with accessory proteins that integrate signals near the fusion site (1, 2). At neuronal synapses, a critical signal for exocytosis is Ca2+ (3), which acts upon tandem C2 domain proteins in the synaptotagmin (syt) (4–8) and Doc2 (9, 10) families to trigger SNARE-catalyzed fusion of vesicular and plasma membranes (10, 11).
Syt1 is a primary Ca2+ sensor for fast, synchronous neurotransmitter release (7, 8). It is activated by relatively large increases (≥1 μm) in cytoplasmic Ca2+ ([Ca2+]i) that trigger the rapid insertion of side chains from each C2 domain into lipid bilayers containing anionic phospholipids (12, 13). It has been rigorously established that penetration of lipid bilayers by syt1 accelerates SNARE-catalyzed fusion in vitro and in cultured neurons (14–16). Doc2β, a closely related protein that lacks a transmembrane domain but contains a munc13-interacting domain at its N terminus (17), regulates asynchronous (18) and spontaneous (10, 19) neurotransmitter release from neurons, synaptic augmentation (20), vesicle priming in chromaffin cells (21, 22), and insulin secretion from β cells (23). Compared with syt1, however, Doc2β–membrane interactions occur with slower kinetics and a much higher sensitivity for [Ca2+] (20–100 nm) (10, 19, 24). Thus, although both syt1 and Doc2β are Ca2+ sensors for exocytosis, their divergent functional paradigms invite a closer comparison to establish common mechanistic principles for Ca2+-sensitive tandem C2 domain proteins. For example, although syt1 must penetrate membranes to stimulate membrane fusion, it has not been established whether—and if so, how—Doc2β penetrates membranes.
Alongside proteins and Ca2+, phospholipid headgroups play key biophysical roles in Ca2+-triggered exocytosis. Of particular note is phosphatidylinositol 4,5-bisphosphate (PIP2), a dynamically regulated (25) polyanionic phospholipid important for exocytosis in chromaffin cells (26), PC12 cells (27), and neurons (28). PIP2 is localized to the plasma membrane and interacts with key components of the vesicular release machinery, including SNARE proteins (29, 30), calcium-activated protein for secretion (CAPS) (31), syt1 (13, 32, 33), and Doc2β (10, 19, 34). In the case of syt1, binding to PIP2 under resting conditions “steers” the C2 domains of this protein, and thus its Ca2+-dependent membrane-penetration activity, toward the plasma membrane to trigger release (13). PIP2 has also been shown to enhance the Ca2+ sensitivity of lipid binding by syt1 cooperatively with phosphatidylserine (PS) (32, 35), the major anionic phospholipid of the cytoplasmic face of the plasma membrane. Similarly, Doc2β binds PIP2-containing membranes in both the presence and absence of Ca2+, and PIP2 is required to localize Doc2β to the plasma membrane (10, 19, 34). Because depletion of PIP2 substantially reduces spontaneous neurotransmitter release in cultured neurons (28), this form of neurotransmission may depend on PIP2 binding by Doc2β, but this interaction has not been studied in detail.
According to current models of tandem C2 domain protein function, Ca2+ is required for membrane penetration and thus the triggering of exocytosis. However, recent findings have challenged this model by demonstrating that rapid uncaging of PIP2 can trigger syt1-dependent exocytosis without a measurable change in [Ca2+]i (36). Given the apparent requirement of Ca2+ for membrane penetration by syt1, how could a stepwise increase in available PIP2 evoke exocytosis?
In the present study, we first demonstrate that, like syt1, Doc2β penetrates lipid bilayers upon binding Ca2+. We report the unexpected finding that, in membranes containing PS, PIP2 drives Ca2+-independent membrane penetration by both syt1 and Doc2β. This interaction stimulates Ca2+-independent fusion mediated by syt1 in vitro. Moreover, in the presence of Ca2+, PIP2 significantly increases the membrane penetration depth of Doc2β but not syt1, thus providing a mechanism by which PIP2 may selectively drive spontaneous release. Our results define key biophysical differences between syt1 and Doc2β and provide a potential molecular mechanism by which PIP2 can directly trigger exocytosis in the absence of increases in [Ca2+]i.
Results
Doc2β penetrates and aggregates membranes in a manner analogous to syt1
We first sought to determine whether the tandem C2 domains of Doc2β share key biochemical properties with syt1 (Fig. 1A). We thus purified the tandem C2 domains (“C2AB”) of both proteins and used a series of assays to define their Ca2+-dependent and -independent interactions with lipid bilayers. To assess whether the Ca2+-binding loops of Doc2β C2AB penetrate membranes in a manner analogous to syt1, residues at the tips of loops 1 and 3 in each C2 domain of Doc2β (His-158 and Phe-222 in C2A and Ala-298 and Gly-361 in C2B) were individually mutated to cysteine and labeled with the environmentally sensitive probe N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine (NBD) (Fig. 1B). Membrane insertion was monitored via fluorescence emission changes after adding liposomes and Ca2+ to NBD-labeled protein. Emission spectra from NBD probes at all four Ca2+-binding loops underwent hypsochromic shifts and substantial intensity increases when both Ca2+ and PS-containing liposomes were present. Ca2+ triggered these fluorescence changes only in the presence of membranes, suggesting that each probe inserts into the bilayer in response to Ca2+ (Fig. 1C and Fig. S1). To confirm a direct interaction between these probes and the acyl chains in the bilayer, we used liposomes containing a membrane-embedded nitroxide (doxyl) spin label on an acyl chain of PC; this moiety quenches fluorescence largely by direct collision with excited-state fluorophores (37). NBD fluorescence, in each loop, was efficiently quenched by a spin label at the 12-position of the acyl chain (12-doxyl-PC, 15 mol %) (Fig. 1C), directly demonstrating that all four Ca2+-binding loops of Doc2β insert into the hydrophobic region of the bilayer.
Figure 1.

Doc2β penetrates membranes in response to Ca2+. A, schematic diagrams of syt1 and Doc2β. MID, munc13-interacting domain; TMD, transmembrane domain. B, model illustrating the putative membrane penetration activity of Doc2 where the distal tip of Ca2+-binding loop 1 was mutated to cysteine and labeled with the fluorescent dye NBD, shown at right. The shaded stripe in the bilayer leaflet depicts the approximate distribution of the quenching nitroxide on 12-doxyl-PC. Ribbon diagrams show C2A (PDB code 4LCV) and C2B (PDB code 4LDC) of Doc2β from Giladi et al. (52). C, NBD emission spectra from each of the four Ca2+-binding loops of Doc2β C2AB. Graph titles indicate the C2 domain and loop labeled (e.g. C2A*(1)-C2B corresponds to loop 1 of C2A, and C2A*(3)-C2B corresponds to loop 3 of C2A). Labeled C2AB was combined with liposomes (15% PS, 30% PC, 20% PE, and 35% cholesterol) in 500 μm EGTA after which Ca2+ was added (250 μm free [Ca2+]). Ca2+ triggered an intensity increase and blue shift in the emission spectra at all four labeling sites, suggesting burial of the probe into the bilayer. Membrane insertion was confirmed with the use of liposomes containing 15% 12-doxyl-PC, which efficiently quenched the fluorescence at each labeled site. Spectra are representative of data from at least four independent trials.
Aggregation of PS-containing liposomes is also a characteristic property of syt1 C2AB in vitro (38). This activity has not been described for Doc2β, a soluble protein whose function may also rely on its ability to juxtapose membranes. To assay for aggregation activity, Doc2β C2AB was mixed with liposomes and Ca2+, and the turbidity of the mixture was monitored by absorbance at 400 nm. As with syt1 C2AB, Doc2β C2AB rapidly and reversibly aggregated liposomes that harbored PS (Fig. S2A). Moreover, this aggregation activity was strongly enhanced when copies of Doc2β C2AB were bound to separate liposomes and thus available to interact in trans (Fig. S2, B and C). This behavior, which was also observed for syt1 (38) (Fig. S2), suggests a common mechanism of aggregation in which C2AB molecules, bound to liposomes via their Ca2+-binding loops, subsequently interact with other liposome-bound C2AB molecules.
PIP2 triggers Ca2+-independent membrane penetration by Doc2β and syt1
We next focused on the role of PIP2 in driving specific modes of membrane binding by syt1 and Doc2β. Because previous studies of membrane penetration by syt1 were performed using nonphysiologic mixtures of phospholipids (13, 15, 39), we assayed the membrane penetration activity of syt1 and Doc2β in the presence of model plasma membranes that included 15 mol % PS and 1 mol % PIP2, a composition that reflects the PIP2 content of neuronal and neuroendocrine cell plasma membranes (29, 40). Indo-1 was used to verify that [Ca2+]free remained very low, i.e. ≤10 nm, upon addition of PS:PIP2 lipids (Fig. S3). Surprisingly, under these conditions, we observed not only Ca2+-independent binding but also Ca2+-independent penetration of the bilayer by both proteins (Fig. 2, A and C). In each case, this activity, at 1 mol % PIP2, was limited to the Ca2+-binding loops of the C2B domain. Inclusion of 12-doxyl-PC in the liposomes resulted in quenching of NBD fluorescence, confirming a direct interaction of C2B loop 3 with the interior of the membrane in the case of each protein (Fig. 2, A and C). Syt1 and Doc2β C2AB diverged in terms of the behavior of C2B loop 1, which failed to penetrate in the case of syt1 but engaged in shallow penetration in the case of Doc2β (Fig. 2, A and C). PS and PIP2 were both required for Ca2+-independent penetration of membranes by both syt1 and Doc2β (Fig. 2, B and D). Previous studies of membrane penetration by syt1 included liposomes containing either PS or PIP2 but not both, thus explaining why this novel interaction was not observed previously (13, 15, 39).
Figure 2.

PS and PIP2 synergistically drive Ca2+-independent membrane penetration by syt1 and Doc2β but exert different effects on each protein. A, emission spectra of NBD-labeled Doc2β C2AB before and after the addition of liposomes containing 15 mol % PS and 1 mol % PIP2 in 500 μm EGTA (≤10 nm [Ca]free). Under these conditions, loops 1 and 3 of C2B demonstrate robust increases in emission intensity. Emission from loop 3 is efficiently quenched by 12-doxyl-PC, indicating Ca2+-independent insertion into the bilayer. Spectra are representative of data from at least four independent trials. B, NBD-labeled Doc2β C2AB was combined with the indicated liposomes, and the NBD emission intensity was measured before and after the addition of Ca2+. For each replicate, emission intensity was normalized to the signal from NBD-labeled protein prior to liposome addition. For Doc2β, PS and PIP2 each support Ca2+-dependent penetration activity. However, when combined, PS and PIP2 drove a marked Ca2+-dependent increase in the emission from C2A loop 3. Both PS and PIP2 were required for Ca2+-independent penetration by loops 1 and 3 of C2B (arrows). C and D, same as above but using syt1 C2AB. In contrast to Doc2β C2AB, PS drives penetration of syt1 C2A more efficiently than PIP2 in the presence of Ca2+. The combination of PIP2 and PS did not drive any additional NBD signal increases in C2A but marginally increased NBD signals in C2B. As with Doc2β, both PIP2 and PS were required for robust Ca2+-independent penetration by Syt1 C2B loop 3 (arrow). Error bars, S.E. of four independent trials; *, p < 0.05; **, p < 0.005; ns, p > 0.5; all by Welch's t test.
These data suggest that syt1 and Doc2β contain at least partially distinct binding sites for PS and PIP2 that, when occupied simultaneously, drive Ca2+-independent insertion of C2B into the bilayer. To confirm that these findings hold true for full-length syt1, we formulated nanodiscs containing the full-length, labeled protein (ND-syt1) (Fig. 3A). As in the case for C2AB, ND-syt1 underwent Ca2+-independent penetration of membranes containing PS and PIP2 but not PS alone (Fig. 3B) This result is of particular significance because, in chromaffin cells, optical uncaging of PIP2 drives a small, syt1-dependent exocytotic burst even in the absence of measurable changes in Ca2+ levels (36) (see “Discussion”).
Figure 3.

Membrane penetration by full-length syt1 reconstituted into nanodiscs. A, experimental scheme. Full-length syt1 was purified, labeled with NBD on loop 3 of C2A or loop 3 of C2B, and reconstituted into 13-nm-diameter nanodiscs comprising membrane scaffolding protein and POPC (ND-syt1). ND-syt1 was combined with liposomes containing acidic phospholipids in EGTA (500 μm) followed by the addition of Ca2+ to assay Ca2+-independent and Ca2+-dependent membrane penetration activity. B, representative spectra for penetration experiments with ND-syt1. As with syt1 C2AB, Ca2+ and acidic phospholipids caused an increase and blue shift in NBD fluorescence. Likewise, in the presence of both PS and PIP2, Ca2+-independent penetration by C2B, but not C2A, was observed. Spectra are representative of results from four independent trials.
PIP2 exhibits differential effects on Doc2β and syt1
Our penetration experiments (Fig. 2) also revealed striking, lipid-dependent differences between syt1 and Doc2β in the presence of Ca2+. In particular, Doc2β C2A loop 3 demonstrated a unique increase in fluorescence only when both PS and PIP2 were present (Fig. 2B). In contrast, we observed no such changes in the analogous position in syt1, which displayed equivalent Ca2+-dependent NBD fluorescence increases upon binding PS-bearing liposomes whether or not PIP2 was included (Fig. 2D). We explored this issue further by examining the impact of PS and PIP2 on the disassembly kinetics of Ca2+ sensor–lipid complexes. In this assay, preassembled C2AB–Ca2+–liposome complexes were rapidly mixed with EGTA to remove free [Ca2+] while FRET was monitored between protein and liposomes using a stopped-flow rapid-mixing instrument (Fig. 4). The inclusion of 1 mol % PIP2 in PS-bearing liposomes had no effect on the disassembly kinetics of syt1 complexes (mean ± S.E.: PS, 73.7 ± 10.0 s−1; PS:PIP2, 79.1 ± 7.8 s−1; p > 0.5, Welch's t test) (Fig. 4B). In striking contrast, PIP2 slowed the disassembly of Doc2 complexes nearly 10-fold (PS, 4.90 ± 0.21 s−1; PS:PIP2, 0.49 ± 0.03 s−1; p = 0.0002, Welch's t test) (Fig. 4C). In combination with data from NBD-labeled penetration assays (Fig. 2), these findings further support a specific role for PIP2 in stabilizing the Ca2+-dependent activated state of Doc2β.
Figure 4.
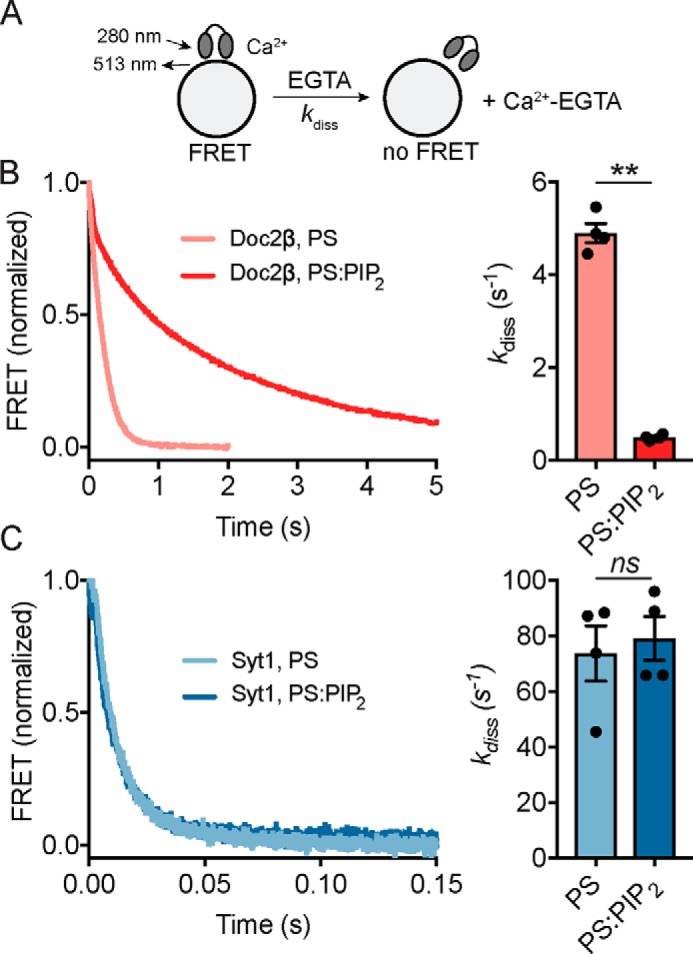
PIP2 slows the disassembly kinetics of C2AB–Ca2+–liposome complexes containing Doc2β but not syt1. A, schematic of disassembly assay. C2AB–Ca2+–liposome complexes were preassembled and then rapidly mixed with EGTA while monitoring FRET between tryptophan residues in C2AB and dansyl-PE acceptors on the liposomes. B, representative traces (left) and rate constants derived from single-exponential fits (right) for disassembly of Doc2β–Ca2+–membrane complexes. The inclusion of 1 mol % PIP2 in liposomes containing 15% PS slowed the observed rates of disassembly by ∼10-fold. C, as above but for syt1 C2AB. In contrast to the case of Doc2β, membrane complex disassembly rates (Kdiss) for syt1 were unchanged with the inclusion of 1 mol % PIP2. Error bars, S.E. of four independent trials; **, p < 0.005; ns, p > 0.5; both by Welch's t test.
Quantitative analysis of membrane penetration activity
Our initial NBD fluorescence results (Figs. 1 and 2) motivated a more quantitative comparison of membrane penetration by syt1 and Doc2β. We thus used the parallax method of London and co-workers (37, 41) to determine the insertion depth of NBD on each loop of syt1 and Doc2β in the presence and absence of Ca2+ and PIP2. We used doxyl-PC labeled at either the 5- or 12-positions of the acyl chain as well as on the choline headgroup (HG-doxyl; Fig. 5A; also known as TEMPO-PC). Quenching efficiencies in the presence of Ca2+ are shown in Fig. 5, C and D, whereas quenching efficiencies in the absence of Ca2+ are shown in Fig. S7. Increased quenching by deeper doxyls and decreased quenching by shallower doxyls indicate deeper insertion of the NBD probe. By comparing the quenching efficiencies of spin labels at various points on the alkyl chains, we quantitatively estimated the depth to which the NBD labels penetrate the membrane. For this analysis, we improved on previous implementations of the parallax analysis by using published molecular dynamics simulations of doxyl-PC quenchers (42) to determine the uncertainty in the measured penetration depth for each probe (see “Experimental procedures”). Calculated depth parameters are shown in Table 1 and represented visually in Fig. 7.
Figure 5.

PIP2 markedly deepens membrane penetration by Doc2β but not syt1. A, illustration depicting membrane-bound C2AB and the approximate distributions of nitroxide quenchers. The yellow star represents NBD label, and green spheres represent Ca2+ ions. B, representative emission spectra for nonquenching liposomes along with liposomes containing the indicated doxyl quencher. Relative quenching efficiencies at different probe locations correspond to the average location of the NBD label in the bilayer. Deeper insertion results in stronger quenching by 12-doxyl versus 5-doxyl and HG-doxyl liposomes, whereas shallower insertion results in stronger quenching by HG-doxyl and 5-doxyl liposomes. C, NBD-labeled Doc2β C2AB was combined with liposomes and Ca2+ (250 μm), and quenching efficiencies of doxyl-PC liposomes with and without PIP2 were quantified. Inclusion of PIP2 drives both loops of C2A deeper into the bilayer as evinced by reduced shallow quenching and increased deep quenching. This effect is also apparent for Doc2β C2B. D, same as in C but using NBD-labeled syt1. In contrast to Doc2β, syt1 C2A penetrates deeply in the absence of PIP2 as shown by relatively efficient quenching by 12-doxyl liposomes. In contrast to the case of Doc2β, PIP2 exhibits only a weak tendency to drive additional penetration by syt1. Error bars, S.E. of four independent trials; *, p < 0.05; **, p < 0.005; ns or unmarked, p > 0.5; all by Welch's t test.
Table 1.
Calculated depth parameters from doxyl quenching experiments
Membrane insertion depth was calculated by measuring the relative quenching efficiencies of doxyl spin labels located at different positions on the lipid acyl chains according to the methods described by Chattopadhyay and London (37). Values of z indicate distance from the center of the bilayer, in Å, and subscripts denote the doxyl pair used to calculate this distance. Each value represents the calculated average distance of the NBD label from the center of the bilayer. Errors represent half-widths of the calculated depth distributions based on molecular dynamics simulations of doxyl quenchers by Kyrychenko and Ladokhin (42). Penetration depths were determined in the presence of 250 μM [Ca2+] free, except in conditions labeled “EGTA.” In these cases, dashes indicate distances not determined, as no penetration was observed in EGTA without PIP2 present. See “Experimental procedures” for details on calculations and error propagation.

Figure 7.

Model of Ca2+-dependent and -independent membrane penetration by Doc2β and syt1 in the presence and absence of PIP2. Calculated membrane penetration depths are illustrated, to scale, for Doc2β and syt1. Models of syt1 and Doc2 were created by rendering the molecular surfaces of the corresponding X-ray or NMR structures (Doc2, as above; syt1, PDB codes 1RSY (C2A) and 1K5W (C2B) from Sutton et al. (53) and Fernandez et al. (54), respectively). The polybasic patch of C2B is rendered cyan in each model. Shaded areas in the bilayer represent the calculated half-widths of the penetration depth measurements for each probe. A, scale drawing of membrane penetration by Doc2β. Prior to binding Ca2+, C2B shallowly penetrates bilayers in the presence of 1% PIP2. After binding Ca2+, all four loops penetrate the bilayer. However, both loops in C2A are relegated to a shallow position unless PIP2 is also present, which enables C2A loop 3 to penetrate 3.7 Å deeper on average into the membrane. In the absence of Ca2+, increases in mol % PIP2 in the target membrane can drive partial penetration of the bilayer by all four loops of Doc2β. B, scale drawing of membrane penetration by syt1. As with Doc2β, 1% PIP2 enables Ca2+-independent penetration by C2B loop 3, and increasing [PIP2] drives penetration by C2A loop 3. Upon binding Ca2+, however, all four loops of syt1 penetrate deeply into the membrane even in the absence of PIP2.
In the presence of Ca2+, PIP2 exerted strikingly different effects on membrane penetration by Doc2β versus syt1 (Figs. 5 and 7). Although PIP2 drove all four loops of both syt1 and Doc2β deeper into the bilayer, this effect was far more pronounced for Doc2β. In particular, loop 3 of Doc2β C2A penetrated, on average, 3.7 Å deeper into the bilayer in the presence of PIP2. In contrast, PIP2 increased the average penetration depth of the loops of syt1 by, at most, 1 Å. Remarkably, Doc2β C2A penetrates only shallowly into PS-bearing membranes lacking PIP2 but penetrates approximately as deeply as syt1 if PIP2 is present (Figs. 5C and 7). Syt1, by contrast, penetrates PS-bearing membranes to nearly its full extent even in the absence of PIP2 (Figs. 5D and 7). These results provide direct evidence that PIP2 substantially deepens Ca2+-dependent membrane penetration by Doc2 but has relatively subtle effects on syt1. Our findings define a mechanistic divergence between syt1 and Doc2β and a biophysical mechanism by which Doc2β acts specifically as a PIP2-dependent Ca2+ sensor.
Elevation of PIP2 drives additional membrane penetration to stimulate membrane fusion
Physiologic [PIP2] in the plasma membrane is ∼1 mol % but can reach >5 mol % at sites of vesicle docking and fusion (29, 40). Even at 1% PIP2, we noted some doxyl quenching of NBD probes on the C2A domains of syt1 and Doc2β, suggesting that further increases in PIP2 might drive additional membrane penetration by these sensors (Fig. S7). To assess how elevating [PIP2] might drive alternative membrane-penetration modes by syt1 and Doc2β, we measured emission from NBD-labeled syt1 and Doc2β C2AB in the presence of liposomes containing increasing mol % PIP2 (Fig. 6, A and B). We observed significant, dose-dependent increases in NBD emission intensity for labels on C2A in both Doc2β and syt1 as PIP2 was increased from 1 to 5 mol %. In the case of Doc2β, elevation of [PIP2] drove penetration by all four loops, with this effect approaching saturation at 5% PIP2 (Fig. 5A). In the case of syt1, increasing [PIP2] drove penetration by C2A loop 3, demonstrating that elevation of [PIP2] can trigger activation of both C2 domains of this protein (Fig. 6B). These results support a specific role for PIP2 in activating both Doc2β and syt1 at physiologically relevant concentrations (illustrated in Fig. 7). Moreover, these findings suggest a mechanism by which syt1, under certain circumstances, may be partially activated by PIP2 to trigger Ca2+-independent vesicle fusion (36).
Figure 6.
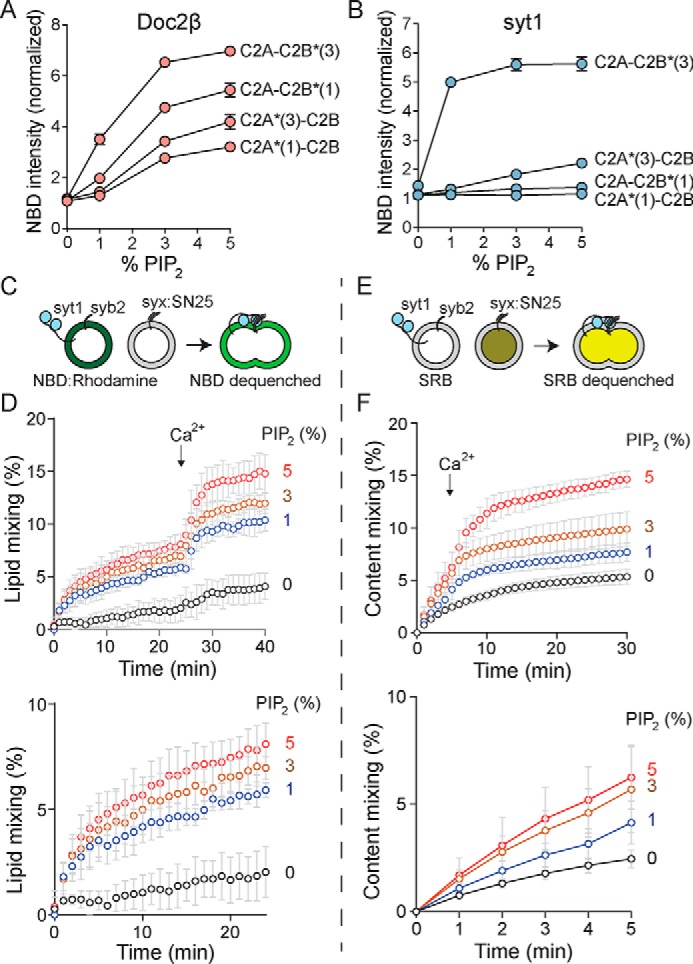
Increasing [PIP2] drives Ca2+-independent penetration by both C2 domains of Doc2β and syt1 and potentiates Ca2+-independent and -dependent vesicle fusion. A, NBD-labeled Doc2β C2AB was combined with liposomes harboring PS and increasing concentrations of PIP2 in the absence of Ca2+, and NBD emission intensity was quantified. Increasing [PIP2] drove substantial intensity increases from NBD labels on all four loops of Doc2β. This effect appeared to reach near-saturation at 5 mol % PIP2. B, as in A but for syt1 C2AB. In addition to robust penetration by C2B loop 3, increasing [PIP2] drove partial penetration by C2A loop 3. C–F, v-SNARE liposomes containing full-length syb2 and full-length syt1 were combined with t-SNARE liposomes containing full-length syntaxin-1A:SNAP-25B (syx:SN25) heterodimer and increasing mol % PIP2. C, scheme of lipid-mixing assay. Fusion of vesicles was monitored by dequenching of NBD. D, results of lipid-mixing assays conducted with increasing mol % PIP2 in the t-SNARE vesicles. Above, full traces; below, Ca2+-free portion of the trace shown on an expanded timescale. PIP2 drove Ca2+-independent and -dependent lipid mixing in a dose-dependent manner. E, scheme of content-mixing assay. Fusion of vesicles was monitored by dequenching of sulforhodamine B. F, results of content-mixing assays conducted with increasing mol % PIP2 in the t-SNARE vesicles. Above, full traces; below, Ca2+-free portion of the trace shown on an expanded timescale. As with lipid-mixing experiments, a dose-dependent effect of PIP2 on Ca2+-independent and -dependent fusion was observed. Error bars, S.E. of four independent trials.
Because PIP2-dependent, Ca2+-independent penetration by syt1 was less extensive than that of Doc2β, we sought to determine whether this novel penetration activity can enhance vesicle fusion in the absence of Ca2+. We thus performed in vitro fusion assays using v-SNARE vesicles containing syb2 and full-length syt1 with t-SNARE vesicles containing syntaxin-1A:SNAP-25B heterodimer and increasing amounts of PIP2 (Fig. 6, C–F). In both lipid and content-mixing assays, elevation of [PIP2] enhanced fusion of v- and t-SNARE vesicles prior to the addition of Ca2+, consistent with the capacity of PIP2 to drive Ca2+-independent activation of syt1 (36). Increasing PIP2 likewise enhanced membrane fusion after the addition of Ca2+, consistent with published findings using in vitro fusion assays (43) and PIP2 uncaging in chromaffin cells (36).
Discussion
Taken together, our results demonstrate key similarities and unanticipated differences between syt1 and Doc2, tandem C2 domain Ca2+ sensors specialized for distinct physiologic functions. Our results reveal that, like syt1, Doc2β aggregates and penetrates membranes containing anionic phospholipids in response to Ca2+ (Figs. 1 and S2). This activity, which likely results in a Ca2+-dependent deformation of the membrane due to the space occupied by the tips of the Ca2+-binding loops (44, 45), thus appears to be a core feature of tandem C2 domain Ca2+ sensors. Given that Doc2β is not anchored to vesicles by a transmembrane domain and that vesicular membranes contain anionic phospholipids, it is possible that this aggregation activity may, in part, underlie the ability of this protein to promote membrane fusion.
Our findings establish a new role for PIP2 in exocytosis by showing that PIP2 directly stimulates penetration of the target membrane by syt1 and Doc2β. Although PIP2 has been understood as a key factor in defining exocytotic sites and priming vesicles for release, our work defines an additional downstream function, i.e. direct activation of Ca2+ sensors that trigger SNARE-catalyzed membrane fusion. Critically, although Ca2+ stimulates membrane penetration, we show that it is not strictly required for this activity when PIP2 is present (Figs. 2, 3, 6, and 7). To our knowledge, this is the first evidence that a C2 domain can penetrate (and thus presumably deform (44, 45)) a membrane without an elevation in [Ca2+]i. Thus, a rapid increase in [PIP2], e.g. via optical uncaging of caged PIP2 as performed by Walter et al. (36), might trigger syt1-dependent release via two nonexclusive mechanisms: recruitment of additional Ca2+ sensors that penetrate the plasma membrane or driving deeper penetration by Ca2+ sensors that are already present at release sites (Figs. 6 and 7). Furthermore, because [PIP2] can reach >5 mol % at release sites (29) and the plasma membrane contains ∼10–15 mol % PS (46), it is likely that the docked and/or primed configurations of syt1 and Doc2β involve some degree of insertion into the plasma membrane. However, although the PIP2 uncaging technique of Walter et al. (36) provides useful mechanistic insights, we note that we are not aware of studies showing such rapid up-regulation of PIP2 at exocytotic sites in endogenous systems.
Strikingly, the Ca2+-independent penetration activity of Doc2β reached near-saturation at 5 mol % PIP2, a dose-response that is well-tuned to the physiologic range of PIP2 levels at sites of fusion. We also note that, although Ca2+ plays key roles in physiologic exocytosis, multiple studies have demonstrated varying degrees of residual exocytosis after dramatically reducing [Ca2+]i (47, 48). Because [Ca2+]i increases lead to activation of phospholipase C and the cleavage of plasma membrane PIP2, the Doc2β–PIP2 interactions defined here may serve to maintain baseline spontaneous fusion rates during quiescent periods. Additionally, cAMP- and GTP-dependent signaling pathways have been shown to potentiate exocytosis in an apparently Ca2+-independent fashion (49). Further studies, in which cellular ATP, PIP2, and Ca2+ are all carefully controlled, may more quantitatively define the role of PIP2 in driving Ca2+-independent exocytosis in live cells.
This work lends key support to the physiologic relevance of PIP2 as a crucial biophysical regulatory factor for Doc2β (Figs. 4–7). Syt1, by contrast, appears to rely on PIP2 for preadsorption onto the plasma membrane (13) rather than full membrane penetration per se. Our results correspond well to those reported by Pérez-Lara et al. (35), who found that PIP2 did not substantially enhance the penetration depth of syt1 in the presence of PS. The divergence between syt1 and Doc2β is readily reconciled with the specialized functions of these proteins. Doc2β operates at near resting [Ca2+] on slow timescales and is thus well-poised to respond to the dynamic (but relatively slow) regulation of PIP2 levels at release sites. Syt1, in contrast, must respond in microseconds to transient Ca2+ elevations. Additional lipid requirements for full penetration by syt1 might come at a kinetic cost that would impair its capacity for triggering rapid membrane fusion. Our stopped-flow data (Fig. 4) support this interpretation, demonstrating that PIP2 robustly stabilizes the active state of Doc2β but not syt1. The findings reported here provide an example of how two highly homologous protein sequences (the tandem C2 domains of syt1 and Doc2β) can retain core mechanistic principles while evolving highly specialized, lipid-dependent regulatory mechanisms. Other tandem C2 domain Ca2+ sensors may be tuned to respond to other lipid headgroups, acyl chain compositions, regulatory proteins, or even small molecules. These regulatory functions, and how they influence the corresponding physiologic processes, remain the focus of ongoing and future studies.
Experimental procedures
Protein purification
Constructs encoding syt1 C2AB (amino acids 140–421) and Doc2β C2AB (amino acids 126–412) were expressed as N-terminal GST fusion proteins (pGEX4T-1 vector, GE Healthcare) in Escherichia coli, purified via GSH-Sepharose affinity chromatography, and cleaved with thrombin in reconstitution buffer (100 mm KCl, 25 mm HEPES-NaOH, pH 7.4) plus 5% glycerol. Full-length synaptobrevin-2 was likewise expressed as a GST fusion protein, purified, and cleaved in a similar buffer containing 400 mm KCl and 1% n-octyl d-glucopyranoside. Full-length syt1 in the pTrcHis vector (Invitrogen) and full-length syntaxin-1A:SNAP-25B heterodimer in the pRSF Duet vector (EMD Millipore) were expressed as N-terminal His6 fusion tags, purified via nickel-NTA-Sepharose affinity chromatography, and eluted in elution buffer (500 mm imidazole, 400 mm KCl, 25 mm HEPES-NaOH, pH 7.4, 1% n-octyl d-glucopyranoside). DTT (2 mm) was added to syntaxin-1A:SNAP-25B heterodimer and full-length syt1 to prevent aggregation. Membrane scaffolding protein MSP1E3D1 (50) was likewise purified by Ni2+-NTA-Sepharose chromatography and eluted in elution buffer without detergent. For full-length syt1, endogenous cysteines were substituted with alanines, and the protein was expressed as an N-terminal His6-SUMO fusion construct in pET28. Purified protein was subjected to on-bead labeling (see below) and eluted in elution buffer containing 0.05% n-β-dodecyl maltoside (Gold Biotechnology). Imidazole and residual free dye were removed by ultrafiltration, and the N-terminal tag was cleaved off with recombinant SENP2 protease followed by removal with Ni2+-NTA-Sepharose resin. During purification, all lysates were treated with DNase and RNase, and beads bearing each Doc2β or syt1 construct were washed extensively with 1 m NaCl, 1 mm Mg2+ to remove any bound nucleic acid contaminants.
Protein mutagenesis and labeling
Native cysteines (Cys-277 in Syt1 and Cys-145, Cys-217, Cys-249, Cys-290, Cys-337, and Cys-387 in Doc2β) were removed and replaced with alanines, and exogenous cysteines were introduced at the indicated positions using site-directed mutagenesis. All mutagenesis was confirmed by Sanger sequencing. For labeling, protein was diluted to 10 μm in 600 μl of reconstitution buffer plus 5% glycerol containing 100 μm tris(2-carboxyethyl)phosphine. Iodoacetamidyl-NBD-amide (Thermo Fisher; 2 mm in DMSO) was added dropwise to this solution for a final dye:protein ratio of 10:1 (mol:mol), and the labeling reaction was allowed to proceed for 2 h at room temperature with rotation. The reaction was then quenched with DTT, and the free dye was removed by desalting on a column (PD MidiTrap, GE Healthcare) equilibrated in reconstitution buffer plus 5% glycerol. Protein concentrations and labeling stoichiometry were determined by UV-visible absorption spectroscopy using an empirically determined extinction coefficient for NBD. Labeling efficiency ranged from 0.8 to 1.2 dye molecules per protein. Full-length syt1 was labeled during purification by incubating protein-bearing Ni2+-NTA-Sepharose resin in 1 ml containing 10% DMSO and 0.5 mg of iodoacetamidyl-NBD amide overnight at 4 °C with rotation. Beads were washed extensively prior to elution.
Liposome preparation
Liposomes were prepared from POPC, POPS, POPE, brain PIP2, and cholesterol (all from Avanti Polar Lipids) stored individually as chloroform stocks except for brain PIP2 (stored in 20:9:1 CHCl3:MeOH:H2O). Unless noted otherwise, liposomes contained 30% POPC, 15% POPS, 20% POPE, and 35% cholesterol (all % mol/mol). For membrane-embedded quenching studies, 15% 5-doxyl-, 12-doxyl-, or headgroup-doxyl-PC replaced POPC in equimolar quantity. In liposomes lacking PS, this lipid was replaced with the same mole fraction of POPC. For stopped-flow rapid-mixing experiments, 5% dansyl-PE replaced an equimolar amount of POPE. For liposome formulation, lipids were combined, and two to three drops of methanol were added. The solvent was evaporated under a stream of nitrogen, and the films were dried under vacuum for at least 2 h. Films were rehydrated in reconstitution buffer at a final concentration of 5 or 10 mm [lipid] and extruded 29 times through a single 100-nm polycarbonate filter (Whatman).
Proteoliposome reconstitution for aggregation assays
Proteoliposomes were formed using 15% PS, 30% PE, and 55% PC, all mol %. Lipids in chloroform stocks were combined, dried under vacuum, rehydrated in reconstitution buffer, and subjected to five freeze–thaw cycles. Protein-free unilamellar vesicles were prepared from this mixture by extrusion through a 50-nm polycarbonate filter (Whatman). Syntaxin-1A:SNAP-25B heterodimer (for t-SNARE–bearing liposomes) or synaptobrevin-2 (for v-SNARE–bearing liposomes) were mixed with protein-free vesicles at a protein:lipid molar ratio of 1:200 with ∼0.8 weight % octyl glucoside in the buffer at 4 °C for 15 min. The mixture was diluted two times with reconstitution buffer, and this diluted mixture was then dialyzed against 2 liters of reconstitution buffer with 5 g of Bio-beads SM2 (Bio-Rad) at 4 °C overnight. For aggregation studies, protein-free liposomes were prepared in the same fashion but with the protein omitted.
Nanodisc reconstitution
POPC (100 nmol), MSP1E3D1 (10 nmol), and full-length labeled syt1 (2 nmol) were combined in reconstitution buffer containing 5% glycerol and 0.05% n-β-dodecyl maltoside. Bio-beads SM2 were added (80 μl of a ∼95% slurry in reconstitution buffer), and the mixture was incubated overnight with rotation to remove n-β-dodecyl maltoside and permit nanodisc self-assembly.
Aggregation assays
C2AB (1 μm) and liposomes (113 μm lipid) were combined in 100 μl of reconstitution buffer containing 200 μm EGTA, and absorbance at 400 nm was monitored in a spectrophotometer (Eppendorf) at room temperature. Ca2+ was added at the indicated points for a total of 1 mm free Ca2+. EGTA was subsequently added for a final concentration of 2 mm [EGTA]. Independent experiments were defined as replicates performed with a unique combination of separately prepared batches of protein and lipid.
Stopped-flow rapid mixing
C2AB (4 μm), liposomes (1 mm lipid), and CaCl2 (250 μm for syt1 and 40 μm for Doc2β) were combined in reconstitution buffer. This mixture was loaded into one syringe of an SX-18.MV stopped-flow spectrometer (Applied Photophysics) at room temperature (23 °C) and rapidly mixed with an equal volume of 2 mm EGTA in the same buffer. Samples were allowed to equilibrate in the spectrometer for 5 min prior to mixing. Excitation at 285 nm was provided via a xenon arc lamp and monochromator (Applied Photophysics), and emission was monitored via photomultiplier tube through a 470-nm long-pass filter (KV470, Schott). Single-exponential decays were fitted using Applied Photophysics Pro-Data SX software prior to normalization, with the first 2 ms of each trace omitted from analysis to account for instrument dead time. Independent experiments were defined as replicates performed with a unique combination of separately prepared batches of protein and lipid.
Penetration assays
NBD-C2AB (0.25 μm) or ND-syt1 (0.15 μm syt1), liposomes (117 μm total lipid), and Ca2+ (250 μm [Ca]free) were combined in 600 μl of reconstitution buffer containing 500 μm EGTA. Spectra (λex = 390 nm; λem = 470–630 nm) were acquired at room temperature (23 °C) in a quartz cuvette using a QM-1 fluorimeter (Photon Technology International) after the addition of each component. In all cases, protein was added first followed by liposomes and finally CaCl2. CaCl2 was added for a total [Ca2+] of 750 μm of which 500 μm was chelated by EGTA, leaving a [Ca2+]free of 250 μm. A buffer blank was subtracted from all traces. For quantification, traces were integrated by taking the average background-subtracted fluorescence intensity between 510 and 610 nm. Averaged traces were normalized to the background-subtracted, integrated signals from labeled C2AB prior to the addition of lipids or Ca2+ for each replicate. Independent experiments were defined as replicates performed with a unique combination of separately prepared batches of protein and lipid. Example spectra for penetration assays are shown in the supporting information.
Depth calculations
Measurements of bilayer penetration depths were performed according to the parallax method of London and co-workers (37, 41) with slight modifications used to estimate distribution widths for each probe location. This method relies on 1) a hard-sphere approximation of quenching by nitroxide radicals and 2) the relative quenching efficiencies of two quenchers at known depths in the bilayer to estimate the position of a fluorescent probe. The final equation used to derive penetration depths is as follows,
| (Eq. 1) |
where ZcF is the distance of probe from the bilayer center, Lc1 is the distance from the bilayer center to the shallow quencher, L21 is the difference in depth between the two quenchers, F1 is the relative fluorescence intensity of the shallow quencher, F2 is the relative fluorescence intensity of the deeper quencher, and C is the concentration of quencher in molecules per Å2, assuming 20 mol % quencher and an area of 70 Å2 per lipid molecule. Both F1 and F2 are expressed as a fraction of the NBD-C2AB emission intensity obtained in the absence of doxyl-PC quencher. For values corresponding to the positions of quenchers in the bilayer, we used the results of the recent molecular dynamics simulations (42) as these data matched previous experimental results well and also provided estimated distribution widths for the location of doxyl-PC quenchers in the bilayer. The half-widths of these distributions were propagated as errors across all mathematical operations in Equation 1 to estimate half-widths for the location of each probe. Errors in F1 and F2 were also propagated, although the errors in these measurements were small compared with the errors corresponding to the quencher distribution widths. Distances from bilayer center were calculated using two pairs of doxyls (5- and 12-doxyl and headgroup- and 12-doxyl). We note that the deviation in measured depth between the two pairs of doxyls used tended to increase with more deeply located NBD probes. These deviations were <2 Å in almost all cases, however, and we speculate that they occurred due to depth-dependent changes in the mobility of the NBD fluorophore and/or deviation from the hard-sphere approximation for quenching by nitroxide radicals. The average calculated depth of each NBD-labeled probe using this method was shallow enough (minimum 8.6 Å from bilayer center) that quenching by 12-doxyl-PC from the opposite leaflet of the bilayer was ignored in our calculations.
Lipid-mixing assays
For preparation of v-SNARE liposomes, full-length syt1 and full-length synaptobrevin-2 were diluted in elution buffer, added to dried lipid films (15% PS, 7% PE, 20% cholesterol, 55% PC, 1.5% NBD-PE, and 1.5% rhodamine-PE, all % mol/mol) at 1:2000 protein:lipid ratio, incubated for 40 min on ice, and dialyzed extensively against reconstitution buffer containing 1 g/liter Bio-beads SM2. The dialyzed liposome suspension was then purified by buffer exchange into reconstitution buffer using a PD-10 column (GE Healthcare). t-SNARE liposomes were prepared similarly by adding t-SNARE heterodimer in elution buffer to lipid films of the same composition (1:2000 protein:lipid ratio) but without NBD-PE or rhodamine-PE and with 0, 1, 3, or 5% PIP2 substituted for an equimolar amount of PC. For lipid-mixing assays, v-SNARE liposomes (0.5 μm) were mixed with t-SNARE liposomes (5 μm) in 100 μl of reconstitution buffer. Fluorescence (460-nm excitation/520-nm emission) was monitored in a plate reader (BioTek) while incubating the reaction at 37 °C with Ca2+ (500 μm) added at the indicated time point. Independent experiments were defined as replicates performed with a unique combination of separately prepared batches of protein and lipid.
Content-mixing assays
v-SNARE liposomes for content-mixing assays were prepared as for lipid-mixing assays but without NBD-PE or rhodamine-PE. t-SNARE liposomes containing PIP2 and sulforhodamine B were prepared as above but with 10 mm sulforhodamine B (Acros Organics) in the elution buffer containing t-SNAREs. For content-mixing assays, v-SNARE liposomes (5 μm) were mixed with t-SNARE liposomes (1 μm) in 100 μl of reconstitution buffer. Fluorescence (530-nm excitation/590-nm emission) was monitored in a plate reader (BioTek) while incubating the reaction at 37 °C with Ca2+ (500 μm) added at the indicated time point. Ca2+ was added earlier in these experiments than in lipid-mixing experiments because longer incubations yielded content mixing that was almost entirely Ca2+-independent in the presence of PIP2. Incubation of dye-containing t-SNARE vesicles in the absence of v-SNARE vesicles did not result in dequenching (data not shown), indicating that this phenomenon was not due to leakage of dye from these vesicles. Independent experiments were defined as replicates performed with a unique combination of separately prepared batches of protein and lipid.
Indo-1 measurements
Indo-1 (0.33 μm) was added to 600 μl of reconstitution buffer containing 500 μm EGTA followed by PS:PIP2 liposomes (0.117 μm) and Ca2+ (250 μm) with spectra taken (λex = 332 nm) after each addition. [Ca2+]free was estimated by comparison with reference spectra (51).
Author contributions
M. M. B. and E. R. C. conceptualization; M. M. B. data curation; M. M. B. formal analysis; M. M. B. and E. R. C. funding acquisition; M. M. B., H. B., and X. L. investigation; M. M. B. visualization; M. M. B. and H. B. methodology; M. M. B. and E. R. C. writing-original draft; M. M. B., H. B., and E. R. C. writing-review and editing; E. R. C. resources; E. R. C. project administration.
Supplementary Material
Acknowledgment
We thank members of the Chapman laboratory for comments and support.
This work was supported by National Institute of Mental Health Grants R01 MH061876, R35 NS097362 (to E. R. C.), and F30 MH116580 (to M. M. B.) and a Howard Hughes Medical Institute investigator grant (to E. R. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
- SNARE
- soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- PS
- phosphatidylserine
- syt1
- synaptotagmin-1
- [Ca2+]i
- cytoplasmic Ca2+
- NBD
- N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine
- Doc2
- double C2 domain
- HG
- headgroup
- ND
- nanodisc
- TEMPO-PC
- 1,2-dipalmitoyl-sn-glycero-3-phospho(tempo)choline
- v-SNARE
- vesicle SNARE
- t-SNARE
- target SNARE
- syb2
- synaptobrevin-2
- GST
- glutathione S-transferase
- NTA
- nitrilotriacetic acid
- SUMO
- small ubiquitin-like modifier
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- doxyl
- N-oxy-4′,4′-dimethyloxazolidine
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- PDB
- Protein Data Bank.
References
- 1. Südhof T. C., and Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chapman E. R. (2008) How does synaptotagmin trigger neurotransmitter release? Annu. Rev. Biochem. 77, 615–641 10.1146/annurev.biochem.77.062005.101135 [DOI] [PubMed] [Google Scholar]
- 3. Katz B., and Miledi R. (1967) The timing of calcium action during neuromuscular transmission. J. Physiol. 189, 535–544 10.1113/jphysiol.1967.sp008183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brose N., Petrenko A. G., Südhof T. C., and Jahn R. (1992) Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025 10.1126/science.1589771 [DOI] [PubMed] [Google Scholar]
- 5. Bhalla A., Chicka M. C., and Chapman E. R. (2008) Analysis of the synaptotagmin family during reconstituted membrane fusion: uncovering a class of inhibitory isoforms. J. Biol. Chem. 283, 21799–21807 10.1074/jbc.M709628200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hui E., Bai J., Wang P., Sugimori M., Llinas R. R., and Chapman E. R. (2005) Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc. Natl. Acad. Sci. U.S.A. 102, 5210–5214 10.1073/pnas.0500941102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Littleton J. T., Stern M., Schulze K., Perin M., and Bellen H. J. (1993) Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca2+-activated neurotransmitter release. Cell. 74, 1125–1134 10.1016/0092-8674(93)90733-7 [DOI] [PubMed] [Google Scholar]
- 8. Geppert M., Goda Y., Hammer R. E., Li C., Rosahl T. W., Stevens C. F., and Südhof T. C. (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- 9. Orita S., Sasaki T., Naito A., Komuro R., Ohtsuka T., Maeda M., Suzuki H., Igarashi H., and Takai Y. (1995) Doc2: a novel brain protein having two repeated C2-like domains. Biochem. Biophys. Res. Commun. 206, 439–448 10.1006/bbrc.1995.1062 [DOI] [PubMed] [Google Scholar]
- 10. Groffen A. J., Martens S., Díez Arazola R., Cornelisse L. N., Lozovaya N., de Jong A. P., Goriounova N. A., Habets R. L., Takai Y., Borst J. G., Brose N., McMahon H. T., and Verhage M. (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327, 1614–1618 10.1126/science.1183765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tucker W. C., Weber T., and Chapman E. R. (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 10.1126/science.1097196 [DOI] [PubMed] [Google Scholar]
- 12. Chapman E. R., and Davis A. F. (1998) Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem. 273, 13995–14001 10.1074/jbc.273.22.13995 [DOI] [PubMed] [Google Scholar]
- 13. Bai J., Tucker W. C., and Chapman E. R. (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44 10.1038/nsmb709 [DOI] [PubMed] [Google Scholar]
- 14. Wang P., Chicka M. C., Bhalla A., Richards D. A., and Chapman E. R. (2005) Synaptotagmin VII is targeted to secretory organelles in pc12 cells, where it functions as a high-affinity calcium sensor. Mol. Cell. Biol. 25, 8693–8702 10.1128/MCB.25.19.8693-8702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bai H., Xue R., Bao H., Zhang L., Yethiraj A., Cui Q., and Chapman E. R. (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat. Commun. 7, 10971 10.1038/ncomms10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Evans C. S., Ruhl D. A., and Chapman E. R. (2015) An engineered metal sensor tunes the kinetics of synaptic transmission. J. Neurosci. 35, 11769–11779 10.1523/JNEUROSCI.1694-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orita S., Naito A., Sakaguchi G., Maeda M., Igarashi H., Sasaki T., and Takai Y. (1997) Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. J. Biol. Chem. 272, 16081–16084 10.1074/jbc.272.26.16081 [DOI] [PubMed] [Google Scholar]
- 18. Yao J., Gaffaney J. D., Kwon S. E., and Chapman E. R. (2011) Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell 147, 666–677 10.1016/j.cell.2011.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Courtney N. A., Briguglio J. S., Bradberry M. M., Greer C., and Chapman E. R. (2018) Excitatory and inhibitory neurons utilize different Ca2+ sensors and sources to regulate spontaneous release. Neuron. 98, 977–991.e5 10.1016/j.neuron.2018.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xue R., Ruhl D. A., Briguglio J. S., Figueroa A. G., Pearce R. A., and Chapman E. R. (2018) Doc2-mediated superpriming supports synaptic augmentation. Proc. Natl. Acad. Sci. U.S.A. 115, E5605–E5613 10.1073/pnas.1802104115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pinheiro P. S., de Wit H., Walter A. M., Groffen A. J., Verhage M., and Sørensen J. B. (2013) Doc2b synchronizes secretion from chromaffin cells by stimulating fast and inhibiting sustained release. J. Neurosci. 33, 16459–16470 10.1523/JNEUROSCI.2656-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Houy S., Groffen A. J., Ziomkiewicz I., Verhage M., Pinheiro P. S., and Sørensen J. B. (2017) Doc2B acts as a calcium sensor for vesicle priming requiring synaptotagmin-1, munc13-2 and SNAREs. Elife 6, e27000 10.7554/eLife.27000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Cantley J., Burchfield J. G., Meoli C. C., Stöckli J., Whitworth P. T., Pant H., Chaudhuri R., Groffen A. J., Verhage M., and James D. E. (2014) Doc2 isoforms play dual roles in insulin secretion and insulin-stimulated glucose uptake. Diabetologia 57, 2173–2182 10.1007/s00125-014-3312-y [DOI] [PubMed] [Google Scholar]
- 24. Groffen A. J., Friedrich R., Brian E. C., Ashery U., and Verhage M. (2006) DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J. Neurochem. 97, 818–833 10.1111/j.1471-4159.2006.03755.x [DOI] [PubMed] [Google Scholar]
- 25. Micheva K. D., Holz R. W., and Smith S. J. (2001) Regulation of presynaptic phosphatidylinositol 4,5-biphosphate by neuronal activity. J. Cell Biol. 154, 355–368 10.1083/jcb.200102098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eberhard D. A., Cooper C. L., Low M. G., and Holz R. W. (1990) Evidence that the inositol phospholipids are necessary for exocytosis. Biochem. J. 268, 15–25 10.1042/bj2680015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hay J. C., Fisette P. L., Jenkins G. H., Fukami K., Takenawa T., Anderson R. A., and Martin T. F. (1995) ATP-dependent inositide phosphorylation required for Ca2+-activated secretion. Nature 374, 173–177 10.1038/374173a0 [DOI] [PubMed] [Google Scholar]
- 28. Di Paolo G., Moskowitz H. S., Gipson K., Wenk M. R., Voronov S., Obayashi M., Flavell R., Fitzsimonds R. M., Ryan T. A., and De Camilli P. (2004) Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422 10.1038/nature02896 [DOI] [PubMed] [Google Scholar]
- 29. James D. J., Khodthong C., Kowalchyk J. A., and Martin T. F. (2008) Phosphatidylinositol 4,5-bisphosphate regulates SNARE-dependent membrane fusion. J. Cell Biol. 182, 355–366 10.1083/jcb.200801056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., and Jahn R. (2011) Membrane protein sequestering by ionic protein–lipid interactions. Nature 479, 552–555 10.1038/nature10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grishanin R. N., Kowalchyk J. A., Klenchin V. A., Ann K., Earles C. A., Chapman E. R., Gerona R. R., and Martin T. F. (2004) CAPS acts at a prefusion step in dense-core vesicle exocytosis as a PIP2 binding protein. Neuron 43, 551–562 10.1016/j.neuron.2004.07.028 [DOI] [PubMed] [Google Scholar]
- 32. van den Bogaart G., Meyenberg K., Diederichsen U., and Jahn R. (2012) Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J. Biol. Chem. 287, 16447–16453 10.1074/jbc.M112.343418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tucker W. C., Edwardson J. M., Bai J., Kim H. J., Martin T. F., and Chapman E. R. (2003) Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J. Cell Biol. 162, 199–209 10.1083/jcb.200302060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Michaeli L., Gottfried I., Bykhovskaia M., and Ashery U. (2017) Phosphatidylinositol (4,5)-bisphosphate targets double C2 domain protein B to the plasma membrane. Traffic 18, 825–839 10.1111/tra.12528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pérez-Lara Á., Thapa A., Nyenhuis S. B., Nyenhuis D. A., Halder P., Tietzel M., Tittmann K., Cafiso D. S., and Jahn R. (2016) PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. Elife 5, e15886 10.7554/eLife.15886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walter A. M., Müller R., Tawfik B., Wierda K. D., Pinheiro P. S., Nadler A., McCarthy A. W., Ziomkiewicz I., Kruse M., Reither G., Rettig J., Lehmann M., Haucke V., Hille B., Schultz C., et al. (2017) PIP2 optical uncaging potentiates exocytosis. Elife 6, e30203 10.7554/eLife.30203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chattopadhyay A., and London E. (1987) Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26, 39–45 10.1021/bi00375a006 [DOI] [PubMed] [Google Scholar]
- 38. Hui E., Gaffaney J. D., Wang Z., Johnson C. P., Evans C. S., and Chapman E. R. (2011) Mechanism and function of synaptotagmin-mediated membrane apposition. Nat. Struct. Mol. Biol. 18, 813–821 10.1038/nsmb.2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hui E., Bai J., and Chapman E. R. (2006) Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J. 91, 1767–1777 10.1529/biophysj.105.080325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenk M. R., Lucast L., Di Paolo G., Romanelli A. J., Suchy S. F., Nussbaum R. L., Cline G. W., Shulman G. I., McMurray W., and De Camilli P. (2003) Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat. Biotechnol. 21, 813–817 10.1038/nbt837 [DOI] [PubMed] [Google Scholar]
- 41. Abrams F. S., and London E. (1993) Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry 32, 10826–10831 10.1021/bi00091a038 [DOI] [PubMed] [Google Scholar]
- 42. Kyrychenko A., and Ladokhin A. S. (2013) Molecular dynamics simulations of depth distribution of spin-labeled phospholipids within lipid bilayer. J. Phys. Chem. B 117, 5875–5885 10.1021/jp4026706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Z., Liu H., Gu Y., and Chapman E. R. (2011) Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J. Cell Biol. 195, 1159–1170 10.1083/jcb.201104079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hui E., Johnson C. P., Yao J., Dunning F. M., and Chapman E. R. (2009) Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell 138, 709–721 10.1016/j.cell.2009.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martens S., Kozlov M. M., and McMahon H. T. (2007) How synaptotagmin promotes membrane fusion. Science 316, 1205–1208 10.1126/science.1142614 [DOI] [PubMed] [Google Scholar]
- 46. Cotman C., Blank M. L., Moehl A., and Snyder F. (1969) Lipid composition of synaptic plasma membranes isolated from rat brain by zonal centrifugation. Biochemistry 8, 4606–4612 10.1021/bi00839a056 [DOI] [PubMed] [Google Scholar]
- 47. Kochubey O., and Schneggenburger R. (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69, 736–748 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- 48. Vyleta N. P., and Smith S. M. (2011) Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium-sensing receptor. J. Neurosci. 31, 4593–4606 10.1523/JNEUROSCI.6398-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hille B., Billiard J., Babcock D. F., Nguyen T., and Koh D. S. (1999) Stimulation of exocytosis without a calcium signal. J. Physiol. 520, 23–31 10.1111/j.1469-7793.1999.00023.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Denisov I. G., Grinkova Y. V., Lazarides A. A., and Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 10.1021/ja0393574 [DOI] [PubMed] [Google Scholar]
- 51. Grynkiewicz G., Poenie M., and Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 52. Giladi M., Michaeli L., Almagor L., Bar-On D., Buki T., Ashery U., Khananshvili D., and Hirsch J. A. (2013) The C2B domain is the primary Ca2+ sensor in DOC2B: a structural and functional analysis. J. Mol. Biol. 425, 4629–4641 10.1016/j.jmb.2013.08.017 [DOI] [PubMed] [Google Scholar]
- 53. Sutton R. B., Davletov B. A., Berghuis A. M., Südhof T. C., and Sprang S. R. (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 10.1016/0092-8674(95)90296-1 [DOI] [PubMed] [Google Scholar]
- 54. Fernandez I., Araç D., Ubach J., Gerber S. H., Shin O., Gao Y., Anderson R. G., Südhof T. C., and Rizo J. (2001) Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 10.1016/S0896-6273(01)00548-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.