

Abstract
BRAKER is a pipeline for highly accurate and fully automated gene prediction in novel eukaryotic genomes. It combines two major tools: GeneMark-ES/ET and AUGUSTUS. GeneMark-ES/ET learns its parameters from a novel genomic sequence in a fully automated fashion; if available, it uses extrinsic evidence for model refinement. From the protein-coding genes predicted by GeneMark-ES/ET we select a set for training AUGUSTUS, one of the most accurate gene finding tools that, in contrast to GeneMark-ES/ET, integrates extrinsic evidence already into the gene prediction step. The first published version, BRAKER1, integrated genomic footprints of unassembled RNA-Seq reads into the training as well as into the prediction steps. The pipeline has since been extended to the integration of data on mapped cross-species proteins, and to the usage of heterogeneous extrinsic evidence, both RNA-Seq and protein alignments. In this book chapter, we briefly summarize the pipeline methodology and describe how to apply BRAKER in environments characterized by various combinations of external evidence.
Keywords: protein-coding genes, gene prediction, AUGUSTUS, GeneMark-ES/ET, RNA-Seq reads, protein mapping to genome, genome annotation pipeline, BRAKER
1. Introduction
BRAKER [1] is a pipeline for the fully automated prediction of protein coding genes with GeneMark-ES/ET [2, 3, 4] and AUGUSTUS [5, 6, 7, 8, 9, 10] in novel eukaryotic genomes. In contrast to other genome annotation pipelines, such as e.g. MAKER [11, 12], BRAKER trains both gene finders in a fully automated fashion before applying them to the genome.
For gene prediction, both GeneMark-ES/ET and AUGUSTUS use statistical models with a large number of parameters. Optimal parameters are species specific. While the same parameters can be used for clades of closely related species, the use of parameters from more distant species typically leads to low prediction accuracy. Thus, a training step for parameter optimization is required. Most gene prediction tools, including AUGUSTUS, must be trained on a previously generated (expert curated) set of gene structures, the procedure is called ‘supervised training’. GeneMark-ES/ET has the extraordinary property to generate parameters by self-training or ‘unsupervised training’. Therefore, an expert curated training set is not required prior to running GeneMark-ES/ET, which can either train itself on an genomic sequence that GeneMark-ES/ET had not previously been trained for, or, if an additional, extrinsic evidence is available for intron intervals, incorporate this information into self-training.
AUGUSTUS is one of the most accurate tools for predicting genes, as shown by the independent EGASP [13, 14, 15], nGASP [16] and RGASP [17] assessments. AUGUSTUS not only has an elaborate statistical model, but also has the capacity to integrate extrinsic evidence from various sources. In contrast to GeneMark-ES/ET, AUGUSTUS is not self-training but requires the training gene set. Compiling such a set often poses problems for users not experienced enough in bioinformatics, as this step requires to execute special tools and tests. The task itself would also take quite a significant amount of time even for experienced users.
Creating a high quality training set can be helped by the availability of extrinsic data. For example, alignments of protein sequences of a closely related species against the new genome can be used to derive gene structures (e.g. using Scipio [18] or GenomeThreader [19]). Inferring gene models from expressed sequence tags (ESTs) against genome alignments (e.g. with PASA [20]) is also an option. If proteins of closely related species or ESTs are available, we refer the reader to WebAUGUSTUS [8], a user friendly web service that runs Scipio or PASA in conjunction with training AUGUSTUS (local installation of the underlying AutoAug.pl pipeline, part of AUGUSTUS, is possible but difficult).
However, with progress made in transcriptome sequencing technology, ESTs have been replaced by short-read RNA-Seq (e.g. with Illumina sequencing).
A possible use of short read RNA-Seq data in context of gene prediction would be to first assemble the short reads into longer transcript sequences, and, subsequently, to map the longer transcripts to genomes and infer genes suitable for a gene finder training. However, the RGASP assessment showed convincingly that gene identification methods that use unassembled RNA-Seq reads and statistical models in the prediction step are superior to methods that assemble the short reads into transcripts [17]. AUGUSTUS, the gene finder inferring gene structure evidence from aligned raw RNA-Seq reads, did perform particularly well in the RGASP assessment.
The principle of BRAKER (Fig. 1) is to execute self-training GeneMark-ES/ET to produce an initial set of predicted genes. GeneMark-ES/ET can run either in ab initio unsupervised mode (ES mode) or in semi-supervised mode (ET mode) if additional evidence for putative splice sites is available in the form of short RNA-seq reads to genome alignments spanning splice junctions. Out of the genes predicted by GeneMark-ES/ET, BRAKER selects those having support in all introns in the extrinsic data (in the absence of extrinsic data, BRAKER selects all genes longer than 800 nt in spliced form). Besides multi-exon genes, a number of single-exon genes, proportional to the number of single-exon genes in the initial GeneMark-ES/ET gene set, is selected at random and added to the whole set of selected genes.
Figure 1:
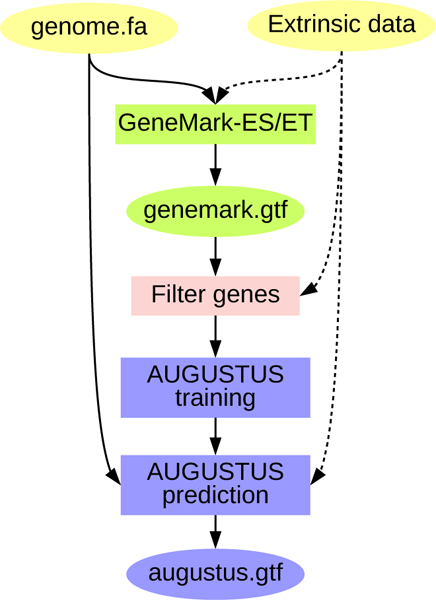
Schematic view of the BRAKER approach to gene prediction: GeneMark-ES/ET is trained (using extrinsic data upon availability) and predicts a first gene set (genemark.gtf). This gene set is filtered. AUGUSTUS is trained on the filtered gene set. AUGUSTUS predictions with species-specific parameters are performed, using extrinsic data upon availability.
Genes with few exons contribute less to improve AUGUSTUS’ accuracy during training; however, a certain number of single exon genes must remain in the training set. The genes with no or few exons usually outnumber the genes with a large number of introns in the initial GeneMark-ES/ET gene set. We apply the following approach to sample genes with a small number of introns. A gene with n ≤ 5 introns is removed from the list if n < N, where N is a random variable sampled from a Poisson distribution with parameter λ = 2. Accordingly, the chance that a gene is kept in training increases with the number of introns.
In the next step, the gene set - represented by genomic coordinates and genomic sequences - is translated into protein sequences. Duplicates are pruned from this set using NCBI BLAST [21, 22] with a similarity threshold of maximum pairwise percent identity of 80%. This set is used to train AUGUSTUS, which uses the newly trained species-specific parameters and extrinsic evidence, if available, to make the final round of gene predictions.
Both, GeneMark-ES/ET and AUGUSTUS are tools capable of using unassembled RNA-Seq information. Self-training GeneMark-ES/ET uses this data in the training step, while AUGUSTUS uses it in the prediction step. The BRAKER1 pipeline was designed to chain both tools in an optimal way with and without splice-site information from mapped RNA-Seq reads. Since its original publication, BRAKER1 has been extended in several aspects: currently it can run with and without any extrinsic evidence; it can use hints on splice-sites obtained from alignments of homologous proteins to the genome of interest, instead of or in addition to the hints from the RNA-Seq read alignments; it can use alignments of proteins from closely or remotely related species; it can use RNA-Seq coverage information for prediction of genes with UTRs, instead of CDS-only prediction. In this book chapter, we refer to this pipeline as the BRAKER pipeline and describe how to apply the pipeline in various circumstances defined by the presence or absence of different types of data.
2. BRAKER software and input files
In this section, we describe required computational resources, software and input files for executing BRAKER.
2.1. Computational resources
BRAKER can in principle be executed on a modern desktop computer with 8 GB RAM (per core). Many subprocesses that are initiated by BRAKER can be parallelized. We recommend a workstation with 8 cores. Please note that many steps in BRAKER that are parallelized use data parallelization, i.e. large files are split into smaller files, and each smaller file is then processed on a separate core. Choosing a very large number of cores may lead to scenarios where one or several smaller files do not contain sufficient data for processing, anymore. BRAKER has therefore been limited to run with at most 48 cores. Also, k-fold cross-validation of optimize_augustus.pl is by default executed with k = 8 on 8 cores, only. Please note that if you set BRAKER to run with more than 8 and up to 48 cores, k will be adapted to the number of cores.
2.2. Software
BRAKER is available for download from GitHub at https://github.com/Gaius-Augustus/BRAKER. You can clone the repository with:

The repository also contains example input files that will be used in this book chapter to demonstrate the usage of BRAKER.
Running BRAKER requires a Linux system with Bash and Perl. Furthermore, BRAKER requires the following CPAN-Perl modules to be installed:
File::Spec::Functions
Hash::Merge
List::Util
Logger::Simple
Module::Load::Conditional
Parallel::ForkManager
POSIX
Scalar::Util::Numeric
YAML
BRAKER calls external bioinformatics software. The called software depends on the input file combination. All input file combinations require the following software to be installed:
AUGUSTUS 3.3.1 or newer. Please use the latest AUGUSTUS version distributed by the original authors from GitHub at https://github.com/Gaius-Augustus/Augustus (see Note 1).
GeneMark-ES/ET 4.33 or newer
If you run BRAKER with RNA-Seq alignments in BAM-format, the following software is required:
If you run BRAKER with protein data from a closely related species, a protein alignment tool is required. We recommend GenomeThreader 1.7.0 or newer.
2.3. Files
BRAKER is a pipeline that can be run with different input file combinations (as depicted in Figures 2 and 3). Here, we describe the formats of files that serve as input to BRAKER.
Figure 2:
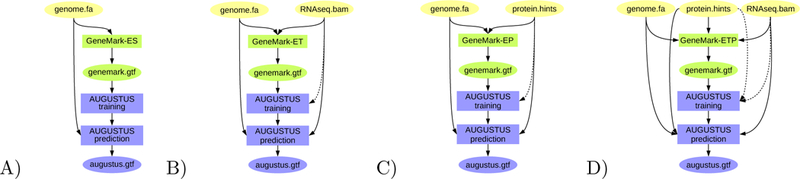
BRAKER pipeline: A) Training GeneMark-ES on genome data only; ab initio gene prediction with AUGUSTUS. B) Training GeneMark-ET supported by RNA-Seq spliced alignment information, prediction with AUGUSTUS with that same spliced alignment information. C) Training GeneMark-EP on protein spliced alignment information, prediction with AUGUSTUS with that same spliced alignment information. D) Training GeneMark-ETP supported by RNA-Seq alignment information and protein spliced alignment information, prediction with AUGUSTUS using the same alignment information. Introns supported by both RNA-Seq and protein alignment information are trusted - their prediction in gene structures by GeneMark-ETP and AUGUSTUS is enforced. Proteins used for C) and D) can be of longer evolutionary distance.
Figure 3:
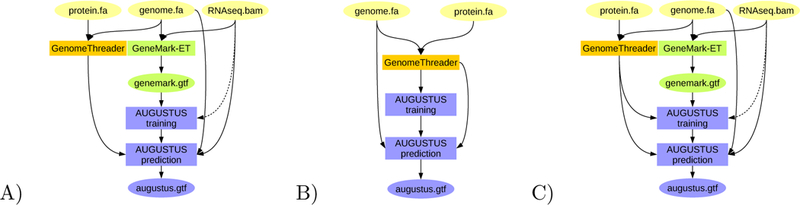
BRAKER pipelines for integration of protein information from closely related species: A) Training GeneMark-ET supported by RNA-Seq spliced alignment information, prediction with AUGUSTUS with spliced alignment information from RNA-Seq data and with gene features determined by alignments from proteins of a very closely related species against the target genome. B) Training AUGUSTUS on the basis of spliced alignment information from proteins of a very closely related species against the target genome. C) Training GeneMark-ET on the basis of RNA-Seq spliced alignment information, training AUGUSTUS on a set of training gene structures compiled from RNA-Seq supported gene structures predicted by GeneMark-ET and spliced alignment of proteins of a very closely related species.
2.3.1. Genome file
Running BRAKER always requires a genome file in FASTA-format. Ideally, you should provide a genome file that contains all (longer) contigs of a genome assembly to BRAKER, and not parts of a single or few chromsomes. Very short contigs often carry only partial gene structures, if any. While AUGUSTUS is capable of predicting partial genes in short sequences, AUGUSTUS training is performed on complete gene structures, only. Including very short contigs in a BRAKER run usually does not improve parameter training but increases runtime because all contigs still have to be processed in the prediction step. We recommend to exclude short contigs (i.e. <3000 nt) if runtime is an issue.
Most eukaryotic genomes contain repetitive elements. Repeats may pose problems to gene prediction, e.g. the prediction of many copies of a transposon may lead to an extremely high number of genes. We strongly recommend that you mask the genome rigorously prior to running BRAKER. RepeatMasker (usage is described in [25], Unit 4.10) is a good starting point. Consider that you might have to create a species-specific repeat library (e.g. with RepeatScout [26]).
There are two implicit ways of storing the repeat locations for a genome sequence: soft-masking and hard-masking. In a soft-masked genomic FASTA file, all parts of the genome that are identified as repeats are written with lower case letters, while all other sequence parts are written in upper case letters. In a hard-masked file, each repeat nucleotide is replaced by the letter N. BRAKER will perform best if repeats are soft-masked.
BRAKER processes RNA-Seq and protein alignment files. Such files contain the name of the genomic sequence that an alignment corresponds to. The name of genomic sequences is stored in the FASTA-header of the genome file. If the FASTA-header is long or contains spaces, some alignment tools will truncate the header. Thus, the sequence name in the alignment file will not be identical with the original sequence name in the genome file. For BRAKER, it is crucial that the names are identical. We therefore recommend that you check the FASTA-headers of your genome file for complexity prior to running any alignment tool and BRAKER. If the headers are long or complex, rename them. You can use e.g. the script http://bioinf.uni-greifswald.de/bioinf/downloads/simplifyFastaHeaders.pl to produce a new file with simple headers and a mapping table with old and new headers.

2.3.2. RNA-Seq alignment file
BRAKER accepts alignment files of RNA-Seq reads mapped against the target genome in BAM-format. Since BRAKER uses the information of how many reads cover a particular potential splice site in the genome, it is crucial, that only RNA-Seq data are used that produce high read coverage, and that only aligners are used that produce spliced alignments. RNA-Seq data produced by sequencing methods that generate a high number of (short) reads from mRNAs (e.g. Illumina) are therefore particularly useful for running BRAKER. Suitable RNA-Seq alignment tools (the list is not exhaustive) are STAR [27], Tophat2 [28] and GSNAP [29].
RNA-Seq data are often not generated in order to enhance gene prediction accuracy, but for other experimental purposes, such as comparing gene expression in different tissues or under different conditions. Often, several biological and/or technical replicates are generated for statistical purposes. It usually does not do any harm to use all available RNA-Seq data, but it increases alignment and BRAKER runtime and memory requirements. You may obtain highly similar results if you restrict yourself to using e.g. one randomly selected replicate of each library type.
If the alignment tool of your choice produces files in SAM-instead of BAM-format, you can easily convert the files with SAMTOOLS:

2.3.3. Protein sequence file
BRAKER accepts files with protein sequences in FASTA-format. Please be aware that BRAKER will align those proteins to the target genome with GenomeThreader or other protein spliced aligners in order to use the alignment information for gene prediction purposes. Only protein sequences from species that are rather closely related align well to the target genome. It will not improve gene prediction results if you include proteins from very distantly related species in the protein sequence file. If you intend to use the protein information to generate training genes for AUGUSTUS, protein sequences must be full-length, i.e. not sequences of partial proteins.
2.3.4. Hints file
BRAKER accepts AUGUSTUS-specific hints files. Hints files contain extrinsic evidence information that indicate certain features of protein coding genes in certain positions in the genome. Hints files can contain information from RNA-Seq, protein alignments, manual annotation, and possibly many other sources. Hints files are in a tabulator-separated 9-column GFF-format. The following format example has been generated by the AUGUSTUS tool bam2hints that is used within BRAKER to generate hints from RNA-Seq BAM-files.
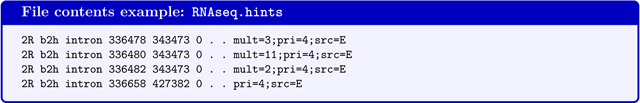
Note that the last column contains a field mult=INT. This indicates the coverage information for a feature, e.g. the given intron in line 1 has support from 3 RNA-Seq reads. The field src=E indicates that this feature was extracted from expression data. The source tags correspond to an AUGUSTUS configuration file that contains weights on how to treat evidence from this particular source. Within BRAKER, 4 types of sources are handled by default:
E for spliced read information from RNA-Seq,
P for information from proteins,
W for coverage information from RNA-Seq (from ‘wiggle’ files),
M for manual hints; BRAKER flags hints with M if they have support from the sources E and P. The prediction of genes with hints of type M is practically enforced by the corresponding parameters.
The third column of the hints file contains the feature name of a hint. The following features are currently supported by BRAKER: intron, start, stop, ass, dss, exonpart, exon, CDSpart, nonexonpart (Repeats). The most important feature is intron, because it is the only feature that GeneMark-ES/ET uses for training. The features ass and dss are automatically derived from intron hints by AUGUSTUS. exon and exonpart features should only be used if UTR-training of AUGUS-TUS has been enabled because if no UTR parameters are available, such hints may lead to false positive CDS/CDSpart predictions by AUGUSTUS. CDSpart and CDS features are typically generated from protein data of close homology when running BRAKER. nonexonpart hints are implicitly provided by using a soft-masked genome.
There are two typical use cases in which hints files are provided to BRAKER:
-
a)Instead of providing RNA-Seq alignments in BAM-format to braker.pl, the first product that braker.pl will produce from a BAM-file with a tool called bam2hints, a hints file with information from the BAM-file, can be provided to BRAKER. Users run bam2hints to extract hints from BAM-files prior calling BRAKER for two reasons:
- Parallelization: BRAKER runs bam2hints in parallel for provided BAM-files, but only on the number of cores that are provided to BRAKER. If the number of BAM-files is large, and additional cores are available but should not be allocated to BRAKER itself, running bam2hints separately on more cores may reduce computational time.
- File size: hints files are much smaller than BAM-files, depending on the computational environment, a small file size may be desired (e.g. in virtual environments).
Please note that BRAKER cannot train UTR parameters for AUGUSTUS if no BAM-file is provided.
-
b)
For running BRAKER with evidence from proteins of remote homology, such as generated by the GaTech protein mapping pipeline (see Fig. 4). GeneMark-ES/ET requires that in this case the hints file contains information on how many protein alignments cover a particular splice-site pair in column 6 (the value should be identical with the mult=INT value):
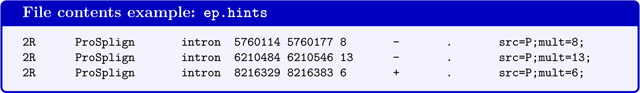
In contrast to hints files from RNA-Seq alignments, the hints file for running BRAKER with intron evidence from proteins of remote homology must contain strand information in column 7.
Figure 4:
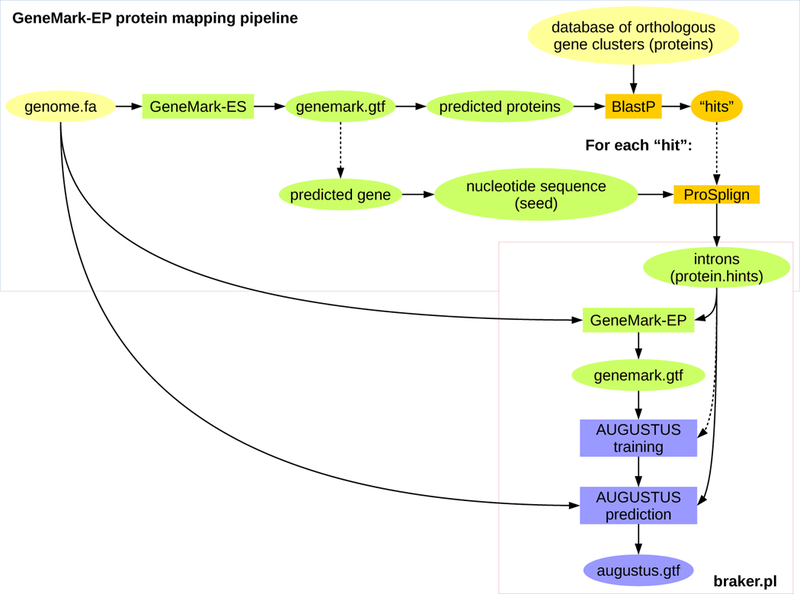
Current outline of the GaTech protein mapping pipeline that can be used to generate evidence for running GeneMark-EP.
3. BRAKER gene prediction, step by step
3.1. Installing and configuring BRAKER
The BRAKER repository contains three directories:
BRAKER/docs/ contains documentation on BRAKER, e.g. the file userguide.pdf that provides detailed installation and configuration instructions,
BRAKER/scripts/ contains the BRAKER Perl scripts and modules, most importantly the script that executes BRAKER: braker. P1,
-
BRAKER/examples/ contains example data for testing BRAKER. A RNA-Seq alignment file in BAM-format (134 MB) that is required for some testing scenarios needs to be downloaded separately. It is available at http://bioinf.uni-greifswald.de/bioinf/braker/RNAseq.bam, you can download it e.g. using the command line tool wget:

The example data set has been generated in order to demonstrate in rather short runtime that all software components work. It was not chosen to lead to good GeneMark-ES/ET and AUGUSTUS parameters or highly accurate gene predictions.
In this book chapter, we will assume that you are working on an Ubuntu system in bash. If you need to install dependencies on another system, Ubuntu/Debian specific package installation commands (sudo apt install ...) might be different.
BRAKER requires Perl 5 (or newer). On Ubuntu, Perl is installed by default upon system installation. For installing the CPAN dependencies, we recommend the installation and usage of cpanminus:

You can subsequently install the required CPAN-modules with a bash loop as follows:

The easiest way to run and configure BRAKER is to add all programs and scripts that are called by BRAKER to your $PATH variable in a bash configuration script, such as the ~/.bashrc file. This will ensure that BRAKER automatically finds all required dependencies. In order to add any software to your $PATH, add or extend the PATH line at the bottom or your ~/.bashrc file. We here demonstrate it for the path to braker.pl only, but you can easily add the paths to all other executables in a similar fashion; separate different paths by colons (:). You need to change your_path_to_braker to the actual path where braker.pl and the other scripts reside:

When you start a new bash session, changes in the ~/.bashrc file are automatically loaded. If you continue to work in a session that had been opened before changing ~/.bashrc, you have to load the new configuration:

You can test whether your changes have taken effect by
-
printing the $PATH variable in bash:

The result will look similar to this, you should find the directory that you just added to the $PATH definition:

-
checking whether the system finds the software that you just added to the $PATH, in our example braker.pl:

This command should return the full path to the executable, e.g.

If there is an empty return value, you most likely made a spelling mistake when extending the $PATH.
The bioinformatics software tools that are called by BRAKER all have their own installation documentation. In case of doubt, we recommend that you read the individual documentation. In the following, we give short instructions and commands for a ‘typical installation’ on Ubuntu that will work in most cases.
3.1.1. GeneMark-ES/ET
Download GeneMark-ES/ET from http://exon.gatech.edu/GeneMark/license_download.cgi. Unpack GeneMark-ES/ET:

The resulting uncompressed folder contains a subdirectory gm_et_linux_64/gmes_petap/, where executables reside. Add this directory to your $PATH.
Move the file gm_key (separate download link on the website) to your home directory and make it a hidden file:

3.1.2. AUGUSTUS, SAMTOOLS, BAMTOOLS
AUGUSTUS consists of the actual binary program augustus and several small auxiliary tools, referred to as auxprogs that need to be compiled from source. Install the Ubuntu package dependencies of AUGUSTUS and third-party software that needs to be compiled in order compile the auxprogs:

The tool bam2wig requires htslib, bcftools, samtools from GitHub (the Makefile is currently not compatible with the Ubuntu package version of SAMTOOLS). Download and install these tools as follows:
Export an environment variable TOOLDIR that points to the directory where the above mentioned tools reside (e.g. ~/):

Obtain AUGUSTUS from GitHub and compile (default configuration is sufficient for BRAKER):

Binaries will be stored to a directory Augustus/bin/. Add the path to the AUGUSTUS binaries, the path to Augustus/scripts/ and the path to samtools (e.g. /usr/local/bin/) to your $PATH.
AUGUSTUS looks for configuration files (species specific paramter files and others) in a directory Augustus/config/. The path to that location must be stored in an environment variable $AUGUSTUS_CONFIG_PATH. In case of BRAKER, the $AUGUSTUS_CONFIG_PATH must be a writable directory because BRAKER will store newly trained species parameter sets there. Add the following line to your ~/.bashrc file:

Confirm that important executables can be found:

3.1.3. NCBI BLAST+
Install via the Ubuntu package system:

3.1.4. GenomeThreader
Download GenomeThreader from http://genomethreader.org/.Unpack it:

Add the path to the directory containing the executable gth, which is located in gth-1.7.0-Linux_x86_64–64bit/bin/, to your $PATH. In addition, add the following lines to your ~/.bashrc file:
Replace ${HOME} by the location of GenomeThreader if it resides elsewhere. Confirm that the executable can be found:

3.1.5. Configuration options
In addition to storing tool locations and the $AUGUSTUS_CONFIG_PATH in the $PATH variable, BRAKER offers two more ways to determine which binary from external bioinformatics tools should be executed:
Command line options. All paths to tools can be provided as command line options when calling braker.pl. If the command line options are provided, they will be used, despite all other maybe co-existing configurations. The options are:
--AUGUSTUS_CONFIG_PATH=/path/ --AUGUSTUS_BIN_PATH=/path/ - only required if the AUGUSTUS binaries do not reside in the default location relative to $AUGUSTUS_CONFIG_PATH --AUGUSTUS_SCRIPTS_PATH=/path/ - only required if the AUGUSTUS scripts do not reside in the default location relative to $AUGUSTUS_CONFIG_PATH --BAMTOOLS_PATH=/path/ --GENEMARK_PATH=/path/ --SAMTOOLS_PATH=/path/ --ALIGNMENT_TOOL_PATH=/path/ - this is the path to GenomeThreader --BLAST_PATH=/path/
Environment variables. If environment variables have been exported and no corresponding command line option is used when calling braker.pl, the environment variables will be used instead of the location in $PATH. The environment variables can be added to your ~/.bashrc, similar to the $AUGUSTUS_CONFIG_PATH:

3.2. Running BRAKER
BRAKER is executed by calling the script braker.pl. The following command line options can be relevant for running BRAKER:
--genome=genome.fa assigns the FASTA file with genomic sequences of the target species.
--species=speciesname allows to specify the species name that should be used to store species-specific parameters; in most modes, this is an optional argument. If it is not provided, BRAKER will generate a species name with the pattern Sp_INT where INT is an integer that has not previously been used on your system to name AUGUSTUS parameter sets. We recommend setting a descriptive name because once trained, the parameter set can be reused for running AUGUSTUS.
--softmasking should be specified if the genome has been softmasked. It must be specified if UTRs shall be trained from RNA-Seq data. We recommend using softmasked genomes and enabling this flag for all BRAKER runs.
--gff3 stores BRAKER output gene models in GFF3-format.
--cores=INT specifies the maximum number of cores that can be used during computation. Be aware: Reserving a very large number of cores might be a waste of resources, because most cores will be idle during a large proportion of run time We recommend the usage of 8 cores (because optimize_augustus.pl carries out a k-fold cross validation with k = 8).
--fungus GeneMark-ES/ET option: run algorithm with fungal branch point model
--crf execute discriminative training using conditional random fields (CRF) within AUGUSTUS; resulting parameters are only kept for final predictions if they show higher accuracy than hidden Markov model (HMM) parameters. The additional step of CRF training increases run time.
--keepCrf keep and use CRF parameters even if they are not better than HMM parameters.
--AUGUSTUS_ab_initio will - if extrinsic evidence is provided and used for predicting genes with AUGUSTUS - execute an additional AUGUSTUS run without the evidence. Results are stored in an output file augustus.ab_initio.gtf
BRAKER will create a directory braker/speciesname/ relative to where BRAKER was called. This directory will contain all results and a log file braker.log that lists all commands and subprocesses initiated by BRAKER. Instead of braker/speciesname/, you may specify a different location to store results of your BRAKER run with --workingdir=DIRECTORY.
The most important results files in the output folder are:
-
augustus.hints.gtf contains genes predicted by AUGUSTUS with extrinsic evidence in GTF-format (the file will not be produced if BRAKER is executed with --esmode and no extrinsic evidence). AUGUSTUS reports gene and transcript as separate features in the gtf-file. AUGUSTUS may predict alternative transcripts, i.e. in addition to the transcript g20.t1 in below example, a transcript g20.t2 could be reported.
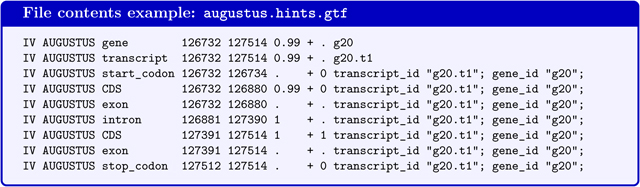
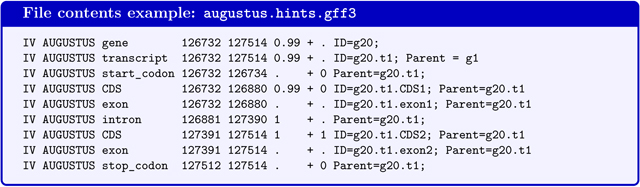
If the command line option --gff3 has been used, a file augustus.hints.gff3 (or in --esmode augustus.ab_initio.gff3) with the same content in gff3-format will be available.
-
GeneMark-E*/genemark.gtf - Genes predicted by GeneMark-ES/ET in GTF-format (the file will not be produced if BRAKER is executed with --trainFromGth)
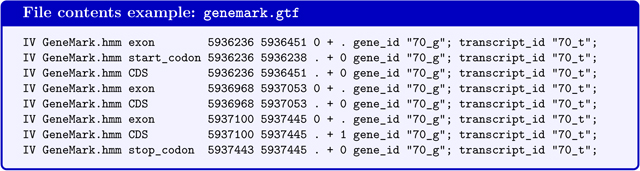
GeneMark-E*/is a subdirectory in the BRAKER output folder. The star will be replaced by the parcitular version of GeneMark-ES/ET that was executed by BRAKER (e.g. GeneMark-ET/).
hintsfile.gff The extrinsic evidence data extracted from RNA-Seq and/or protein data. The introns are used for training GeneMark-ES/ET, while all features are used for predicting genes with AUGUSTUS. The file is in GFF-format (example given in Subheading 2.3.4).
The new species-specific AUGUSTUS parameters are stored in a directory ${AUGUSTUS_CONFIG_PATH}/species/speciesname/ and can be re-used for running AUGUSTUS (also independent from BRAKER).
Concerning the accuracy of results, see Note 2.
3.2.1. Genome file only
If only the genome sequence is available but no extrinsic data that can be used by GeneMark-ES/ET for model refinement during training, self-training GeneMark-ES is executed with the genome as sole input. Genes predicted by GeneMark-ES with a coding sequence longer than 800 nt are selected for training AUGUSTUS. AUGUSTUS predicts genes in the genomic sequences ab initio (see Fig. 2A).
This approach has low accuracy compared to all other modes of running BRAKER. Before choosing this approach, consider that running BRAKER with hints from proteins of remote homology (see Subheading 3.2.3) can be expected to improve prediction accuracy. Also consider that RNA-Seq data for your species might be available in the Sequence Read Archive (GenBank, NCBI). Running BRAKER with such data might therefore also be an option (see Subheading 3.2.2). The genome file only approach is suitable when no suitable evidence is available or if low prediction accuracy is not a problem or if computational time for alignment is a limiting factor.
The accuracy of the ab initio self-training depends on the clade. It is best for genomes with homogeneous genes, such as those from fungi and protists. On the other end of the spectrum, in mammalian genomes, the current self-training algorithm does not produce reliable results due to genome inhomogeneity (about 40% variance in the gene GC-content). In plants and animals with a more narrow range of inhomogeneity (e.g. insects where 90% of genes vary in GC-content by no more than 10%) the self-training produced gene predictions with decent accuracy.
The command line option for running this pipeline is --esmode (derived from the tool name GeneMark-ES). A minimal command would be:
braker.pl --genome=genome.fa --esmode
The pipeline can be applied to the softmasked example genome sequence as follows:

If BRAKER is run with --esmode then the AUGUSTUS output file is not named augustus.hints.gtf but augustus.ab_initio.gtf.
3.2.2. With evidence from RNA-Seq alignment data
If a genome sequence and corresponding RNA-Seq alignments (from the same species) are available, GeneMark-ET is executed. GeneMark-ET uses information about putative splice sites from spliced RNA-Seq read alignments in order to enhance training. In particular, GeneMark-ET uses the information of how many alignments support an individual splice-site. For this reason, BRAKER should not be executed with alignments of assembled RNA-Seq data: The information how many reads support a putative splice site will be lost during the assembly step. After training on the filtered GeneMark-ET gene set, AUGUSTUS predicts genes using RNA-Seq spliced alignments as extrinsic evidence for introns (see Fig. 2B). If the AUGUSTUS training of untranslated regions is enabled, RNA-Seq coverage information will additionally be integrated. Please note that this is the only mode that currently allows training and prediction of untranslated regions of genes with BRAKER.
In order to run BRAKER with RNA-Seq data supplied as BAM-file(s) (in case of multiple files, separate them by comma), call BRAKER with the following minimal set of options:
braker.pl --genome=genome.fa --bam=file1.bam,file2.bam
The pipeline is applicable to the example data set as follows:

If you wish to incorporate RNA-Seq coverage information into AUGUSTUS predictions, the command line option --UTR=on will lead to an attempt to construct UTR training examples from information in the RNA-Seq BAM-file. If a sufficient number of training structures can be generated, speciesspecific UTR parameters will be trained for AUGUSTUS. Subsequently, AUGUSTUS will predict genes including coverage information and with UTRs. The file augustus.hints_utr.gtf will contain the final gene models. Note: UTR training will fail for the provided example data set because it does not contain sufficient information for constructing a large number of training UTRs.
Depending on local computational resources and the number of BAM-files, some users prefer to carry out bam2hints conversion before running BRAKER as follows:

The example data set contains a prepared RNA-Seq hints file and the pipeline can be tested as follows:

The training of UTR parameters is not possible on the basis of hints files.
3.2.3. With evidence generated by mapping cross-species proteins
If RNA-Seq data is not available, spliced alignments of protein families can provide evidence that is formally similar to the information about introns from RNA-Seq alignments: genomic coordinates and a count how may alignments support a particular splice junction. Proteins of closely related species serve well as informants about splice junctions, but this approach is also suitable if the phylogenetic distance between target and informant species increases. Full-length alignability of informant proteins and target genome is not required.
Gene prediction accuracy of BRAKER with this type of evidence is lower than with RNA-Seq evidence.
Constructing spliced alignments for a large number of proteins with a large genome is computationally expensive. In order to reduce run time, GeneMark-ES can be used to generate predicted proteins that can be searched for similarity to protein family members with BLAST. Genomic sequences that were predicted to carry proteins with resulting BLAST hits can be aligned to their hit proteins with a spliced aligner, such as ProSplign [30]. Intron evidence can be extracted from the spliced alignments. A possible pipeline is outlined in Figure 4 (Tomas Bruna, Alexandre Lomsadze and Mark Borodovsky, available for download at http://exon.gatech.edu/GeneMark/Braker/protein_mapping_pipeline.tar.gz). It is important that the protein database contains many representatives of a single gene. Suitable databases with orthologous gene clusters are e.g. EggNogg [31] or OrthoDB [32]. One can in principle use larger databases, such as RefSeq, provided the computational time is acceptable.
A protein mapping pipeline for generating hints from proteins of remote homology for BRAKER is not part of BRAKER. Instead, BRAKER runs with the externally generated hints file. Experienced BRAKER users have reported that they generated suitable hints files with their own mapping pipelines.
The conceptual design of the BRAKER pipeline with evidence from proteins of remote homology is depicted in Figure 2C.
For calling BRAKER with a hints file from proteins of remote homology, provide the option --epmode, which will ensure that GeneMark-EP from the GeneMark-ES/ET tool suite is called:
braker.pl --genome=genome.fa --hints=ep.hints --epmode
The example data set contains a suitable hints file, BRAKER can be called with it as follows:

3.2.4. With evidence by mapping cross-species proteins and RNA-Seq alignments
Using remotely related proteins in addition to RNA-Seq can increase the accuracy somewhat, albeit the running time increases significantly. If both data sources are used, we refer to the GeneMark-ES/ET tool as GeneMark-ETP.
Intron information that is present in both data sources is weighted as reliable evidence and prediction of genes with this information is enforced both in GeneMark-ETP and in AUGUSTUS. The BRAKER pipeline for this mode is shown in Figure 2D.
From the AUGUSTUS point of view, two separate gene prediction runs are performed after training (see Fig. 5):
In one run, AUGUSTUS runs with evidence from RNA-Seq and with evidence provided by RNA-Seq and proteins (evidence from proteins, only, is not used).
In another run, AUGUSTUS runs with protein and RNA-Seq evidence, and protein evidence is given higher priority.
Figure 5:
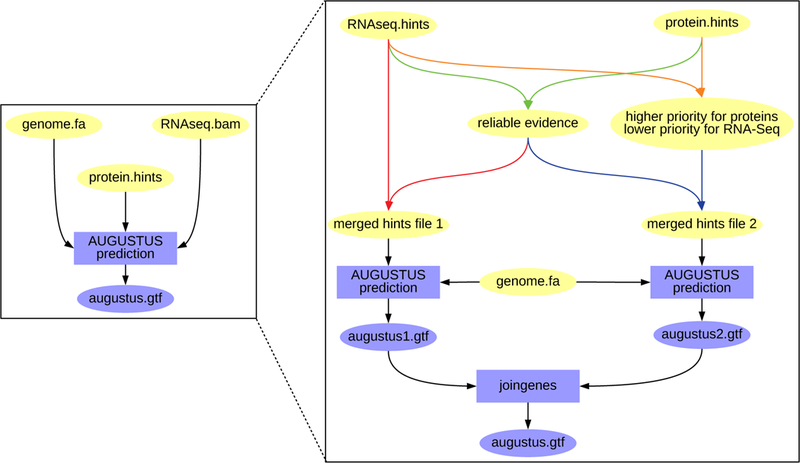
The AUGUSTUS runs of the BRAKER pipeline that are above depicted like on the left as a single box, actually comprise two separate runs (right side) if both RNA-Seq and protein evidence is provided. First, evidence that occurs in both sources is filtered and weighted as reliable, i.e. manual hints for AUGUSTUS (green arrows). This reliable evidence is merged with the remaining RNA-Seq hints for a first AUGUSTUS run (red arrows). For another run, priority 5 is assigned to protein hints, and priority 4 is assigned to RNA-Seq hints (orange arrows), and both hints are merged with the reliable hints (blue arrows). These hints are used for a second AUGUSTUS run. The results of both runs are merged by the AUGUSTUS tool joingenes in a non-redundant fashion.
In both runs, introns provided by both evidence sources are enforced. Subsequently, the gene models of both runs are merged with the AUGUSTUS tool joingenes.
The reason for running AUGUSTUS twice is to increase sensitivity. In practice, we observed that a small proportion of gene models that has support from RNA-Seq data only, gets lost if AUGUSTUS is run with both evidence sources in one run.
For calling BRAKER with both sources of evidence, provide evidence from both sources and specify the option --etpmode. A minimal call would look like this:
braker.pl --genome=genome.fa --hints=ep.hints --bam=RNAseq.bam --etpmode
Alternative to providing the RNA-Seq evidence in a BAM-file, it can also be provided in a hints file (separately or merged with other hints):
braker.pl --genome=genome.fa --hints=ep.hints,RNAseq.hints --etpmode
The pipeline can be tested with the example data set as follows:

3.2.5. Evidence from proteins of close homology
It is well-established that alignments of proteins of closely related species to the target genome are helpful to genome annotation. The general approach is employed by many tools (e.g. Scipio [18], GenomeThreader [19]) and pipelines (e.g. MAKER [11, 12], WebAUGUSTUS [8]). From the BRAKER perspective, using this type of extrinsic evidence is merely a side-project because other resources in principle satisfy the needs of users for accomplishing this task already. Nevertheless, three different pipeline modes that incorporate proteins of close homology are implemented in BRAKER (see Fig. 3).
If a file with protein sequences in FASTA-format is provided with the argument --prot_seq=FILE, BRAKER executes alignment of those proteins against the target genome. BRAKER in principle supports GenomeThreader (--prg=gth), Exonerate [33] (--prg=exonerate) and Spaln2 (--prg=spaln) [34, 35, 36]. We recommend GenomeThreader because in comparison to Exonerate, it is fast, and in comparison to Spaln2, it is currently available for download. BRAKER is routinely tested with GenomeThreader only. The argument --prg=TOOLNAME (TOOLNAME can be gth for GenomeThreader, exonerate or spaln) must be provided if a protein sequence file is given. BRAKER will generate hints for introns, parts of CDS, start codons and stop codons from protein alignments.
Please be aware that GenomeThreader will only confidently align proteins that are fairly closely related to the target species. The return in additional accuracy can be expected to diminish for informant species whose protein homologs are less than 80% identical on average (approximately the distance between Drosophila melanogaster and Drosophila pseudoobscura). In our experience, it increases run time, but not prediction accuracy if a large number of protein sequences from several rather distantly related species are provided to BRAKER for running GenomeThreader.
If both RNA-Seq alignment and protein sequence evidence is provided, AUGUSTUS is run twice, as described in Subheading 3.2.4 and depicted in Figure 5.
In the following, we describe three different ways to call BRAKER with proteins of close homology.
-
A)
Evidence from RNA-Seq alignments for training, additional evidence from protein alignments for prediction
If RNA-Seq evidence is available, GeneMark-ET usually performs very well and produces a high quality training gene set for AUGUSTUS. Evidence from proteins of close homology can be added to the RNA-Seq evidence during the prediction step with AUGUSTUS in order to increase prediction accuracy; in this setup proteins are not used for training AUGUSTUS (see figure 3A).
A minimal BRAKER call with proteins of close homology, the aligner GenomeThreader and RNA-Seq data with the example data looks like this:

-
B)
Evidence from proteins of close homology only
GenomeThreader produces complete gene structures when aligning proteins to the genome. In lack of RNA-Seq data, and if proteins of a very closely related species are available, using the protein alignment derived gene models for training of AUGUSTUS and predicting genes with AUGUSTUS and protein evidence in BRAKER is an alternative to WebAUGUSTUS or pipelines such as GeMoMa [37, 38] (GeMoMa uses the genome and gene coordinates of an informant species rather than its protein sequences). The BRAKER pipeline with GenomeThreader and AUGUSTUS for proteins of close homology is illustrated in Figure 3B.
If this mode is chosen, specified by the command line argument --trainFromGth, GeneMark-ES/ET will not be executed. A minimal call for running BRAKER in this mode is:

-
Evidence from RNA-Seq alignments and evidence from proteins of close homology for training and prediction
In addition to combining evidence from RNA-Seq and proteins of close homology as described, BRAKER can combine the GeneMark-ET RNA-Seq gene set with the gene structures produced by GenomeThreader protein alignment and use a combined set for training AUGUSTUS. Both sources of evidence are then used in the AUGUSTUS gene prediction step, too. The approach is shown in Figure 3C.
The command line option to add GenomeThreader produced genes to the gene set for training AUGUSTUS is --gth2traingenes. It can be applied to the example data set as follows:

In principle, training gene structures derived from RNA-Seq data with GeneMark-ET and training gene structures from the alignment of proteins of close homology with GenomeThreader could complement each other to improve AUGUSTUS parameters during training. However, in the current implementation of BRAKER, this is in practice often not the case. It appears that the genes that most valuably contribute to training AUGUSTUS are included in both sets, and the genes that can be added from proteins do not add positively to training. The observation has been confirmed and reported to us by independent users. We advise users who choose this approach to carefully compare results of their BRAKER run with the results of a BRAKER run that excludes GenomeThreader genes from the training step as described above.
3.2.6. Using BRAKER to execute AUGUSTUS with pre-trained parameters
If a high quality AUGUSTUS parameter set for a particular species already exists (produced by BRAKER or other sources), BRAKER can be used to process and integrate extrinsic evidence from RNA-Seq alignments, from proteins of remote homology and from protein sequences of close homology with AUGUSTUS. Running GeneMark-ES/ET and training AUGUSTUS is skipped, in this case.
The existing parameter set must be specified with --species=speciesname (speciesname = parameter set name), and --skipAllTraining will bypass execution of GeneMark-ES/ET and training AUGUSTUS. This option can be applied to all BRAKER running modes.
You may test this with e.g. the fly parameter set and the example RNA-Seq BAM-file:

BRAKER by default creates the output directory braker/speciesname This can be disadvantageous if you wish to run similar tasks for the same parameter set with different extrinsic evidence combinations or genomes. We therefore recommend specifying an output directory specific to the particular BRAKER run with --workingdir=DIRECTORY.
3.2.7. Training and predicting UTRs on the basis of an existing BRAKER run
Since training UTR parameters for AUGUSTUS is a functionality that has been added to BRAKER rather recently, it might currently be a common use case to update an existing BRAKER run with UTR training and AUGUSTUS predictions that integrate coverage information from RNA-Seq.
In order to do this, the existing parameter set must be specified with --species=speciesname, genome file and RNA-Seq BAM-file must be provided. The option --useexisting will tell BRAKER to modify the existing species parameter set. The argument —AUGUSTUS_hints_preds= augustus.hints.gtf must point to the already existing BRAKER output file of AUGUSTUS in GTF-format. --UTR=on enables UTR training. The argument --flanking_DNA=INT refers to the size of the genomic noncoding flanking region around training genes. It must be provided. In a full BRAKER run, a suitable size is determined automatically. You can extract it from the old braker.log file:

The return value might look similar to the one below:

A call for running training UTR parameters and performing gene predictions with UTR parameters and coverage information could look like this:
braker.pl --species=Sp_1 --useexisting --genome=genome.fa --bam=RNASeq.bam \--AUGUSTUS_hints_preds=augustus.hints.gtf --UTR=on --flanking_DNA=1778
4. Notes
The most frequently reported problems with running BRAKER have their source in using outdated AUGUSTUS scripts. Please always use up-to-date AUGUSTUS (from https://github.com/Gaius-Augustus/Augustus) with BRAKER. BRAKER may not be compatible with AUGUSTUS versions provided from other sources.
The accuracy of results always depends on the input files and on the properties of the individual species. We strongly advise to inspect the results file augustus.hints.gtf (or augustus.hints_utr.gtf) in context with the available extrinsic evidence in a visualizing genome browser, e.g. the UCSC genome browser [39], JBrowse [40] or Artemis [41].
5 Acknowledgement
This work is supported in part by the US National Institutes of Health grant HG000783 to MB and by German Research Foundation grant 1009/12–1 to MS.
6 References
- [1].Hoff KJ, Lange S, Lomsadze A, Borodovsky M, and Stanke M BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics, 32(5):767–769, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, and Borodovsky M Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Research, 33(20):6494–6506, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, and Borodovsky M Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Research, pages gr–081612, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lomsadze A, Burns PD, and Borodovsky M Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Research, 42(15):e119, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stanke M, Schoffmann O, St. Dahms, B. Morgenstern, and S. Waack. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 7:62, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stanke M, Keller O, Gunduz I, Hayes A, Waack S, and Morgenstern B AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research, 3(34):W435–W439, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stanke M, Steinkamp R, Waack S, and Morgenstern B AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research, 32:W309–W312, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hoff KJ and Stanke M WebAUGUSTUS – a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Research, 41(W1):W123–W128, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].König S, Romoth LW, Gerischer L, and Stanke M Simultaneous gene finding in multiple genomes. Bioinformatics, 32(22):3388–3395, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stanke M, Diekhans M, Baertsch R, and Haussler D Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24(5):637–644, 2008. [DOI] [PubMed] [Google Scholar]
- [11].Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, and Yandell M MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research, 18(1):188–196, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Holt C and Yandell M MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics, 12(1):491, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Abbott A Competition boosts bid to find human genes. Nature, 435:134, 2005. [DOI] [PubMed] [Google Scholar]
- [14].Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ash-burner M, Bajic VB, Birney E, Castelo R, Eyras E, Ucla C, Gingeras TR, Harrow J, Hubbard T, Lewis SE, and Reese MG EGASP: the human ENCODE Genome Annotation Assess¬ment Project. Genome Biology, 7(1):S2, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stanke M, Tzvetkova A, and Morgenstern B AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biology, 7(1):S11, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Coghlan A, Fiedler T, McKay S, Flicek P, Harris T, Blasiar D, the nGASP Consortium, and L. Stein. nGASP - the nematode genome annotation assessment project. BMC Bioinformatics, 9(1):549, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Steijger T, Abril JF, Engstrom PG, Kokocinski F, Akerman M, Alioto T, Ambrosini G, Antonarakis SE, Behr J, Bohnert R, Bucher P, Cloonan N, Derrien T, Djebali S, Du J, Dudoit S, Gerstein M, Gingeras TR, Gonzalez D, Grimmond SM, Habegger L, Iseli C, Jean G, Kahles A, Lagarde J, Leng J, Lefebvre G, Lewis S, Mortazavi A, Niermann P, Rätsch G, Reymond A, Ribeca P, Richard H, Rougemont J, Rozowsky J, Sammeth M, Sboner A, Schulz MH, Searle SMJ, Solorzano ND, Solovyev V, Stanke M, Steijger T, Stevenson BJ, Stockinger H, Valsesia A, Weese D, White S, Wold BJ, Wu J, Wu TD, Zeller G, Zerbino D, Zhang MQ, Hubbard TJ, Guigo R, Harrow J, and Bertone P Assessment of transcript reconstruction methods for RNA-seq. Nature Methods, 10(12):1177–1184, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Keller O, Odronitz F, Stanke M, Kollmar M, and Waack S Scipio: Using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinformatics, 9(1):278, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gremme G Computational Gene Structure Prediction. PhD thesis, Universität Hamburg, 2013. [Google Scholar]
- [20].Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, Salzberg SL, and White O Improving the Arabidopsis genome annotation using maximal transcipt alignment assemblies. Nucleic Acids Research, 31(19):5654–5666, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ A basic local alignment search tool. Journal of Molecular Biology, 215(3):403–410, 1990. [DOI] [PubMed] [Google Scholar]
- [22].Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL BLAST+: architecture and applications. BMC Bioinformatics, 10(1):421, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, and Marth GT BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics, 27(12):1691–1692, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R The sequence alignment/map format and SAMtools. Bioinformatics, 25(16):2078–2079, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen N Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics, 5(1):4.10. 1–4.10. 14, 2004. [DOI] [PubMed] [Google Scholar]
- [26].Price AL, Jones NC, and Pevzner PA De novo identification of repeat families in large genomes. Bioinformatics, 21(suppl_1):i351–i358, 2005. [DOI] [PubMed] [Google Scholar]
- [27].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1):15–21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Daehwan K, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4):R36, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu TD and Nacu S Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics, 26(6):873–881, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kapustin Y, Souvorov A, Tatusova T, and Lipman D Splign: algorithms for computing spliced alignments with identification of paralogs. Biology Direct, 3(1):20, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, et al. eggNOG v3. 0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Research, 40(D1):D284–D289, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, and Kriventseva EV OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Research, 41(D1):D358–D365, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Slater GSC and Birney E Automated generation of heuristics for biological sequence compar¬ison. BMC Bioinformatics, 6(1):31, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gotoh O Direct mapping and alignment of protein sequences onto genomic sequence. Bioinformatics, 24(21):2438–2444, 2008. [DOI] [PubMed] [Google Scholar]
- [35].Gotoh O A space-efficient and accurate method for mapping and aligning cdna sequences onto genomic sequence. Nucleic Acids Research, 36(8):2630–2638, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Iwata H and Gotoh O Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Research, 40(20):e161–e161, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, and Hartung F Using intron position conservation for homology-based gene prediction. Nucleic Acids Research, 44(9):e89–e89, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Keilwagen J, Hartung F, Paulini M, Twardziok SO, and Grau J Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics, 19(1):189, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D, et al. The UCSC genome browser database: 2018 update. Nucleic Acids Research, 46(D1):D762–D769, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Skinner ME, Uzilov AV, Stein LD, Mungall CJ, and Holmes IH JBrowse: A next-generation genome browser. Genome Research, gr–094607, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Carver T, Harris SR, Berriman M, Parkhill J, and McQuillan JA Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics, 28(4):464–469, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]