

Abstract
A series of zinc tetraphenylporphyrin photosensitizers furnished with three different anchoring groups, benzoic acid, phenylphosphonate and coumarin-3-carboxylic acid, were prepared using ‘click’ methodology. All three gave modest performances in liquid junction devices with I3–/I– as the electrolyte. The distinct spectroscopic properties of the porphyrins allowed a detailed investigation of the adsorption behaviour and kinetics for charge transfer at the NiO|porphyrin interface. The adsorption behaviour was modelled using the Langmuir isotherm model and the phosphonate anchoring group was found to have the highest affinity for NiO (6.65 × 104 M−1) and the fastest rate of adsorption (2.46 × 107 cm2 mol−1 min−1). The photocurrent of the p-type dye-sensitized solar cells increased with increasing dye loading and corresponding light harvesting efficiency of the electrodes. Coordinating the zinc to a pyridyl-functionalized fullerene (C60PPy) extended the charge-separated state lifetime from ca 200 ps to 4 ns and a positive improvement in the absorbed photon to current conversion efficiency was observed. Finally, we confirmed the viability of electron transfer from the appended C60PPy to phenyl-C61-butyric acid methyl ester, a typical electron transporting layer in organic photovoltaics. This has implications for assembling efficient solid-state tandem solar cells in the future.
This article is part of a discussion meeting issue ‘Energy materials for a low carbon future’.
Keywords: nickel oxide, photocathodes, artificial photosynthesis, dye-sensitized solar cells, tandem cells, transient absorption spectroscopy
1. Introduction
A significant part of the scientific community focuses on developing new approaches to meet the demand of global energy thirst which avoid damaging the environment. Hydrocarbons will continue to play the major role in the energy portfolio for the coming decades, but the rising cost, diminishing availability and concerns over CO2 emissions adds increasing pressure to develop sustainable and secure alternatives. Solar energy utilization has been the subject of intense research, development and deployment efforts that have accelerated during the past decade [1]. Chemistry has played a fundamental role in the design of materials and molecular systems for solar energy conversion into electricity. One example which has attracted attention is the dye-sensitized solar cell (DSC), which offers a versatile, printable alternative to conventional crystalline silicon technology [2]. The efficiencies reached by the state-of-the-art DSC devices under full-sunlight conditions are only half of that reached by silicon [3,4]. Although, much higher efficiencies are possible under ambient light [5].
State-of-the-art DSCs contain a single photoactive electrode (i.e. photoanodes for n-type DSCs). The efficiency can potentially be improved by constructing tandem cells in which a second sensitized photocathode is incorporated in place of the platinized counter electrode found in n-DSCs [6,7]. In a tandem cell, the light transmitted by the photoanode can be converted by the photocathode, increasing the theoretical efficiency to around 45% [8]. Despite an increasing amount of work devoted to developing p-type DSCs, efficiency which matches the state-of-the-art n-type devices has not been reported yet [9]. In p-DSCs, the light absorption by the dye is followed by rapid electron transfer from the valence band of the semiconductor, typically NiO, to the dye, followed by electron transfer from the dye to the redox mediator (typically I3–/I–). For an efficient p-DSC, the dye must be strongly adsorbed onto the semiconductor surface, to promote an efficient charge transfer. Therefore, the dye should possess an anchoring group which can react with surface hydroxyl groups on the metal oxide to form covalent bonds. For n-type devices, the most commonly studied anchoring units are carboxylic and cyanoacetic acids, but a wide range of others including phosphonates, sulfonates and catechol have been used [2,10]. Conversely, for p-type devices, the range of anchors which have been studied is much smaller, with carboxylic acids predominantly used [11–13]. Furthermore, the dyes employed for p-DSCs which have performed the most efficiently were synthesized through linear synthetic routes [9,14,15]. Following this approach, it is difficult to tune individual components (such as the anchor, donor and acceptor groups) without following separate linear routes for each new dye. This is particularly true for the anchoring group, which is generally synthesized either via oxidation of a carbonyl introduced using the Vilsmeier formylation [16–18] or the hydrolysis of tert-butyl benzoate [9]. A more modular approach would be desirable, where the chromophore and anchoring group could be synthesized separately and then assembled, using the minimum number of reactions possible.
The aim of this work is to use click chemistry [19–22] to introduce a range of anchoring groups, employing a porphyrin as the chromophore. Porphyrins are often derivatized using ‘click’ reactions [23], are typically robust to anchor group hydrolysis [24] and provide a distinct spectroscopic handle for anchoring studies. On the other hand, previous work with porphyrins as sensitizers (figure 1) has given modest results (for ZnP, JSC = 0.19 mA cm−2, VOC = 98 mV, FF = 35, η = 0.006%; for ZnTCPP, JSC = 0.5 mA cm−2, VOC = 120 mV, FF = 40%, η = 0.02%) [26,27]. It has been postulated that the limiting factor was fast electron-hole recombination at the interface between the dye and the NiO and by appending an naphthalene diimide (NDI) electron acceptor to ZnP the p-DSC performance was improved (ZNP-NDI JSC = 1.38 mA cm−2, VOC = 127 mV, FF = 32%, η = 0.012%) [28]. The hypothesis that the increased performance arose from a longer-lived charge-separated state when NDI was present was supported by transient absorption spectroscopy, which showed that recombination between NiO and the excited dye occurred on a 50–100 ps time scale for NiO/ZnPref, whereas for NiO/ZNP-NDI most of the excited species decayed on a < 2 ns time scale. The longer lifetime for ZNP-NDI permits more efficient regeneration of the dye by the redox electrolyte which manifested in a higher JSC. The higher VOC for ZNP-NDI can also be explained by reduced recombination, as fewer electrons in the NiO valence band will increase the difference between the NiO Fermi-level and the redox potential of the electrolyte. Likewise, Tian also tested a supramolecular system with ZnTCPP coordinated to N-methyl-2-(4′-pyridyl)-3,4-fulleropyrrolidine (C60PPy) [26]. This was inspired by previous reports of supramolecular interactions between porphyrins and C60 derivatives, which showed a fast porphyrin to C60 charge transfer process and a long lifetime for the reduced fullerene [29,30]. NiO films sensitized using ZnTCPP/C60PPy performed more efficiently than the porphyrin alone (JSC = 1.5 mA cm−2, VOC = 158 mV, FF = 38%, η = 0.09%). The authors attributed this increased performance to a suppression of recombination between the reduced dye and NiO. However, time-resolved experiments were not reported to confirm an increase in charge-separated state lifetime.
Figure 1.

Chemical structure of porphyrins synthesized and tested in p-DSCs previously; ZnPref and ZNP-NDI reported by Odobel et al. [25]; ZnTCPP and C60PPy reported by Tian et al. [26].
In this study, three porphyrins (1–3, figure 2) were functionalized with a carboxylic acid, a phosphonate and a coumarin-based anchoring system. The effect of the anchoring group on dye adsorption and p-DSC performances has been studied. The carboxylic acid was chosen as this is the most widely used anchor for p-DSCs. The phosphonate has been used as a direct comparison, as it is widely used for TiO2 and is reported to be much more stable toward desorption than its carboxylate counterparts [31]. Finally, the coumarin anchor was chosen, which permits a multidentate binding mode between the carboxylic acid and the ketone, which should improve the anchoring stability. Charge transfer from NiO to coumarin-based dyes has been previously reported and occurs on the femtosecond timescale [32,33]. Therefore, this anchoring group should facilitate efficient photoinduced charge transfer from NiO to the porphyrin. A C60 acceptor was coordinated through the Zn following the approach by Tian et al. [26]. The dynamics of charge-separation have been investigated using transient absorption spectroscopy to evaluate the effect of the electron acceptor on the charge-separated state lifetime and the results compared with the performance of the corresponding p-type DSCs.
Figure 2.

Chemical structure of the porphyrins studied and a schematic of the adsorbed supramolecular porphyrin-C60PPy conjugate. (Online version in colour.)
2. Results and discussion
(a). Synthesis and characterization of the porphyrins
1–3 were prepared via a copper-catalysed azide-alkyne cycloaddition reaction (details available in the electronic supplementary material). The alkyne fragment chosen was 5-ethynyl-10,15, 20-tri-p-tolyl-porphyrin and the anchors were functionalized with the azide group. The porphyrin was coordinated with zinc to stop copper insertion occurring during the click reaction and the tolyl groups were used to help reduce porphyrin stacking. This was intended to improve solubility and suppress aggregation when the dye was adsorbed onto NiO. The alkyne group was synthesized as a direct meso-substituent using trimethylsilyl (TMS)-propynal, rather than using TMS-ethynylbenzaldehyde, which is more commonly used [34–36]. This was intended to improve electronic coupling between the porphyrin core and the anchor, as meso-phenyl rings in tetra-aryl porphyrins are non-planar [37].
The identity of the products was confirmed by 1H NMR spectroscopy and the spectra were in general agreement with typical tetraphenylporphyrin derivatives. However, for 1, the triazole was shifted upfield in comparison with 2 and 3 and appeared as two 1H doublets (with J = 1.5 Hz) at 6.97 and 6.49 ppm. This is not consistent with a mixture of regioisomers, due to the 1 : 1 integral ratio and absence (or broadening) of any other aromatic peaks. The weak splitting of these peaks suggests a 4JHH coupling, possibly indicative of a protonated triazole, which could arise due to a zwitterionic interaction (electronic supplementary material, figure S6). The chemical shift of these peaks (6.97 and 6.49 ppm) is similar to a porphyrin appended to an unfunctionalized meso-triazole group (6.57 ppm) reported by the group of Osuka [38]. This zwitterionic interaction could also explain why the 1H NMR spectrum of 1 (when M = Zn) is clearly defined, whereas for 2 and 3 the peaks were considerably broadened compared to the free-base derivatives (electronic supplementary material, figure S7). This broadening has been observed for triazole-functionalized porphyrins due to self-assembly of the triazole subunits with zinc [39]. The dynamic exchange between the monomeric and self-assembled species occurs on a shorter time scale with respect to the NMR measurement [40], leading to peak averaging, which causes the broadened signals. If the triazole is protonated (as is possibly the case with 1), then the loss of one nitrogen lone-pair and a decrease in the electron density of the ring will hinder this self-assembly. This self-assembly of 2 and 3 was not observed when coordinating solvents (tetrahydrofuran (THF) or CH3CN) were employed which will compete with the triazole for coordination to the zinc porphyrin.
(b). Optical studies
The UV–visible absorption and emission spectra of 1–3 are reported in the electronic supplementary material, figures S8–S11 and corresponding parameters are summarized in the electronic supplementary material, table S2. The absorption spectra of 1–3 are typical of metalloporphyrins, with three major bands in the visible region [41–43]. The Soret bands of 1 and 2 were very similar, with a small decrease in intensity and a slight broadening for 2. Otherwise, 3 presented a larger decrease in the peak intensity and an increased broadening compared to 1 and 2.
The steady-state emission spectra of 1–3 show two bands at 606–610 nm and 657–659 nm. For 1 and 2, the emission spectra were mostly independent from the excitation wavelength; however for 3, the emission from the second band was more intense when excited at 550 nm (relative intensity 1.0 versus 0.95). Quantum yields were not determined, but for samples measured under the same instrumental settings with identical absorptions at the excitation wavelength, the relative intensities of the band at ca 610 nm follows the trend 1 (1.0) > 2 (0.64) > 3 (0.59). Compared to the absorption spectra, the most emissive porphyrin was 1, which had the narrowest absorption band, while the least emissive porphyrin was 3, which had the broadest band. The uncorrected excitation spectra were recorded for both emission bands as shown in the electronic supplementary material, figures S12–S14. A good agreement of the absorption and excitation spectra was obtained for both the Soret and Q-bands, indicating that the emissive species for both bands was the same. Zero-zero energy of the molecules, which is used to estimate the electrochemical driving force for the electron transfer process in DSC, was calculated by taking the intercept of the normalized absorption and emission spectra. For porphyrins that do not have a clear overlap of the absorption and emission spectra, different methods have been used to calculate E0-0 [44]. Here, it has been calculated using either the median wavelength values between corresponding absorption/emission bands (method A) or from the intercept of Lorentzian fits of the absorption/emission bands (method B). The values obtained from both methods were similar and the average excited state energies were mostly independent on the nature of the anchoring group (electronic supplementary material, table S3).
(c). Electrochemistry
The redox potentials of 1–3 were determined using cyclic voltammetry and differential pulse voltammetry (DPV) and are summarized in the electronic supplementary material, table S4 and cyclic voltammograms of the oxidation and reduction processes for 1–3 are provided in the electronic supplementary material, figures S18–S20. Concerning porphyrin 1, two quasi-reversible oxidation peaks were observed, with the peak separation dependent on scan rate. Upon increasing the oxidative scan window a third irreversible process was revealed, with a peak current five times higher than the first two processes. For porphyrin 2, the same pattern was observed; however, quasi-reversible oxidation peaks presented a smaller separation (ca 100 mV). Similarly, upon increasing the oxidative scan window a third process was revealed, but the peak current of this wave was roughly equal to the first two processes. As for 1, only one reductive process (electronic supplementary material, figure S19) was observed, which was quasi-reversible, as a return wave was present at higher scan rates. At a scan rate of 100 mV s−1, a shoulder peak was seen on the reductive wave, but this was not resolved at slower or faster scan rates. Cyclic voltammograms of the oxidation process for 3 show two quasi-reversible processes and upon increasing the potential window an irreversible oxidation process was observed, with a peak current roughly twice the size of the first two processes. The reduction process could not be resolved using cyclic voltammetry, as a broad reduction was recorded that overlapped with the solvent window.
DPV was used to resolve the peak potentials of the oxidation (electronic supplementary material, figure S21) and reduction (electronic supplementary material, figure S22) processes of 1–3. In each case, there were three well-resolved oxidation processes and a fourth that is generally broader and less well-defined. There was a little variation in the first oxidation potential; all three porphyrins were oxidized between +0.29 and + 0.32 V versus Fc, with 3 the easiest to oxidize (+0.29 V versus Fc) and 2 the hardest (+0.32 V versus Fc). The reduction processes occur within a potential range of −1.74 to −1.87 V versus Fc, with 2 reduced at lower potentials (−1.74 V versus Fc) and 3 requiring the highest potential (−1.87 V versus Fc).
These redox potentials can be used to estimate the driving force for electron transfer from the valence band of NiO to the photoexcited dye and regeneration of the reduced dye by the redox mediator, which are summarized in the electronic supplementary material, table S5 and represented schematically in the electronic supplementary material, figure S23. ΔGreg is reasonably large in all cases, with the highest driving force of −1.05 eV for 3 and the lowest of −0.92 eV for 1. This suggests that regeneration will not be a limiting factor to device performances. The estimated value of ΔGinj was highest for 2 (−0.33 eV) and lowest for 3 (−0.20 eV), as the HOMO was higher in potential. It is worth noting that these driving forces are similar to a porphyrin studied by Zhang et al. (ZnPref ΔGinj = −0.30 eV and ΔGreg = −1.09 eV) [27]. Liu et al. [45] proposed that ca −0.8 eV driving force is needed for a high quantum yield for hole injection. Consequently, the low ΔGinj for 1–3 (and those of Odobel et al.) could contribute to the low performances typically obtained from porphyrin sensitized p-DSCs [27].
(d). Computational studies
Theoretical energy levels and the corresponding molecular orbital distributions were calculated using the hybrid B3LYP [46] exchange functional and a 6-311G(d,p) basis set. Solvation was treated using the polarizable continuum model [47]. Initially, the ground state geometries were optimized (in THF) then frequency calculations were performed to check if this was a true energy minimum. Time-dependent density functional theory (TDDFT) calculations were then used to determine the vertical excitation energies.
The orbital distributions for HOMO-1 through to LUMO + 2, alongside the calculated energy levels, are shown in the electronic supplementary material, figures S25 and S26 and the energies are summarized in the electronic supplementary material, table S6. In each case, there were four orbitals localized on the porphyrin core: the HOMO-1 to LUMO + 1 for 1 and 2 and the HOMO-1/HOMO and LUMO + 1/+2 for 3. The calculated energies of these orbitals were within 0.05 eV. The electron density of the HOMO was located on the porphyrin core and the four meso-substituents (with electron density on the triazole, but not on the anchor) for all three porphyrins. For the unoccupied porphyrin orbitals, the lower energy orbital was localized on the core and the meso-substituents in the x-axis, while the higher energy orbital was distributed across the core and y-axis substituents. The major difference between the three porphyrins was the unoccupied orbital associated with the anchor. For 1 and 2, this was the LUMO + 2 which was higher in energy for the phosphonate than the carboxylic acid, but for 3, this orbital was lower in energy and was the LUMO. For 1 and 2, this orbital had electron density on the triazole, thus the anchor and porphyrin are coupled. However, for 3, there was no electron density on the triazole in the LUMO, therefore, the anchor and porphyrin are seemingly decoupled.
TDDFT was used to compare the experimentally obtained absorption spectra and the calculated electronic transitions. The calculated energies are summarized in the electronic supplementary material, table S7 and these transitions, overlaid with the experimental UV–visible absorption spectra, are shown in the electronic supplementary material, figures S27–29. There was a reasonable agreement between the calculated and experimental absorption spectra, with lower intensity transitions calculated at lower energy (corresponding to the Q-bands) and higher intensity transitions at higher energy (the Soret band). In each case, the calculated Q-band transitions were predominantly from HOMO to LUMO (or LUMO + 1, the lowest porphyrin orbital, for 3) and HOMO to LUMO + 1 (+2 for 3) transitions. The Soret band transitions were primarily due to excitations to the higher energy porphyrin orbitals. This agrees with the theory that the Soret band is from S0–S2 transitions and the Q-bands from S0–S1. For 1 and 3, the orbital localized on the anchoring group was not involved with any of the excitations, but for 2, there was a small charge-transfer contribution to the Soret band from a porphyrin to anchor transition.
(e). Dye adsorption studies
To investigate how the chemical structure of the different anchoring groups affects the dye's affinity to NiO, dye loading studies were performed (figure 3). NiO films were prepared employing the same procedure used for solar cell tests (provided in the electronic supplementary material). The concentration dependence on dye loading was investigated by measuring the equilibrium surface concentration from a range of initial dye bath concentrations (20–300 µM). Surface concentration at full monolayer coverage (Γmax) and binding constants (K), as determined using the Langmuir adsorption isotherm model, are summarized in table 1.
Figure 3.

(a) Equilibrium surface concentration of three-layer (1.5 µm thick) NiO films as a function of dye bath concentration (films immersed for 18 h in a THF solution of the dye). The surface concentration was determined using the absorbance of the Soret band (ca 420 nm) at lower dye bath concentrations and the highest energy Q-band (ca 550 nm) at higher concentrations. (b) Surface concentration of a three-layer (1.5 µm thick) NiO film as a function of time (film immersed in a 300 µM THF solution of the dye) for 2. The surface concentration was determined using the absorbance of the Soret band (ca 420 nm) at lower surface concentrations and the highest energy Q-band (ca 550 nm) at higher concentrations. (c) Surface concentration of a three-layer (1.5 µm thick) NiO films as a function of time (film immersed in a 300 µM THF solution of the dye) for 3. The surface concentration was determined using the absorbance of the Soret band (ca 420 nm) at lower surface concentrations and the highest energy Q-band (ca 550 nm) at higher concentrations. (d) UV–visible absorption spectra of 1-3 adsorbed onto NiO (18 h adsorption from a 300 µM THF dye bath).
Table 1.
Binding constants (KL) and surface concentration at full monolayer coverage (Γmax) as determined using the Langmuir adsorption isotherm model (nonlinear fitting. Data are reported as an average value with standard errors in parenthesis.
| 1 | 2 | 3 | |
|---|---|---|---|
| KL (M−1) | —a | 6.65 × 104 | 2.81 × 104 |
| (5.35 × 103) | (8.04 × 103) | ||
| Γmax (mol cm−2)b | 8.41 × 10−10 | 1.07 × 10−8 | 1.30 × 10−8 |
| (7.65 × 10−12) | (2.16 × 10−10) | (4.47 × 10−10) |
aCould not be fit to the Langmuir model.
bΓmax reported as the surface concentration from adsorption with a dye bath concentration of 300 µM.
For 2 and 3, the dye loading depended on the concentration of the dye bath, with the absorption of the film (and corresponding surface concentration) increasing as the dye bath concentration increased (figure 3a). The surface concentration reached a plateau between 75 and 150 µM. Giovannetti et al. [48] observed changes in the shape of the Q-bands at higher dye bath concentrations, for the adsorption of a porphyrin onto TiO2, indicating aggregation is occurring. Here, there was no change to the shape of the absorption spectra upon increasing dye bath concentration. This suggests there was little interaction between the adsorbed dye molecules and that only a monolayer had formed. The dye loading was higher for 3 (1.30 × 10−8 mol cm−2) than 2 (1.07 × 10−8 mol cm−2), possibly due to the larger footprint of the phosphonate with respect to the coumarin. There was a much lower dye loading for 1 than 2 or 3, which was surprising as there were only small steric differences between the three dyes (with 1 being the smallest). As carboxylic acids are so prevalent in the literature, it is intriguing that 1 had the lowest dye loading. It is possible that there were alternative anchoring modes competing with the acid for 1, which were not present for 2 and 3. For 2 and 3, the equilibrium data could be analysed using isotherm models, but for 1, the film absorbances were comparatively much lower at each dye bath concentration studied (figure 3). The low dye uptake and high error associated with the measurements performed at lower dye bath concentrations made an accurate analysis of the data impossible. All further thermodynamic/kinetic analysis of the adsorption process has been carried out for just 2 and 3. The fitting is described in detail in the electronic supplementary material. The data were found to be most accurately modelled using the Langmuir isotherm. The Langmuir adsorption isotherm is one of the simplest models for the adsorption of molecules on a solid surface. It has been routinely used to describe dye adsorption onto semiconductor surfaces [49–51] and is based on four assumptions:
-
1.
Only a monolayer of adsorbent forms on the solid surface.
-
2.
All adsorption sites are uniform and equally accessible.
-
3.
There is no interaction between adsorbed molecules.
-
4.
All adsorption occurs through the same mechanism.
The binding constants, as determined from the various methods (see electronic supplementary material, table S9), were in the range of 6.03–6.81 × 104 M−1 and 2.61–3.02 × 104 M−1 for 2 and 3, respectively, with the values agreeing within error. This indicates that the phosphonate anchoring group has a higher affinity for NiO than the coumarin anchor. The binding constant for 2 is larger than for the phosphonate-anchored ruthenium trisbipyridine complex studied by Pellegrin et al. [11] (K = 1.7 × 104 M−1). In the same report, a larger binding constant of 1.8 × 105 M−1 was calculated for a ruthenium complex anchored using carboxylic acid groups. Wu and colleagues studied the adsorption behaviour of the benchmark dye P1 onto NiO and reported a binding constant of 3.54 (± 0.16) × 105 M−1 [51]. As the two reported binding constants for dyes containing carboxylic anchoring groups are on the order of 105 M−1, it is surprising that the binding of 1 was so poor. It is possible that 1 was instead bound through the triazole instead of the acid. This could explain the lower dye loading compared to 2 and 3, because anchoring through the triazole could cause the porphyrin to lie flat onto the NiO surface, reducing the amount of dye that would be able to adsorb. The surface area occupied by each dye molecule on NiO (details provided in the electronic supplementary material) was estimated to be 1.81 and 1.34 nm2 for 2 and 3, respectively, which corresponds to 11 and 13 dye molecules per NiO crystal, respectively.
Studying the kinetics of adsorption provides additional insight into the adsorption process. NiO films were immersed in a 300 µM solution of the dye (the same concentration used for the solar cell studies) and the surface concentration was determined at regular intervals from the film absorbance (using electronic supplementary material, equation S1). The evolution of surface concentration over time is provided in figure 3b,c and the kinetic parameters for dye uptake, fit to various kinetic models, are summarized in the electronic supplementary material, table S10. For the adsorption of both 2 and 3 onto NiO, there was an initial fast uptake of dye followed by a slower uptake, until a plateau is reached. For the uptake of 2 on NiO, the surface concentration reached 75% of the final saturated value in the first 10 min. A saturated surface concentration was then reached after 40 min. The uptake of 3 on NiO was much slower, with the surface concentration reaching 50% of the final value over 20 min in the initial fast uptake process. A saturated surface concentration was then reached after 280 min, which was seven times slower than for 2. The adsorption kinetics for the uptake of 2 and 3 onto NiO have been modelled using pseudo-first and second-order rate equations (electronic supplementary material, figures S36–S39 and table S10).
A pseudo-first-order model has been previously used to model the uptake of N719 [52] onto TiO2 and the uptake of P1 onto NiO [53]. However, in this case, the pseudo-first-order model did not accurately describe the kinetic data (electronic supplementary material, figures S36 and S37). If the reaction does have a first-order dependence on a reactant, then a plot of ln ((qe − q)/qe) versus time should be linear. Two linear relationships can be seen here; an initial part with a larger gradient at shorter times (t < 10 min), then another at longer times with a lower gradient. The linear regressions were lower here than for the second-order model (R2 = 0.904–0.926 versus 0.998–0.999).
The pseudo-second-order model has been used to describe the uptake of catechol-thiophene sensitizers onto TiO2 [54]. This was successfully used to model the adsorption of 2 and 3 onto NiO (electronic supplementary material, figures S36 and S37), as plots of t/qe versus time were linear, with high R2 values. Pseudo-second-order rate constants of 2.46 × 107 and 2.97 × 106 cm2 mol−1 min−1 were calculated for 2 and 3, respectively. This indicates that the phosphonate anchor has both a higher affinity for NiO (as determined by the Langmuir adsorption studies) and that the adsorption of the phosphonate occurred with a higher rate. It has been suggested that a second-order kinetic process for dye adsorption indicates the rate-limiting step is a chemisorption process, where an adsorbate-adsorbent electron exchange takes place [54].
(f). Device performances
Devices were constructed from 1 to 3 by soaking the 1.5 µm thick NiO films for 18 h in a solution of the dye in THF (0.3 mM). The electrolyte used was 1 M LiI and 0.1 M I2 in CH3CN. The performances of the champion cells and representative standard deviations are summarized in table 2 and compared to previously reported values for ZnPref as a benchmark [27].
Table 2.
Photovoltaic performance of p-DSCs constructed using 1–3. Standard deviations as determined for five devices constructed from the same batch of cells for JSC, VOC, FF and η are provided in parenthesis. JSC is the short-circuit current density at the V = 0 intercept, VOC is the open-circuit voltage at the J = 0 intercept, FF is the device fill factor, η is the power conversion efficiency; IPCE is the monochromatic incident photon-to-current conversion efficiency.
| JSC/mA cm−2 | VOC/mV | FF | η/% | IPCE/% (λ nm) | |
|---|---|---|---|---|---|
| 1 | 0.28 (0.05) | 57 (3) | 0.35 (0.016) | 0.006 (9 × 10−4) | 5.4 (430), 1.0 (570), 0.6 (610) |
| 2 | 0.45 (0.02) | 74 (3) | 0.35 (0.014) | 0.012 (6.8 × 10−4) | 8.2 (430), 2.9 (570), 1.8 (610) |
| 3 | 0.51 (0.04) | 76 (6) | 0.37 (0.006) | 0.014 (8.1 × 10−4) | 9.8 (430), 3.5 (570), 2.2 (610) |
| ZnPref | 0.19 | 98 | 0.35 | 0.006 | — |
The J-V curves for the champion devices were measured under illumination (figure 4a) and in the dark (figure 4b). Only modest performances were achieved for any of the dyes, with the highest VOC and JSC (76 mV and 0.51 mA cm−2, respectively) obtained for 3. While the performances of 1-3 were relatively low, these results are similar to previously reported p-DSCs that use porphyrins which do not contain a secondary electron acceptor, such as ZnP and ZnTCPP [26,27]. The trend in both VOC and JSC followed the order of 3 > 2 > 1, which was almost the same trend followed for dye loading and thus film absorbance. The trend in JSC is consistent with the differences in optical density, as the films with a higher absorbance will have an increased light harvesting efficiency (LHE) and therefore produce a higher JSC. Examination of the J-V curves measured in the dark (figure 4b) shows that the dark current was higher for 1 than 2 or 3, which were almost identical. This also helps to explain the trend in JSC, as net currents are equal to the photocurrent produced under illumination minus the dark current. The dark currents are due to recombination between the redox electrolyte and NiO. As the dark currents have an inverse relationship with dye loading, it is likely that the reduced surface coverage of 1 did not protect the NiO surface from recombination to the same extent as 2 or 3. The higher dark current for 1 leads to a lower VOC, as increased recombination causes the Fermi-level of NiO to be less positive (thereby reducing the energy gap between EF-NiO and the redox potential of the electrolyte). The IPCE spectra of the highest-performing devices are shown in figure 4c. The IPCE at 430 nm, which corresponds to the porphyrin Soret band, is indicative of contributions to the photocurrent from dye excitation. This contribution is lowest for 1 and almost masked by the IPCE from the photolysis of triiodide. For 2 and 3, the IPCE at 430 nm is higher (8.2 and 9.8%, respectively) and contribution from the Q-bands can also be observed.
Figure 4.

(a) J-V curves of p-DSCs constructed using 1-3 measured under illumination (AM 1.5, 100 mW cm−2). (b) J-V curves of p-DSCs constructed using 1-3 measured in the dark. (c) IPCE spectra of p-DSCs constructed using NiO films sensitized with 1-3. (d) Charge lifetime as a function of extracted charge density for p-DSCs constructed using 1-3.
The devices were also analysed using small amplitude square wave modulated photovoltage (electronic supplementary material, figure S40) and charge extraction (electronic supplementary material, figure S41) experiments [55]. From the charge extraction measurement (electronic supplementary material, figure S41), the VOC at the same extracted charge density follows the trend 1 > 2 > 3. This indicates a positive shift in the valence band position. Additionally, it appears that the charge lifetime of 1 is higher (electronic supplementary material, figure S40). These two factors would be expected to reduce the dark current and increase JSC. However, care must be taken in comparing devices with different charge densities, as the amount of injected charge will effect EF,NiO.
Combining these measurements gives the charge lifetime as a function of extracted charge density (figure 4d), which corrects for any shifts in the valence band. For each dye, the charge lifetimes were longer at lower charge densities, as there was a lower concentration of holes in the NiO valence band to recombine with the electrolyte. Charge lifetimes (at the same charge density) follow the trend of 3 > 2 > 1, suggesting that the suppression of recombination, as dye coverage increases, leads to a higher JSC.
The supramolecular system, reported in figure 2, was employed to try to improve the performances of 3 (figure 2). Porphyrin-fullerene dyad systems have been thoroughly investigated elsewhere and electron transfer from the chromophore to the acceptor is typically highly efficient [56–58]. C60PPy was synthesized following a literature procedure via the Prato reaction; [59] 4-pyridinecarboxaldehyde and N-methylglycine were reacted with C60 in refluxing toluene to form the pyrrolidinofullerene. This was coordinated to the porphyrin through a sequential adsorption process, where the porphyrin was first adsorbed onto NiO and then the dyed film was immersed in a toluene solution of C60PPy overnight. C60PPy cannot be directly observed using UV–visible absorption spectroscopy, as the major absorption band is in the UV-region (λmax = 310 nm) which overlaps with the absorption from FTO/NiO, while a less intense absorption (λmax = 431 nm) overlaps with the porphyrin Soret band. However, a red-shift of the porphyrin Soret and Q-bands has been previously reported as evidence for the formation of supramolecular interactions between a porphyrin and C60 (electronic supplementary material, figure S15) [60]. The photovoltaic performances of p-DSCs sensitized using 3 with and without the presence of C60PPy are summarized in table 3.
Table 3.
Photovoltaic performance of p-DSCs constructed using 3 with and without cheno/C60PPy. Standard deviations as determined for five devices constructed from the same batch of cells for JSC, VOC, FF and η are provided in parenthesis. JSC is the short-circuit current density at the V = 0 intercept, VOC is the open-circuit voltage at the J = 0 intercept, FF is the device fill factor, η is the power conversion efficiency.
| Jsc/mA cm−2 | Voc/mV | FF | η/% | |
|---|---|---|---|---|
| 3 | 0.51 (0.04) | 76 (6) | 0.37 (0.006) | 0.014 (8.1 × 10−4) |
| 3/C60PPy | 0.42 (0.08) | 46 (0.5) | 0.32 (0.007) | 0.006 (1.2 × 10−3) |
| 3/cheno//C60PPy | 0.55 (0.02) | 80 (4) | 0.35 (0.011) | 0.015 (1.3 × 10−3) |
The J-V curves measured under illumination are shown in figure 5. While Tian and co-workers observed improved performances for the supramolecular porphyrin-C60PPy system [26], in this case, 3 performed worse when the fullerene was coordinated. There was a 0.09 mA cm−2 decrease in JSC and a 30 mV decrease in VOC, combined with a reduced fill factor for the optimum device. This produced an efficiency that was less than half the value of 3-sensitized NiO alone. The J-V measurements performed in the dark (figure 5b) show a large increase in the dark current when C60PPy is present. This suggests that the reduced VOC is due to an increase in recombination, possibly as C60PPy has introduced an additional recombination pathway. As pyridine rings can anchor to semiconductor surfaces, [61,62] it is possible that C60PPy is both coordinated to the porphyrin and anchored to free binding sites on NiO. If this is the case, then the fullerenes anchored to the surface could intercept electrons from the reduced porphyrin and recombine with NiO, instead of being regenerated by the redox electrolyte, which could explain the decreased JSC.
Figure 5.

(a) J-V curves of p-DSCs constructed using 3 with and without cheno/C60PPy measured under illumination (AM 1.5, 100 mW cm−2). (b) J-V curves of p-DSCs constructed using 3 with and without cheno/C60PPy measured in the dark. (c) IPCE spectra of p-DSCs constructed using NiO films sensitized with 3 (THF, 0.3 mM, 18 h) with and without the sequential adsorption of cheno (ethanol, 0.3 mM, 16 h) and then C60PPy (toluene, 0.3 mM, 16 h). (d) Charge lifetime as a function of extracted charge density for p-DSCs constructed using 3 and 3/cheno/C60PPy.
The porphyrins developed in this work have only one anchoring group, but ZnTCPP contains four carboxylic acids capable of binding to NiO. Tian et al. provided evidence, in the form of XPS and FTIR measurements, to indicate that the majority of ZnTCPP adsorbs to NiO ‘lying down’ in a flat orientation [26]. This would effectively cover the NiO surface with porphyrin rings, blocking C60PPy from binding directly to the surface. As the porphyrin is only anchored through one group for 3, it could possibly ‘stand up’ and point away from the surface, thereby leaving binding sites accessible to C60PPy. To try and replicate this concept of surface coverage, the 3-sensitized NiO films were dyed overnight in an ethanolic solution of chenodeoxycholic acid (cheno, electronic supplementary material, figure S16) prior to coordination with C60PPy. Cheno is routinely added to dye baths as a co-adsorbent to prevent dye aggregation as its carboxylic acid group allows efficient adsorption onto NiO. The smaller size of cheno compared to 3 could allow it to bind to sites that the larger porphyrin could not reach. The NiO films were sensitized in a sequential process; first with the porphyrin, then cheno and finally C60PPy. The UV–visible absorption spectra of a NiO film after immersion in each solution is provided in the electronic supplementary material, figure S17. There was a large reduction in the porphyrin film absorptivity after immersion in the cheno dye bath, indicating that cheno displaced some of the porphyrin molecules. A red-shift of the Soret band was again seen after immersion in the solution of C60PPy, which indicated the successful coordination of the fullerene to the porphyrin. The photovoltaic properties of p-DSCs constructed using 3-sensitized NiO with and without treatment with cheno/C60PPy are summarized in table 3.
The J-V curves measured under illumination are shown in figure 5a. Despite the reduction in film absorptivity after treatment with cheno (and corresponding reduction in LHE), devices made using the cheno/C60PPy treated films slightly outperformed the untreated films. There was an increase in both JSC and VOC which, despite a reduced FF, gave a slightly higher efficiency. The J-V measurements performed in the dark (figure 5b) show a reduced dark current for 3/cheno/C60PPy. This suggests that the high dark current (and reduced JSC and VOC) for 3/C60PPy was due to C60PPy anchored onto the NiO surface facilitating recombination. The reduced dark current for 3/cheno/C60PPy indicates that cheno effectively blocks the redox electrolyte from recombining with the NiO surface.
The IPCE spectra of the highest-performing devices for 3 and 3/cheno/C60PPy are shown in figure 5c. In general, the IPCE profile was lower for 3/cheno/C60PPy than 3, except for at the maximum absorption of the Soret band. This is to be expected, as the IPCE of a device is dependent on its LHEs and the film absorption was reduced after treatment with cheno. The IPCE is the product of the LHE, the injection efficiency and the charge collection efficiency. For 3/cheno/C60PPy, the LHE was lower and the injection efficiency was assumed to be similar for both systems, as the dye is the same for both. Therefore, the charge collection efficiency must be higher for the supramolecular system. The charge collection efficiency is dependent on the probability of the holes in NiO reaching the conducting substrate, rather than recombining with the electrolyte or the reduced dye. It is possible that both factors have been improved in this system, as cheno is expected to reduce the electrolyte/NiO recombination and C60PPy is expected to reduce the dye/NiO recombination.
Comparing the shape of the IPCE for both systems, the Soret band is more clearly pronounced for 3/cheno/C60PPy than 3 alone. For 3/cheno/C60PPy, the IPCE between 330 and 400 nm, which is caused by the photolysis of triiodide, [63] is reduced. This was likely due to cheno blocking the electrolyte from reaching the NiO surface, thereby hindering electron transfer from NiO to the diiodide radical. Therefore, for 3/cheno/C60PPy, the porphyrin provided a larger contribution to the photocurrent than in devices made using only 3. As the LHE was lower for 3/cheno/C60PPy, but the IPCE are similar, the absorbed photon to current conversion efficiency (APCE) would be higher. Indeed, the APCE at 440 nm = 10% for 3 and 12% for 3/cheno/C60PPy.
Charge extraction measurements (electronic supplementary material, figure S42) show a higher extracted charge at the same photovoltage for the system without cheno/C60PPy, suggesting reduced recombination. This contradicts the dark current measurements (figure 5b), where the current is lower for cheno/C60PPy, suggesting a reduction in recombination. However, the dark current measurement is a combination of any shifts in EVB,NiO and EF,NiO and any reduction in recombination. Charge lifetime measurements (electronic supplementary material, figure S43) show that the lifetimes were mostly similar for both systems, which would suggest that recombination rates were similar. On the other hand, it is difficult to compare systems with different dye loadings, as different amounts of charge injection from NiO will affect the charge density within the NiO valence band. Therefore, it is necessary to compare the charge lifetimes as a function of charge density (figure 5d). Charge lifetimes were shorter for 3/cheno/C60PPy than 3 at the same charge density, which suggests there was more recombination for 3/cheno/C60PPy. As for 3/C60PPy, it is possible that the presence of C60PPy introduced a new recombination pathway that was not fully blocked by using cheno.
In conclusion, while using C60PPy alone caused a reduction in p-DSC performances, the combination of cheno/C60PPy produced a small improvement, despite a reduction in film absorption. From the IPCE measurements (figure 5c), this improvement appears to be due to a higher APCE compensating for the reduced LHE. Further improvements can be imagined by optimizing the porphyrin:cheno ratio, to block the surface sites not occupied by porphyrin molecules without displacing the dye. This would lead to a higher LHE, which coupled with the higher charge collection efficiency would improve the device IPCE/JSC, leading to a higher power conversion efficiency.
(g). Kinetics
The charge transfer dynamics of dyes 1, 2 and 3 were investigated by femtosecond time-resolved transient absorption spectroscopy in order to evaluate the charge separation at the dye|NiO interface. Details concerning the apparatus employed to carry out measurements are reported in the electronic supplementary material. Firstly, the excited states of 1, 2 and 3 in THF solution were recorded, then the experiments were repeated with the porphyrins adsorbed on NiO (NiO|1, NiO|2 and NiO|3) to explore the formation and decay of the NiO+|dye– charge-separated state. The transient absorption spectra of 1, 2 and 3 in solution are reported in figure 6 and the electronic supplementary material, figure S44 and the transient absorption spectra of porphyrins on NiO are presented in figure 7 and the electronic supplementary material, figure S45. Global analysis was carried out using the OPTIMUS software and corresponding spectra are reported in the electronic supplementary material, figures S46–S51 [64].
Figure 6.

(a) Femtosecond transient absorption spectra of 1 in THF solution after excitation at 550 nm, the inset kinetic trace is probed at 461 nm. (b) Femtosecond transient absorption spectra of 2 in THF solution after excitation at 550 nm, the inset kinetic trace is probed at 474 nm. (c) Femtosecond transient absorption spectra of 3 in THF solution after excitation at 550 nm, the inset kinetic trace probed at 475 nm.
Figure 7.

(a) Femtosecond transient absorption spectra of 1 on NiO after excitation at 550 nm, the inset kinetic trace is probed at 458 nm. (b) Femtosecond transient absorption spectra of 2 on NiO after excitation at 550 nm, the inset kinetic trace is probed at 463 nm. (c) Femtosecond transient absorption spectra of 3 on NiO after excitation at 550 nm, the inset kinetic trace is probed at 461 nm. Arrows highlight the weak transient at 660 nm, which corresponds to the porphyrin radical anion.
The spectral shape of 1, 2 and 3 excited states at 550 nm are generally similar. The bleach centred at ca 420 nm corresponds to the loss of the ground state, while the large positive absorption between 440 and 500 nm was attributed to the formation of the singlet excited state, which evolves over ca 2 ns to be replaced by the triplet excited state (figure 6). The minimum number of time components required to fit the decays of the porphyrin transient absorption using global analysis of the solution data was three (for 1) or four (for 2 and 3). Short components (within 200 ps) are consistent with vibrational and solvent relaxation and the major component was of the order of 1000 ps which is consistent with S1–T1 intersystem crossing [27]. The last component in the time scale of microseconds is associated with the decay of the porphyrin triplet state, and the lifetime was resolved by fitting the transient at around 460 nm (see figure 6 insets; electronic supplementary material, table S11) with a monoexponential function. All porphyrins presented a triplet lifetime around 1 µs (see electronic supplementary material, table S11), with 3 presenting the longest lifetime. The lifetimes obtained by fitting single-point traces at 460 nm were in excellent agreement with the kinetic constants obtained by global analysis.
The spectral features between 430 and 500 nm in the transient spectra for NiO|1, NiO|2 and NiO|3 (figure 7) were similar to those of the dyes in solution and formed within the time resolution of the instrument. All spectra contain a bleaching of the ground state, along with the formation of the excited state. For NiO|1 and NiO|2, the weak negative signal at around 650 nm is consistent with stimulated emission. The excited states of NiO|1, NiO|2 and NiO|3 decayed three orders of magnitude faster than the dyes in solution, which suggests that, in all cases, the excited state is rapidly quenched by electron transfer from the NiO to the dye followed by formation of charge-separated state. To exclude that the rapid excited state quenching is due to dye aggregation, experiments were performed with samples in which chenodeoxycholic acid (cheno), which is known to break aggregates, was used as co-adsorbent [65]. As the decay of the transient absorption was slightly faster in the presence of this additive (electronic supplementary material, figure S55), we can exclude aggregation. Consistent with this, a broad, weak absorption was observed for 1 at wavelengths longer than 600 nm, which is characteristic of holes introduced in NiO. Furthermore, for NiO|2, a weak feature at 660 nm, which can be attributed to the porphyrin radical anion, was present after about 10 ps. Changes associated with charge transfer from NiO to 3 were more significant. The broad stimulated emission band (centred around 560–600 nm) was quenched, and, at the same time, a weak absorption similar to that detected for NiO|2 was identified around 660 nm, which is also consistent with the porphyrin radical anion.
Global analysis of the time evolution of the TA spectra measured upon 550 nm excitation was performed, using three (for NiO|1) and five (NiO|2 and NiO|3) exponential functions to determine the numbers of time constants required to represent the data. As previously reported by Zhang et al., these time constants should not be interpreted as unique processes, but they represent an approximate description of the multi-exponential kinetics [27]. The decay associate spectra (DAS) contain shorter components (Ʈ1= 15 ps for NiO|1, Ʈ1= 0.75 ps and Ʈ2= 3 ps for NiO|2, Ʈ1= 0.8 ps and Ʈ2= 4 ps for NiO|3) which were similar to decay of the S1 excited state in solution (electronic supplementary material, figures S49–51a). Likewise, the species associated spectra (SAS; electronic supplementary material, figures S50 and 51b) of NiO|2 and NiO|3 show that the bleach/stimulated emission at ca 650 nm for Ʈ2 components shift to ca 640 nm for Ʈ3 components, which suggests that the initial three components (Ʈ1 – Ʈ3) for NiO|2 and NiO|3 and the first component (Ʈ1) for NiO|1 mainly reflect hole injection [24]. The ca 200 ps time constants (Ʈ2= 190 ps for NiO|1, Ʈ3= 200 ps for NiO|2 and Ʈ3= 210 ps for NiO|3) were associated with the rapid charge-recombination at the NiO+|porphyrin– interface. Very little signal survived on a time scale greater than 1 ns. Time constants obtained by fitting of decay traces around 460 nm were in excellent agreement with the time constant assigned to the rapid recombination step from global analysis (see inset figure 7; electronic supplementary material, table S12) and in both cases, the transient lifetimes were longer for 3. In summary, the results from global analysis of the data (DAS and SAS) allows us to conclude that hole injection into NiO occurred following excitation of porphyrins with time constants around 1–40 ps and this was followed by rapid charge recombination, mainly on the 200 ps time scale, with consequent recovery of 70% of the signal.
The kinetics of the supramolecular system (NiO|cheno|3-C60PPy), where the C60 acceptor was employed to improve the solar cell performance of 3 (figure 2), were also analysed, and the associate spectra are reported in the electronic supplementary material, figure S52. It is worth mentioning that we obtained generally longer-lived signals for NiO|cheno|3-C60PPy with respect to NiO|3 (figure 8). Global analysis of the 600–700 nm wavelength region was carried out by fitting kinetic traces with four time constants (electronic supplementary material, figure S53). The results were similar to those for NiO|3, with short components (Ʈ1, Ʈ2) that are consistent with vibrational relaxation and simultaneous decay of the stimulated emission (around 660 nm). The Ʈ3= 320 ps and Ʈ4= 4300 ps components did not contain a stimulated emission feature and the presence of a more prominent band around 670 nm is consistent with a combination of the 3 radical anion and the C60PPy radical anion. Ʈ3 is on the order of hundreds of picoseconds and is consistent with charge-recombination. This component was also resolved by fitting the decay of NiO|cheno|3-C60PPy at 660 nm with a monoexponential function, unless otherwise states. Results are reported in the electronic supplementary material, figure S54 and table S13, in which the time constant associated with NiO|cheno|3-C60PPy was higher with respect to NiO|3.
Figure 8.
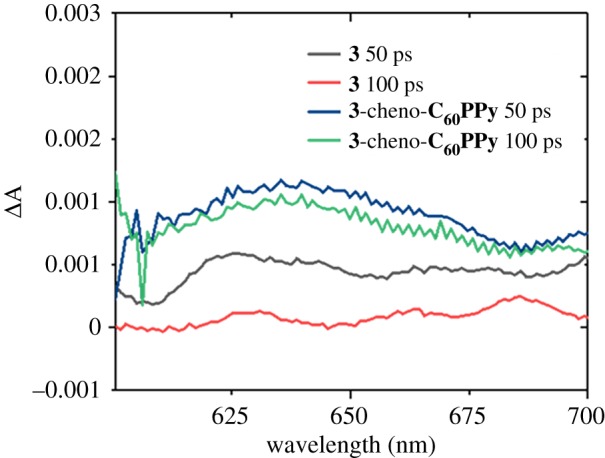
Femtosecond transient absorption spectra of NiO|3 and NiO|cheno|3-C60PPy after excitation at 550 nm at two different delays.
The positive effect of the C60 acceptor on the charge-separated state lifetime prompted us to explore the possibility of applying this approach in solid-state p-type DSCs. In previous work, it has been shown that poor electron transfer from the dye to PCBM limited the photocurrent and engineering the dye|PCBM interface is key to increase the device performance [66,67]. In this work, a layer of phenyl-C61-butyric acid methyl ester (PCBM) was applied by spin coating on top of NiO|cheno|3-C60PPy, in order to investigate the effect of this electrode structure (NiO|cheno|3-C60PPy|PCBM) on the charge-separated state lifetime. The results were investigated by employing global analysis, fitting kinetic traces with four time constants. Results for NiO|cheno|3-C60PPy|PCBM are reported in the electronic supplementary material, figure S56 and are quite similar to those obtained for NiO|cheno|3-C60PPy with short components (Ʈ1, Ʈ2) that can be associated with vibrational relaxation and simultaneous decay of the stimulated emission. In the presence of PCBM, the components Ʈ3= 700 ps and Ʈ4= ∞ ps did not contain a stimulated emission feature. At the same time, the SAS for NiO|cheno|3-C60PPy|PCBM reveal that the porphyrin fine structure was lost in the Ʈ2= 50 ps component and the signal centred at around 640 nm was broader with respect to the equivalent component observed for NiO|cheno|3-C60PPy. The lifetime constant attributed to rapid recombination ( Ʈ3 ), was longer in the presence of PCBM than the corresponding lifetime observed with NiO|cheno|3-C60PPy. Furthermore, a long-lived component ( Ʈ4 ) was present in the spectra for NiO|cheno|3-C60PPy|PCBM, which survived beyond the duration of the experiment. This suggests that PCBM intercepts the electrons from the dye/C60 conjugate and so recombination with holes in the NiO valence band is slower.
3. Conclusion
We have described a simple ‘click’ chemistry approach to functionalize dyes with different anchoring groups and systematically studied their adsorption behaviour on mesoporous NiO and their performance in p-type DSCs. We observed a limitation of the triazole linker, which appeared to form a zwitterion in the benzoic acid derivative, 1. This affected the dye loading. The adsorption behaviour agreed with the Langmuir isotherm model, which indicates that a monolayer of dye binds to the surface. The adsorption kinetics were fitted to a pseudo-second-order rate model and adsorption occurred more rapidly for the anchor that had a higher affinity for NiO (2). IPCE measurements confirmed that excitation of the porphyrin was contributing toward the photocurrent for each device and the characteristics of the p-DSCs incorporating 1–3 were comparable or higher to other porphyrins tested in the literature, which do not contain a secondary electron acceptor. p-DSC performances followed the trend 3 > 2 > 1, which has been partially attributed to higher dye loading increasing the LHE, thereby increasing the JSC. p-DSCs made using NiO|3-C60PPy had a lower power conversion efficiency than the dyes alone, possibly due to the fullerene anchoring directly to NiO and introducing an alternative charge-recombination pathway. Cheno was used as a co-adsorbent to block the free surface sites prior to the coordination of C60PPy, to try and block the anchoring of C60PPy to the surface. The addition of cheno reduced the concentration of the porphyrins on the NiO surface; however, devices made using NiO|cheno|3-C60PPy slightly outperformed NiO|3 alone, leading to an improved APCE.
The distinct spectroscopic properties of the porphyrins and C60 radical permitted us to monitor the charge-separation and recombination processes in the photocathodes. The rate of electron transfer from the NiO to the photoexcited porphyrins occurred rapidly (τ = 1–40 ps) and was followed by rapid charge recombination (≈70% of the signal decayed with τ < 200 ps), which is consistent with our previous work on organic dyes adsorbed on NiO. The structure of the anchoring group or the presence of cheno did not appear to greatly affect the dynamics. The coordination of the electron acceptor, C60, to the porphyrin extended the charge-separated state lifetime, however. Furthermore, adding an electron transport layer (PCBM) to the film provided a pathway to intercept the charge and extend charge-separation on greater than microsecond time scales. This has implications for solid-state tandem DSSCs and the information provided by these experiments will enable us to engineer optimized dyes which bind strongly to the NiO surface and drive electron transfer from the metal oxide towards a solid-state electron transport layer in tandem solar cells.
Supplementary Material
Acknowledgements
We thank Newcastle University for a PhD studentship for G.H.S.
Data accessibility
Raw data supporting the publication can be found at https://doi.org/10.25405/data.ncl.c.4539992.
Competing interests
We declare we have no competing interests.
Funding
The North East Centre for Energy Materials EP/R021503/1, STFC for access to the CLF ULTRA facility for transient spectroscopy and ERC starting grant, p-TYPE 715354.
References
- 1.Lewis NS. 2016. Research opportunities to advance solar energy utilization. Science 351, 1920 ( 10.1126/science.aad1920) [DOI] [PubMed] [Google Scholar]
- 2.Hagfeldt A, Boschloo G, Sun L, Kloo L, Pettersson H. 2010. Dye-sensitized solar cells. Chem. Rev. 110, 6595–6663. ( 10.1021/cr900356p) [DOI] [PubMed] [Google Scholar]
- 3.Green MA, Emery K, Hishikawa Y, Warta W, Dunlop ED. 2014. Solar cell efficiency tables (version 44). Prog. Photovoltaics Res. Appl. 22, 701–710. ( 10.1002/pip.2525) [DOI] [Google Scholar]
- 4.Mathew S, et al. 2014. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 6, 242–247. ( 10.1038/nchem.1861) [DOI] [PubMed] [Google Scholar]
- 5.Freitag M, et al. 2017. Dye-sensitized solar cells for efficient power generation under ambient lighting. Nat. Photonics 11, 372–378. ( 10.1038/nphoton.2017.60) [DOI] [Google Scholar]
- 6.He J, Lindström H, Hagfeldt A, Lindquist S-E. 2000. Dye-sensitized nanostructured tandem cell-first demonstrated cell with a dye-sensitized photocathode. Sol. Energy Mater. Sol. Cells 62, 265–273. ( 10.1016/s0927-0248(99)00168-3) [DOI] [Google Scholar]
- 7.Odobel F, Pellegrin Y, Gibson EA, Hagfeldt A, Smeigh AL, Hammarström L. 2012. Recent advances and future directions to optimize the performances of p-type dye-sensitized solar cells. Coord. Chem. Rev. 4, 2551–2564. ( 10.1021/jz400861v) [DOI] [Google Scholar]
- 8.Bremner SP, Levy MY, Honsberg CB. 2008. Analysis of tandem solar cell efficiencies under AM1.5G spectrum using a rapid flux calculation method. Prog. Photovoltaics Res. Appl. 16, 225–233. ( 10.1002/pip.799) [DOI] [Google Scholar]
- 9.Nattestad A, Mozer AJ, Fischer MKR, Cheng Y-B, Mishra A, Bäuerle P, Bach U. 2010. Highly efficient photocathodes for dye-sensitized tandem solar cells. Nat. Mater. 9, 31–35. ( 10.1038/nmat2588) [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Cole JM. 2015. Anchoring groups for dye-sensitized solar cells. ACS Appl. Mater. Interfaces 7, 3427–3455. ( 10.1021/am507334m) [DOI] [PubMed] [Google Scholar]
- 11.Pellegrin Y, et al. 2011. Ruthenium polypyridine complexes as sensitizers in NiO based p-type dye-sensitized solar cells: effects of the anchoring groups. J. Photochem. Photobiol. A Chem. 219, 235–242. ( 10.1016/j.jphotochem.2011.02.025) [DOI] [Google Scholar]
- 12.Brunner F, et al. 2016. Modular synthesis of simple cycloruthenated complexes with state-of-the-art performance in p-type DSCs. J. Mater. Chem. C 4, 9823–9833. ( 10.1039/C6TC03874C) [DOI] [Google Scholar]
- 13.Cui J, et al. 2014. Organic sensitizers with pyridine ring anchoring group for p-type dye-sensitized solar cells. J. Phys. Chem. C 118, 16 433–16 440. ( 10.1021/jp410829c) [DOI] [Google Scholar]
- 14.Alemu G, Cui J, Cao K, Li J, Shen Y, Wang M. 2014. Investigation of the regeneration kinetics of organic dyes with pyridine ring anchoring groups by scanning electrochemical microscopy. RSC Adv. 4, 51 374–51 380. ( 10.1039/C4RA07908F) [DOI] [Google Scholar]
- 15.Click KA, Beauchamp DR, Garrett BR, Huang Z, Hadad CM, Wu Y. 2014. A double-acceptor as a superior organic dye design for p-type DSSCs: high photocurrents and the observed light soaking effect. Phys. Chem. Chem. Phys. 16, 26 103–26 111. ( 10.1039/c4cp04010d) [DOI] [PubMed] [Google Scholar]
- 16.Qin P, Zhu H, Edvinsson T, Boschloo G, Hagfeldt A, Sun L. 2008. Design of an organic chromophore for p-type dye-sensitized solar cells. J. Am. Chem. Soc. 130, 8570–8571. ( 10.1021/ja8001474) [DOI] [PubMed] [Google Scholar]
- 17.Qin P, Wiberg J, Gibson EA, Linder M, Li L, Brinck T, Hagfeldt A, Albinsson B, Sun L. 2010. Synthesis and mechanistic studies of organic chromophores with different energy levels for p-type dye-sensitized solar cells. J. Phys. Chem. C 114, 4738–4748. ( 10.1021/jp911091n) [DOI] [Google Scholar]
- 18.Zhang Q-Q, et al. 2015. A push–pull thienoquinoidal chromophore for highly efficient p-type dye-sensitized solar cells. J. Mater. Chem. A 3, 7695–7698. ( 10.1039/C5TA01348H) [DOI] [Google Scholar]
- 19.Kolb HC, Finn MG, Sharpless KB. 2001. Click chemistry: diverse chemical function from a few good reactions. Angew. Chemie Int. Ed. 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- 20.Liang L, Astruc D. 2011. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) ‘click’ reaction and its applications. An overview. Coord. Chem. Rev. 255, 2933–2945. ( 10.1016/j.ccr.2011.06.028) [DOI] [Google Scholar]
- 21.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. 2002. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective ‘Ligation’ of Azides and terminal alkynes. Angew. Chemie Int. Ed. 41, 2596–2599. () [DOI] [PubMed] [Google Scholar]
- 22.Tornøe CW, Christensen C, Meldal M. 2002. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064. ( 10.1021/jo011148j) [DOI] [PubMed] [Google Scholar]
- 23.Ladomenou K, Nikolaou V, Charalambidis G, Charisiadis A, Coutsolelos AG. 2017. Porphyrin–BODIPY-based hybrid model compounds for artificial photosynthetic reaction centers. C. R. Chim. 20, 314–322. ( 10.1016/j.crci.2016.07.009) [DOI] [Google Scholar]
- 24.Nikolaou V, Karikis K, Farré Y, Charalambidis G, Odobel F, Coutsolelos AG. 2015. Click made porphyrin–corrole dyad: a system for photo-induced charge separation. Dalt. Trans. 44, 13 473–13 479. ( 10.1039/C5DT01730K) [DOI] [PubMed] [Google Scholar]
- 25.Maufroy A, Favereau L, Anne FB, Pellegrin Y, Blart E, Hissler M, Jacquemin D, Odobel F. 2015. Correction: Synthesis and properties of push–pull porphyrins as sensitizers for NiO based dye-sensitized solar cells. J. Mater. Chem. A 3, 3908–3917. ( 10.1039/C4TA05974C) [DOI] [Google Scholar]
- 26.Tian H, et al. 2014. Enhancement of p-type dye-sensitized solar cell performance by supramolecular assembly of electron donor and acceptor. Sci. Rep. 4, 4282 ( 10.1038/srep04282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, et al. 2016. Molecular-structure control of electron transfer dynamics of push–pull porphyrins as sensitizers for NiO based dye sensitized solar cells. RSC Adv. 6, 77 184–77 194. ( 10.1039/C6RA15195G) [DOI] [Google Scholar]
- 28.Maufroy A, Favereau L, Anne FB, Pellegrin Y, Blart E, Hissler M, Jacquemin D, Odobel F. 2015. Synthesis and properties of push–pull porphyrins as sensitizers for NiO based dye-sensitized solar cells. J. Mater. Chem. A 3, 3908–3917. ( 10.1039/C4TA05974C) [DOI] [Google Scholar]
- 29.Yamada H, Imahori H, Nishimura Y, Yamazaki I, Fukuzumi S. 2002. Enhancement of photocurrent generation by ITO electrodes modified chemically with self-assembled monolayers of porphyrin–fullerene dyads. Adv. Mater. 14, 892 () [DOI] [Google Scholar]
- 30.Yamada H, Imahori H, Nishimura Y, Yamazaki I, Ahn TK, Kim SK, Kim D, Fukuzumi S. 2003. Photovoltaic properties of self-assembled monolayers of porphyrins and porphyrin−fullerene dyads on ITO and gold surfaces. J. Am. Chem. Soc. 125, 9129–9139. ( 10.1021/ja034913f) [DOI] [PubMed] [Google Scholar]
- 31.Guerrero G, Alauzun JG, Granier M, Laurencin D, Mutin PH. 2013. Phosphonate coupling molecules for the control of surface/interface properties and the synthesis of nanomaterials. Dalt. Trans. 42, 12 569 ( 10.1039/c3dt51193f) [DOI] [PubMed] [Google Scholar]
- 32.Morandeira A, Boschloo G, Hagfeldt A, Hammarström L. 2008. Coumarin 343−NiO films as nanostructured photocathodes in dye-sensitized solar cells: ultrafast electron transfer, effect of the I3−/I− redox couple and mechanism of photocurrent generation. J. Phys. Chem. C 112, 9530–9537. ( 10.1021/jp800760q) [DOI] [Google Scholar]
- 33.Morandeira A, Boschloo G, Hagfeldt A, Hammarström L. 2005. Photoinduced ultrafast dynamics of coumarin 343 sensitized p-type-nanostructured NiO films. J. Phys. Chem. B 109, 19 403–19 410. ( 10.1021/jp053230e) [DOI] [PubMed] [Google Scholar]
- 34.Fazio MA, Lee OP, Schuster DI. 2008. First triazole-linked porphyrin−fullerene dyads. Org. Lett. 10, 4979–4982. ( 10.1021/ol802053k) [DOI] [PubMed] [Google Scholar]
- 35.Shetti VS, Ravikanth M. 2010. Synthesis of triazole-bridged unsymmetrical porphyrin dyads and porphyrin-ferrocene conjugates. Eur. J. Org. Chem. 2010, 494–508. ( 10.1002/ejoc.200901070) [DOI] [Google Scholar]
- 36.de Miguel G, et al. 2011. Triazole bridges as versatile linkers in electron donor–acceptor conjugates. J. Am. Chem. Soc. 133, 13 036–13 054. ( 10.1021/ja202485s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silvers SJ, Tulinsky A. 1967. The crystal and molecular structure of triclinic tetraphenylporphyrin. J. Am. Chem. Soc. 89, 3331–3337. ( 10.1021/ja00989a036) [DOI] [PubMed] [Google Scholar]
- 38.Maeda C, Yamaguchi S, Ikeda C, Shinokubo H, Osuka A. 2008. Dimeric assemblies from 1,2,3-triazole-appended Zn(II) porphyrins with control of NH-tautomerism in 1,2,3-triazole. Org. Lett. 10, 549–552. ( 10.1021/ol7028299) [DOI] [PubMed] [Google Scholar]
- 39.Roberts DA, Schmidt TW, Crossley MJ, Perrier S. 2013. Tunable self-assembly of triazole-linked porphyrin-polymer conjugates. Chem. - A Eur. J. 19, 12 759–12 770. ( 10.1002/chem.201301133) [DOI] [PubMed] [Google Scholar]
- 40.Stibrany RT, Vasudevan J, Knapp S, Potenza JA, Emge T, Schugar HJ. 1996. Two modes of self-coordinating edge-over-edge Zn(II) porphyrin dimerization: a structural and spectroscopic comparison. J. Am. Chem. Soc. 118, 3980–3981. ( 10.1021/ja9536734) [DOI] [Google Scholar]
- 41.Harriman A, Richoux MC, Neta P. 1983. Redox chemistry of metalloporphyrins in aqueous solution. J. Phys. Chem. 87, 4957–4965. ( 10.1021/j150642a038) [DOI] [Google Scholar]
- 42.Nappa M, Valentine JS. 1978. The influence of axial ligands on metalloporphyrin visible absorption spectra. Complexes of tetraphenylporphinatozinc. J. Am. Chem. Soc. 100, 5075–5080. ( 10.1021/ja00484a027) [DOI] [Google Scholar]
- 43.Dorough GD, Miller JR, Huennekens FM. 1951. Spectra of the Metallo-derivatives of α,β,γ,δ-Tetraphenylporphine. J. Am. Chem. Soc. 73, 4315–4320. ( 10.1021/ja01153a085) [DOI] [Google Scholar]
- 44.Manke A-M, Geisel K, Fetzer A, Kurz P. 2014. A water-soluble tin(IV) porphyrin as a bioinspired photosensitiser for light-driven proton-reduction. Phys. Chem. Chem. Phys. 16, 12 029–12 042. ( 10.1039/C3CP55023K) [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, et al. 2014. Modulated charge injection in p-type dye-sensitized solar cells using fluorene-based light absorbers. ACS Appl. Mater. Interfaces 6, 3448–3454. ( 10.1021/am405610b) [DOI] [PubMed] [Google Scholar]
- 46.Becke AD. 1993. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. ( 10.1063/1.464913) [DOI] [Google Scholar]
- 47.Miertuš S, Scrocco E, Tomasi J. 1981. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55, 117–129. ( 10.1016/0301-0104(81)85090-2) [DOI] [Google Scholar]
- 48.Giovannetti R, Zannotti M, Alibabaei L, Ferraro S. 2014. Equilibrium and kinetic aspects in the sensitization of monolayer transparent TiO2 thin films with porphyrin dyes for DSSC applications. Int. J. Photoenergy 2014, 834269 ( 10.1155/2014/834269) [DOI] [Google Scholar]
- 49.Kim T-Y, Lee J-W, Jin E-M, Park J-Y, Kim J-H, Park K-H. 2013. In situ measurement of dye adsorption on TiO2 thin films for dye-sensitized solar cells. Measurement 46, 1692–1697. ( 10.1016/j.measurement.2013.01.008) [DOI] [Google Scholar]
- 50.Holliman PJ, Vaca Velasco B, Butler I, Wijdekop M, Worsley DA. 2008. Studies of dye sensitisation kinetics and sorption isotherms of Direct Red 23 on Titania. Int. J. Photoenergy 2008, 1–7. ( 10.1155/2008/827605) [DOI] [Google Scholar]
- 51.Natu G, Huang Z, Ji Z, Wu Y. 2012. The effect of an atomically deposited layer of alumina on NiO in p-type dye-sensitized solar cells. Langmuir 28, 950–956. ( 10.1021/la203534s) [DOI] [PubMed] [Google Scholar]
- 52.Lee C-R, Kim H-S, Jang I-H, Im J-H, Park N-G. 2011. Pseudo first-order adsorption kinetics of N719 dye on TiO2 surface. ACS Appl. Mater. Interfaces 3, 1953–1957. ( 10.1021/am2001696) [DOI] [PubMed] [Google Scholar]
- 53.Zannotti M, Wood CJ, Summers GH, Stevens LA, Hall MR, Snape CE, Giovanetti R, Gibson EA. 2015. Ni Mg mixed metal oxides for p-type dye-sensitized solar cells. ACS Appl. Mater. Interfaces 7, 24 556–24 565. ( 10.1021/acsami.5b06170) [DOI] [PubMed] [Google Scholar]
- 54.An B-K, Hu W, Burn PL, Meredith P. 2010. New Type II catechol-thiophene sensitizers for dye-sensitized solar cells. J. Phys. Chem. C 114, 17 964–17 974. ( 10.1021/jp105687z) [DOI] [Google Scholar]
- 55.Duffy N, Peter L, Rajapakse RM., Wijayantha KG. 2000. A novel charge extraction method for the study of electron transport and interfacial transfer in dye sensitised nanocrystalline solar cells. Electrochem. Commun. 2, 658–662. ( 10.1016/S1388-2481(00)00097-7) [DOI] [Google Scholar]
- 56.Guldi DM. 2002. Fullerene–porphyrin architectures; photosynthetic antenna and reaction center models. Chem. Soc. Rev. 31, 22–36. ( 10.1039/b106962b) [DOI] [PubMed] [Google Scholar]
- 57.D'Souza F, et al. 2002. Spectroscopic, electrochemical, and photochemical studies of self-assembled via axial coordination zinc porphyrin−fulleropyrrolidine dyads †. J. Phys. Chem. A 106, 3243–3252. ( 10.1021/jp013165i) [DOI] [Google Scholar]
- 58.D'Souza F, Deviprasad GR, Rahman MS, Choi J. 1999. Self-assembled porphyrin−C 60 and porphycene−C 60 complexes via metal axial coordination. Inorg. Chem. 38, 2157–2160. ( 10.1021/ic981358n) [DOI] [PubMed] [Google Scholar]
- 59.Prato M, Maggini M, Giacometti C, Scorrano G, Sandona G, Farnia G. 1996. Synthesis and electrochemical properties of substituted fulleropyrrolidines. Tetrahedron 52, 5221–5234. ( 10.1016/0040-4020(96)00126-3) [DOI] [Google Scholar]
- 60.D'Souza F, Ito O. 2009. Supramolecular donor-acceptor hybrids of porphyrins/phthalocyanines with fullerenes/carbon nanotubes: electron transfer, sensing, switching, and catalytic applications. Chem. Commun., 2009 4913–4928. ( 10.1039/b905753f) [DOI] [PubMed] [Google Scholar]
- 61.Dong Y, Wei L, Fan R, Yang Y, Wang P. 2016. Influence of anchoring group numbers in an efficient pyridine-anchor co-adsorbent of pyridinecarboxaldimine substituted aminonaphthalene on the performance of N719 sensitized solar cells. RSC Adv. 6, 39 972–39 981. ( 10.1039/C6RA06017J) [DOI] [Google Scholar]
- 62.Martini LA, Moore GF, Milot RL, Cai LZ, Sheehan SW, Schmuttenmaer CA, Brudvig GW, Crabtree RH. 2013. Modular assembly of high-potential zinc porphyrin photosensitizers attached to TiO2 with a series of anchoring groups. J. Phys. Chem. C 117, 14 526–14 533. ( 10.1021/jp4053456) [DOI] [Google Scholar]
- 63.Gibson EA, Le Pleux L, Fortage J, Pellegrin Y, Blart E, Odobel F, Hagfeldt A, Boschloo G.. 2012. Role of the triiodide/iodide redox couple in dye regeneration in p-type dye-sensitized solar cells. Langmuir 28, 6485–6493. ( 10.1021/la300215q) [DOI] [PubMed] [Google Scholar]
- 64.Slavov C, Hartmann H, Wachtveitl J. 2015. Implementation and evaluation of data analysis strategies for time-resolved optical spectroscopy. Anal. Chem. 87, 2328–2336. ( 10.1021/ac504348h) [DOI] [PubMed] [Google Scholar]
- 65.Favereau L, et al. 2017. Engineering processes at the interface of p-semiconductor for enhancing the open circuit voltage in p-type dye-sensitized solar cells. Adv. Energy Mater. 7, 1601776 ( 10.1002/aenm.201601776) [DOI] [Google Scholar]
- 66.Zhang L, Boschloo G, Hammarström L, Tian H. 2016. Solid state p-type dye-sensitized solar cells: concept, experiment and mechanism. Phys. Chem. Chem. Phys. 18, 5080–5085. ( 10.1039/C5CP05247E) [DOI] [PubMed] [Google Scholar]
- 67.Pham TTT, et al. 2016. Toward efficient solid-state p-type dye-sensitized solar cells: the dye matters. J. Phys. Chem. C, 121, 129–139. ( 10.1021/acs.jpcc.6b10513) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data supporting the publication can be found at https://doi.org/10.25405/data.ncl.c.4539992.