

Abstract
Objectives
Focal cortical dysplasia (FCD) is a major cause of drug‐resistant focal epilepsy in children, and the clinicopathological classification remains a challenging issue in daily practice. With the recent progress in DNA methylation–based classification of human brain tumors we examined whether genomic DNA methylation and gene expression analysis can be used to also distinguish human FCD subtypes.
Methods
DNA methylomes and transcriptomes were generated from massive parallel sequencing in 15 surgical FCD specimens, matched with 5 epilepsy and 6 nonepilepsy controls.
Results
Differential hierarchical cluster analysis of DNA methylation distinguished major FCD subtypes (ie, Ia, IIa, and IIb) from patients with temporal lobe epilepsy patients and nonepileptic controls. Targeted panel sequencing identified a novel likely pathogenic variant in DEPDC5 in a patient with FCD type IIa. However, no enrichment of differential DNA methylation or gene expression was observed in mechanistic target of rapamycin (mTOR) pathway–related genes.
Significance
Our studies extend the evidence for disease‐specific methylation signatures toward focal epilepsies in favor of an integrated clinicopathologic and molecular classification system of FCD subtypes incorporating genomic methylation.
Keywords: diagnostic biomarker, epigenetic, focal cortical dysplasia, focal epilepsy, mTOR pathway
Key Points.
Clinicopathologic classification of focal cortical dysplasia (FCD) is challenging in everyday praxis
DNA methylation distinguished between human FCD types Ia, IIa, and IIb; temporal lobe epilepsy; and nonepileptic controls
DNA methylation signatures may constitute a clinically useful molecular biomarker for FCD subtypes
1. INTRODUCTION
Focal cortical dysplasia (FCD) is a frequent pathoanatomic substrate of severe drug‐resistant focal epilepsy particularly in children.1 FCDs present with variable and difficult‐to‐classify histopathology patterns, including architectural, cytoarchitectural, and white matter abnormalities.2, 3 The International League Against Epilepsy (ILAE) proposed a clinicopathologic consensus classification based on microscopic review of surgical specimens.4
The most frequent subtype is FCD II, which is localized mainly in the frontal lobe and often constrained to the depth of a sulcus.5 FCD type II was histopathologically characterized by megalocytic neurons (FCD type IIa) and “opalesque” balloon cells (FCD type IIb). Another FCD subtype has been described in young children with a multilobar onset in the posterior quadrant, daily seizures without focal neurologic deficits, intellectual disabilities, and drug‐resistance from onset.6 Their histopathologic hallmark is abundance of neuronal microcolumns across cortical layers 3‐5, that is, FCD Ia.7 Other FCD subtypes still lack comprehensive characterization of clinical phenotypes and often associate with a principal brain lesion, that is, FCD type IIIa in the temporolateral neocortex of patients with hippocampal sclerosis.
Continuous implementation of advanced diagnostic tools for presurgical evaluation has proven helpful in identifying FCD, for example, intracerebral electroencephalography (EEG) recording and high‐resolution magnetic resonance imaging (MRI).8 Nevertheless, detection of subtle FCD, diagnostic distinction of FCD subtypes, and large FCD patterns not compromising gross brain anatomy remain difficult in routine diagnostics. Prediction of postsurgical seizure freedom varies between FCD subtypes and is another issue demanding clarification.6, 9, 10 Thus, identification of pathogenetic signatures to develop useful diagnostic, predictive, or prognostic biomarkers and targeted therapies is an unmet clinical need.
The impact of integrating molecular genotypes with histopathologic phenotypes has recently been recognized by the World Health Organization (WHO) panel for the classification of brain tumors in 2016.11 Particularly, genomic DNA methylation classifiers have been identified as a valuable source in the decision making process for disease diagnosis, prognosis, and treatment.12 In this study, we examined whether genomic DNA methylation and gene expression could be also used to distinguish human FCD tissue.
2. METHODS
2.1. Patients included in the study
Twenty‐six subjects from the European Epilepsy Brain Bank with available fresh‐frozen human brain samples and adjacent formalin‐fixed paraffin‐embedded (FFPE) tissue blocks were included in the present study (Figure 1A).1, 13 All patients were recruited between 2002 and 2014 and underwent extensive presurgical evaluation including video‐EEG monitoring, neuropsychological examination, and MRI (1.5 or 3 Tesla; Siemens). One patient received computerized tomography (CT) imaging only. In selected cases, PET and SPECT analysis were applied. En bloc resections were carried out depending on the presurgical characterization of the seizure focus. Resected tissue samples were sliced and alternating sections were either formalin‐fixed paraffin‐embedded or immediately frozen (in liquid nitrogen or over isopentane) and stored at −80°C until further use.13 FFPE‐ and cryosections from all surgical specimens underwent systematic evaluation by two experienced neuropathologists (I.B. and R.C.) with regard to the presence of seizure‐associated lesions and even representation of gray and white matter.4, 14 Histopathologically, 15 patients displayed FCD type Ia (microcolumns in Figure 1B; red arrow heads; n = 4; meanAge ± standard error of mean (SEM): 5.3 ± 1.0), type IIa (dysmorphic neurons in Figure 1B; black arrows; n = 6; meanAge ± SEM: 5.5 ± 1.4), or type IIb (dysmorphic neurons and balloon cells; black arrows and asterisks in Figure 1B; n = 5; meanAge ± SEM: 10.6 ± 3.3). Five patients had temporal lobe epilepsy (TLE) associated with hippocampal sclerosis. In the present study, we used the resected temporal neocortex, which showed no histopathologic signatures of FCD (TLE; Figure 1B, upper right panel; meanAge ± SEM: 30.60 ± 7.12 years). This group was used to control for aberrant methylation patterns that could have been induced by chronic seizures.15, 16 Autopsy cases and surgical samples from nonepilepsy patients served as controls (CTRL; n = 6; meanAge ± SEM: 15.5 ± 2.0 years). Criteria for including these specimens as control tissue were age <25 years, for autopsies a postmortem range <24 hours, no prior history of seizures, and no signs of autolysis upon histopathologic brain examination. Patients with FCD type III (ie, FCD and an associated second lesion, eg, hippocampal sclerosis) were excluded from the study to avoid misinterpretation with regard to which lesion would be associated primarily with any molecular findings. Informed and written consent was obtained from all patients, their parents, or legal representatives if patients were underage. All studies were conducted in accordance with the Declaration of Helsinki and were approved by the local ethics committee of the Friedrich‐Alexander University Erlangen‐Nürnberg (FAU) Medical Faculty (Ref. No. 92_14 B).
Figure 1.
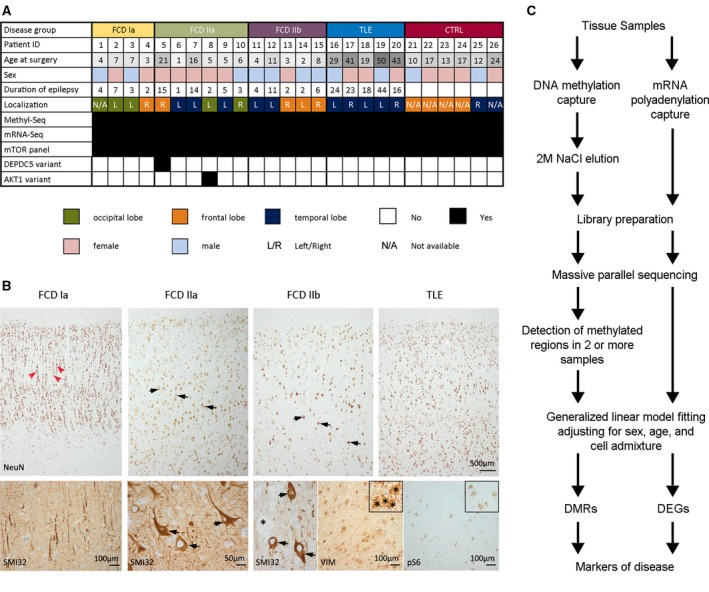
A, Clinical data summary. B, Histology overview shows NeuN immunohistochemical staining of representative neocortical specimens obtained from epilepsy patients with either focal cortical dysplasia (FCD) Ia, IIa, IIb, or temporal lobe epilepsy (TLE) (from left to right). Upon histopathologic examination, FCD subtype Ia was characterized by abundant microcolumnar organization (red arrow heads) and reduced cortical thickness. FCD type IIa and IIb showed no identifiable cortical layering except for layer 1. Hallmark of FCD II variants was the presence of dysmorphic neurons (black arrows), which presented with a significantly enlarged cell body and nucleus, malorientation, and cytoplasmic accumulation of neurofilament protein (SMI32). Histopathologic detection of Vimentin (VIM)–positive balloon cells further classified subtype IIb (2011 International League Against Epilepsy [ILAE] classification). FCD II cases showed varying degrees of pS6 immunoreactivity. In contrast to FCDs, temporal neocortex specimen obtained from patients with TLE and included in the present study showed no signs of cortical dyslamination or any cytomorphological changes similar to autopsy controls. C, Project design to investigate the association of FCD subtypes with genome‐wide DNA methylation and gene expression
2.2. DNA methylation profiling
For DNA methylation profiling, 500 ng of human DNA from each sample was fragmented to a median size of 200‐300 bp and subjected to methylated DNA capture according to the MethylMiner protocol (Invitrogen), enabling exclusive capture of methylated double‐stranded DNA. Fragmented and enriched DNA was eluted at high salt concentrations (2 mol/L NaCl). Ten nanograms of enriched DNA was used in library preparation using the NEB Next DNA Library Prep Reagent Set for Illumina (New England Biolabs). In parallel, 10 ng of sonicated DNA without enrichment also underwent library preparation to act as an input (background) control for each patient sample. Quality of sequencing libraries was assayed using the Shimadzu MultiNA capillary electrophoresis system (Shimadzu). Libraries were sequenced at a concentration of 13 pmol/L on the Illumina HiSeq2500 (Illumina) with a 100 bp single‐read length. Base calling was performed with RTA v1.18.61 software. Sequenced tags were aligned to the human reference genome hg19 using BWA‐ALN.17 Duplicate reads that aligned to the same location and strand in a given sample were removed using Samtools.18 Profiles of DNA methylation were compared between each sample and its respective input, using the MACS peak calling software.19 Identified peaks that occurred in at least two samples were used for further analysis. The numbers of read tags aligning to each region were extracted using featureCounts20 producing a matrix of counts (tags per region per sample). Genes with less than 100 sequence reads across all samples were removed from further analysis. All external data sources used for methylation analysis are summarized in Table S1.
2.3. Gene expression profiling
Ten milligrams of snap‐frozen human brain tissue was used for total RNA extraction using Trizol reagent (Thermo Fisher Scientific), followed by DNAse digestion. RNA quality was verified on the Shimadzu MultiNA capillary electrophoresis system (Shimadzu). Following Dynabead Oligo(dT) enrichment (Invitrogen), messenger RNA (mRNA) was prepared into sequence‐ready libraries with the NEBNext mRNA Library Prep Reagent Set for Illumina (New England Biolabs). Libraries were sequenced as described earlier with a 100 bp single‐read length. Sequence tags were aligned to the human reference genome hg19 and Ensembl transcript reference (Homo Sapiens 75) using STAR aligner.21 The numbers of read tags aligning to each region were extracted using featureCounts20 producing a matrix of counts (tags per gene per sample). Genes with fewer than 100 sequence reads across all samples were removed from further analysis.
2.4. Identification of confounding factors
To estimate sequence abundance, we used generalized linear modeling (GLM) with a negative binomial link function implemented in the edgeR package.22 Read abundance values were normalized using trimmed mean of M values (TMM) normalization.23 Disease groups were modeled along with potential confounding factors of sex, age, and cell admixture. With regard to cellular composition of the tissue, we focused on normalizing for neuronal cell content, as this has been shown to strongly affect differential results and be a large source of unwanted variance.24, 25 To decide if the modeling method would successfully account for sources of uninformative variation, the GLM methylation model was fitted on neuronal nuclear antigen (NeuN) promoter methylation, whereas in the RNA set the expression of this gene was used. The rank of the resulting beta values was tested for an association with gene sets derived from previous experiments using a Wilcox gene set test (X Genes, genes located on the X chromosome; Y Genes, genes located on the Y chromosome; Horvath (up/down), genes identified as having methylation that is correlated or anticorrelated with age (derived from Horvath et al26), and Neuron Marker, the top 500 neuron‐specific genes compared to astrocyte and endothelial cells (derived from Zhang et al27). Notably, all major markers of sex, age, and neuron specificity behaved as expected (Figure S1). The beta value for NeuN promoter methylation was correlated significantly with neuronal marker genes P <1.2e‐5, indicating that the NeuN methylation beta values captured a significant amount of the neuron‐specific signature. For differential methylation analysis, pairwise comparisons between all diagnosis groups were performed (ie, CTRL; FCD Ia, IIa, and IIb; TLE), generating P values and log2‐fold changes used for filtering and downstream analysis. Group sizes of all subtype pathologies and controls were equally matched ranging from four to six samples per group with a mean of five samples per group.
2.5. Cluster analysis and genomic distribution of differentially methylated regions
Clustering was performed by taking the library size normalized values for differential regions (P < 1e‐4), which were unique to any pairwise comparison and had absolute log2‐fold change (FC) for confounding covariates of <0.5. These values were scaled to the normal distribution before performing Manhattan distance‐based hierarchical clustering on both regions and samples using the heatmap.2 function in the R package gplots.
The circos plot was generated using circlize28 package. Methylated regions with P < 1e‐4 for comparisons of epilepsy groups vs controls are shown. An equal number (958) of top‐enriched regions are shown for mRNA‐seq. Proportions for pie charts were calculated by assigning differentially methylated regions (DMRs) to exactly one feature.
Differentially methylated regions were assigned to the first features in the following priority: promoters, enhancers, and gene bodies. Any remaining DMRs were assigned as intergenic. When calculating the significance of the overlap of DMRs with genomic features, the Fisher's exact test was used with a random sample of 1000 regions of the same genomic width as the original set.
2.6. Correlation of DNA methylation and mRNA expression
To explore the relationship between DNA methylation and mRNA expression when comparing disease groups, the signed rank of the −log10 (P value) of the change in the promoter DNA methylation (within 2 kbp from TSS) was plotted vs the signed rank of the −log10 (P value) of the change in RNA transcription. This was transformed into a density distribution using a two‐dimensional kernel. This density distribution was then colored as a heatmap for visualization.
2.7. Gene panel sequencing
Ten milligrams of snap‐frozen human brain tissue was used for genomic DNA extraction using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer's instructions. For panel sequencing, all exons and flanking intronic regions of target genes were captured using 200 ng of genomic DNA and the HaloPlex target enrichment system (Agilent Technologies), according to the manufacturer's protocol. Quality check was performed using the 2100 Bioanalyzer (Agilent Technologies). DNA libraries were sequenced on a Genome Analyzer IIx (GAIIx, Illumina) in 100 bp reads. Bioinformatics analysis was performed using BWA17 to map sequencing reads to the human reference genome UCSC GRCh37/hg19 assembly, Picard (V1.109, http://broadinstitute.github.io/picard/), GATK (V3.1, https://www.broadinstitute.org/gatk/) to call variants, and Annovar29 to perform variants annotation. All possible sequencing and alignment artifacts were excluded from further analysis. Upon their identification and validation through conventional Sanger sequencing, we classified variants in patients and controls according to the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines.30 Genes included in the panel are summarized in Table S2.
To validate DEPDC5 gene expression from a newly identified likely pathogenic DEPDC5 variant, we used custom‐designed primers mapping on DEP domain containing 5 (DEPDC5) exons 7 and 10. Following real‐time polymerase chain reaction (RT‐PCR) using fastStart Taq DNA Polymerase (Roche), RT‐PCR products were checked on 1% agarose gel (Experteam). DNA bands were excised, purified with the Nucleospin Extract II agarose gel extraction kit (Macherey‐Nagel), and sequenced afterward using BigDye Terminator V1.1 chemistry (Life Technologies). Primers and RT‐PCR conditions are available upon request.
2.8. Data access
Methyl‐Seq and mRNA‐Seq data were deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) and are available under GEO accession number GSE128301.
3. RESULTS
Sequencing was used to assess mRNA expression and DNA methylation changes in 26 samples (Figure 1A‐B). The experimental design using methyl‐CpG and mRNA capture followed by massive parallel sequencing to characterize DNA methylation (Methyl‐Seq) and gene expression (mRNA‐Seq) patterns is shown in Figure 1C.
3.1. Multidimensional scaling of DNA methylation profiles and confounding factors
DNA methylation was detected in 505 Mbp of the human genome, and we assessed differential methylation in FCD subtypes, non‐FCD epilepsy (ie, TLE), and nonepilepsy age‐matched controls. Analysis of DNA methylation in clinical tissue samples may be subject to multiple confounding factors that are technical (eg, batch effects) or biological (eg, age) in origin. We adjusted for known confounders in our analysis by assessing sex,31 age,32 and heterogeneous cell composition24, 33 using generalized linear modeling (Figure S1). DMRs associated with confounding variables (β > 0.5) were removed, and a total of 1333 DMRs were identified at a threshold of P <1e‐4 (Figure 2A, left panel). DMRs were categorized showing 675 regions with elevated (hyper‐) methylation and 658 regions with reduced (hypo) methylation. Multidimensional scaling (MDS) was used to identify patterns in DNA methylation by integrating phenotypic variables (age, sex, and cellular composition). This analysis was important because it allowed us to assess DNA methylation changes in sampled brain regions and tissue obtained from biopsy or autopsy. We observed no association for DMRs with anatomic lobe (Figure 2B), or sample origin (autopsy or biopsy; Figure 2C). However, we did observe DMR clustering by disease group (Figure 2D), which was not associated with copy number variation or genomic input (Figure 2E). Taken together, these results suggest that the DNA methylation differences identified between disease groups were unlikely to be a result of genetic background or phenotypic confounding variables.
Figure 2.
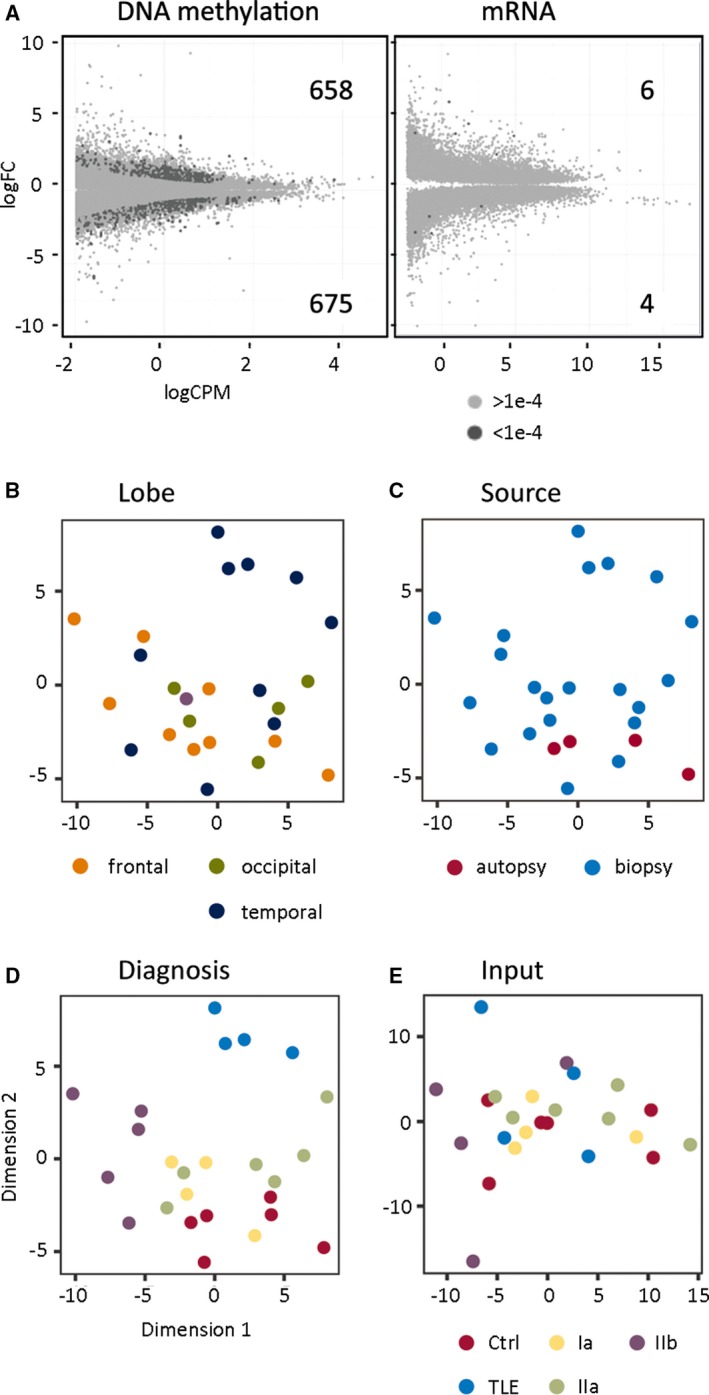
A, Genome‐wide sequencing data identified strong differences in DNA methylation but not messenger RNA (mRNA) expression in epilepsy subtypes compared to a nonepilepsy control group. Plots compare the sequence abundance (logCPM) and fold change (log2FC) for epilepsy subtypes (P < 1e‐4, dark gray; P > 1e‐4, light gray). The number of differentially methylated regions (DMRs) and differentially expressed genes (DEGs) is shown. (B‐E), Identified DMRs are associated with disease groups rather than covariates. A multidimensional scaling (MDS) plot was generated from DMRs (P < 1e‐4) with low confounding variable influence (β < 0.5). Samples were labeled with the covariates: (B) Lobe and (C) Sample source showed no association with the identified DMRs. D, Epilepsy subtypes and nonepilepsy control groups showed the strongest clustering. E, Input DNA (genetic background) did not drive clustering according to disease groups
Multidimensional scaling analysis of differential gene expression data from the same samples identified “lobe” as strong additional phenotypic variable (Figure S2). Adjusted mRNA comparisons of FCD and non‐FCD epilepsy groups with the nonepilepsy control group identified 10 differentially expressed genes (DEGs; six increased and four decreased) at a threshold of P < 1e‐4 (Figure 2A, right panel). When compared to methylation sequencing results, mRNA had a much higher variance, with a median excess dispersion of 0.159 (compared to 0.008 for Methyl‐Seq). The results imply that because of variance, gene expression was less reliable for FCD classification.
3.2. Differential hierarchical cluster analysis
To determine whether genomic DNA methylation can be used to identify FCD subtypes we applied differential hierarchical cluster analysis. Intergroup comparisons showed that FCD subtypes were distinguishable by DNA methylation highlighting separation of FCD groups from other epilepsy phenotypes (TLE), and no‐seizure controls (Figure 3A). To define methylation with genomic location, we assigned DMRs with promoter (±2 kb from TSS), gene body, or enhancer sequences of annotated genes. From the total of 1333 DMRs, 465 were successfully annotated with one of these features. Promoters represented only 12% of all the differential methylation, whereas gene bodies and intergenic regions accounted for the majority of the changes (29% and 30%, respectively). Among the top associations of differentially methylated genes (DMGs), calcium/calmodulin‐dependent protein kinase 1D (CAMKD1) was increased for methylation in FCD type Ia. In contrast, alpha‐internexin (INA) was decreased for methylation in FCD type IIa compared to non‐FCD epilepsy. Other DMGs appeared highly specific for FCD type IIb or TLE (Figure 3B). Our data support the concept of using DNA methylation signatures as diagnostic biomarkers to support classification of, for example, FCD subtypes in clinical praxis.
Figure 3.
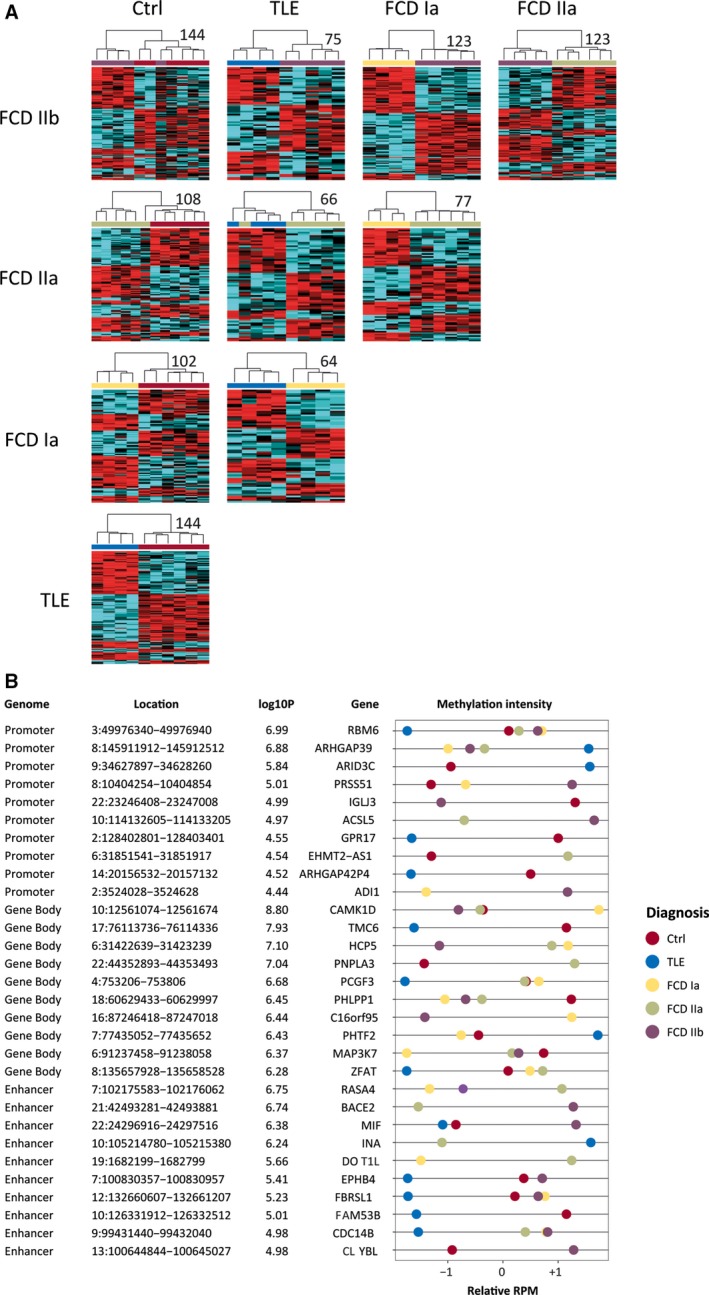
A, Differentially methylated regions (DMRs) distinguished focal cortical dysplasia (FCD) Ia, FCD IIa, FCD IIb, temporal lobe epilepsy (TLE), and nonepilepsy control from each other in differential cluster analyses. Heatmaps of DMRs (P < 1e‐4) with low confounding variable influence (β < 0.5) are shown. The numbers of DMRs for each comparison are displayed next to each heatmap. B, Examples of differentially methylated genes (DMGs), which were identified through DMRs that are located at a gene's promoter, gene body, or enhancer. Genome, annotated part of the genome; Location, position of DMG on the hg19 human reference genome; log10P, the highest log10 P value for the DMG; Gene, annotated gene name; methylation intensity, scaled sequence abundance, showing comparisons with cutoff of P < 1e‐4; RPM, reads per million
3.3. Genomic distribution of DMRs and correlation with gene expression
Genome‐wide visualization of all differential methylation signals demonstrated even distribution on all autosomes, with no evidence for clustering at particular chromosomes or gross chromosomal regions (eg, toward central autosome domains or ends; Figure 4A). DMRs were observed at genomic features such as gene promoters (±2 kb from TSS), exons, introns, enhancers, and CpG islands (CGIs) together with their shores (±1 kb from CGIs) and shelves (±1‐9 kb from CGIs; Figure 4B). Promoters represented only 12% of all the differential methylation, whereas gene bodies and intergenic regions accounted for the majority of the changes (29% and 30%, respectively). Differential methylation at CGIs, shores, and shelves accounted for <22% of total DMRs, with the majority observed in CpG shelves (Figure 4C).
Figure 4.
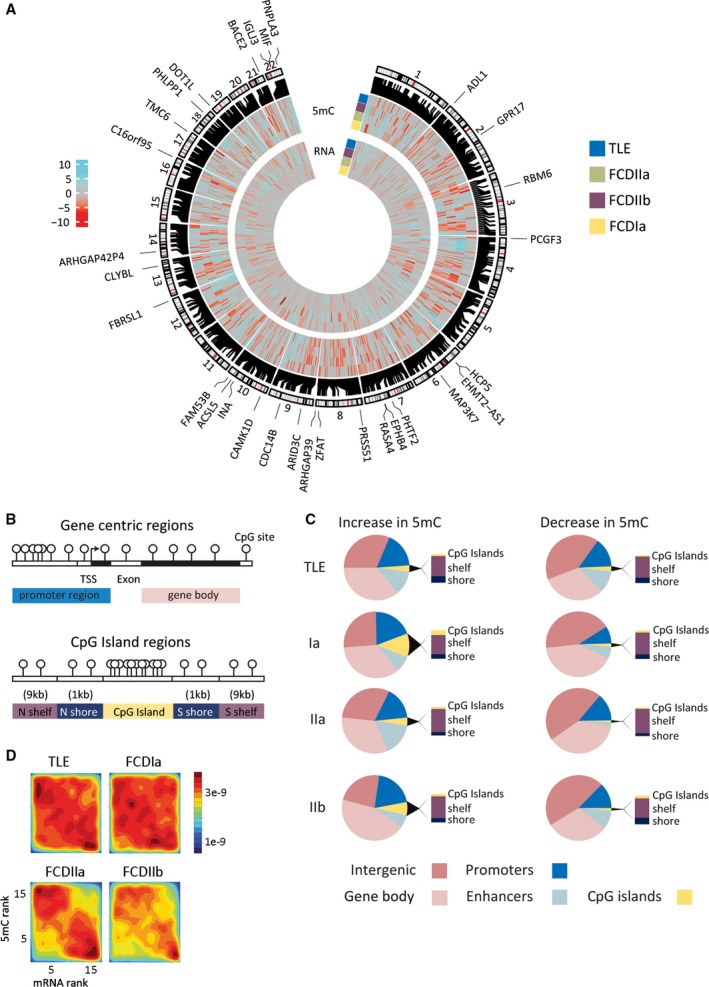
A, Whole genome sequencing detected strong changes in DNA methylation and messenger RNA (mRNA) expression that distinguished epilepsy subtypes. Differential regions are shown on the human genome autosomes. The outer subtype ring summarizes hypermethylation (cyan) and hypomethylation events (red) in the individual epilepsy groups, that is, focal cortical dysplasia (FCD) Ia, FCD IIa, FCD IIb, and temporal lobe epilepsy (TLE), compared to the nonepilepsy control group showing 958 unique loci with a cutoff of P < 1e‐4. The inner subtype ring summarizes mRNA changes in the epilepsy groups compared to the nonepilepsy control group. Genomic locations of differentially methylated regions (DMRs) and differentially methylated genes (DMG)s are shown. Heatmap scale shows differential methylation and expression changes between epilepsy and nonepilepsy groups expressed as the –log10 P value. The outer circle of genes is the top 30 DMG. B, Graphical representation of genomic features and CpG Islands, shelves, and shores. Regions are: promoter (±2 kb from TSS), gene body, CpG islands (CGIs), and CpG island shores (±1 kb from CGI) and shelves (±1‐9 kb from CGI). C, The majority of subtype DMRs were colocated with functional genomic elements. Distribution of identified DMRs clustered across genomic features including gene promoter region, gene bodies, CGIs and CGI shores and shelves. D, Rank‐rank density plot showing significant inverse correlation of DNA promoter methylation and gene expression in all subtype pathologies. Hypermethylation was associated with reduced gene expression (upper left corner), whereas hypomethylation was primarily found in upregulated genes (lower right corner)
Because DNA methylation may functionally regulate gene expression, we assessed whether pathology‐specific DMRs from Methyl‐Seq were associated with gene expression changes from mRNA‐Seq. Comparing ranked differentials we observed an inverse correlation for promoter methylation with gene expression in all subtype pathologies, that is, hypermethylation was associated with reduced gene expression, whereas hypomethylation was found primarily in upregulated genes (Spearman correlation, p TLE = 2.3e‐15, p FCD_Ia = 3.2e‐05, p FCD_IIa = 1.1e‐77, p FCD_IIb = 9.6e‐38; Figure 4D).
3.4. Mechanistic target of rapamycin pathway analysis
We next specifically explored the possibility of molecular alterations in the mechanistic target of rapamycin (mTOR) pathway, because mutations in genes associated with mTOR were described recently with the pathogenesis of FCD II subtypes.34, 35, 36 To understand the possible genetic burden in our cohort, we performed panel sequencing of 54 mTOR pathway–related genes. We obtained a mean coverage ranging from 76× to 1539x. Tissue DNA from a patient histopathologically classified as FCD IIa carried a novel, likely germline, and likely pathogenic variant affecting the donor splice site of DEPDC5 exon 8 (DEP Domain Containing 5; NM_001242896.1): c.483 + 1G>A, nine reference reads and 20 alternative reads (heterozygous at Sanger sequencing; Figure 1A). In silico analysis for the mutation predicted a frame shift with the insertion of a premature stop codon. If translated, the truncated protein is predicted to contain 153 instead of 1603 amino acids (Figure S3). To demonstrate that the identified variant affected mRNA splicing, we amplified cDNA synthetized from mRNA extracted from the dysplastic brain tissue and blood of that patient using primers mapping on DEPDC5 exons 7 and 10. Agarose gel electrophoresis revealed two RT‐PCR products in both tissues, and Sanger sequencing confirmed that exon 8 was deleted (Figure S3). Due to our retrospective study design, we had no access to parents’ blood to verify whether this constitutive mutation occurred de novo. Another patient, also classified with FCD IIa, carried a missense variant of uncertain significance in AKT1 exon 11 (AKT serine/threonine kinase 1; NM_005163): c.1099C>T; p.Arg367Cys, 576 reference reads and 475 alternative reads (heterozygous at Sanger sequencing), frequency in gnomAD database =0.00001625, one occurrence in the Catalogue of Somatic Mutations in Cancer = COSM1235769 in brain tissue (Figure 1A). No blood sample of that patient was available for further genetic studies. In the control group, we identified variants in the following genes: MTOR (NM_004958.3): c.3113T>C; p.Met1038Thr, 1076 reference reads, 717 alternative reads (heterozygous at Sanger sequencing), not reported in literature or in public databases; TSC2 (tuberous sclerosis complex 2; NM_000548.3): c.4524_4526delCTT; p.1508_1509del, 2043 reference reads, 1656 alternative reads (heterozygous at Sanger sequencing), frequency in gnomAD databases = 0.005092; DEPDC5 (NM_001242896.1): c.4510 C>T; p.His1504Tyr, 3308 reference reads, 1414 alternative reads (heterozygous at Sanger sequencing), not reported in literature or in public databases. These variants were considered of uncertain significance as they were either absent or present with low frequency (<1%) in public databases and have not been associated previously with human disease. Furthermore, in silico pathogenicity prediction results were inconclusive, segregation studies were not possible in the families, and none of the control cases exhibited seizures at any point before death/sampling. Activation of mTOR signaling has been postulated in FCD II,37 but analysis of mRNA profiling data for our panel of mTOR pathway–related genes did not identify changes in gene expression. However, we found seven genes (FOXO6, GNAQ, PHLPP1, PDPK1, TSC2, DEPDC5, and CNTNAP2) to be differentially methylated either in their promoter region or gene body (Table 1, Figure S4). Notably, PHLPP1 gene body and FOXO6 promoter hypermethylation distinguished FCDs from TLE and nonepilepsy controls, but we did not observe differential methylation of mTOR pathway–related genes in FCD subtypes. Taken together, our data provided no evidence that genes implicated in the mTOR pathway could be used as molecular classifiers to distinguish FCD subtypes.
Table 1.
Top 10 differentially promoter or gene body methylated mTOR pathway–related genes
| DataSet | GeneID | logFC | P value | Adj. P value | Contrast | GeneSymbol |
|---|---|---|---|---|---|---|
| Promoter | ENSG00000204060 | 1.75 | 1.61E‐05 | 0.05176 | Ctrl_vs_FCDIIa | FOXO6 |
| Promoter | ENSG00000156052 | 1.13 | 4.17E‐05 | 0.03882 | Ctrl_vs_FCDIIb | GNAQ |
| Promoter | ENSG00000204060 | 1.82 | 9.97E‐05 | 0.04797 | Ctrl_vs_FCDIIb | FOXO6 |
| Promoter | ENSG00000132024 | −0.26 | 0.00014 | 0.07459 | Ctrl_vs_FCDIa | CC2D1A |
| Promoter | ENSG00000204060 | 1.54 | 0.00033 | 0.10911 | Ctrl_vs_FCDIa | FOXO6 |
| Promoter | ENSG00000118689 | 0.72 | 0.00069 | 0.13200 | Ctrl_vs_FCDIa | FOXO3 |
| Promoter | ENSG00000167965 | −0.32 | 0.00140 | 0.52418 | FCDIa_vs_FCDIIb | MLST8 |
| Promoter | ENSG00000164327 | 0.37 | 0.00211 | 0.14265 | Ctrl_vs_FCDIIb | RICTOR |
| Promoter | ENSG00000040199 | −0.24 | 0.00310 | 0.16109 | Ctrl_vs_FCDIIb | PHLPP2 |
| Promoter | ENSG00000100150 | 0.28 | 0.00480 | 0.58119 | FCDIIb_vs_TLE | DEPDC5 |
| Genomic region | ||||||
| GeneBody | 18:60629433‐60629997 | 1.16 | 3.54E‐07 | 0.06283 | Ctrl_vs_FCDIa | PHLPP1 |
| GeneBody | 18:60629433‐60629997 | 0.97 | 2.62E‐05 | 0.21187 | Ctrl_vs_FCDIIb | PHLPP1 |
| GeneBody | 16:2629631‐2630231 | −1.11 | 2.70E‐05 | 0.26838 | Ctrl_vs_TLE | PDPK1 |
| GeneBody | 16:2110009‐2110609 | 0.63 | 2.81E‐05 | 0.23720 | FCDIa_vs_FCDIIb | TSC2 |
| GeneBody | 22:32220413‐32221013 | 1.32 | 5.38E‐05 | 0.37167 | FCDIIb_vs_TLE | DEPDC5 |
| GeneBody | 18:60629433‐60629997 | 0.82 | 6.32E‐05 | 0.41850 | Ctrl_vs_FCDIIa | PHLPP1 |
| GeneBody | 22:32220413‐32221013 | 1.12 | 7.01E‐05 | 0.55320 | FCDIIa_vs_TLE | DEPDC5 |
| GeneBody | 7:146547868‐146548468 | −1.11 | 8.22E‐05 | 0.55320 | FCDIIa_vs_TLE | CNTNAP2 |
| GeneBody | 17:78698196‐78698796 | −0.74 | 0.00017 | 0.40836 | Ctrl_vs_TLE | RPTOR |
| GeneBody | 22:32220413‐32221013 | −0.90 | 0.00017 | 0.34157 | Ctrl_vs_FCDIIb | DEPDC5 |
Table summarizes differential methylation of mTOR pathway–related genes. Differentials meeting our significance criteria (P < 1e‐4) were marked in bold.
Abbreviations: Ctrl, control; FCD, focal cortical dysplasia; TLE, temporal lobe epilepsy; logFC, log2‐fold change; CNTNAP2, contactin‐associated protein‐like 2; DEPDC5, DEP domain containing 5; FOXO6, forkhead box O6; GNAQ, G protein subunit alpha Q; PHLPP1, ph domain and leucine rich repeat protein phosphatase 1; PDPK1, 3‐phosphoinositide dependent protein kinase 1; TSC2, tuberous sclerosis complex 2.
4. DISCUSSION
Hierarchical cluster analysis identified disease‐specifying DNA methylation signatures in three common FCD subtypes, that is, FCD Ia, IIa, and IIb. This study is first of its kind showing that human FCD subtypes can be distinguished based on DNA methylation. Methylation sequencing in FCDs showed intergenic regions, gene bodies, and enhancers to be subject to differential methylation, which seemed to be distinct from promoter‐centered DNA methylation changes described in human brain tumors.12 These findings have implications for the understanding and scientific approach to FCD classification and may pave the way toward an integrated clinicopathologic and molecular diagnosis of focal epilepsies.
FCD IIa and IIb are the most common malformations of cortical development in children with drug‐resistant focal epilepsy.1 Despite the growing body of studies addressing FCD, with more than 740 papers listed in PubMed since the release of the first international consensus classification in 2011, consistent information about their origin or timing of the lesion during brain development is still lacking.38, 39 Somatic mutations in genes belonging to the mTOR pathway have been shown to occur in the range of 15.6% to 46% of patients with FCD according to different studies.34, 36, 38, 40, 41, 42, 43 The genetic variants represent somatic mosaicisms, affecting predominantly MTOR or DEPDC5, with allele fractions of 1%‐12.6 %. We identified variants in mTOR pathway genes in 2 of 15 patients with FCD (14.2%) including a novel likely pathogenic germline splicing variant in DEPDC5 and a single nucleotide variant of uncertain significance in AKT1. The sequencing coverage of 76× to 1539× may have been too low to identify somatic mutations with low mutant allele fraction in the other FCD samples. Of interest, our DNA methylation and RNA sequencing analysis did not reveal the mTOR pathway as a principal target. The large gap of noninformative genetic data in approximately 60%‐80% of patients with FCD II, in particular those with FCD IIb, calls for extended molecular‐genetic investigations integrating single‐cell genome‐wide DNA38 and RNA sequencing44 or alternative sequencing strategies45, 46 to potentially identify the pathogenic cause of FCD II. Future progress in precision medicine will build on such analysis to develop a targeted drug treatment, in particular when epilepsy surgery is not an option for a given patient. With increasingly available pharmacological compounds regulating the epigenomic machinery,47 our findings will add to the knowledge pool for searching these new drug targets.
Clinicopathologic classification of non–FCD‐II subtypes remains a matter of debate.48 Lesions in this category are histopathologically described by dyslamination and disrupted organization of cortical architecture, but with normal neurons and glial cells.4 MRI often shows no changes in signal intensity. Hence, these epilepsies are often regarded as “nonlesional.” An inherent example is that of FCD type Ia, histopathologically defined by excessive microcolumns that are visible in some amount also in normal brain.49 Despite the severe clinical phenotype in children with neurodevelopmental delay, daily seizures and drug‐resistance from onset, an MRI specific signature and biomarker of FCD Ia, has not been established.4 In addition, published clinicopathologic series of this subtype are limited.48 Our methylation data, although highly limited with regard to sample numbers, suggests FCD Ia to be a distinct molecular entity, which may be highly relevant with regard to the development of an integrated clinicopathologic and genetic classification of FCD.
Our protocol distinguished the most common FCD I and II subtypes, that is, FCD Ia, IIa, and IIb, from each other, but also from epileptogenic neocortex without architectural abnormalities obtained from patients with chronic temporal lobe seizures as well as from nonepileptic neocortex obtained from postmortem and biopsy controls. This is an additional important finding of our study and in line with previously published data showing that methylation patterns in various epilepsy animal models associate with seizure phenotype and etiology.15, 16 However, the functional role of aberrant DNA methylation in the development of FCD will require further investigation.
Tissue heterogeneity is an important confounding factor in this type of study. Different ratios of cell types may contribute to variable molecular signatures of complex brain tissue samples.25 In this study, we used state of the art statistical methods to correct for cellular heterogeneity–based DNA methylation. Wilcox gene set testing and multidimensional scaling analysis showed that neuronal methylation was not the primary driver of FCD subtype clustering. Future studies will need to dissect the methylation profiles of all major cell types contributing to the epileptic network in FCD.
In conclusion, a comprehensive approach integrating genotype‐phenotype analysis will be key to better understand FCD pathogenesis and classify clinically meaningful FCD subtypes. Despite the tremendous advances in genetic mapping used by many groups and consortia, it seems that genetic factors alone cannot fully explain FCD susceptibility. Our data for the first time suggest that DNA methylation signatures may help to classify FCD subtypes. Further studies are needed to validate the present findings and further address the large spectrum of histopathologically described FCD variants and hitherto unclassifiable specimens clinically suspected as FCD (which was beyond the purpose of this study). Characterizing the methylome in epilepsy tissue will also add to the growing demand for targeted pharmacologic treatment, which is needed urgently for approximately 17 million people with therapy‐resistant epilepsy worldwide.50 The results of this study strengthen the evidence base against DNA methylation merely being an epiphenomenon of FCD development.
DISCLOSURE
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
We kindly thank B. Rings for her expert technical assistance. Our work was supported by the European Union's Seventh Framework Program (FP7 DESIRE project, grant agreement #602531) and the NHMRC – European Union Research Support grant APP1075563. A.E. is supported by The Netherlands Organization for Health Research and Development (ZonMw; Translationeel Onderzoek Project 95105004). We thank and acknowledge the Australian Genome Research Facility (AGRF) for high throughput sequencing, and the support it receives from the Commonwealth. Requests for methodologies for epigenetic profiling, sequencing, and computational epigenomics should be addressed to sam.el-osta@monash.edu or antony.kaspi@monash.edu.
Kobow K, Ziemann M, Kaipananickal H, et al. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia. 2019;60:1091–1103. 10.1111/epi.14934
REFERENCES
- 1. Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377:1648–56. [DOI] [PubMed] [Google Scholar]
- 2. Tassi L, Colombo N, Garbelli R, Francione S, Lo Russo G, Mai R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain. 2002;125:1719–32. [DOI] [PubMed] [Google Scholar]
- 3. Palmini A, Najm I, Avanzini G, Babb T, Guerrini R, Foldvary‐Schaefer N, et al. Terminology and classification of the cortical dysplasias. Neurology. 2004;62:S2–8. [DOI] [PubMed] [Google Scholar]
- 4. Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinico‐pathological spectrum of Focal Cortical Dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harvey AS, Mandelstam SA, Maixner WJ, Leventer RJ, Semmelroch M, MacGregor D, et al. The surgically remediable syndrome of epilepsy associated with bottom‐of‐sulcus dysplasia. Neurology. 2015;84:2021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krsek P, Pieper T, Karlmeier A, Hildebrandt M, Kolodziejczyk D, Winkler P, et al. Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia. 2009;50:125–37. [DOI] [PubMed] [Google Scholar]
- 7. Blümcke I, Pieper T, Pauli E, Hildebrandt M, Kudernatsch M, Winkler P, et al. A distinct variant of focal cortical dysplasia type I characterised by magnetic resonance imaging and neuropathological examination in children with severe epilepsies. Epileptic Disord. 2010;12:172–80. [DOI] [PubMed] [Google Scholar]
- 8. Najm IM, Tassi L, Sarnat HB, Holthausen H, Russo GL. Epilepsies associated with focal cortical dysplasias (FCDs). Acta Neuropathol. 2014;128:5–19. [DOI] [PubMed] [Google Scholar]
- 9. Krsek P, Maton B, Korman B, Pacheco‐Jacome E, Jayakar P, Dunoyer C, et al. Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol. 2008;63:758–69. [DOI] [PubMed] [Google Scholar]
- 10. Lerner JT, Salamon N, Hauptman JS, Velasco TR, Hemb M, Wu JY, et al. Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia. 2009;50:1310–35. [DOI] [PubMed] [Google Scholar]
- 11. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella‐Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 12. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blümcke I, Aronica E, Miyata H, Sarnat HB, Thom M, Roessler K, et al. International recommendation for a comprehensive neuropathologic workup of epilepsy surgery brain tissue: a consensus Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2016;57:348–58. [DOI] [PubMed] [Google Scholar]
- 14. Blümcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013;54(7):1315–29. [DOI] [PubMed] [Google Scholar]
- 15. Dębski KJ, Pitkanen A, Puhakka N, Bot AM, Khurana I, Harikrishnan KN, et al. Etiology matters ‐ genomic dna methylation patterns in three rat models of acquired epilepsy. Sci Rep. 2016;6:25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013;126(5):741–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al.; 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 21. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montaño CM, Irizarry RA, Kaufmann WE, Talbot K, Gur RE, Feinberg AP, et al. Measuring cell‐type specific differential methylation in human brain tissue. Genome Biol. 2013;14:R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115–R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosc. 2014;34:11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2. [DOI] [PubMed] [Google Scholar]
- 29. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J, Morgan M, Hutchison K, Calhoun VD. A Study of the influence of sex on genome wide methylation. PLoS One. 2010;5:e10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol. 2015;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Titus AJ, Gallimore RM, Salas LA, Christensen BC. Cell‐type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet. 2017;26:R216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–86. [DOI] [PubMed] [Google Scholar]
- 35. Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marsan E, Baulac S. Review: mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol. 2018;44:6–17. [DOI] [PubMed] [Google Scholar]
- 38. D'Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017;21:3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blumcke I, Sarnat HB. Somatic mutations rather than viral infection classify focal cortical dysplasia type II as mTORopathy. Curr Opin Neurol. 2016;29:388–95. [DOI] [PubMed] [Google Scholar]
- 40. Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 41. Mirzaa GM, Campbell CD, Solovieff N, Goold C, Jansen LA, Menon S, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 2016;73:836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Møller RS, Weckhuysen S, Chipaux M, Marsan E, Taly V, Bebin EM, et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet. 2016;2:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–7. [DOI] [PubMed] [Google Scholar]
- 44. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rand AC, Jain M, Eizenga JM, Musselman‐Brown A, Olsen HE, Akeson M, et al. Mapping DNA methylation with high‐throughput nanopore sequencing. Nat Methods. 2017;14:411–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. 2017;14:407–10. [DOI] [PubMed] [Google Scholar]
- 47. Hahnen E, Hauke J, Tränkle C, Eyüpoglu IY, Wirth B, Blümcke I. Histone deacetylase inhibitors: possible implications for neurodegenerative disorders. Expert Opin Investig Drugs. 2008;17:169–84. [DOI] [PubMed] [Google Scholar]
- 48. Najm IM, Sarnat HB, Blümcke I. Review: the international consensus classification of Focal Cortical Dysplasia – a critical update 2018. Neuropathol Appl Neurobiol. 2018;44:18–31. [DOI] [PubMed] [Google Scholar]
- 49. Hildebrandt M, Pieper T, Winkler P, Kolodziejczyk D, Holthausen H, Blümcke I. Neuropathological spectrum of cortical dysplasia in children with severe focal epilepsies. Acta Neuropathol. 2005;110:1091–11. [DOI] [PubMed] [Google Scholar]
- 50. WHO . Epilepsy Factsheet No. 999. 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials