

Abstract
Emerging evidence has revealed that noradrenaline (NA), the main neurotransmitter of the sympathetic nervous system (SNS), regulates a variety of immune functions via binding to adrenergic receptors present on immune cells. In this study, we examined the role of NA in the regulation of neutrophil functions. Neutrophils were isolated from the bone marrow of naïve mice and treated with NA at various concentrations to assess the effect on various neutrophil functions. Additionally, we performed cremaster intravital microscopy to examine neutrophil‐endothelial cell interactions following NA superfusion in vivo. In a separate group of animals, mice were subjected to an experimental model of stroke and at 4 and 24 h neutrophils were isolated for assessment on their ability to migrate toward various chemokines. Treatment of neutrophils with NA for 4 h significantly impaired neutrophil chemotaxis and induced an N2 neutrophil phenotype with reduced expression of the genes critical for cytoskeleton remodeling and inflammation. Prolonged NA administration promoted neutrophils to release myeloperoxidase and IL‐6, but suppressed the production of interferon‐γ and IL‐10, reduced neutrophil activation and phagocytosis. Superfusion of NA over the cremaster muscle almost completely inhibited fMLP‐induced neutrophil adhesion/arrest and transmigration. Furthermore, using a mouse model of stroke, a pathological condition in which SNS activation is evident, neutrophils isolated from poststroke mice showed markedly reduced chemotaxis toward all of the chemokines tested. The findings from our study indicate that neutrophil chemotaxis, activation, and phagocytosis can all be negatively regulated in an NA‐dependent manner. A better understanding of the relationship between sympathetic activation and neutrophil function will be important for the development of effective antibacterial interventions.
Keywords: neutrophil, noradrenaline, sympathetic nervous system
Abbreviations
- AR
adrenergic receptor
- C5a
complement component 5a
- DHR
dihydrorhodamine
- FOV
field of view
- KC
chemokine (C‐X‐C motif) ligand 1
- MCA
midcerebral artery
- MCAO
middle cerebral artery occlusion
- MFI
median fluorescence intensity
- MPO
myeloperoxidase
- NA
noradrenaline
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- SNS
sympathetic nervous system
- TTC
2,3,5‐triphenyltetrazolium chloride
1. INTRODUCTION
The nervous system and the immune system are two integrative systems that work collaboratively to detect threats, provide host defense, restore homeostasis, and their crosstalk is crucial for maintaining the health and well‐being of the host. One of the major neural pathways responsible for regulating host defense against injury and foreign pathogens is the sympathetic nervous system (SNS). Research in the past decade revealed that noradrenaline (NA), the main neurotransmitter of the SNS, regulates a variety of immune functions via adrenergic receptors (ARs) present on immune cells. In particular, NA is able to regulate immune cell development, survival, and proliferative capacity,1 as well as their level of gene expression for cytokines and antibodies.2, 3, 4 Additionally, sympathetic neural outflow can influence leukocyte trafficking for immune surveillance and recruitment and directs the cell surface expression of molecules important for the cell‐to‐cell interactions that are necessary for a coordinated immune response.5 Historically, NA was believed to have immunosuppressive effects on the immune system.6 In fact, chronic stress studies demonstrated that activation of the SNS impairs immune cell function, reduces the size of a number of primary immune organs, and inhibits leukocyte functions or shift them toward an anti‐inflammatory, wound‐healing phenotype.7, 8, 9
Earlier studies focused mainly on the effect of SNS activation on the function of the adaptive immune system, primarily due to the presence and anatomical distribution of sympathetic fibers in the spleen, various lymph nodes, and bone marrow.10, 11, 12 In a mouse model of stroke, we have previously shown that targeted intervention to inhibit SNS signaling significantly reduced the incidence of poststroke infections.13, 14 Clinically, blockade of adrenergic signals via the use of propranolol was found to be efficacious as an intervention for poststroke infection.55 However, the effect of SNS activation on innate immune regulation, particularly neutrophil function, and the mechanisms underlying any changes are currently unknown. Neutrophils are an essential part of the innate immune system. Specifically, circulating neutrophils in the presence of chemotactic stimuli promptly adhere to the endothelial cells and transmigrate in a directed fashion into the extravascular space.15 Moreover, localized remodeling of the cytoskeleton that ultimately leads to directed migration enables neutrophils to arrive at the site of chemoattractant release/production to undergo phagocytosis, generate a respiratory burst, and secrete a multitude of antimicrobial enzymes and cytokines.16 Being highly motile, neutrophils are one of the first responders to be rapidly recruited to sites of infection where their release of antimicrobial factors and soluble mediators can in turn amplify the inflammatory response.17 Therefore, neutrophils are not only required for the initial response to pathogens following an infection, but are indispensable for coordinating further recruitment of other leukocytes to establish an appropriate immune response.18
There is accumulating evidence to indicate that neutrophils are able to take on different phenotypes and functions during various diseases.19, 20 In fact, it is becoming clear that simply removing neutrophils or halting their recruitment following inflammation has the potential to impact on inflammatory responses in a more complex fashion than originally believed. Concepts such as “neutrophil heterogeneity” and “neutrophil plasticity” have begun to emerge, with evidence indicating that under pathological conditions, neutrophils may differentiate into discrete subsets defined by distinct phenotypic and functional characteristics, namely of “N1” proinflammatory or “N2” anti‐inflammatory nature.21 Given the abundance of neutrophils within the human body and their critical role in fighting infection, it is likely that functionally impaired neutrophils contribute toward an increased susceptibility to bacterial infection. However, whether the activation of SNS results in neutrophil functional impairment is currently unknown. In this study, we aimed to examine the role of NA in the regulation of neutrophil function, including their ability to migrate toward chemotactic stimuli in both in vitro and in vivo settings.
2. MATERIALS AND METHODS
2.1. Animals
Six‐to‐twelve‐week‐old male C57BL/6J wild‐type mice were obtained from the Monash Animal Research Platform and housed in a specific pathogen‐free facility (Monash Medical Centre Animal Facility, Clayton, VIC, Australia). Following transportation, mice were acclimatized for a minimum period of 7 days before use. LysM‐eGFP mice on a C57BL/6J background were bred and kept under specific pathogen‐free conditions at the Monash Animal Research Platform. Mice were housed in a 12‐h light‐dark cycle in a temperature‐controlled environment with free access to food and water. All procedures were approved by the Monash University Animal Ethics Committee under regulations which comply with the National Institute of Health guidelines for the care and use of laboratory animals.
2.2. Isolation of bone marrow neutrophils
Neutrophils were isolated from the bone marrow of wild‐type mice using the Ficoll method. Briefly, mice were culled via cervical dislocation, and the femurs and tibia of both hind legs removed. The distal end of each bone was cut using sterile scissors, and bone marrow cells were dislodged by 10 s centrifugation at 1000 g room temperature. Bone marrow cells were suspended in chemotaxis assay buffer (47.5% Hank's balanced salt solution, 47.5% RPMI, 1% penicillin streptomycin, 2% l‐glutamine, and 2% fetal calf serum) and filtered through a 70 μm mesh filter. Cells were resuspended at 5 × 106 cells per 6 ml and overlayed on 3 ml of Ficoll‐Paque Plus solution (GE Healthcare, Chicago, Illinois), and centrifuged for 20 min with no brakes at 600 g, at room temperature. Cell pellets were washed with PBS, and red blood cell were lysed for 1 min using 1 ml of Ammonium‐Chloride‐Potassium (ACK) lysis buffer. Cells were then resuspended in chemotaxis assay buffer or media and filtered again using a 70 μm filter. This method of bone marrow neutrophil (BMN) isolation enrichment yielded a Ly6G+ purity of 63.3 ± 1.2%.
2.3. Noradrenaline treatment
The catecholamine was prepared by the dissolution of crystalline NA (Sigma, St. Louis, Missouri) in PBS to produce a final concentration of 100 nM (10−7 M), 1 μM (10−6 M), or 10 μM (10−5 M). In this study, neutrophils were incubated with NA for a total of 4 or 24 h.
2.4. MTT assay for neutrophil metabolic activity
Neutrophils suspended in chemotaxis assay buffer were seeded into a 96‐well flat‐bottom plate at 1 × 105 cells per well and treated with increasing concentrations of NA for 4 and 24 h. For the 4 h treatment, 0.5 mg/ml MTT (Sigma) was added to each well 1 h prior to NA treatment for a total of 5 h. For the 24 h treatment, MTT was added into the wells and incubated with the cells during the last 5 h of NA treatment. Following incubation, the media was removed and replaced with 100 μl of DMSO and left in the dark at room temperature, until the crystals were dissolved. After this time, a Sunrise standard 96‐well microplate reader (Tecan, Männedorf, Switzerland) was used to obtain absorbance readings for each well. The reference absorbance, measured at 690 nm, was subtracted from the absorbance value, which was read at 570 nm. Magellan data analysis software (Tecan) was then used to compute a final absorbance reading.
2.5. Neutrophil chemotaxis assays
Using the Ficoll method, neutrophils were isolated from the bone marrow of wild‐type mice. Neutrophils were treated with various concentrations of NA and incubated in a 37°C, 5% CO2‐supplemented incubator for 4 h before being washed and resuspended in chemotaxis assay buffer at 2.5 × 106 cells/ml. Cells were then plated into the upper chamber of a Transwell membrane plate (Corning) and the chemokine fMLP (Sigma), KC (CXCL1; BioLegend), or complement component 5a (C5a; R&D Systems, Minneapolis, Minnesota) were loaded into the bottom chamber of the plate at increasing concentrations. The plate was then incubated in a 37°C, 5% CO2‐supplemented incubator for 3 h. Following incubation, the number of migrated Ly6G+ neutrophils (∼90% of the entire population) was determined by analysis using a Navios flow cytometer (Beckman‐Coulter, Brea, California) and FlowJo analysis software (version 10).
2.6. RNA extraction
RNA was extracted from BMNs using TRI reagent (Sigma) as per the manufacturer's instructions. Isolated neutrophils, untreated or treated with NA for either 4 or 24 h, were collected, centrifuged, and the supernatant aspirated and stored for quantification of cytokines and myeloperoxidase (MPO). Cell pellets were then lysed for 5 min at room temperature using TRI reagent. 1‐Bromo‐3‐chloropropane (Sigma) was then added to each sample and shaken vigorously before being allowed to stand for 5 min. The mixture was then centrifuged at 500 g at 4°C for 15 min, and the aqueous phase was collected. 2‐Propanol (Sigma) was then added to each sample and allowed to stand for 10 min before being centrifuged for 10 min at 500 g at 4°C. The supernatant was then removed, and the sample was washed with 75% (v/v) ethanol. The supernatant was again removed, the pellet was dried for 10–15 min in a laminar flow hood, and the pellet was resuspended in RNase‐free water.
2.7. Reverse transcription
cDNA was synthesized using the Superscript III first strand synthesis kit (InVitrogen, Carlsbad, California) and RQ1 DNase (Promega, Madison, Wisconsin) to ensure no genomic DNA contamination. Approximately 500 ng of RNA was incubated at 37°C for 60 min with 10× Superscript III RT buffer, magnesium chloride, and RQ1 RNase‐free DNase. DNase STOP solution (Promega) was then added to each sample and incubated at 65°C for a further 10 min. Subsequently, random hexamers and 10 mM dNTP mix were added and incubated at 65°C for 5 min and chilled on ice. A cDNA synthesis mix was then prepared using Superscript III RT buffer, 25 mM magnesium chloride, 0.1 M DTT, RNaseOUT, and Superscript III RT. After addition of the cDNA synthesis mix, the samples were incubated at 25°C for 10 min, 50°C for 50 min, and 85°C for 5 mins and chilled on ice. The samples were stored at –80°C until use.
2.8. Real‐time quantitative PCR
Real‐time quantitative polymerase chain reaction (RT‐qPCR) of AR and CXC chemokine receptor expression in neutrophils was performed using Power SYBR Green Master Mix (Applied Biosystems, Foster City, California) as per the manufacturer's instructions. Briefly, a master mix was prepared for each gene using 2× Power SYBR Green PCR Master Mix, RNase‐free H2O, and 500 nM each of the respective forward and reverse primers (Bioneer, Kew East, Victoria, Australia) for the receptor genes Adra1a, Adra1b, Adra1d, Adra2a, Adra2b, Adra2c, Adrb1, Adrb2, Adrb3, Cxcr1, Cxcr2, Cxcr4, and 18s (Table 1). Nine microliters of master mix was then added to 1 μl of each cDNA sample in a 384‐well PCR plate (Applied Biosystems). All cDNA samples were performed in triplicate. RT‐qPCR was performed using a 7900HT Fast Real‐Time PCR system (Applied Biosystems). An initial 10 min denaturation phase at 95°C was performed, followed by an annealing stage consisting of 45 cycles of a 15 s step at 95°C and a 1 min step at 60°C. A final dissociation stage was performed consisting of a 15 min step at 95°C, a 1 min step at 60°C, and a final 15 s step at 95°C. Expression of each sample was determined via comparison to the housekeeping gene 18s.
Table 1.
Primer sequences used for RT‐qPCR using 7900HT fast real‐time PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Adra1a | 5′‐GCCCGGGGCTTTTATCCATGA‐3′ | 5′‐GAAGATGTGGCCTCAGCCAG‐3′ |
| Adra1b | 5′‐CTGGTCTTAGCTTCGTGGCA‐3′ | 5′‐CTCGCTCGCCTCTAATGGG‐3′ |
| Adra1d | 5′‐CAGGGACACAGAGTAGCAAGG‐3′ | 5′‐TGAGGGAACAGAGAACCCAGAG‐3′ |
| Adra2a | 5′‐TTTCCCCTGTGCCTAACTGC‐3′ | 5′‐TGGCTTTATACACGGGGCTG‐3′ |
| Adra2b | 5′‐GAGTCCAAGAAGCCCCATCC‐3′ | 5′‐GGTGTCCATTAGCCTCTCCG‐3′ |
| Adra2c | 5′‐AGGACTTCAGGCGCTCTTTC‐3′ | 5′‐AGAGGGTCATTGCCTGAAGC‐3′ |
| Adrb1 | 5′‐TCATCGTGGTGGGTAACGTG‐3′ | 5′‐ACCAGCAATCCCATGACCAG‐3′ |
| Adrb2 | 5′‐CAATAGCAACGGCAGAACGG‐3′ | 5′‐TCAACGCTAAGGCTAGGCAC‐3′ |
| Adrb3 | 5′‐TCCACCGCTCAACAGGTTTG‐3′ | 5′‐TGGGGCAACCAGTCAAGAAG‐3′ |
| Cxcr1 | 5′‐GCTGGTGCCTCAGATCAAA‐3′ | 5′‐CCAAGAAGGGCAGGGTCAAT‐3′ |
| Cxcr2 | 5′‐CTTCCAGTTCAACCAGCCCT‐3′ | 5′‐CTTAATCCTGCAGTAGTTCTACGA‐3′ |
| Cxcr4 | 5′‐TGCAGCAGGTAGCAGTGAAA‐3′ | 5′‐TGTATATACTCACACTGATCGGTTC‐3′ |
| 18s | 5′‐CTTAGAGGGACAAGTGGCG‐3′ | 5′‐ACGCTGAGCCAGTCAGTGTA‐3′ |
2.9. RT‐qPCR using Fluidigm dynamic array
RT‐qPCR was also performed by the Medical Genomics Facility at Monash Health Translational Precinct using the Fluidigm Biomark system. Target genes underwent 14 cycles of preamplification using Taqman gene expression assays, including a no template control, and Biomark real‐time PCR was completed on 48.48 dynamic array integrated fluidic circuits. Prior to commencement of the PCR, a uracil N‐glycosylase step was used to prevent carryover of PCR products for 2 min at 50°C and a further hot start for 10 min at 96.5°C. Forty cycles of denaturation and annealing were then performed at 96°C for 15 s and 60°C for 60 s, respectively. Gene expression was quantified relative to the housekeeping gene Gapdh and normalized relative to untreated cells.
2.10. Quantitation of MPO activity
To measure the activity of MPO within the neutrophils and released into the media during 24 h NA challenge, samples were immediately frozen in liquid nitrogen and stored at –80°C until the MPO activity assay was performed. MPO activity was measured using an assay previously described and modified to be used in a 96‐well plate.14 Briefly, cells were homogenized by continuous pipetting for 15 s in 100 μl of hexadecyltrimethylammonium bromide (HTAB) buffer before being centrifuged for 4 min at 5000 g. The samples were then placed on ice. Following recording of blank absorbance values of a 96‐well flat‐bottom plate, 7 μl from each of the homogenized cell samples and 70 μl of the collected media were added directly to the wells of the plate. An o‐dianisidine hydrochloride solution was added and incubated for 5 min before reading with an absorbance reader at wavelength 405 nm.
2.11. Quantification of cytokines using a cytometric bead array
After 24 h of NA treatment, the supernatant from neutrophils plated at 2 × 106 cells/well was aspirated and quantified using the Mouse Th1/Th2/Th17 Cytometric Bead Array assay (BD Biosciences, San Jose, California). A 1:2 serial dilution for each mouse Th1/Th2/Th17 cytokine standards, reconstituted in assay diluent, was prepared in a 96‐well V‐bottom plate, beginning with a concentration of 5000 pg/ml. Fifty μlmicroliters of each supernatant sample was also added to the plate. A mixture of capture beads was then prepared using 3 μl of each bead per sample (Mouse IL‐2, IL‐4, IL‐6, IFN‐γ, TNF, IL‐17A, and IL‐10) and 14 μl of assay diluent per sample. Twenty‐five microliters of bead mix and 25 μl of PE detection reagent were then added to each of the standard wells. The plate was then incubated in the dark at room temperature for 2 h. The samples were then washed and resuspended in wash buffer. The cytokines in each sample were detected using a Navios flow cytometer, and their concentrations were calculated using FCAP Array software (BD Biosciences).
2.12. Phagocytosis
Neutrophil phagocytosis was assessed using the pHrodo Bioparticles Phagocytosis Kit (Life Technologies, Carlsbad, California). Briefly, cells were isolated and incubated for 24 h with NA before being washed and resuspended in chemotaxis assay buffer. Cells were then seeded into a 96‐well V‐bottom plate at 1 × 105 cells per well. The samples were incubated for 15 min with 2.5 μl of PE‐conjugated Escherichia coli at either 4 or 37°C. After incubation, cells were placed immediately on ice and washed with PBS before staining with Brilliant Violet 510‐conjugated anti‐mouse Ly6G (1A8, BioLegend, San Diego, California). Cells were centrifuged and resuspended in FACs buffer (PBS supplemented with 2% fetal calf serum and 0.8% EDTA) and examined using a BD FACSCanto flow cytometer (BD Biosciences) and analyzed using FlowJo software.
2.13. Respiratory burst
Respiratory burst in isolated neutrophils was assessed using the Neutrophil/Monocyte Respiratory Burst Assay Kit (Cayman Chemical, Ann Arbor, Michigan). Following NA treatment, the cells were washed and resuspended in RPMI supplemented with 50 μl of 1 M calcium and 2% fetal calf serum. Cells were then seeded into a 96‐well V‐bottom plate at 2 × 106 cells/ml and incubated with 50 ng of dihydrorhodamine (DHR) 123 for 15 min in a 37°C, 5% CO2‐supplemented incubator. Two hundred nanomolal phorbol myristate acetate (PMA) assay reagent was then added to each well and incubated for 30 min in a 37°C, 5% CO2‐supplemented incubator. Following incubation, the cells were immediately placed on ice before being washed and resuspended in respiratory burst assay buffer. Cells were then stained with Brilliant Violet 510‐conjugated anti‐mouse Ly6G (1A8, BioLegend) and FITC‐conjugated anti‐mouse CD11b (M1/70, BioLegend). A BD FACSCanto II (BD Biosciences) flow cytometer was used to examine the fluorescence of cells. FlowJo software was used to gate the neutrophil cell population, determine the median fluorescence intensity (MFI) of DHR 123 for this population, and examine the expression of CD11b on neutrophils.
2.14. Intravital microscopy
Migration and function of neutrophils following exposure to NA was examined using intravital microscopy of the cremaster muscle in LysM‐eGFP mice, as previously described.22 Mice were anesthetized via intraperitoneal injection of an anesthetic cocktail of 150 mg/kg of ketamine and 10 mg/kg of xylazine. The left jugular vein was cannulated to administer additional anesthesia. The animal was placed on a thermal‐controlled heating pad, regulating the core temperature to 37°C. The cremaster muscle was dissected free of tissues and exteriorized onto an optically clear viewing pedestal. The muscle was cut longitudinally with a cautery and held flat against the pedestal by attaching silk sutures to the corners of the tissue. The muscle was then superfused with bicarbonate‐buffered saline and covered with a coverslip held in place with vacuum grease (Thermo Fisher Scientific, Waltham, Massachusetts).
The cremasteric microcirculation was visualized using a spinning disk confocal microscope system (Ultraview VoX; PerkinElmer, Waltham, Massachusetts) mounted on an upright microscope (Axioplan2 Imaging, Oberkochen, Germany) as previously described.23 The cremasteric microvasculature was visualized through a 20× immersion objective lenses. Images were detected using a 512 × 512‐pixel electron‐multiplying charge‐coupled device camera (C9100‐13; Hamamatsu, Shizuoka, Japan). Data acquisition was controlled via Volocity (Perkin Elmer). Brightfield, and 488 nm of laser excitation was used in rapid succession with an exposure time of 0.120–0.317 s for each channel.
Two postcapillary venules (25–40 μm in diameter) were selected in each experiment, and images were recorded at the maximum rate for 3 min at 0 min (i.e., baseline before superfusion) and at 30 and 60 min following superfusion of 30 μM fMLP with or without the presence of 10−5 M NA in the superfusion buffer. In addition, to examine the behavior and migration of transmigrated neutrophils, we imaged and recorded continuously the activity of neutrophils at 15 frames/min between 30 and 60 min after superfusion. Venular diameter, velocity of noninteracting passing neutrophils, and the number of adherent/arrested and transmigrated neutrophils were determined off‐line via video playback analysis. Neutrophils were considered adherent/arrested to the venular endothelium if they remained stationary for 30 s or longer. Neutrophil transmigration was defined as the number of extravascular neutrophils visible per microscopic field centered on a postcapillary venule and was determined by averaging data derived from two fields per animal.
2.15. Mouse model of focal ischemia
The midcerebral artery occlusion (MCAO) was performed as previously described.24 Briefly, mice were anesthetized via intraperitoneal injection of an anesthetic cocktail of 150 mg/kg of ketamine and 10 mg/kg of xylazine, and an incision was made into the neck anterior to the sternum. The common, external, and internal arteries were isolated, and a monofilament of defined diameter was inserted into the internal carotid artery, coming to rest at the origin of the midcerebral artery (MCA). At this stage, a laser Doppler perfusion monitor (Perimed AB, Järfälla‐Stockholm, Sweden) on the cranium of each mouse was used to verify occlusion of the MCA. A drop in the preocclusion perfusion reading of more than 70% was considered a successful occlusion. Mice underwent MCAO for 30 or 60 min before removal of the filament and were monitored following reperfusion. Twenty‐four hours following MCAO, mice were culled and cells were isolated for study. A sham procedure involved anesthesia and incision into the neck before parting the salivary glands and replacing them in position and suturing the wound.
At 24 h after stroke onset, 2,3,5‐triphenyl‐tetrazolium chloride (TTC; Sigma) was used to visualize brain infarct following MCAO. The brain of each mouse was excised and placed in iced‐cold PBS for approximately 60 s following cervical decapitation. The brain was then sliced into 2 mm sections in the coronal plane and incubated in 50 mg/ml TTC dissolved in PBS, in a 37°C water bath for 5–10 min. The TTC was then removed, and the brain sections were resuspended in cold PBS. Both sides of the brain slices were photographed, and the infarct size was quantified using ImageJ analysis software.
2.16. Flow cytometry
The surface expression of CXC chemokine receptors on neutrophils isolated from blood and bone marrow were analyzed. Blood was collected into heparinized syringes via cardiac puncture, and red blood cells were lysed and washed with FACs buffer (PBS supplemented with 2% fetal calf serum and 0.8% EDTA). Cells were then stained with APC.Cy7‐conjugated anti‐mouse CD45 (30‐F11; eBioscience), Brilliant violet 510‐conjugated anti‐mouse Ly6G (1A8, BioLegend) and PE‐conjugated anti‐mouse CXCR2 (SA044G4, BioLegend) or CXCR4 (2B11, BD Pharmingen) for 30 min on ice. Cells were then washed and resuspended in FACs buffer. Cell populations were examined using a BD FACSCanto II and further analyzed using FlowJo software.
2.17. Statistical analysis
Data were statistically analyzed using GraphPad Prism 6.0b using multiple t‐tests, or one‐ or two‐way ANOVA tests with Tukey's or Bonferroni multiple comparisons tests. All results are represented by mean ± sem. Statistical significance was accepted at P < 0.05.
3. RESULTS
3.1. β2‐adrenergic receptors are highly abundant on neutrophils
Recent studies have implicated a role for SNS activation in the suppression of immune cell function; however, whether sympathetic output can directly mediate functional changes in neutrophils is unknown. First, using RT‐qPCR, we confirmed that BMNs express multiple AR subtypes, with β2‐adrenergic receptor (β2‐AR) (Adrb2) expression being significantly higher (Figure 1A). Strikingly, there was a 1000‐fold greater expression of Adrb2 in BMNs compared to other ARs. Circulating NA represents only a minor fraction (5–10%) of the amount of NA secreted from sympathetic nerve endings.25 It is highly influenced by NA reuptake, metabolism, and clearance from the circulation26; hence the precise physiological or pathological levels of NA following SNS activation has been elusive. Therefore, in this study, we utilized a range of NA concentrations when assessing its effect on BMN function in vitro. Additionally, the cytotoxicity potential of NA at various concentrations on BMNs was examined to ensure that any observed functional impairment is not merely due to cell death. Treatment of BMNs with NA concentrations of 100 nM (10−7 M), 1 μM (10−6 M), or 10 μM (10−5 M) for 4 h did not cause cytotoxicity, and this was confirmed using propidium iodide staining showing lack of difference in cell viability between the untreated and NA‐treated BMNs (data not shown). Intriguingly, BMNs treated with 10 μM (10−5 M) demonstrated elevated metabolic activity (Figure 1B).
Figure 1.
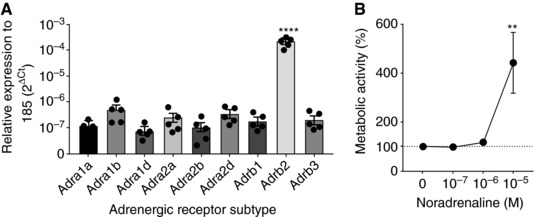
β2‐AR regulation and metabolic activity in neutrophils. (A) Raw expression of mRNA for various ARs in untreated BMNs, relative to the housekeeping gene 18s. Data represent mean ± sem; N ≥ 4; ****P < 0.0001 compared to all other receptor subtypes, One‐way ANOVA with Tukey's multiple comparisons test. (B) Quantitation of neutrophil metabolic activity following 4 h NA treatment, using an MTT assay. Data represent mean ± sem; N ≥ 4; **P < 0.01 vs. 0 M of NA, One‐way ANOVA with Tukey's multiple comparisons test
3.2. NA impairs neutrophil chemotaxis and negatively regulates proinflammatory gene expression
Next, to examine the effect of SNS activation on neutrophil function, we treated BMNs with various concentrations of NA and assessed their chemotactic function in vitro. We observed a significant reduction in the number of cells migrating toward the potent chemokine fMLP following 4 h treatment with NA (Figure 2A). Specifically, neutrophil migration toward fMLP was significantly impaired even at the lowest NA concentration examined (10−7 M NA). Furthermore, this NA‐induced chemotaxis impairment was not a result of reduced expression of the fMLP receptor, Formyl peptide receptor 1, by the BMNs (Fpr1; Figure 2B). In fact, a 4‐fold increase in Fpr1 mRNA expression was observed following 4 h of 10−7 M NA compared to untreated BMNs. Similarly, the expression of another chemoattractant receptor, Ccr1, demonstrated significant upregulation following NA administration, whereas Ccr2 reminded unchanged (Figure 2C). In addition, NA induced marked upregulation of Cxcr1 but no change in the expression of Cxcr2 (Figure 2D). These data suggest that the impairment of neutrophil chemotaxis after NA treatment is unlikely to be attributed to reduced expression of chemoattractant receptors.
Figure 2.
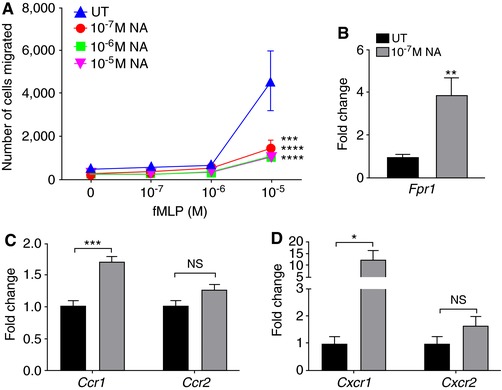
NA suppresses neutrophil chemotaxis to fMLP independently of chemokine receptor expression. (A) Migration toward fMLP of untreated BMNs and BMNs treated with increasing concentrations of NA for 4 h. Data represent mean ± sem; N = 5. ***P < 0.001, ****P < 0.0001 compared to UT, Two‐way ANOVA with Tukey multiple comparisons test. (B) Gene expression of Fpr1 in untreated BMNs and those treated with 10−7 M NA for 4 h, relative to the housekeeping gene 18s, and normalized to untreated neutrophils. Data represent mean ± sem; N = 5. **P < 0.01, unpaired t‐test. (C) Fold change in the gene expression of chemokine receptors, Ccr1 and Ccr2. Data represent mean ± sem; N ≥ 5; ***P < 0.001, unpaired t‐test. (D) Fold change in the gene expression of chemokine receptors, Cxcr1 and Cxcr2. Data represent mean ± sem; N ≥ 5; *P < 0.05, unpaired t‐test. NS: No statistical significance. NA: noradrenaline; UT: untreated
To explore alternative mechanisms by which NA may suppress neutrophil chemotaxis, we examined the expression of 45 genes in BMNs using RT‐qPCR following a 4 h in vitro challenge with 10−7 M NA. We observed significant changes in several groups of genes involved in N2 neutrophil characterization (Figure 3A), cytoskeleton remodeling (Figure 3B), and proinflammatory responses (Figure 3C). Specifically, there was a ∼8‐fold increase in the expression of N2 gene Arg1 following 10−7 M NA challenge (Figure 3A). Conversely, NA downregulated genes involved in the Akt/PI3K pathway (Akt1 and Pten; Figure 3B), which are critical for cells to undergo cytoskeletal rearrangement and therefore effective migration.27 It is conceivable that the NA‐induced reduction of Akt1 and Pten expression contributes to the impairment in the capacity of BMNs to migrate toward fMLP in vitro (Figure 2A), as well as in vivo from the vasculature toward the site of infection and into the affected tissue.28, 29 In addition, NA downregulated the expression of Fas (cell surface death receptor) on BMNs as well as proinflammatory genes, including Icam1, Myd88, Mmp9, and Tnf (Figure 3C). Other genes that we investigated were unaffected after NA treatment (see Table 2), suggesting a specific regulation of neutrophil functions rather than a global reduction in gene expression following NA administration. Taken together, these results demonstrate that treatment of BMNs with 10−7 M NA for 4 h significantly impairs neutrophil chemotaxis through a mechanism independent of effects on chemoattractant receptor expression. Instead, the NA‐induced N2 neutrophil phenotype, altered expression of the genes critical for cytoskeleton remodeling, and downregulation of proinflammatory genes are more likely to contribute to this phenotype.
Figure 3.
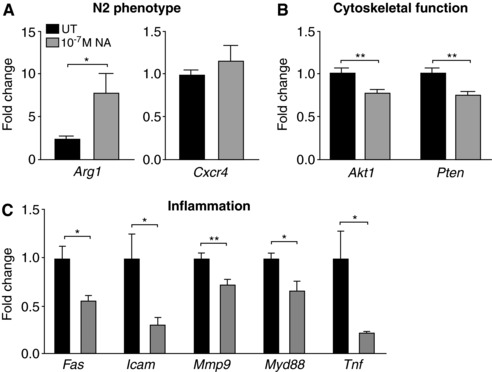
Differential gene expression in neutrophils following NA treatment. Fold changes in the expression of genes related to N2 neutrophil phenotype (A), cytoskeletal function (B), and inflammation (C), relative to the housekeeping gene Gapdh and normalized to untreated neutrophils. Data represent mean ± sem. N ≥ 5. *P < 0.05, **P < 0.01, unpaired t‐test. NA: noradrenaline; UT: untreated
Table 2.
Taqman GE assays used for Fluidigm Dynamic Array
| Gene name | Life Technology Code | Assay number |
|---|---|---|
| 18s | Mm04277571_s1 | M000833 |
| Akt1 | Mm01331626_m1 | M000427 |
| Arg1 | Mm00475988_m1 | M000157 |
| Arp2/3 | Mm00773842_m1 | M000757 |
| Atf3 | Mm00476032_m1 | M000291 |
| Beta‐Actin | Mm01205647_g1 | M000825 |
| Ccl2 | Mm00441242_m1 | M000212 |
| Ccl3 | Mm00441259_g1 | M000373 |
| Ccl5 | Mm01302427_m1 | M000374 |
| Ccr1 | Mm00438260_s1 | M000862 |
| Ccr2 | Mm99999051_gH | M000160 |
| Ccr5 | Mm01216171_m1 | M000381 |
| Ccr7 | Mm00432608_m1 | M000166 |
| Cd44 | Mm01277163_m1 | M000527 |
| Cd49b/Itga2 | Mm00434371_m1 | M000241 |
| Cd62l/Sell | Mm00441291_m1 | M000243 |
| Cdc42 | Mm01194005_g1 | M000754 |
| Chil3 | Mm00657889_mH | M000390 |
| Cx3cr1 | Mm02620111_s1 | M000153 |
| Cxcr2 | Mm99999117_s1 | M000861 |
| Cxcr3 | Mm00438259_m1 | M000213 |
| Cxcr4 | Mm01292123_m1 | M000026 |
| Fas | Mm00433237_m1 | M000164 |
| Gapdh | Mm03302249_g1 | M000054 |
| Icam | Mm00516023_m1 | M000700 |
| IL‐10 | Mm00439614_m1 | M000234 |
| Il12a | Mm00434165_m1 | M000191 |
| Il12b | Mm00434170_m1 | M000195 |
| Il1b | Mm00434228_m1 | M000214 |
| Itgb1 | Mm01253230_m1 | M000067 |
| Itgb2 | Mm00434513_m1 | M000068 |
| Mapk14 | Mm01301009_m1 | M000416 |
| Mmp9 | Mm00442991_m1 | M000706 |
| Mrc1 | Mm00485148_m1 | M000155 |
| Myd88 | Mm00440338_m1 | M000242 |
| Nwasp | Mm01136990_m1 | M000808 |
| Osteopontin/Spp1 | Mm00436767_m1 | M000135 |
| p21/Cdkna | Mm04205640_g1 | M000453 |
| Pparg | Mm01184322_m1 | M000134 |
| Pten | Mm00477208_m1 | M000269 |
| Rac1 | Mm01201653_mH | M000983 |
| RhoA | Mm00834507_g1 | M000805 |
| Rpl13a | Mm01612987_gH | M000223 |
| Tgfb1 | Mm00441724_m1 | M000173 |
| Tlr2 | Mm00442346_m1 | M000252 |
| Tlr4 | Mm00445273_m1 | M000154 |
| Tnf | Mm00443258_m1 | M000174 |
| Vegfa | Mm00437304_m1 | M000187 |
3.3. Prolonged NA challenge stimulates neutrophil release of MPO and IL‐6 but reduces IFN‐γ production
We next examined the effect of prolonged SNS activation on neutrophil function by treating BMNs with the tested concentrations of NA for 24 h in vitro. Similar to the 4 h challenge, we did not observe any cytotoxicity associated with this treatment at 24 h, but instead found significant elevation of neutrophil metabolic activity at NA concentration of 10−6 and 10−5 M compared to untreated BMNs (Figure 4A). To examine the effect of NA on neutrophil function, we assessed neutrophil production and release of antimicrobial mediators at 24 h following NA. MPO is an enzyme abundantly expressed in neutrophils with an important role in antimicrobial function upon its release into the extracellular space.30 In the absence of additional inflammatory or pathogenic stimuli, BMNs exposed to 10−5 M NA for 24 h were found to potently release MPO (Figure 4B). This was evident by the significant reduction of cellular MPO units within BMNs, whereas corresponding elevated MPO levels were found in the cell culture media at 24 h following NA treatment (Figure 4B). Furthermore, the cell culture media of BMNs at 24 h following NA treatment demonstrated significantly increased levels of IL‐ 6 at all NA concentrations examined (Figure 4C), and markedly reduced levels of IFN‐γ (Figure 4D) and IL‐10 (Figure 4E) in response to 10−5 M NA. In contrast, the levels of IL‐2 and IL‐4 were unchanged after NA challenge (Figure 4F–G). These data suggest prolonged NA administration alone promotes neutrophils to release MPO and IL‐6 and suppresses the production of inflammatory mediators including IFN‐γ and IL‐10.
Figure 4.
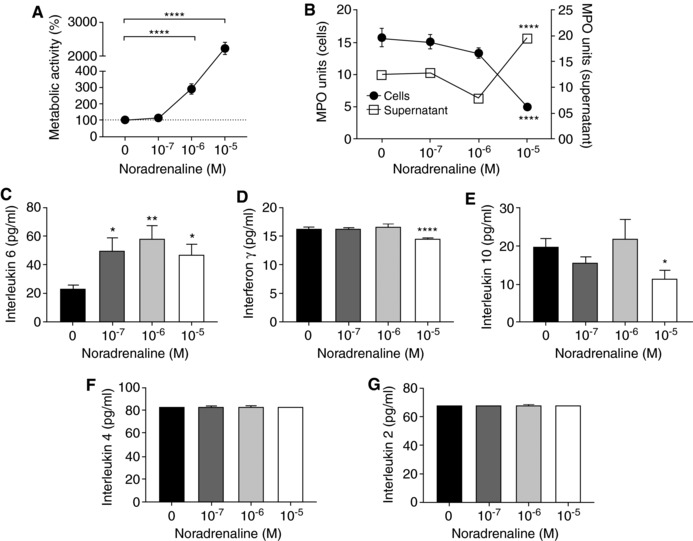
The effect of prolonged NA treatment on neutrophil function. (A) The metabolic activity of neutrophils treated with NA for 24 h as measured by the MTT assay. Data represent mean ± sem. N = 8. ****P < 0.0001 compared to control, One‐way ANOVA with Bonferroni or Tukey's multiple comparisons test. (B) MPO activity of neutrophils (2 × 106) treated with or without NA for 24 h and the supernatant (70 μl) aspirated from these cells were quantified. Data represent mean ± sem. N = 8. ****P < 0.0001 compared to control, One‐way ANOVA with Bonferroni or Tukey's multiple comparisons test. Measurement of interleukin 6 (C), IFN‐γ (D), IL‐10, (E) IL‐4 (F), and IL‐2 (G) released into the cell media following 24 h of NA challenge, quantified using a cytometric bead array. Data represent mean ± sem. N = 8. *P < 0.05, **P < 0.01, ****P < 0.0001, One‐way ANOVA with Tukey's multiple comparisons test
3.4. Activation of SNS diminishes neutrophil activation and phagocytosis
Upon infection, neutrophils rapidly migrate toward the invading pathogens via chemotaxis and undergo phagocytosis to efficiently engulf and kill the bacteria.31 To assess the effect of SNS activation on neutrophil phagocytosis, we pretreated the BMNs at various concentrations of NA for 24 h then challenged these cells with fluorescently labeled E. coli. Neutrophil phagocytosis was not affected at the lower NA concentrations, but a significant impairment in bacterial phagocytosis was observed at 10−5 M NA (Figure 5A). In separate experiments, we examined the effect of NA administration on neutrophil activation and capability to generate respiratory burst upon stimulation with PMA. Our results demonstrated that BMNs expression of CD11b significantly decreased in a dose‐response manner at 24 h following NA challenge (Figure 5B, C). Despite this, PMA‐induced respiratory burst by BMNs was not affected following the activation of SNS (Figure 5D). Taken together, these findings indicate that prolonged exposure of neutrophils with NA markedly impairs their capacity to phagocytose bacteria and undergo activation upon stimulation.
Figure 5.
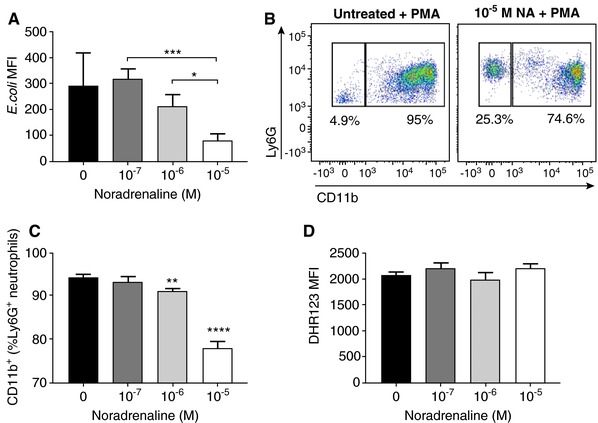
Prolonged exposure of NA suppresses neutrophil phagocytosis and activation, but not PMA‐induced respiratory burst. (A) Quantification of the MFI of E. coli phagocytosed by neutrophils treated with 24 h of various concentrations of NA. Data represent mean ± sem. N ≥ 8. *P < 0.05, ***P < 0.001, unpaired t‐test. (B) Representative flow cytometric plot of PMA‐induced CD11b expression on untreated Ly6G+ neutrophil or Ly6G+ neutrophil pretreated with 10−5 M NA for 24 h. (C) Percentage of Ly6G+ neutrophils expressing CD11b on the cell surface after 24 h pretreatment with NA following 30 min stimulation with PMA. Data represent mean ± sem. N ≥ 4. **P < 0.01, ****P < 0.0001 compared to control. One‐way ANOVA with Tukey's multiple comparisons test. (D) Quantification of PMA‐induced respiratory burst by neutrophils treated with 24 h of various concentrations of NA. Data represent mean ± sem. DHR123: Dihydrorhodamine 123. MFI: mean fluorescence intensity
3.5. Effect of SNS activation on neutrophil trafficking in vivo
Our data thus far suggest NA treatment impairs a number of neutrophil functions, including chemotaxis toward fMLP and activation following PMA challenge. To investigate whether intravascular trafficking of neutrophils is also impaired after SNS activation, we superfused the potent chemoattractant fMLP over the cremaster muscle of the LysM‐eGFP mice, in the presence or absence of NA in the superfusion buffer, and examined the response in cremasteric postcapillary venules via intravital microscopy. LysM‐eGFP mice express GFP in the regulatory region of the lysozyme M gene expressed in myeloid cells in which endogenous neutrophils are easily identifiable.32 We visualized and quantified the number of adherent/arrested neutrophils in at least 2 vessels per mouse during the 1 h of superfusion. Our results indicated that fMLP superfusion significantly increased the number of adherent/arrested neutrophils to the vessel wall at 30 and 60 min. However, this response was almost abolished in the presence of NA (Figure 6A and Movies 1 and 2). We also measured the number of transmigrating neutrophils between 30 and 60 min of fMLP superfusion. Transmigrating neutrophils were clearly visible in mice that were superfused with fMLP only (Figure 6B), and these cells underwent extravascular migration of an average distance of 16.5 ± 2.3 μm (Figure 6C and Movie 1). In contrast, superfusion of fMLP in combination with NA significantly reduced the number of transmigrated neutrophils (Figure 6B and Movie 2). In addition, the only transmigrated neutrophil observed traveled a distance of only 8.5 μm, ∼ 50% of the average distance in the fMLP alone group (Figure 6D). To examine whether superfusion of the cremaster muscle with NA impact on blow flow or vessel constriction, we quantified the speed of noninteracting neutrophils passing through blood vessels (Figure 6E). Neutrophils that have no interaction with the endothelium, and passing through blood vessels can serve as a surrogate marker for blood flow velocity. We also measured the diameter of the blood vessels in the presence or absence of NA superfusion (Figure 6F). We found neither the speed of noninteracting neutrophils nor blood vessel diameter was different following NA challenge, suggesting the reduced neutrophil adhesion/arrest and transmigration after NA are not due to a lack of cells traveling through the region of interest over the set time interval.
Figure 6.
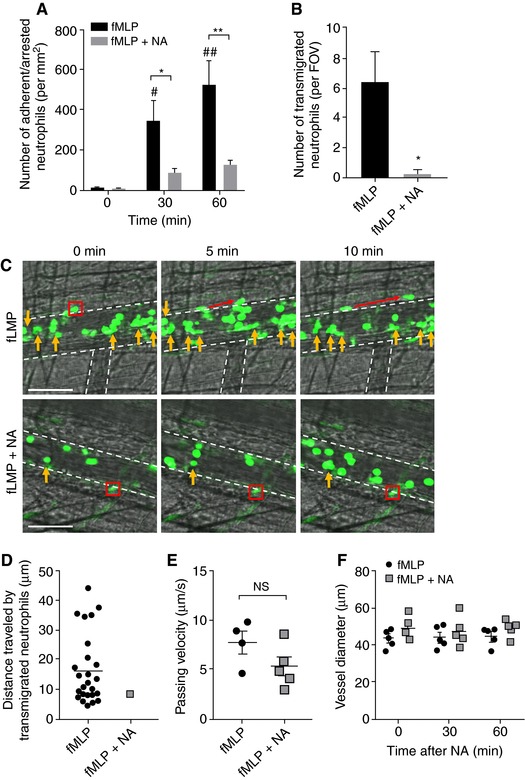
NA superfusion abolishes fMLP‐induced neutrophil adhesion and transmigration in vivo. (A) The number of adherent/arrested GFP+ neutrophils in LysM‐eGFP mice following superfusion of 30 μM fMLP with or without the presence of 10−5 M NA was quantified. Data represent mean ± sem; N ≥ 4 individual mice per group; # P < 0.05, ## P < 0.01 vs. 0 min; *P < 0.05, **P < 0.01 vs. corresponding fMLP only, unpaired t‐test. (B) The number of transmigrated GFP+ neutrophils in LysM‐eGFP mice following superfusion of 30 μM fMLP with or without the presence of 10−5 M NA was quantified. Data represent mean ± sem; N ≥ 4 individual mice per group; *P < 0.05 vs. fMLP only, unpaired t‐test. (C) Representative images of fMLP‐induced neutrophil transmigration with or without the presence of NA. Yellow arrows denote adherent/arrested neutrophils (it should be noted that the majority of neutrophils visible in these images are rolling cells), red box and arrows denote transmigrating neutrophils during 10 min observation periods, and white dotted lines denote the outline of the postcapillary venules. Scale bar = 80 μm. FOV: field of view. (D) The distance traveled by transmigrated GFP+ neutrophils in LysM‐eGFP mice following superfusion of 30 μM fMLP with or without the presence of 10−5 M NA was quantified. Data represent distance traveled for individual neutrophils; N ≥ 4 individual mice per group. (E) The velocity of noninteracting GFP+ neutrophils in LysM‐eGFP mice following superfusion of 30 μM fMLP with or without the presence of 10−5 M NA was quantified. Data represent mean ± sem; N ≥ 4 individual mice per group; NS: no statistical significance. (F) The diameter of postcapillary venules before and during 1 h superfusion of 30 μM fMLP with or without the presence of 10−5 M NA was quantified. Data represent mean ± sem; N ≥ 4 individual mice per group. NA: noradrenaline
Emerging evidence from recent studies indicates that dysregulation of the sympathetic‐immune axis contributes to a number of disease states.33, 34 In fact, patients with stroke exhibit markedly increased plasma NA levels for several days following the ischemic event.35 Moreover, stroke patients with greater increases in plasma NA display higher susceptibility to infectious complications and an increased risk of mortality.36, 37, 38, 39 However, the mechanisms surrounding this are unclear. Therefore, we investigated whether the activation of SNS following stroke impairs the important antibacterial functions of neutrophils, rendering the host at a greater risk of infections. To address this, we investigated neutrophil migration after a mouse model of stroke (MCAO). Consistent with previous reports, a 30 min MCAO elicited a mild brain injury in mice whereas 60 min MCAO induced a more severe cerebral infarct size development at 24 h following stroke onset (Figure 7A). We isolated BMNs at 24 h following the cerebral ischemic‐reperfusion event and examined the neutrophil chemotaxis toward a number of potent chemoattractants. Following MCAO, we observed a significant reduction in the migration of neutrophils toward all chemoattractants tested, including fMLP (Figure 7B), KC (Figure 7C), and C5a (Figure 7D). Moreover, this effect was greater for neutrophils isolated from mice subjected to a 60 min MCAO (severe stroke), compared with those from mice subjected to a 30 min MCAO (mild stroke) in response to KC (Figure 7C). Although we cannot exclude the possibility that reduced neutrophil chemotaxis at 24 h poststroke may be in part attributable to an exhaustion of the primed reservoir, BMNs isolated from sham‐operated and poststroke mice at a much earlier time point (4 h) also demonstrated significantly decreased neutrophil migratory function in vitro (Supplemental Figure 1).
Figure 7.
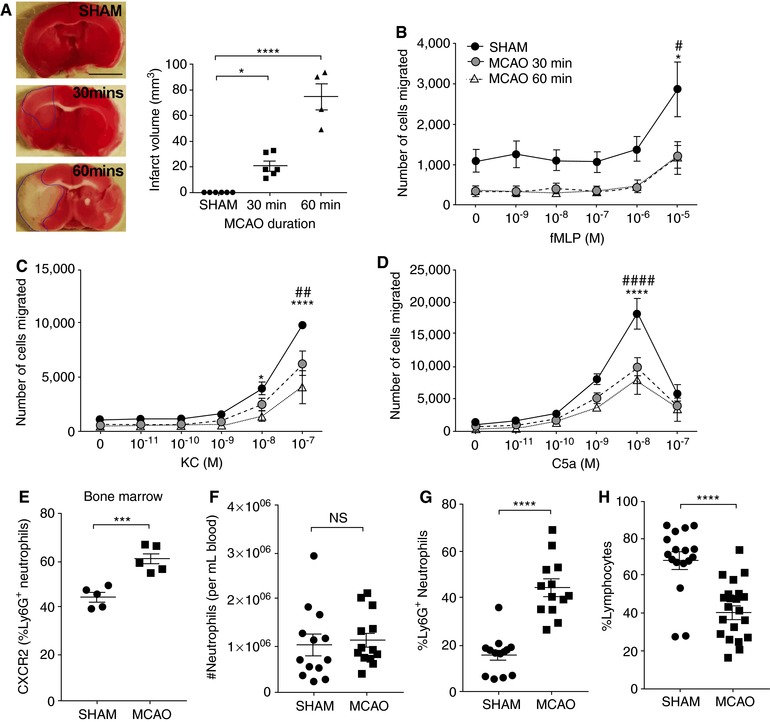
Stroke impairs neutrophil chemotaxis. (A) Representative image of brain infarct size and brain infarct quantification in mice at 24 h after stroke onset. Data represent mean ± sem; N ≥ 3 individual mice per group; *P < 0.05 and ****P < 0.0001 vs. Sham, unpaired t‐test. The migration of BMNs isolated from mice that were sham operated, 30 min MCAO‐, and 60 min MCAO‐operated toward fMLP (B), KC (C), and C5a (D) were assessed. Data represent mean ± sem; N ≥ 3 individual mice per group; # P < 0.05, ## P < 0.01, #### P < 0.0001 for Sham compared with 30 min MCAO; *P < 0.05, ****P < 0.0001 for Sham compared with 60 min MCAO, two‐way ANOVA with Tukey's multiple comparisons test. Cell surface expression of CXCR2 (E) on Ly6G+ neutrophils isolated from bone marrow of sham‐ and MCAO‐operated mice. N ≥ 5 individual mice per group. Data represent mean ± sem. ***P < 0.001, t‐test. The number (F) and percentage (G) of neutrophils from the blood of sham‐ and MCAO‐operated mice. (H) The percentage of lymphocytes from the blood of sham‐ and MCAO‐operated mice. N ≥ 12 individual mice per group. Data represent mean ± sem. ****P < 0.0001, t‐test. MCAO: midcerebral artery occlusion
To investigate whether the reduced neutrophil chemotaxis at 24 h is contributed by altered expression of chemokine receptors, we performed flow cytometric analysis on the surface expression of CXCR2 and CXCR4 in neutrophils isolated from the bone marrow and blood after stroke (Supplemental Figure 2). At 24 h following stroke onset, we found no difference in the expression of CXCR4 on circulating neutrophils or BMNs between sham‐operated and poststroke mice (Supplemental Figure 3). Similarly, circulating neutrophils from sham‐operated and poststroke mice demonstrated comparable CXCR2 expression (Supplemental Figure 3). However, neutrophils isolated from the bone marrow of poststroke mice exhibited significantly elevated CXCR2 surface expression compared to their sham‐operated controls (Figure 7E), suggesting the lack of migration toward the chemokines examined is not attributable to decreased chemokine receptor expression. In fact, we consistently observed unchanged circulating neutrophil number at 24 h after stroke (Figure 7f), whereas the elevated proportion of neutrophils in the blood of poststroke mice (Figure 7g) can be explained by the significant reduction of lymphocytes in the blood (Figure 7H and Supplemental Figure 4). Taken together, our findings demonstrate that neutrophil chemotaxis is severely impaired following in vitro or in vivo following NA challenge, and in a pathological condition whereby SNS activation is known to occur.
4. DISCUSSION
There is a growing body of evidence that suggests dysregulation of the sympathetic‐immune axis contributes to a number of disease states,33, 34 but the underlying processes have been elusive to date. Neutrophils are an essential part of the host innate immune system; however, whether their critical antibacterial defense mechanisms are affected following the activation of SNS is currently unknown. Clearly, elucidating the relationship between sympathetic activation and neutrophil function during physiological and pathological stress will be important for the development of novel therapeutic strategies and to aid in the identification of “at risk” patients for appropriate interventions. In this study, we assessed a number of neutrophil functions following experimental models of sympathetic activation. We demonstrated that the β2‐AR is highly abundant in neutrophils and that administration of NA can directly suppress critical antibacterial functions of neutrophils. In particular, our findings indicated that NA inhibits the migratory function of neutrophils toward potent chemoattractants in vitro and that in an in vivo model of pathological SNS activation similar negative regulatory effects on neutrophil function are observed.
The molecular pathways by which the activation of SNS mediates neutrophil functional impairment are currently unknown. Findings supporting the location of sympathetic nerves and NA release within the vicinity of immune cells prompted the design of studies to determine whether ARs were expressed on the immune cell surface.40 The expression of such receptors would be necessary in order for neural signals to be delivered to immune cells. There are two types of receptors that bind NA, namely the α‐AR and the β‐AR, which are expressed in a tissue‐specific manner and exhibit differing affinities for NA.41, 42 Elegant reviews over a decade ago have detailed that the β2‐AR subtype is the primary receptor expressed on immune cells in both rodents and humans.43 In fact, bulk populations of CD8+ and CD4+ T cells express the β2‐AR, as do naive CD4+ T cells and murine Th1 cells, whereas clones of murine Th2 cells do not.44 However, conflicting data exist for human lymphocytes, with studies suggesting both the presence and absence of expression of β2‐AR on human T cells. Clarity in this area has been hampered by the difficulty in obtaining purified populations of human IFNγ‐ and IL‐4‐producing cells. Nevertheless, our data demonstrated that neutrophils also express primarily β2‐AR (∼1000 fold higher). Moreover, the functional effect of NA binding to this receptor is striking. Acute exposure (4 h) of NA to BMNs significantly impaired neutrophil chemotaxis toward fMLP in a chemokine receptor expression‐independent fashion. Our findings from gene expression study suggest neutrophils treated with NA upregulate a N2 phenotypic marker, Arg1, and downregulate a number of proinflammatory genes as well as genes associated with cytoskeleton remodeling important for migratory behavior. Therefore, consistent with earlier studies examining the effect of sympathetic outflow on adaptive immune system, results from our study indicate the activation of the SNS also induces an immunosuppressive effect on neutrophils.
In addition, we observed elevated neutrophil release of MPO and IL‐6 following prolonged (24 h) exposure of NA alone. Despite previous studies noting that IL‐6 has the capacity to prime the neutrophil to undergo respiratory burst in response to an inflammatory stimulus,45 NA‐stimulated neutrophil production of IL‐6 was not associated with increased PMA‐induced respiratory burst. Instead, neutrophils treated with NA for 24 h showed significantly decreased production of inflammatory mediators including IFN‐γ and IL‐10, impairment of bacteria phagocytosis and a marked reduction of CD11b expression upon PMA challenge. Intriguingly, superfusion of NA was sufficient to abolish fMLP‐induced neutrophil‐endothelial interactions in vivo. IL‐6 signaling via STAT3 has been shown to limit the recruitment of neutrophils and potentially represents a critical event for the termination of the innate immune response.46 In light of our findings showing elevated IL‐6 production following NA exposure, it is feasible that the impaired neutrophil chemotaxis is driven by IL‐6 signaling. Collectively, our findings are supportive of an immunosuppressive modulatory role for NA on neutrophil function. In fact, the immunosuppressive effect of sympathetic activation has been widely demonstrated in the context of both psychological stress models and exercise science, whereby psychological stressors activating the “fight or flight” response and high‐intensity exercise primarily impair immune function and increase susceptibility to infection.47, 48, 49 Numerous studies have observed that elite athletes commonly suffer symptoms of upper respiratory tract infections.50 Moreover, studies examining neutrophil function following exercise have also shown that high‐intensity bouts of exercise and exercise for longer durations result in impaired oxidative burst in response to stimulation.51, 52 These changes can be seen in an instantaneous manner following acute high intensity exercise, whereas regular high‐intensity exercise can lead to chronic suppression of immune function and may therefore lead to immune deficit, predisposing the individual to infection.51, 53
For many years, stroke patients have demonstrated markedly increased plasma NA levels persistent for several days following the ischemic event.35 Patients with greater increases in plasma NA levels display greater alterations in their leukocyte profile, higher susceptibility to infectious complications, and an increased risk of mortality,36, 37, 38, 39 supporting the notion of an immunosuppressive, and potentially pathological, role of NA. In fact, there is a correlation between the circulating levels of NA and impairment of neutrophil respiratory burst in stroke patients.54 A separate research group observed that stroke patients diagnosed with infection have significantly reduced neutrophil respiratory burst compared with patients with no evidence of infection.55 Collectively, these studies suggest that the function of neutrophils may be NA dependent and closely associated with infection incidence after stroke. In a mouse model of stroke, we demonstrate significant impairment of neutrophil chemotaxis to both end target chemoattractants (fMLP and C5a) and intermediary endogenous chemoattractants (KC) in poststroke mice. Interestingly, the impairment of neutrophil migratory behavior toward KC was found to be injury severity dependent. Furthermore, we have shown that BMNs isolated from sham‐operated and poststroke mice at a much earlier time point (4 h) also demonstrated significantly decreased neutrophil migratory function. Despite this, the possibility that the reduced neutrophil chemotaxis poststroke may be in part attributable to an exhaustion of the primed reservoir cannot be excluded. A key defining feature of the immune system is the migratory capacity of its cellular constituents, an attribute which allows the deployment of immune effector cells to the appropriate anatomical location to eliminate pathogen‐bearing cells. We have previously shown the migratory behavior and antibacterial function of invariant natural killer T cells to be impaired after stroke, via a mechanism involving SNS activation.14 In the present study, we provide evidence from three separate models (in vitro NA stimulation, in vivo NA superfusion, and following a mouse model of stroke) to demonstrate that the activation of SNS significantly impairs neutrophil chemotaxis, reduces bacterial phagocytosis, and modulates neutrophil expression and production of inflammatory mediators, changes likely to result in inhibition of critical host defense functions.
The findings of our experiments indicate the negative regulation of neutrophil function after NA is not due to neurotoxicity or NA‐induced cell death. In contrast, we consistently observed enhanced metabolic activity of neutrophils following NA exposure. The interconnecting relationship between immune cell energy metabolism and function was first reported in the early 1960s, but renewed interest in recent years has led to the growing understanding of the importance of metabolic alterations underlying an effective immune response.56 Emerging research indicates that cellular metabolic pathways serve three major functions: generation of energy, production of building blocks necessary for cellular maintenance and proliferation, and modulation of cellular signaling. Evidence also indicates that the metabolic alterations in myeloid cells differ depending on the type of stimulus, immune context, and tissue microenvironment.57 Our findings of NA‐induced metabolic activity in neutrophils at the same time as impaired chemotaxis raise the possibility that activation of ARs on neutrophils results in decreased effector function via effects on cellular metabolism. To our knowledge, this is the first report of enhanced metabolic activity induced by the activation of SNS in the modulation of neutrophil function.
Neutrophils are highly abundant within the human body and perform critical functions to protect the host from infections. The findings from this study indicate that a number of neutrophil antibacterial functions are impaired or negatively regulated in a NA‐dependent manner. We provide evidence to suggest that the activation of SNS weakens neutrophil migration, activation, and phagocytosis, inhibiting their critical host defense functions. Accumulating research now indicates that targeted therapeutics to inhibit SNS signaling may afford host protection against the development of poststroke infections.14, 58, 59, 60, 61 In fact, blockade of adrenergic signals via the use of propranolol, which was found to be efficacious as an intervention for poststroke infection,62 may improve stroke outcome by restoring neutrophil responsiveness to signals of infection. Therefore, a better understanding of the relationship between sympathetic activation and neutrophil function will be important for the development of effective therapeutic interventions.
Supporting information
MOVIE 1. Neutrophil behavior in LysM‐eGFP mice during fMLP superfusion. Intravital spinning‐disk confocal microscopy was utilized to examine the behavior of GFP+ neutrophils in the cremasteric microvasculature of LysM‐eGFP mice. The movie was taken at 60 min following 30 μM fMLP superfusion. The time lapse was recorded at 15 fpm and exported to video at 10 fps. Scale bar = 50 μm
MOVIE 2. Neutrophil behavior in LysM‐eGFP mice during fMLP superfusion in the presence of NA. Intravital spinning‐disk confocal microscopy was utilized to examine the behavior of GFP+ neutrophils in the cremasteric microvasculature of LysM‐eGFP mice. The movie was taken at 60 min following 30 μM fMLP superfusion in the presence of 10−5 M NA. The time lapse was recorded at 15 fpm and exported to video at 10 fps. Scale bar = 50 μm
SUPPLEMENTAL FIGURES
AUTHORS’ CONTRIBUTIONS
C.H.Y.W. conceived, designed, and supervised the project. A.J.N., S.W., P.H., and C.H.Y.W. performed the experiments, collected, analyzed, and conducted interpretation of the data. M.J.H. provided the LysM‐eGFP mice and intellectual input on the project. A.J.N. and C.H.Y.W. wrote the manuscript. All authors have read and critically reviewed the manuscript.
COMPETING INTERESTS
The authors declare no competing financial and nonfinancial interests.
ETHICS APPROVAL STATEMENT
All procedures were approved by the Monash University Animal Ethics Committee (MMCB/2014/29), which comply with the National Institute of Health guidelines for the care and use of laboratory animals.
FUNDING
This work was supported by Heart Foundation Future Leader Fellowship (100863, C.H.Y.W.) and funded by a grant from the Australian National Health and Medical Research Council (NHMRC APP1104036) and the Rebecca L. Cooper Medical Research Foundation and ANZ Trustees. M.J.H is supported by an NHMRC Senior Research Fellowship (1042775). The funding bodies have no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
ACKNOWLEDGMENTS
We would like to acknowledge the Medical Genomics Facility at the Monash Health Translational Precinct for performing Fluidigm Dynamic Array for gene analysis. We would also like to acknowledge Dr. Remy Robert (Monash University) for providing us with anti‐mouse CXCR2 and CXCR4 antibodies.
Nicholls AJ, Wen SW, Hall P, Hickey MJ, Wong CHY. Activation of the sympathetic nervous system modulates neutrophil function. J Leukoc Biol. 2018;103:295–309. 10.1002/JLB.3MA0517-194RR
One sentence summary: Sympathetic activation of neutrophils significantly impairs several critical host defense functions, including chemotaxis, phagocytosis and neutrophil‐endothelial cell interactions.
REFERENCES
- 1. Ordovas‐Montanes J, Rakoff‐Nahoum S, Huang S, Riol‐Blanco L, Barreiro O, von Andrian, UH . The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 2015;36:578–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nance, DM , Sanders, VM. Autonomic innervation and regulation of the immune system (1987‐2007). Brain Behav Immun. 2007;21:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mantovani, A , Sica, Macrophages A., innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. [DOI] [PubMed] [Google Scholar]
- 4. Wirth T, Westendorf AM, Bloemker D, et al. The sympathetic nervous system modulates CD4(+)Foxp3(+) regulatory T cells via noradrenaline‐dependent apoptosis in a murine model of lymphoproliferative disease. Brain Behav Immun. 2014;38:100–10. [DOI] [PubMed] [Google Scholar]
- 5. Bellinger, DL , Lorton, D. Autonomic regulation of cellular immune function. Auton Neurosci. 2014;182:15–41. [DOI] [PubMed] [Google Scholar]
- 6. Sanders, VM , Straub, RH . Norepinephrine, the β‐adrenergic receptor, and immunity. Brain Behav Immun. 2002;16:290–332. [DOI] [PubMed] [Google Scholar]
- 7. Yin D, Tuthill D, Mufson RA, Shi, Y . Chronic restraint stress promotes lymphocyte apoptosis by modulating CD95 expression. J Exp Med. 2000;191:1423–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiang, Y , Kobilka, BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–1532. [DOI] [PubMed] [Google Scholar]
- 9. Wang J, Charboneau R, Barke RA, Loh HH, Roy, S . μ‐Opioid receptor mediates chronic restraint stress‐induced lymphocyte apoptosis. J Immunol. 2002;169:3630–3636. [DOI] [PubMed] [Google Scholar]
- 10. Leposavic G, Pilipovic I, Radojevic K, Pesic V, Perisic M, Kosec, D . Catecholamines as immunomodulators: a role for adrenoceptor‐mediated mechanisms in fine tuning of T‐cell development. Auton Neurosci. 2008;144:1–12. [DOI] [PubMed] [Google Scholar]
- 11. Pilipovic I, Radojevic K, Perisic M, et al. Catecholaminergic signalling through thymic nerve fibres, thymocytes and stromal cells is dependent on both circulating and locally synthesized glucocorticoids. Exp Physiol. 2012;97:1211–1223. [DOI] [PubMed] [Google Scholar]
- 12. Straub, RH. Complexity of the bi‐directional neuroimmune junction in the spleen. Trends Pharmacol Sci. 2004;25:640–646. [DOI] [PubMed] [Google Scholar]
- 13. Stanley D, Mason LJ, Mackin KE, et al. Translocation and dissemination of commensal bacteria in post‐stroke infection. Nat Med. 2016;22:1277–1284. [DOI] [PubMed] [Google Scholar]
- 14. Wong CH, Jenne CN, Lee WY, Leger C, Kubes, P . Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334:101–105. [DOI] [PubMed] [Google Scholar]
- 15. Petri B, Phillipson M, Kubes, P . The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180:6439–6446. [DOI] [PubMed] [Google Scholar]
- 16. Wong CH, Heit B, Kubes, P . Molecular regulators of leucocyte chemotaxis during inflammation. Cardiovasc Res. 2010;86:183–191. [DOI] [PubMed] [Google Scholar]
- 17. Kolaczkowska, E , Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 18. Alard JE, Ortega‐Gomez A, Wichapong K, et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med. 2015;7:317ra196. [DOI] [PubMed] [Google Scholar]
- 19. Ma Y, Yabluchanskiy A, Iyer RP, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Oliveira S, Rosowski EE , Huttenlocher, A . Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beyrau M, Bodkin JV, Nourshargh, S . Neutrophil heterogeneity in health and disease: a revitalized avenue in inflammation and immunity. Open Biol. 2012;2:120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Norman MU, Van De Velde NC, Timoshanko JR , Issekutz A, Hickey, MJ . Overlapping roles of endothelial selectins and vascular cell adhesion molecule‐1 in immune complex‐induced leukocyte recruitment in the cremasteric microvasculature. Am J Pathol. 2003;163:1491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abeynaike LD, Deane JA, Westhorpe CL, et al. Regulatory T cells dynamically regulate selectin ligand function during multiple challenge contact hypersensitivity. J Immunol. 2014;193:4934–4944. [DOI] [PubMed] [Google Scholar]
- 24. Connolly Jr ES, Winfree CJ, Stern DM, Solomon RA, Pinsky, DJ . Procedural and strain‐related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–532. [DOI] [PubMed] [Google Scholar]
- 25. Esler M, Jennings G, Lambert G, Meredith I, Horne M, Eisenhofer, G . Overflow of catecholamine neurotransmitters to the circulation: source, fate, and functions. Physiol Rev. 1990;70:963–985. [DOI] [PubMed] [Google Scholar]
- 26. Esler M, Jennings G, Korner P, et al. Assessment of human sympathetic nervous system activity from measurements of norepinephrine turnover. Hypertension. 1988;11:3–20. [DOI] [PubMed] [Google Scholar]
- 27. Gambardella, L , Vermeren, S . Molecular players in neutrophil chemotaxis—focus on PI3K and small GTPases. J Leukoc Biol. 2013;94:603–612. [DOI] [PubMed] [Google Scholar]
- 28. Stroka KM, Hayenga HN, Aranda‐Espinoza, H . Human neutrophil cytoskeletal dynamics and contractility actively contribute to trans‐endothelial migration. PLoS One. 2013;8:e61377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Worthylake, RA , Burridge, K. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr Opin Cell Biol. 2001;13:569–577. [DOI] [PubMed] [Google Scholar]
- 30. Parker, H , Winterbourn, CC. Reactive oxidants and myeloperoxidase and their involvement in neutrophil extracellular traps. Front Immunol. 2012;3:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson KE, Boyle KB, Davidson K, et al. CD18‐dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood. 2008;112:5202–5211. [DOI] [PubMed] [Google Scholar]
- 32. Devi S, Li A, Westhorpe CL, Lo CY, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. 2013;19:107–112. [DOI] [PubMed] [Google Scholar]
- 33. Abboud FM, Harwani SC, Chapleau, MW . Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension. 2012;59:755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp, R . Differential effects of sympathetic nervous system and hypothalamic–pituitary–adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun. 2014;41:200–209. [DOI] [PubMed] [Google Scholar]
- 35. Myers MG, Norris JW, Hachniski V, Sole, MJ . Plasma norepinephrine in stroke. Stroke. 1981;12:200–204. [DOI] [PubMed] [Google Scholar]
- 36. Walter U, Kolbaske S, Patejdl R, et al. Insular stroke is associated with acute sympathetic hyperactivation and immunodepression. Eur J Neurol. 2013;20:153–159. [DOI] [PubMed] [Google Scholar]
- 37. Rohweder G, Ellekjær H, Ø Salvesen, Naalsund E, Indredavik, B . Functional outcome after common poststroke complications occurring in the first 90 days. Stroke. 2015;46:65–70. [DOI] [PubMed] [Google Scholar]
- 38. Langhorne P, Stott D, Robertson L, et al. Medical complications after stroke a multicenter study. Stroke. 2000;31:1223–1229. [DOI] [PubMed] [Google Scholar]
- 39. Wartenberg KE, Stoll A, Funk A, Meyer A, Schmidt JM, Berrouschot, J . Infection after acute ischemic stroke: risk factors, biomarkers, and outcome. Stroke Res Treat 2011;2011:830614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bellinger, DL , Lorton, D. Autonomic regulation of cellular immune function. Auton Neurosci. 2014;182:15–41. [DOI] [PubMed] [Google Scholar]
- 41. Karaszewski JW, Reder AT, Anlar B, Arnason, GW . Increased high affinity beta‐adrenergic receptor densities and cyclic AMP responses of CD8 cells in multiple sclerosis. J Neuroimmunol. 1993;43:1–7. [DOI] [PubMed] [Google Scholar]
- 42. Prenner L, Sieben A, Zeller K, Weiser D, Haberlein, H . Reduction of high‐affinity beta2‐adrenergic receptor binding by hyperforin and hyperoside on rat C6 glioblastoma cells measured by fluorescence correlation spectroscopy. Biochemistry. 2007;46:5106–5113. [DOI] [PubMed] [Google Scholar]
- 43. Kin, NW , Sanders, VM . It takes nerve to tell T and B cells what to do. J Leukoc Biol. 2006;79:1093–1104. [DOI] [PubMed] [Google Scholar]
- 44. Swanson MA, Lee WT, Sanders, VM . IFN‐gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. 2001;166:232–240. [DOI] [PubMed] [Google Scholar]
- 45. Borish L, Rosenbaum R, Albury L, Clark, S . Activation of neutrophils by recombinant interleukin 6. Cell Immunol. 1989;121:280–289. [DOI] [PubMed] [Google Scholar]
- 46. Fielding CA, McLoughlin RM, McLeod L, et al. IL‐6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol. 2008;181:2189–2195. [DOI] [PubMed] [Google Scholar]
- 47. Arranz L, Guayerbas N, De la Fuente, M . Impairment of several immune functions in anxious women. J Psychosom Res. 2007;62:1–8. [DOI] [PubMed] [Google Scholar]
- 48. Arranz L, de Vicente A, Muñoz M, De la Fuente, M . Impaired immune function in a homeless population with stress‐related disorders. NeuroImmunoModulation. 2009;16:251–260. [DOI] [PubMed] [Google Scholar]
- 49. Gleeson, M. Immune function in sport and exercise. J Appl Physiol. 2007;103:693–699. [DOI] [PubMed] [Google Scholar]
- 50. Nieman, DC. Is infection risk linked to exercise workload? Med Sci Sports Exerc. 2000;32:S406‐S411. [DOI] [PubMed] [Google Scholar]
- 51. Robson P, Blannin A, Walsh N, Castell L, Gleeson, M . Effects of exercise intensity, duration and recovery on in vitro neutrophil function in male athletes. Int J Sports Med. 1999;20:128–130. [DOI] [PubMed] [Google Scholar]
- 52. Smith, J , Pyne, D. Exercise, training, and neutrophil function. Exerc Immunol Rev. 1996;3:96–116. [PubMed] [Google Scholar]
- 53. van de Weert–van PB, de Vrankrijker AM, Fentz J, et al. Effect of long‐term voluntary exercise wheel running on susceptibility to bacterial pulmonary infections in a mouse model. PLoS One. 2013;8:e82869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seki Y, Sahara Y, Itoh E, Kawamura, T . Suppressed neutrophil respiratory burst in patients with haemorrhagic stroke. J Clin Neurosci. 2010;17:187–190. [DOI] [PubMed] [Google Scholar]
- 55. Ruhnau J, Schulze K, Gaida B, et al. Stroke alters respiratory burst in neutrophils and monocytes. Stroke. 2014;45:794–800. [DOI] [PubMed] [Google Scholar]
- 56. Lachmandas E, Boutens L, Ratter JM, et al. Microbial stimulation of different Toll‐like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. 2016;2:16246. [DOI] [PubMed] [Google Scholar]
- 57. Stienstra R, Netea‐Maier RT, Riksen NP, Joosten LAB, Netea, MG . Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. 2017;26:142–156. [DOI] [PubMed] [Google Scholar]
- 58. Chamorro A, Horcajada J, Obach V, Vargas M, Revilla M, Torres F, Cervera A, Planas AM, Mensa, J . The early systemic prophylaxis of infection after stroke study a randomized clinical trial. Stroke. 2005;36:1495–1500. [DOI] [PubMed] [Google Scholar]
- 59. Schwarz S, Al‐Shajlawi F, Sick C, Meairs S, Hennerici, MG . Effects of prophylactic antibiotic therapy with mezlocillin plus sulbactam on the incidence and height of fever after severe acute ischemic stroke. The Mannheim Infection in Stroke Study (MISS). Stroke. 2008;39:1220–1227. [DOI] [PubMed] [Google Scholar]
- 60. Offner H, Vandenbark A, Hurn, P . Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience. 2009;158:1098–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vogelgesang A, Becker K, Dressel, A . Immunological consequences of ischemic stroke. Acta Neurol Scand. 2014;129:1–12. [DOI] [PubMed] [Google Scholar]
- 62. Sykora M, Siarnik P, Diedler J, Collaborators, VA . Beta‐blockers, pneumonia, and outcome after ischemic stroke: evidence from Virtual International Stroke Trials Archive. Stroke. 2015;46:1269–1274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MOVIE 1. Neutrophil behavior in LysM‐eGFP mice during fMLP superfusion. Intravital spinning‐disk confocal microscopy was utilized to examine the behavior of GFP+ neutrophils in the cremasteric microvasculature of LysM‐eGFP mice. The movie was taken at 60 min following 30 μM fMLP superfusion. The time lapse was recorded at 15 fpm and exported to video at 10 fps. Scale bar = 50 μm
MOVIE 2. Neutrophil behavior in LysM‐eGFP mice during fMLP superfusion in the presence of NA. Intravital spinning‐disk confocal microscopy was utilized to examine the behavior of GFP+ neutrophils in the cremasteric microvasculature of LysM‐eGFP mice. The movie was taken at 60 min following 30 μM fMLP superfusion in the presence of 10−5 M NA. The time lapse was recorded at 15 fpm and exported to video at 10 fps. Scale bar = 50 μm
SUPPLEMENTAL FIGURES