

Abstract
The HIV fusion inhibitor T20 has been approved to treat those living with HIV/AIDS, but treatment gives rise to resistant viruses. Using combinatorial phage‐displayed libraries, we applied a saturation scan approach to dissect the entire T20 sequence for binding to a prefusogenic five‐helix bundle (5HB) mimetic of HIV‐1 gp41. Our data set compares all possible amino acid substitutions at all positions, and affords a complete view of the complex molecular interactions governing the binding of T20 to 5HB. The scan of T20 revealed that 12 of its 36 positions were conserved for 5HB binding, which cluster into three epitopes: hydrophobic epitopes at the ends and a central dyad of hydrophilic residues. The scan also revealed that the T20 sequence was highly adaptable to mutations at most positions, demonstrating a striking structural plasticity that allows multiple amino acid substitutions at contact points to adapt to conformational changes, and also at noncontact points to fine‐tune the interface. Based on the scan result and structural knowledge of the gp41 fusion intermediate, a library was designed with tailored diversity at particular positions of T20 and was used to derive a variant (T20v1) that was found to be a highly effective inhibitor of infection by multiple HIV‐1 variants, including a common T20‐escape mutant. These findings show that the plasticity of the T20 functional sequence space can be exploited to develop variants that overcome resistance of HIV‐1 variants to T20 itself, and demonstrate the utility of saturation scanning for rapid epitope mapping and protein engineering.
Keywords: phage display, peptide engineering, HIV‐1, fusion inhibitor, antiviral therapy
Abbreviations
- 5HB
five‐helix bundle
- 6HB
six‐helix bundle
- aMLV
amphotropic murine leukemia virus
- CHR
C‐terminal heptad repeat
- Fab
antigen‐binding fragment
- FP
fusion peptide
- HR
heptad repeat
- MPER
membrane proximal external region
- NHR
N‐terminal heptad repeat
- wt
wild type
Introduction
Attachment and fusion of HIV‐1 to host cells is mediated through its surface envelope glycoprotein (gp160). HIV‐1 gp160 is synthesized as a single polypeptide that assembles as a trimeric structure. Subsequent posttranslational cleavage by the host protease furin produces the non‐covalently associated surface subunit gp120 and the transmembrane subunit gp41 that cooperate to mediate attachment to and fusion with the host cell. Specifically, gp120 facilitates attachment of the virus to the host receptor CD4 and co‐receptor CXCR4 or CCR5, whereas gp41 contains the fusion machinery necessary for the merger of the viral and host membranes. Gp41 is comprised of multiple functional regions, including a hydrophobic fusion peptide (FP), an N‐terminal heptad repeat (NHR), a chain reversal region, a C‐terminal heptad repeat (CHR), a membrane proximal external region (MPER), a highly conserved transmembrane anchor, and a cytoplasmic domain [Fig. 1(a)]. The NHR domain is highly conserved,1 whereas the CHR domain is more diverse among HIV‐1 variants.2
Figure 1.
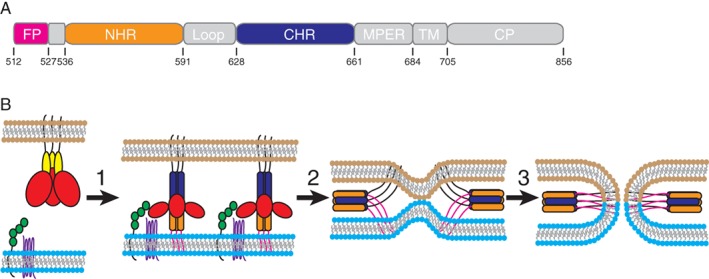
Gp41 and HIV‐1 fusion. (a) Schematic representation of the HIV‐1 gp41 sequence with regions labeled as follows: FP, fusion peptide (magenta); NHR, N‐terminal heptad repeat (orange); CHR, C‐terminal heptad repeat (blue); MPER, membrane proximal external region; TM, transmembrane anchor; CP, cytoplasmic domain. The residue numbers, defining the boundaries of the regions, correspond to positions in gp160 of HIV‐1HXB2. (b) The fusion of viral and host membranes mediated by gp41. The viral (brown) and host membranes (cyan) are shown at the top and bottom, respectively. (1) The binding of gp120 (red) to CD4 (green) and co‐receptor CXCR4 or CCR5 (purple) triggers a conformational change that releases gp120 from gp41 (yellow), which enables the insertion of the gp41 fusion peptides (magenta) into the host cell membrane, resulting in the exposure of the helical NHR (orange) pre‐hairpin intermediate. (2) The CHR helices (blue) zip up along the outside of the NHR coiled‐coil, which leads to the formation of 6HB and pulls the viral and host membranes together. (3) This results in the formation of a fusion pore, which completes the membrane fusion process. T20 competes with CHR for binding to NHR and thus prevents the conformational changes required for 6HB formation and membrane fusion.
According to the current model of HIV‐1 entry, 8–12 gp160 trimers sit on the viral surface in a metastable, high‐energy fusion competent state, in which the gp41 fusion subunit is “clamped” by gp120 [Fig. 1(b)]. Upon binding of gp120 to its receptor and co‐receptor, structural constraints on gp41 are released to allow the insertion of its hydrophobic FP into the host cell membrane to form a central trimeric NHR helical pre‐hairpin intermediate, which acts as a tether between the viral and target cell membranes. Subsequent conformational changes cause CHR to fold back into the central NHR trimer to form the final post‐fusion six‐helix bundle (6HB) structure, and formation of the energetically favorable 6HB drives the fusion process.
Synthetic peptides derived from either the NHR or CHR regions of gp41 were found to have potent antiviral activities.3, 4, 5, 6 These peptides are called fusion or entry inhibitors, because they prevent host‐virus membrane fusion by competitively binding to gp41 and blocking formation of the post‐fusion 6HB structure. T20 (also known as enfuvirtide or Fuzeon), a 36‐amino‐acid synthetic peptide derived from residues 638–673 of gp41, mimics the CHR and parts of the MPER and is an approved drug for HIV‐1 treatment.7 T20 competes with native gp41 CHR for binding to the central NHR trimer and thus prevents the conformational changes required for 6HB formation and fusion.8, 9 Elucidation of the core structure of the HIV‐1 fusion protein gp411, 10 and the structure of the regions linking up to the membrane anchors11 show how T20 associates with its target gp41 inner core mimetic 5HB and thus inhibits viral entry by blocking the formation of the pre‐hairpin fusion intermediate.
In cell culture, T20 is a potent inhibitor of HIV‐1 entry12 that acts synergistically with other fusion inhibitors or different HIV inhibitor classes.13, 14 Moreover, T20 is highly effective against HIV‐1 clinical isolates that are resistant to reverse transcriptase and protease inhibitors.7, 15 However, like other HIV‐1 drugs, prolonged treatment with T20 can induce drug resistance resulting in viral rebound in treated HIV/AIDS patients.16, 17, 18 Also, the short half‐life of T20 necessitated efforts to improve the design and pharmacokinetic profile of HIV peptidyl fusion inhibitors. Since the approval of T20, rational peptide design utilizing sequence information from HIV‐1, HIV‐2, and SIV isolates has shown some success in creating peptides with expanded neutralization properties and longer half‐lives.19, 20 One such peptide, T1249, underwent successful phase I/II clinical trials21, 22 but development was ultimately abandoned due to formulation difficulties. More recent efforts to take advantage of the demonstrated efficacy of these peptidyl fusion inhibitors have shown promise. For instance, conjugation of T20 and T1249 to a fatty acid moiety showed potent fusion inhibition.23 With new peptide delivery strategies and formulations being developed, the relevance of HIV peptidyl fusion inhibition to clinical practice may see a renaissance. Consequently, there continues to be a need for the development of further generations of HIV‐1 fusion inhibitors with improved drug resistance profiles.
To explore the likelihood of developing T20 variants that can inhibit T20‐resistant HIV‐1 strains, we surveyed the degree of structural plasticity within T20 by using a combinatorial saturation scanning approach. This approach has been applied previously to assess the structural and functional effects of all possible point mutations across large surfaces of several proteins, including human growth hormone,24 engineered autonomous heavy‐chain variable domains of human antibodies,25 the paratope of an antibody,26 and ubiquitin variants.27 The saturation scan provided information on the effects of every possible amino acid substitution across every position of T20. The scan results and structural information for gp41 fusion intermediates were utilized to design a tailored combinatorial library that was used to derive T20 variants that bound to 5HB, a mimetic of the gp41 NHR coiled‐coil region.28 One T20 variant was found to be highly effective in inhibiting infection by multiple HIV‐1 variants, including an isolate that was resistant to T20. Our findings show that the sequence space around T20 can be explored to identify variants that can overcome T20 resistance in HIV‐1 isolates, and such variants could be further developed as broadly neutralizing antiviral therapeutics for treatment of multidrug resistant HIV‐1.
Results and Discussion
Production of stable single‐chain 5HB and 6HB proteins
In the post‐fusion gp41 ectodomain core, three NHR helices form a central trimeric coiled‐coil. Three CHR helices pack in an antiparallel fashion into the grooves of the NHR trimer to form 6HB. In order to provide a suitable bait for phage display studies, soluble, single‐chain HIV‐1 gp41 5HB and 6HB were designed, as described,29 based on the simple fold of the native post‐fusion gp41 (Fig. S1). The close proximity of the N‐ and C‐termini from different chains in the native gp41 post‐fusion state allowed linkage to form a single‐chain protein that was readily amenable to expression and purification. 5HB consists of three NHR helices (residues 546–582) connected by short glycine‐serine flexible linkers to two CHR helices (residues 625–661), which leaves one CHR binding site available for interactions with phage‐displayed peptides. 6HB is similar to 5HB, but it contains an extra CHR helix to recapitulate the 6HB structure. 5HB was successfully refolded from inclusion bodies, while 6HB was expressed in the soluble fraction in Escherichia coli, and both proteins were purified to homogeneity in multi‐milligram quantities (Fig. S1).
Phage display of a T20‐Fab fusion protein
As T20 is a short peptide that is prone to degradation inside cells, we decided to display it in a monovalent format by fusion to the N‐terminus of the heavy chain of the antigen‐binding fragment (Fab) of a stable and well‐behaved antibody. We hypothesized that the large Fab protein would act as a chaperone to protect the T20 peptide from degradation and would thus facilitate efficient, monovalent display of T20 on the phage surface. For this purpose, we used a phagemid designed for the display of an anti‐maltose binding protein Fab fused to the N‐terminus of the C‐terminal domain of the M13 gene‐3 minor coat protein.30, 31 A phage ELISA confirmed robust display of the T20‐Fab fusion protein, as evidenced by strong signals for phage binding to immobilized 5HB and low background signals with immobilized 6HB as a post‐fusion mimic or with an irrelevant immobilized protein (Fig. S2).
Saturation scan of T20
To assess the contributions to binding of each of the 36 residues in T20, we utilized a phage display saturation scan to compare the relative abilities of the 20 genetically encoded amino acids to function at each position. Six separate phage‐displayed libraries were designed to collectively cover all 36 positions, with each library covering six positions within a continuous stretch of T20 primary sequence. Each position was diversified with a degenerate codon, NNK (N = A/C/G/T, K = G/C), which encoded for all 20 amino acids. Each constructed library contained at least 109 unique members, which provided reasonable coverage of the theoretical DNA sequence space (326 = 1.07 × 109) and exceeded the number of theoretical amino acid combinations (206 = 6.4 × 107). For quality control, approximately 20 clones from each naive library were sequenced, and the frequency of each amino acid type at each position was used to generate a sequence logo for the naïve libraries (Fig. S3). Although the limited number of sequences did not allow for in‐depth statistical analysis of amino acid frequencies, we did not observe significant occurrence of any particular amino acid at any of the 36 positions that were scanned. Thus, the naïve libraries were constructed as designed and a random distribution of amino acid sequences was achieved at all positions.
To determine the constraints on the T20 sequence imposed by recognition of 5HB, we cycled phage pools representing each library through four rounds of selection for binding to immobilized 5HB. From each library, we sequenced variants that bound specifically to 5HB by phage ELISA and obtained approximately 100 unique sequences in order to tabulate the occurrence of each amino acid type at each position to compile the saturation scan data set (Fig. 2). Cysteine and proline residues were scarce, occurring only 3 and 46 times, respectively, across all 36 positions in 576 unique sequences. The paucity of cysteine residues was likely due to their propensity to form disulfide bonds that could interfere with the native disulfides that are required for the correct folding and stability of the phage gene‐3 minor coat protein. Proline residues only appeared near the ends of the polypeptide and absence in the central region was likely due to the propensity of proline residues to disrupt α‐helices.
Figure 2.

The saturation scan data set for T20 binding to 5HB. At each position of T20 (wt), following selection for binding to 5HB, the percent occurrence of each amino acid was calculated after normalization for codon bias. The wt occurrences are boxed and conserved values (>25%) are shaded gray. The consensus lists the over‐represented amino acids (>25%).
We generated a sequence logo based on the amino acid frequency at each diversified position [Fig. 3(a)] and calculated bit scores to quantify Shannon entropy. The bit score measures the degree of randomness within a population and varies between 0 and 4.32 for completely random or completely conserved positions, respectively.34 Based on bit scores, eight positions were classified as highly conserved (>2), four positions were classified as moderately conserved (1.2–2), and the rest were classified as non‐conserved (<1.2). Sequence conservation was poor at the first four and last 10 positions. The tryptophan‐rich C‐terminal region of T20 binds to lipids and likely interacts with the host membrane during fusion but does not interact with the NHR.28, 35, 36, 37 A recent crystal structure of gp41 including the FP and MPER also suggested that there is no regular coiled‐coil interaction between the C‐terminal region of T20 and its N‐terminal counterpart (FP proximal region), but instead, the aromatic residues in the C‐terminal region are believed to form a hydrophobic patch that facilitates membrane fusion by interacting with host membrane.11 These observations are consistent with the lack of sequence conservation in the saturation scan of the C‐terminal region, as we selected for binding to the 5HB polypeptide but not to lipid membrane.
Figure 3.
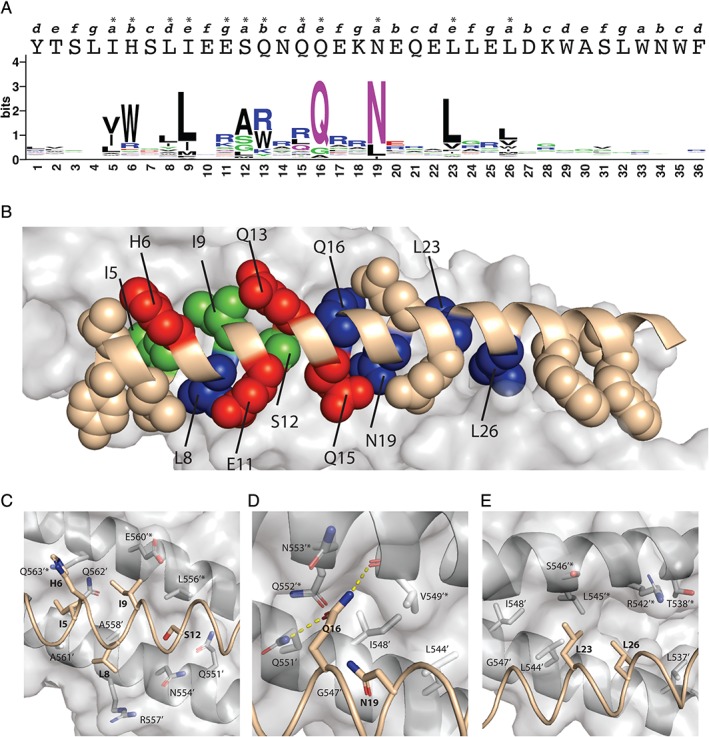
Structural rationalization of the T20 saturation scan. (a) Sequence logo plot of the normalized fraction of residues occurring at each scanned position of T20 following selection for binding to 5HB. In the sequence logo, amino acids are colored according to their chemical properties: green, polar (G, S, T, Y, C, Q, N); blue, basic (K, R, H); red, acidic (D, E); and black, hydrophobic (A, V, L, I, P, W, F, M). The primary sequence of T20 is shown above the sequence logo in upper case letters. The heptad position for each residue is shown above the T20 sequence in lower case italics and asterisks (*) indicate conserved residues with bit values >1.2. (b) Structure of post‐fusion gp41 core (PDB ID code 2X7R). The structure corresponding to 5HB is shown as a gray surface, and that corresponding to the T20 main chain is shown as a ribbon colored in wheat. The side chain atoms of 20 residues in T20 that are within contact distance to 5HB are shown as spheres colored wheat, blue, green, or red if they were non‐conserved or conserved as wt, homologous, or nonhomologous residues, respectively. (c–e) Details of the molecular interactions between T20 and 5HB, with close ups of: (c) the amino‐terminal hydrophobic epitope, (d) central hydrophilic dyad, and (e) carboxyl‐terminal hydrophobic dyad. Hydrogen bonds are represented by dashed yellow lines. Figure 3(a) was generated using WebLogo32 and Figure 3(b‐e) was generated using Pymol.33
The 12 positions that exhibited high or moderate conservation in the saturation scan were located among 22 positions spanning residues 5–26, inclusive [Fig. 3(a)]. The distribution of these conserved positions roughly fits with a 4‐3 heptad repeat (HR) pattern (Fig. S4) that is consistent with the 6HB structure of gp41. In the classic 6HB structure model (Fig. S5), three NHRs pack as an interior parallel three‐helix bundle and residues at the e′ and g′ positions from two adjacent helices form exposed hydrophobic grooves (positions in NHR are labeled as “prime” to distinguish them from positions in CHR). The grooves make intimate contacts with residues at the a and d positions of three outer CHRs, which pack against the NHR coiled‐coil trimer in an antiparallel orientation.1, 5, 36 Residues at NHR positions b′ and c′ interact with residues at CHR position e and g, respectively. However, contacts between inter‐helical positions other than these typical pairs are often observed.1 In the saturation scan of positions 5–26, all a and e positions were conserved and four out of six b and d positions were conserved, while in contrast, no c or f positions were conserved and only one out of three g positions were conserved (Fig. S4). These results suggest that the helix face composed of positions a, b, d, and e is buried in the helical bundle, whereas the helix face composed of positions c, f, and g is exposed to solvent.
Analysis of previous crystal structures indicated that NHRs and CHRs were not in perfect antiparallel alignment, but instead, were positioned with an oblique orientation of CHRs relative to NHRs, so that the CHR is slightly tilted toward the left or right NHR helix at its amino or carboxyl terminus, respectively.1 Consequently, residues at CHR positions b and e are more buried than those in the classic structure, whereas residues at CHR position g are more solvent exposed. As a result, CHR positions b and e can make contact with NHR, and this may explain the sequence conservation at these positions of T20 [Fig. 3(a), S4]. Taken together, our saturation scan results fit well with the HIV‐1 6HB structure.
Structural rationalization of T20 saturation scan data
Inspection of the core structure of gp411, 10 allowed us to rationalize the saturation scanning data. Binding of T20 to 5HB places 20 of 36 T20 residues within 4 Å of 5HB residues and results in burial of 1318 Å2 of surface area, which represents more than one‐third of the total surface area of T20 (3812 Å2).11 The saturation scan revealed 12 conserved positions in the central region of T20 [Fig. 3(a)]. Notably, the residues at these 12 positions reside in the hydrophobic groove formed by two adjacent NHRs in the 5HB. Residues at eight of the 12 conserved positions are highly buried, with an average solvent accessible area of 5%, and five of these (positions 8, 16,19, 23, and 26) are conserved as the wild‐type (wt), whereas the other three are conserved as residues that are homologous to the wt (positions 5, 9, and 12). Residues at the other four conserved positions (6, 11, 13, and 15) are less buried, with an average solvent accessible area of 45%, and these are conserved as residues that are nonhomologous with the wt [Fig. 3(b)]. Mapping of the 12 conserved positions onto the gp41 core structure revealed that they cluster into three distinct epitopes: a hydrophobic epitope near the amino terminus (positions 5, 8, 9, and 12); a hydrophilic dyad at the center of the helix (positions 16 and 19); and a hydrophobic dyad near the carboxyl terminus (positions 23 and 26) [Fig. 3(c–e)].
In the amino‐terminal hydrophobic epitope, residues at positions 5, 8, 9, and 12 reside at the 5HB‐binding surface and are highly buried. By interacting with residues from the two adjacent NHRs, they form a hydrophobic core at the T20–5HB interface. Leu8 is involved in hydrophobic interactions with Ala558′ and the aliphatic portion of the Arg557′ side chain (“′” denotes NHR residues) [Fig. 3(c)]. In the saturation scan, position 8 was conserved as the wt, whereas positions 5 and 9 were conserved as valine and leucine residues, respectively, which were homologous to the wt isoleucine at these positions. These homologous substitutions may fine‐tune the fit within the hydrophobic groove formed by the two NHRs of 5HB. At position 12, alanine is preferred over the wt serine residue, and glycine also occurred with significant frequency. Substitution of a hydrophilic serine residue with a small hydrophobic alanine residue may increase hydrophobicity at this position while retaining small size for hydrophobic packing, suggesting that a small hydrophobic residue in this position may be advantageous to permit close contact. Position 6, occupied by a hydrophilic histidine in the wt, was conserved as a larger and more hydrophobic tryptophan residue in the saturation scan. This strong preference indicated that although position 6 was relatively solvent accessible, an increase in hydrophobicity in this position may enable more extensive intermolecular contacts to expand the hydrophobic patch and thereby increase the interaction energy. Overall, our analysis indicated that the amino‐terminal hydrophobic epitope was formed by three hydrophobic residues that are conserved as wt or homologous counterparts, and this hydrophobic patch could be expanded to improve T20–5HB association by introduction of large or small hydrophobic side chains at positions 6 and 12, respectively.
At the center of the T20 helix, the hydrophilic residues Gln16 and Asn19 form a second cluster at the binding interface. Interestingly, these two residues are the most highly conserved as the wt among all 36 positions surveyed in the saturation scan [Figs. 2 and 3(a)], suggesting precise geometry and chemistry are required at these interface positions. Gln16 is involved in hydrogen bond interactions with residues from both adjacent NHRs [Fig. 3(d)]. The side chain of Gln16 forms hydrogen bond interactions with the side chain of Gln551′ on one NHR and the main chain of Val549′* on the neighboring NHR (“*” denotes the second NHR). Gln16 is also involved in hydrophobic interactions with Gln551′, Q552′*, Asn553′*, and Val549′*. Hydrophobic interactions also occur between Asn19 and NHRs, forming a continuous hydrophobic patch together with Gln16. The fact that Gln16 and Asn19 make multiple precise interactions with 5HB likely explains the high conservation of wt sequence at these positions. Indeed, conservation of Asn19, as well as Leu26 (see below), is consistent with previous reports that Asn19 and Leu26 are critical for gp41 core structure stabilization and membrane fusion, and that substitutions at these positions were detrimental for T20 binding to 5HB.1, 28, 38
Near the carboxyl‐terminal end of the peptide, leucine residues at positions 23 and 26 form a second hydrophobic epitope. These residues are involved in hydrophobic interactions with several residues from the two NHRs, forming a large hydrophobic interface [Fig. 3(e)]. Similar to the hydrophobic epitope near the amino terminus, the carboxyl‐terminal hydrophobic epitope makes favorable but rather nonspecific interactions with 5HB. The hydrophobic epitopes are separated by a central dyad of two hydrophilic side chains approximately one helix turn apart, which make precise interactions that cannot be accommodated by other side chains. Aligning a combination of hydrophobic and hydrophilic epitopes along a helical structure for specific protein recognition is not uncommon in nature, as we observed a similar scenario in the M13 bacteriophage coat.39, 40
The saturation scan also showed preference for nonhomologous substitutions at three other positions (11, 13, and 15). Arg was highly prevalent at these positions, and notably, was also favored at four less conserved positions (14, 17, 18, and 25). These residues reside on the solvent exposed face of the helix, aside for position 15, which resides at the d position of the HR but still shows significant solvent exposure due to the tilted orientation of T20 relative to NHRs. 5HB is overall negatively charged (pI = 5.61), and thus, it is likely that T20 variants harboring positively charged Arg residues at positions that do not directly contact 5HB may nonetheless be enriched due to favorable long‐range electrostatic interactions. Taken together, the saturation scan analysis reveals that the interaction between T20 and 5HB is mediated by three distinct functional epitopes: amino‐ and carboxyl‐terminal hydrophobic epitopes that make nonspecific interactions, separated by a central dyad of hydrophilic residues that make precise interactions with 5HB.
Selection of T20 variants optimized for binding to 5HB
The shotgun scan analysis showed that one face of the central region of the T20 α‐helix is required for binding to 5HB. Although sequence conservation was observed across this face, striking functional plasticity within the T20 structural epitope for 5HB binding was also evident: multiple residue types could be accommodated even at the most conserved buried positions (Figs. 2 and 3), and similar plasticity was observed previously for the binding of human growth hormone to its receptor.24 These results suggest that the T20 interface that binds to 5HB is rather plastic and tolerant to substitutions, and moreover, some substitutions may enhance affinity for 5HB. To explore this possibility, we used the saturation scanning data as a guide to construct a tailored T20 library designed to favor substitutions that could enhance binding to 5HB (Table S1). The first four and last 10 residues were fixed as wt, since no significant consensus was observed in the shotgun scan data set. Across positions 5–26, 19 positions were subjected to tailored randomization with particular degenerate codons chosen to encompass residues that were highly prevalent in the shotgun scan data set (although the degeneracy of the genetic code necessitated the occurrence of other residues at some positions). The theoretical diversity of the designed library was 1.9 × 1014, whereas the actual diversity of the library was 5.6 × 109.
The library was cycled through four rounds of binding selections with immobilized 5HB under stringent conditions by successively lowering the concentration of immobilized antigen and more intensive washing at each subsequent round. 6HB was also included as a soluble competitor to ensure high specificity of potential T20 variants for 5HB relative to 6HB. Sequencing of 96 clones that exhibited strong binding to 5HB but not to 6HB by phage ELISA revealed 18 unique T20 variants (Fig. S6), and 12 of these exhibited 5HB binding comparable to or better than T20 in competitive phage ELISA and were therefore chosen for characterization as synthetic peptides. Synthesis failed for two peptides, and thus, 10 T20 variants were tested as synthetic peptides in HIV‐1 viral neutralization assays.
Neutralization of HIV‐1 by T20 variants
Neutralization assays were performed on a modified reference panel of pseudotyped viruses that represent the diversity present in circulating strains of moderately neutralization‐resistant HIV‐1.41 These viruses express full‐length HIV‐1 gp160 from a circulating recombinant form (CRF, 92TH021) and from Clades A (94UG103), B (92BR020), and C (IAVI C22). The panel also included HIV‐1 laboratory strains that are easy to neutralize (NL43 and JRCSF) and a common T20‐escape mutation on the JRCSF background (JRCSF‐GIA).42 An envelope glycoprotein from amphotropic murine leukemia virus (aMLV) was added as a negative control that is not inhibited by T20.
The 10 T20 variants exhibited varying degrees of neutralization across clades (Table 1, Figs. 4 and S7). For the NL43 lab‐adapted isolate, six T20 variants (T20v1‐6) exhibited improved neutralization compared with T20. The greatest improvements were seen for T20v1 and T20v2, which exhibited approximately fivefold lower IC50 values than T20. Three variants (T20v1‐3) exhibited approximately twofold better neutralization against the clade B isolate 92BR020. Moreover, four variants (T20v1‐4) that showed improved neutralization of NL43 and 92BR020 also displayed improved neutralization activity against the JRCSF‐GIA isolate that contains a common T20‐escape mutation. Strikingly, T20v1 neutralized JRCSF‐GIA more than an order of magnitude more potently than did T20, whereas potencies against JRCSF were virtually identical. Against clade A (94UG103), clade C (IAVI C22), CRF (92TH021), and JRCSF, the variants exhibited similar or poorer neutralization potencies compared with T20.
Table 1.
T20 variant sequences and IC50 values for viral neutralization
| Peptide | Sequencea | IC50 (μg/mL)b | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | NL43 | JRCSF | 94UG103 | 92BR020 | IAVI C22 | 92TH021 | JRCSF‐GIA | |
| T20 | I | H | S | L | I | E | E | S | Q | N | Q | Q | E | K | N | E | Q | E | L | L | E | L | 0.37 | 0.09 | 0.03 | 0.30 | 0.03 | 0.02 | 3.58 |
| T20v1 | · | R | E | I | L | V | · | · | R | I | · | · | · | · | · | · | R | · | · | R | D | I | 0.06 | 0.10 | 0.06 | 0.17 | 0.12 | 0.06 | 0.29 |
| T20v2 | · | · | · | I | · | · | · | · | R | · | R | · | · | · | · | · | · | A | · | · | · | · | 0.06 | 0.06 | 0.03 | 0.17 | 0.09 | 0.03 | 1.05 |
| T20v3 | L | R | · | I | · | · | · | G | R | · | · | · | · | · | · | · | · | A | · | · | · | · | 0.13 | 0.08 | 0.03 | 0.19 | 0.08 | 0.03 | 1.34 |
| T20v4 | · | R | · | I | · | · | · | · | R | · | · | · | · | · | · | · | · | K | · | · | · | V | 0.15 | 0.12 | 0.04 | 0.22 | 0.50 | 0.03 | 1.80 |
| T20v5 | L | R | · | · | · | · | · | · | R | · | L | · | · | · | · | · | · | A | · | · | · | V | 0.17 | 0.15 | 0.14 | 0.50 | 0.29 | 0.13 | 5.69 |
| T20v6 | L | W | · | I | · | · | · | G | R | · | L | · | · | · | · | · | · | K | · | · | · | · | 0.16 | 0.14 | 0.21 | 0.62 | 0.59 | 0.10 | 5.68 |
| T20v7 | · | W | K | V | L | N | D | A | R | E | · | · | · | N | · | Q | E | T | · | V | · | I | 0.37 | 0.16 | 0.45 | 1.12 | 0.23 | 0.07 | 5.10 |
| T20v8 | · | R | E | I | M | N | K | · | W | G | · | · | R | R | · | · | G | T | · | A | · | I | 0.40 | 0.24 | 0.31 | 0.53 | 1.11 | 0.24 | 10.2 |
| T20v9 | · | R | E | · | · | S | N | A | R | T | · | · | T | D | · | · | E | S | · | R | N | V | 1.20 | 0.34 | 0.41 | 0.82 | 1.09 | 0.35 | 16.8 |
| T20v10 | · | W | · | I | · | I | D | G | R | E | · | · | R | D | · | · | G | V | · | A | D | · | 23.3 | 0.50 | 3.35 | 12.0 | 0.45 | 0.11 | 24.4 |
Positions that were conserved as the wt sequence are indicated by dots.
The IC50 of each T20 variant for each pseudotyped virus was defined as the concentration of peptide that inhibited virus infection by 50%. Bold values indicate IC50 < 0.30 μg/mL.
Figure 4.
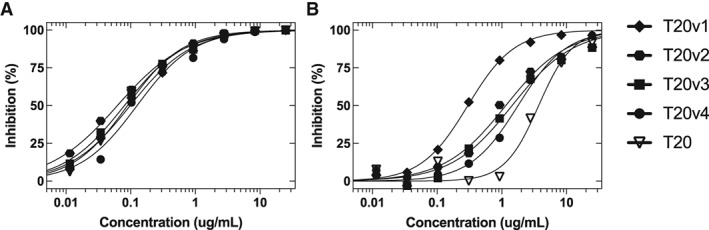
Neutralization of HIV‐1 by T20 and variants. (a) JRCSF or (b) JRCSF‐GIA pseudotyped viruses were preincubated with various concentrations of the indicated inhibitors (x‐axis) prior to measuring infectivity, and percent inhibition (y‐axis) was normalized to infectivity in the absence of inhibitor. Complete inhibition curves for the T20 variants and controls are presented in Figure S7. IC50 values are reported in Table 1.
Binding affinities for 5HB estimated by phage ELISA did not exactly match potencies observed for viral neutralization, and some peptides showed strong binding to 5HB but poor neutralization potencies. The cause of this discrepancy is not clear, but we reason that there may be two issues. First, the region of gp41 that must be targeted for viral inhibition is only exposed transiently,20 and 5HB may not be a perfect mimic of the actual target for T20 activity. Second, fusion of T20 variants to large phage particles may alter binding activity in comparison with free peptides. Nevertheless, despite these caveats, our results highlight the value of exhaustive analysis of functional sequence space for designing biologic inhibitors of highly variable pathogens such as HIV‐1. In particular, the discovery of T20v1, which showed expanded range for inhibition of infection by one T20‐escape strain, demonstrates the power of this comprehensive approach to explore and optimize protein sequence space.
Conclusions
We applied saturation scanning mutagenesis to dissect the functional epitope of peptide T20 for binding to the gp41 prefusogenic mimetic 5HB. The scan revealed three distinct functional epitopes: two hydrophobic epitopes separated by a hydrophilic dyad. The scan also revealed a strikingly plastic interface in which even buried positions could usually accommodate multiple residue types, and we took advantage of this feature to design an affinity maturation library in which diversity at functional residues was restricted to common sequences from the scan. This library yielded multiple highly diverse variants, which nonetheless retained the ability to potently inhibit multiple HIV‐1 isolates. Importantly, the variant T20v1 exhibited broadened specificity, as it was able to neutralize an HIV‐1 isolate that was resistant to T20. Our results open promising avenues for developing peptidic HIV‐1 fusion inhibitors with enhanced potencies and broadened specificities, which in turn could expand the toolbox for treatment of HIV‐1 isolates that resist current treatments. Moreover, our general strategies for comprehensive and systematic exploration of functional sequence space could be applied to understand and optimize many other therapeutic proteins.
Materials and Methods
Expression, refolding, and purification of recombinant HIV‐1 gp41 5HB
Generation of a single‐chain recombinant HIV‐1 gp41 molecule for the 5HB gp41 NHR/CHR helices has been described previously.29 The construct contains alternating copies of residues 546–582 of NHR, and residues 625–661 of CHR from HIV‐1 gp41 [residue numbers corresponding to their positions in gp160 of the HIV‐1HXB2 (GenBank™ accession number: AAB50262)]. Linker regions were inserted between the helices such that NHR was linked to CHR by a GGSGG motif (L1) and CHR was linked to NHR by a GSSGGSGSG motif (L2). The DNA fragment encoding NHR‐L1‐CHR‐L2‐NHR‐L1‐CHR‐L2‐NHR was commercially synthesized, subcloned into a pET46 Ek/LIC vector, and expressed in BL21 (DE3) E. coli cells. Cell cultures were grown to an OD600 = 0.6 at 37°C and induced with a final concentration of 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG). The cultures were harvested 4 h post‐induction at 37°C. The E. coli cells were resuspended in 30 mL of 50 mM Tris–HCl pH 8.0, 300 mM NaCl, and 20 mM imidazole (Buffer A), lysed using a hydraulic cell disruption system (Constant Systems) and centrifuged at 30,000g for 40 min. The resulting pellet was washed once with refolding buffer (6M guanidine‐HCl and 100 mM Tris–HCl, pH 8.0), resuspended in refolding buffer, and incubated overnight at room temperature with constant stirring. A final high‐speed centrifugal clarification step removed any remaining insoluble debris and the resolubilized recombinant HIV‐1 gp41 5HB was stored at −80°C. Prior to initiating the refolding process, recombinant HIV‐1 gp41 5HB concentration was determined by absorbance at 280 nm and purity was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) using the method of Pepinsky.43 HIV‐1 gp41 5HB was diluted to 0.5 mg/mL in refolding buffer and dialyzed (3500 MWCO) against 100 mM glycine pH 3.5 overnight at 4°C with gentle stirring. Refolded recombinant HIV‐1 gp41 5HB was then dialyzed overnight into PBS and further purified using a Superdex‐200 XK 16/40 prep grade column equilibrated in PBS.
Expression and purification of HIV‐1 gp41 6HB
A single‐chain gp41 6HB was generated in a similar fashion to the HIV‐1 gp41 5HB. Here, the NHR and CHR helices have slightly extended boundaries, with 6HB‐NHR spanning residues 530–582 and the 6HB‐CHR spanning residues 625–669. DNA encoding for NHR‐L1‐CHR‐L2‐NHR‐L1‐CHR‐L2‐NHR‐L1‐CHR was synthesized, subcloned into pET46 Ek/LIC, and expressed in BL21 (DE3) cells. Cell cultures were grown to an OD600 = 0.6 at 37°C and protein expression was induced with a final concentration of 0.5 mM IPTG overnight at 18°C. Pelleted cells were resuspended in Buffer A prior to lysis using a hydraulic cell disruption system. Clarified lysate was applied over Ni‐NTA affinity resin. HIV‐1 gp41 6HB was stepwise eluted with 100–500 mM imidazole in Buffer A and further purified by size‐exclusion chromatography using a Superdex‐200 XK 16/40 prep grade column equilibrated in PBS. Protein concentration and purity were determined by absorbance at 280 nm and SDS‐PAGE.
Construction of T20 display phagemid
T20 is fused to a well‐behaved Fab fragment and displayed on the surface of phage particles. The parental Fab display phagemid used displays an anti‐maltose binding protein Fab fused to the N‐terminus of the C‐terminal domain of the gene‐3 minor coat protein (cP3).30 The T20‐Fab fusion was constructed by fusing T20 to N‐terminus of the Fab VH domain (T20‐Fab). For generation of the T20‐Fab phagemid, DNA encoding T20 peptide was assembled by self‐priming PCR44 using oligonucleotides and cloned into HP153 using the MluI and BglII restriction sites. In this T20‐Fab construct, the T20 peptide is flanked with spacer linkers: ASSG at the N‐terminus and GSSGSGSG at the C‐terminus.
Saturation scanning mutagenesis
Phage‐displayed T20 saturation scanning libraries were constructed, sorted, and analyzed by using a hexahistidine‐containing 6HB as the pre‐absorption reagent, and 5HB as the immobilized bait for biopanning, as described previously45 with minor modifications. Briefly, for each saturation scanning library, a stop template, which contains TAA stop codons at the region targeting for mutation, was constructed based on the T20‐Fab phagemid and used as the template for site‐directed mutagenesis method.46, 47 Each of the saturation scanning phage libraries was designed by using one mutagenic oligonucleotide to replace the stop codons with degenerate codons, NNK (N = A/C/G/T, K = G/C), which encoded for all 20 natural amino acid residues at each position. Six libraries were designed with each covering a region of six consecutive residues for introducing the mutations into the T20 sequence. The libraries representing diversity at each region were handled separately and each was independently pretreated with immobilized 6HB to remove nonspecific binders followed by biopanning against the immobilized target 5HB. After four rounds of selections for binders, individual clones were picked and grown in a 96‐well plate format. Culture supernatants were used directly in phage ELISA45 to identify T20 variant‐phage that bound to HIV‐1 gp41 5HB. Direct phage ELISAs and competitive phage ELISAs were performed as described previously.47 ELISA‐positive binding clones from each selection were sequenced. At least 100 unique clones were obtained for each selection and subjected to statistical analysis. The occurrence of each amino acid type at each position was normalized for codon bias in the degenerate codon NNK (e.g., the occurrence of a particular amino acid was divided by the number of unique codons encoding this amino acid in the degenerate codon NNK). The normalized amino acid fraction at each position was plotted graphically as a sequence logo32 and tabulated as described previously.25
Affinity optimization library of T20 based on saturation scanning mutagenesis
Based on the results from saturation scanning mutagenesis, one library was constructed for affinity maturation of the parental T20. In the library, tailored mutations (Table S1) were introduced into in total 19 positions from positions 5 to 26 excluding positions 16, 19, and 23. Binding selections with this library was performed with 5HB immobilized on Nunc 96‐well Maxisorp immunoplates under more stringent conditions. The concentrations of immobilized antigen were successively lowered from 5 μg/mL in round 1, to 2 μg/mL in rounds 2 and 3, and to 1 μg/mL in round 4. The number of washes with PT buffer after phage incubation was increased from 8 in round 1, 10 in rounds 2 and 3, and to 12 in round 4. In rounds 2–4, 10 μg/mL of gp41 6HB was added to the phage solution prior to incubation with immobilized 5HB as a competitor for removal of nonspecific clones. After four rounds of phage selection, a single‐point competitive phage ELISA47 was used to rapidly estimate the affinities of phage‐displayed anti‐5HB clones obtained from the last round of selection. The fraction of T20 variant‐displayed phage uncomplexed with solution phase 5HB was calculated by dividing the A450 signal in the presence of 50 nM of solution phase 5HB by the A450 in the absence of 5HB.
Peptide synthesis
T20 variants identified from phage selection (Table 1) were commercially synthesized by GenScript (Piscataway, NJ). All peptides were acetylated at the N termini and amidated at the C‐termini. The peptides were purified to homogeneity (>95% purity) by high‐performance liquid chromatography and peptide identities were confirmed by ESI mass spectrometry. Peptide concentrations were determined by ultraviolet absorption at 280 nm. A T20 (enfuvirtide/Fuzeon) synthesized directly by Trimeris/Roche was obtained through the NIH AIDS Reagent Program (Cat. No. #9845) and used as a control.
HIV‐1 neutralization assays
HIV‐1 neutralization by T20 variants was measured using a recombinant virus assay with a single cycle of infectivity.48, 49 Briefly, a replication‐defective retroviral vector containing a firefly luciferase reporter gene is co‐transfected in HEK293T cells with a second plasmid that encodes a HIV‐1 envelope gp160 sequence. HIV‐1 gp160 from five different isolates were utilized in this study (94UG103, 92BR020, IAVI‐C22, 92TH021, and JRCSF‐GIA). The supernatant containing the pseudotyped viruses were harvested and incubated with serial dilutions of T20 variants for 1 h and subsequently used to infect human U87 cells expressing CD4, CCR5, and CXCR4. The ability of T20 and T20 variants to neutralize HIV‐1 infectivity was assessed by measuring luciferase activity 72 h post‐viral infection. Moreover, two panels of controls were performed: a virus control and T20 control. The virus control panel consists of two neutralization‐sensitive lab strain gp160 (NL4‐3 and JRCSF), and negative control retroviral envelope glycoprotein from aMLV. The aMLV envelope glycoprotein is used as a specificity control, as this envelope glycoprotein is not lentiviral in origin and is not inhibited by T20. T20 obtained directly from Roche/Trimeris was used as positive control. Neutralization efficacy was presented as the concentrations of the peptides that inhibited the viral infection by 50%. The assays were performed by Monogram Biosciences (South San Francisco, CA).
Supporting information
Figure S1 Purification and characterization of HIV‐1 gp41 5HB and 6HB. (A) Schematic of the HIV‐1 gp41 5HB single chain construct. GGSGG and GSSGGSGSG linkers are used between the NHR‐CHR and CHR‐NHR, respectively. Superdex‐200 XK 16/40 prep grade size‐exclusion chromatograms of (B) 5HB and (C) 6HB. Coomassie‐stained SDS‐PAGE analysis of the eluted protein peaks is shown in the inset panel.
Figure S2: Assessment of T20‐Fab display by direct binding phage ELISA. T20 displayed as an amino terminal fusion to Fab VH (T20‐Fab) was tested in phage ELISA for binding to 5HB, 6HB and an irrelevant control protein (CTRL, a carboxyl terminally hexahistidine tagged human SIRPa IgV protein). The display with Fab only was utilized in phage ELISA as a control. The test was performed in triplicate and repeated three times independently. Data are presented as average values ± SD.
Figure S3: The distribution of amino acid types at each position of T20 in the naïve libraries. Normalized fraction of residues occurring at each randomized position of T20 in the naive libraries is plotted as a sequence logo. Amino acids are colored according to their chemical properties: green for polar residues (G,S,T,Y,C,Q,N), blue for basic residues (K,R,H), red for acidic residues (D,E) and black for hydrophobic residues (A,V,L,I,P,W,F,M). The primary sequence of T20 is shown above the sequence logo.
Figure S4: Helical wheel projection of T20 viewed down the helix axis from the N‐ to C‐terminus. Buried and partially buried positions according to classic 6HB are colored as follows: positions a and d, yellow; positions e and g, blue. Residue numbers 5–26 are shown at the corresponding heptad positions and are colored according to bit values derived from amino acid frequencies (Fig. 2), as follows: <1.2, gray; 1.2–2, green; >2, red.
Figure S5: Helical wheel projection of classic 6HB structure model viewed down the NHR axis from the N‐ to C‐terminus. Buried and partially buried positions are colored yellow and blue, respectively. Three NHRs pack together to form a central trimeric coiled coil with interactions between positions a’ and d’. Three CHRs pack in an antiparallel fashion within hydrophobic grooves lined by NHR positions e’ and g’, resulting in the burial of positions a and d and interactions between positions e and b’ and between positions g and c’.
Figure S6: The binding specificity and affinity estimation of T20 variants in phage ELISA. (A) T20 variants displayed phage bound to 5HB, 6HB as well as BSA. (B) 5HB binding affinity estimation of T20 variants by single point competitive phage ELISA. The sub‐saturated T20 variant‐Fab fusion‐phage solution was tested for 5HB binding in the presence of 50 nM solution phase 5HB. The ELISA signal was normalized to the signal obtained without any competitor in solution for each clone. The test was performed in duplicate and shown is the average of the two tests.
Table S1. Design of T20 optimization library based on T20 saturation scanning mutagenesis.
Acknowledgments
We thank Hua Long for performing some of the phage selection experiments in this work. This work was supported by a Canadian Institutes of Health Research (CIHR) Operating Grant (MOP‐115066) and Canada Research Chair in Structural Virology to J.E.L. Moreover, support for stipends was provided by an Ontario Graduate Scholarship and CIHR‐Vanier Canada Graduate Scholarship to J.D.C. The T20 fusion inhibitor was obtained from Roche (Cat. No. #9845) through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
Significance statement: Prolonged therapeutic use of the HIV‐1 fusion inhibitor T20 leads to the evolution of resistant viral strains. We surveyed comprehensively the functional contributions of all T20 residues for binding to a fusion intermediate mimic and engineered a variant T20v1 that neutralized many diverse HIV‐1 strains, including a strain that was resistant to T20. These results show that combinatorial protein engineering can be applied to develop T20 variants with enhanced potencies and broadened specificities.
References
- 1. Chan DC, Fass D, Berger JM, Kim PS (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. [DOI] [PubMed] [Google Scholar]
- 2. Dorn J, Masciotra S, Yang CF, Downing R, Biryahwaho B, Mastro TD, Nkengasong J, Pieniazek D, Rayfield MA, Hu DJ, Lal RB (2000) Analysis of genetic variability within the immunodominant epitopes of envelope gp41 from human immunodeficiency virus type 1 (HIV‐1) group M and its impact on HIV‐1 antibody detection. J Clin Microbiol 38:773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wild C, Oas T, McDanal C, Bolognesi D, Matthews T (1992) A synthetic peptide inhibitor of human‐immunodeficiency‐virus replication ‐ correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A 89:10537–10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang SB, Lin K, Strick N, Neurath AR (1993) HIV‐1 inhibition by a peptide. Nature 365:113. [DOI] [PubMed] [Google Scholar]
- 5. Lu M, Blacklow SC, Kim PS (1995) A trimeric structural domain of the HIV‐1 transmembrane glycoprotein. Nat Struct Biol 2:1075–1082. [DOI] [PubMed] [Google Scholar]
- 6. Chan DC, Chutkowski CT, Kim PS (1998) Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 95:15613–15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lalezari JP, Henry K, O'Hearn M, Montaner JSG, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M, TORO 1 Study Group (2003) Enfuvirtide, an HIV‐1 fusion inhibitor, for drug‐resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. [DOI] [PubMed] [Google Scholar]
- 8. Pierson TC, Doms RW, Pohlmann S (2004) Prospects of HIV‐1 entry inhibitors as novel therapeutics. Rev Med Virol 14:255–270. [DOI] [PubMed] [Google Scholar]
- 9. Briz V, Poveda E, Soriano V (2006) HIV entry inhibitors: mechanisms of action and resistance pathways. J Antimicrob Chemother 57:619–627. [DOI] [PubMed] [Google Scholar]
- 10. Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC (1997) Atomic structure of the ectodomain from HIV‐1 gp41. Nature 387:426–430. [DOI] [PubMed] [Google Scholar]
- 11. Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W (2010) Crystal structure of HIV‐1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog 6:31000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR, Merutka G (1996) HIV‐1 membrane fusion mechanism: structural studies of the interactions between biologically‐active peptides from gp41. Biochemistry 35:13697–13708. [DOI] [PubMed] [Google Scholar]
- 13. Pan CG, Cai LF, Lu H, Qi Z, Jiang SB (2009) Combinations of the first and next generations of human immunodeficiency virus (HIV) fusion inhibitors exhibit a highly potent synergistic effect against enfuvirtide‐sensitive and ‐resistant HIV type 1 strains. J Virol 83:7862–7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jenabian MA, Saidi H, Charpentier C, Van Herrewege Y, Son JC, Schols D, Balzarini J, Vanham G, Belec L, Grp AMPS (2009) In vitro synergistic activity against CCR5‐tropic HIV‐1 with combinations of potential candidate microbicide molecules HHA, KRV2110 and enfuvirtide (T20). J Antimicrob Chem 64:1192–1195. [DOI] [PubMed] [Google Scholar]
- 15. Lazzarin A, Clotet B, Cooper D, Reynes J, Arasteh K, Nelson M, Katlama C, Stellbrink H, Delfraissy J, Lange J, Huson L, DeMasi R, Wat C, Delehanty J, Drobnes C, Salgo M, TORO 2 Study Group (2003) Efficacy of enfuvirtide in patients infected with drug‐resistant HIV‐1 in Europe and Australia. N Engl J Med 348:2186–2195. [DOI] [PubMed] [Google Scholar]
- 16. Perez‐Alvarez L, Carmona R, Ocampo A, Asorey A, Miralles C, de Castro SP, Pinilla M, Contreras G, Taboada JA, Najera R (2006) Long‐term monitoring of genotypic and phenotypic resistance to T20 in treated patients infected with HIV‐1. J Med Virol 78:141–147. [DOI] [PubMed] [Google Scholar]
- 17. Chinnadurai R, Rajan D, Munch J, Kirchhoff F (2007) Human immunodeficiency virus type 1 variants resistant to first‐ and second‐version fusion inhibitors and cytopathic in ex vivo human lymphoid tissue. J Virol 81:6563–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eggink D, Baldwin CE, Deng YQ, Langedijk JPM, Lu M, Sanders RW, Berkhout B (2008) Selection of T1249‐resistant human immunodeficiency virus type 1 variants. J Virol 82:6678–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cai L, Jiang S (2010) Development of peptide and small‐molecule HIV‐1 fusion inhibitors that target gp41. ChemMedChem 5:1813–1824. [DOI] [PubMed] [Google Scholar]
- 20. Yi HA, Fochtman BC, Rizzo RC, Jacobs A (2016) Inhibition of HIV entry by targeting the envelope transmembrane subunit gp41. Curr HIV Res 14:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lalezari JP, Bellos NC, Sathasivam K, Richmond GJ, Cohen CJ, Myers RA, Henry DH, Raskino C, Melby T, Murchison H, Zhang Y, Spence R, Greenberg ML, DeMasi RA, Miralles GD (2005) T‐1249 retains potent antiretroviral activity in patients who had experienced virological failure while on an enfuvirtide‐containing treatment regimen. J Infect Dis 191:1155–1163. [DOI] [PubMed] [Google Scholar]
- 22. Eron JJ, Gulick RM, Bartlett JA, Merigan T, Arduino R, Kilby JM, Yangco B, Diers A, Drobnes C, DeMasi R, Greenberg M, Melby T, Raskino C, Rusnak P, Zhang Y, Spence R, Miralles GD (2004) Short‐term safety and antiretroviral activity of T‐1249, a second‐generation fusion inhibitor of HIV. J Infect Dis 189:1075–1083. [DOI] [PubMed] [Google Scholar]
- 23. Zhu YM, Zhang XJ, Ding XH, Chong HH, Cui S, He JS, Wang XQ, He YX (2018) Exceptional potency and structural basis of a T1249‐derived lipopeptide fusion inhibitor against HIV‐1, HIV‐2, and simian immunodeficiency virus. J Biol Chem 293:5323–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pal G, Kouadio JLK, Artis DR, Kossiakoff AA, Sidhu SS (2006) Comprehensive and quantitative mapping of energy landscapes for protein‐protein interactions by rapid combinatorial scanning. J Biol Chem 281:22378–22385. [DOI] [PubMed] [Google Scholar]
- 25. Barthelemy PA, Raab H, Appleton BA, Bond CJ, Wu P, Wiesmann C, Sidhu SS (2008) Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem 283:3639–3654. [DOI] [PubMed] [Google Scholar]
- 26. Fisher RD, Ultsch M, Lingel A, Schaefer G, Shao L, Birtalan S, Sidhu SS, Eigenbrot C (2010) Structure of the complex between HER2 and an antibody paratope formed by side chains from tryptophan and serine. J Mol Biol 402:217–229. [DOI] [PubMed] [Google Scholar]
- 27. Leung I, Dekel A, Shifman JM, Sidhu SS (2016) Saturation scanning of ubiquitin variants reveals a common hot spot for binding to USP2 and USP21. Proc Natl Acad Sci U S A 113:8705–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Champagne K, Shishido A, Root MJ (2009) Interactions of HIV‐1 inhibitory peptide T20 with the gp41 N‐HR coiled coil. J Biol Chem 284:3619–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frey G, Rits‐Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC (2006) Small molecules that bind the inner core of gp41 and inhibit HIV envelope‐mediated fusion. Proc Natl Acad Sci U S A 103:13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Persson H, Ye W, Wernimont A, Adams JJ, Koide A, Koide S, Lam R, Sidhu SS (2013) CDR‐H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol 425:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen G, Sidhu SS. Design and generation of synthetic antibody libraries for phage display In: Ossipow V, Fischer N, Eds, 2014. Monoclonal antibodies: methods and protocols, Methods in molecular biology. Volume 1131 2nd ed. Totowa, NJ:Humana Press; p. 113–131. [DOI] [PubMed] [Google Scholar]
- 32. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: A sequence logo generator. Genome Res 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schrodinger, LLC (2010) The PyMOL Molecular Graphics System, Version 1.5.0.4.
- 34. Schneider TD, Stephens RM (1990) Sequence logos ‐ a new way to display consensus sequences. Nucleic Acids Res 18:6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu SW, Jing WG, Cheung B, Lu H, Sun J, Yan XX, Niu JK, Farmar J, Wu SG, Jiang SB (2007) HIV gp41 C‐terminal heptad repeat contains multifunctional domains ‐ relation to mechanisms of action of anti‐HIV peptides. J Biol Chem 282:9612–9620. [DOI] [PubMed] [Google Scholar]
- 36. Markosyan RM, Leung MY, Cohen FS (2009) The six‐helix bundle of human immunodeficiency virus Env controls pore formation and enlargement and is initiated at residues proximal to the hairpin turn. J Virol 83:10048–10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. He YX, Cheng JW, Lu H, Li JJ, Hu J, Qi Z, Liu ZH, Jiang SB, Dai QY (2008) Potent HIV fusion inhibitors against Enfuvirtide‐resistant HIV‐1 strains. Proc Natl Acad Sci U S A 105:16332–16337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cao J, Bergeron L, Helseth E, Thali M, Repke H, Sodroski J (1993) Effects of amino‐acid changes in the extracellular domain of the human‐immunodeficiency‐virus type‐1 gp41 envelope glycoprotein. J Virol 67:2747–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roth TA, Weiss GA, Eigenbrot C, Sidhu SS (2002) A minimized M13 coat protein defines the requirements for assembly into the bacteriophage particle. J Mol Biol 322:357–367. [DOI] [PubMed] [Google Scholar]
- 40. Held HA, Sidhu SS (2004) Comprehensive mutational analysis of the M13 major coat protein: improved scaffolds for C‐terminal phage display. J Mol Biol 340:587–597. [DOI] [PubMed] [Google Scholar]
- 41. Simek MD, Rida W, Priddy FH, Pung P, Carrow E, Laufer DS, Lehrman JK, Boaz M, Tarragona‐Fiol T, Miiro G, Birungi J, Pozniak A, McPhee DA, Manigart O, Karita E, Inwoley A, Jaoko W, Dehovitz J, Bekker LG, Pitisuttithum P, Paris R, Walker LM, Poignard P, Wrin T, Fast PE, Burton DR, Koff WC (2009) Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high‐throughput neutralization assay together with an analytical selection algorithm. J Virol 83:7337–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei XP, Decker JM, Liu HM, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu XY, Shaw GM, Kappes JC (2002) Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T‐20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pepinsky RB (1991) Selective precipitation of proteins from guanidine hydrochloride‐containing solutions with ethanol. Anal Biochem 195:177–181. [DOI] [PubMed] [Google Scholar]
- 44. Dillon PJ, Rosen CA (1990) A rapid method for the construction of synthetic genes using the polymerase chain‐reaction. Biotechniques 9:298. [PubMed] [Google Scholar]
- 45. Sidhu SS, Lowman HB, Cunningham BC, Wells JA (2000) Phage display for selection of novel binding peptides. Applications of chimeric genes and hybrid proteins, Pt C. Methods Enzymol 328:333–363. [DOI] [PubMed] [Google Scholar]
- 46. Kunkel TA, Roberts JD, Zakour RA (1987) Rapid and efficient site‐specific mutagenesis without phenotypic selection. Methods Enzymol 154:367–382. [DOI] [PubMed] [Google Scholar]
- 47. Fellouse FA, Sidhu SS. Making antibodies in bacteria In: Howard GC, Kaser MR, Eds , 2006. Making and using antibodies: a practical handbook. Boca Raton, FL: CRC Press; p. 157–180. [Google Scholar]
- 48. Richman DD, Wrin T, Little SJ, Petropoulos CJ (2003) Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc Natl Acad Sci U S A 100:4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, Chappey C, Kiss LDB, Paxinos EE, Petropoulos CJ (2007) Development and characterization of a novel single‐cycle recombinant‐virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob Agents Chemother 51:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Purification and characterization of HIV‐1 gp41 5HB and 6HB. (A) Schematic of the HIV‐1 gp41 5HB single chain construct. GGSGG and GSSGGSGSG linkers are used between the NHR‐CHR and CHR‐NHR, respectively. Superdex‐200 XK 16/40 prep grade size‐exclusion chromatograms of (B) 5HB and (C) 6HB. Coomassie‐stained SDS‐PAGE analysis of the eluted protein peaks is shown in the inset panel.
Figure S2: Assessment of T20‐Fab display by direct binding phage ELISA. T20 displayed as an amino terminal fusion to Fab VH (T20‐Fab) was tested in phage ELISA for binding to 5HB, 6HB and an irrelevant control protein (CTRL, a carboxyl terminally hexahistidine tagged human SIRPa IgV protein). The display with Fab only was utilized in phage ELISA as a control. The test was performed in triplicate and repeated three times independently. Data are presented as average values ± SD.
Figure S3: The distribution of amino acid types at each position of T20 in the naïve libraries. Normalized fraction of residues occurring at each randomized position of T20 in the naive libraries is plotted as a sequence logo. Amino acids are colored according to their chemical properties: green for polar residues (G,S,T,Y,C,Q,N), blue for basic residues (K,R,H), red for acidic residues (D,E) and black for hydrophobic residues (A,V,L,I,P,W,F,M). The primary sequence of T20 is shown above the sequence logo.
Figure S4: Helical wheel projection of T20 viewed down the helix axis from the N‐ to C‐terminus. Buried and partially buried positions according to classic 6HB are colored as follows: positions a and d, yellow; positions e and g, blue. Residue numbers 5–26 are shown at the corresponding heptad positions and are colored according to bit values derived from amino acid frequencies (Fig. 2), as follows: <1.2, gray; 1.2–2, green; >2, red.
Figure S5: Helical wheel projection of classic 6HB structure model viewed down the NHR axis from the N‐ to C‐terminus. Buried and partially buried positions are colored yellow and blue, respectively. Three NHRs pack together to form a central trimeric coiled coil with interactions between positions a’ and d’. Three CHRs pack in an antiparallel fashion within hydrophobic grooves lined by NHR positions e’ and g’, resulting in the burial of positions a and d and interactions between positions e and b’ and between positions g and c’.
Figure S6: The binding specificity and affinity estimation of T20 variants in phage ELISA. (A) T20 variants displayed phage bound to 5HB, 6HB as well as BSA. (B) 5HB binding affinity estimation of T20 variants by single point competitive phage ELISA. The sub‐saturated T20 variant‐Fab fusion‐phage solution was tested for 5HB binding in the presence of 50 nM solution phase 5HB. The ELISA signal was normalized to the signal obtained without any competitor in solution for each clone. The test was performed in duplicate and shown is the average of the two tests.
Table S1. Design of T20 optimization library based on T20 saturation scanning mutagenesis.