

Abstract
Suppressor of IKKepsilon (SIKE) is a 207 residue protein that is implicated in the TLR3‐TANK‐binding kinase‐1‐mediated response to viral infection. SIKE's function in this pathway is unknown, but SIKE forms interactions with two distinct cytoskeletal proteins, α‐actinin and tubulin, and SIKE knockout reduces cell migration. As structure informs function and in the absence of solved structural homologs, our studies were directed toward creating a structural model of SIKE through biochemical and biophysical characterization to probe and interrogate SIKE function. Circular dichroism revealed a primarily (73%) helical structure of minimal stability (<T m > =32°C) but reversibly denatured. Limited proteolysis (LP) and chemical modification identified the N‐terminal 2/3 of the protein as dynamic and accessible, whereas size exclusion chromatography (SEC) confirmed three homo‐oligomeric species. SEC coupled to chemical crosslinking characterized the primary species as dimeric, a secondary hexameric species, and a higher order aggregate/polymer. Fluorescence polarization using intrinsic tryptophan fluorescence contextualized the anisotropy value for the SIKE dimer (molecular weight 51.8 kDa) among proteins of known structure, bovine serum albumin (BSA; 66 kDa), and glutamate dehydrogenase (GDH; 332 kDa). Radii of gyration for BSA and GDH provided exclusionary values for SIKE tertiary and dimeric quaternary models that otherwise conformed to secondary structure, LP, and modification data. Dimeric quaternary models were further culled using acrylamide quenching data of SIKE's single tryptophan that showed a single, protected environment. The low cooperativity of folding and regions of dynamic and potentially disordered structure advance the hypothesis that SIKE forms a conformational ensemble of native states that accommodate SIKE's interactions with multiple, distinct protein‐binding partners.
Keywords: SIKE, innate immunity, circular dichroism, fluorescence polarization, crosslinking, molecular modeling
Introduction
Innate immunity forms the first line of defense against damage‐associated molecular patterns (endogenous origin) and pathogen‐associated molecular patterns. Pattern recognition receptors (PRRs) recognize evolutionarily conserved components of endogenous or exogenous danger signals and upregulate signaling cascades. At the center of these signaling cascades are catalytic hubs, typically kinases, that rapidly activate multiple substrates to form a host response. TANK‐binding kinase 1 (TBK1) serves as a catalytic hub downstream of several PRRs to phosphorylate transcription factors, upregulate autophagosome formation, and sequester pathogen.1 Delineating the TBK1 targets responsible for these responses uncovered suppressor of IKKepsilon (SIKE), a protein of unknown structure or function.2
SIKE is a 207 residue ubiquitously expressed protein that shares no sequence similarity with any structurally characterized protein. Initially, SIKE was characterized as an endogenous inhibitor of TBK1,2 but kinetic characterization reclassified it as a high‐affinity substrate.3 Recent studies have shown that SIKE associates with cytoskeletal structures within the cell.4 Nonphosphorylated SIKE formed a complex with α‐actinin, an actin‐binding protein that crosslinks actin filaments,5 whereas phosphorylated SIKE formed a complex with tubulin, a primary component of microtubules. Loss of SIKE reduced cellular migration as measured by a wound‐healing assay in a CRISPR/Cas9 SIKE knockout cell line.4 These biochemical data implicated SIKE in cytoskeletal rearrangements downstream of a TBK1‐mediated response, although the functional role of SIKE in these complexes remains unknown.
To garner further insight into SIKE's function, we employed biochemical and biophysical characterization to assess a predictive SIKE model. Our experimental data suggest that SIKE is an alpha helical protein of minimal stability with the N‐terminal half of the protein readily susceptible to tryptic cleavage and chemical modification. Fluorescence polarization, size exclusion chromatography (SEC), and crosslinking experiments show that SIKE self associates to form a stable dimer that can further interact to form a hexamer. The computational model and predicted dimer interface conform to all experimental data and provide a platform to map interactions and predict the functional consequences of these interactions.
Results
Circular dichroism
Circular dichroism (CD) spectra of SIKE were collected to assess the secondary structure of SIKE. Minima at 222 and 208 nm dominated the spectrum, indicative of primarily alpha helical content [Fig. 1(A)]. To estimate SIKE secondary structural content, data were submitted to SELCON36 and CONTINLL7, 8 analyses (DichroWeb9), which, when the results were averaged, predicted ~70% α‐helix and the remaining ~30% split between β‐turns and unordered regions (Table 1). No β‐strand structure was predicted. To assess the structural stability of SIKE, thermal melts of the protein were measured as a function of mean residue ellipticity (MRE) at 222 nm [Fig. 1(C)] with full spectra collected every 5°C [Fig. 1(C), inset]. This process was readily reversible [Fig. 1(D)]. A T m value of 35.1°C ± 0.6°C was calculated by fitting the thermal melt data to a four‐parameter sigmoidal equation (see Materials and Methods section). This value is coincident with a shift of the π→π*|| transition (208 nm minima) from 207.4 ± 0.2 to 206.2 nm [Fig. 1(C), inset]. This equation assumes that the two states of the protein, folded and unfolded, are well defined in the data. As SIKE's folded state appears minimally stable, as noted by the gradual increase in MRE at 222 nm even between 10°C and 20°C, the T m was also calculated from the maximum of the first derivative for the thermal melt curve (data not shown). This approach yielded a T m value of 29.1°C ± 0.06°C, which, when averaged with the value from the curve fit, would give a T m value of ~32°C. The thermal melt shows a gradual increase in slope from the presumed folded state at 10°C to the unfolded state at 80°C, reflected in the cooperativity factor value of 13.8 ± 0.5 derived from the curve fit. Cooperativity observed in folding and unfolding reactions arises from either the formation or destabilization, respectively, of coordinated interaction networks within the three‐dimensional (3‐D) structure.10 Reduced cooperativity is observed for proteins where local interactions, formed by residues nearby in sequence, predominate in the 3‐D structure.11
Figure 1.
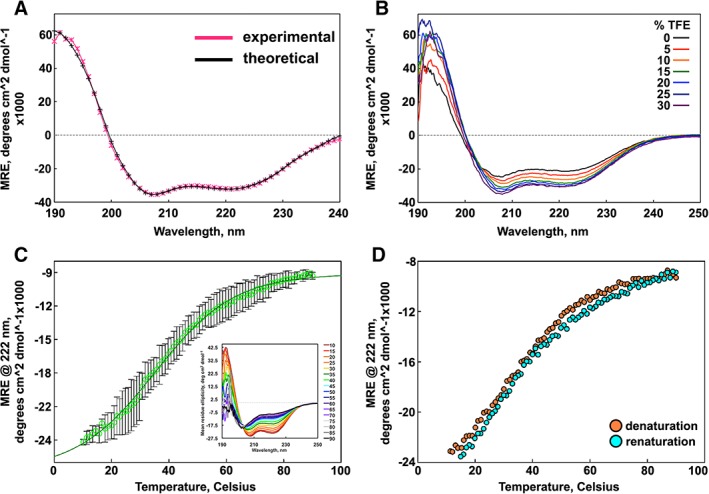
Secondary structure and stability of SIKE. (A) CD spectra of 386 nM SIKE ( ) in 5 mM NaH2PO4, pH 8, and theoretical fit to data (*; −) from DichroWeb CONTINLL analysis are indicative of alpha helical content. (B) Titration of TFE (0%–30%) into 464 nM SIKE delineate an increase in alpha helical content (208 and 222 nm minima) up to 25% TFE. (C) Thermal melt of SIKE at 773 nM (
) in 5 mM NaH2PO4, pH 8, and theoretical fit to data (*; −) from DichroWeb CONTINLL analysis are indicative of alpha helical content. (B) Titration of TFE (0%–30%) into 464 nM SIKE delineate an increase in alpha helical content (208 and 222 nm minima) up to 25% TFE. (C) Thermal melt of SIKE at 773 nM ( ) monitored at 222 nm shows a gradual loss of alpha helical content. Average of three independent measurements is shown with error depicted as standard deviation. Fit to a sigmoidal four‐parameter model (
) monitored at 222 nm shows a gradual loss of alpha helical content. Average of three independent measurements is shown with error depicted as standard deviation. Fit to a sigmoidal four‐parameter model ( ) yielded a cooperativity factor of 13.8 ± 0.5 consistent with gradual loss of structure rather than a sharp transition and a T
m of 35.1°C ± 0.6°C. First derivative analysis of the curve yielded a maxima (T
m) of 29.7°C (not shown). Inset: CD spectra of SIKE at increasing T, colored red to black. (D) Thermal denaturation was reversible. All data were collected in 5 mM NaH2PO4, pH 8 buffer. Spectra are representative of a minimum of three independent measurements.
) yielded a cooperativity factor of 13.8 ± 0.5 consistent with gradual loss of structure rather than a sharp transition and a T
m of 35.1°C ± 0.6°C. First derivative analysis of the curve yielded a maxima (T
m) of 29.7°C (not shown). Inset: CD spectra of SIKE at increasing T, colored red to black. (D) Thermal denaturation was reversible. All data were collected in 5 mM NaH2PO4, pH 8 buffer. Spectra are representative of a minimum of three independent measurements.
Table 1.
Secondary Structural Content of SIKE
To investigate stabilization of the secondary structure, CD spectra with an increasing percentage of 2,2,2‐trifluorethanol (TFE) were collected [Fig. 1(B)]. Secondary structural content at 25% TFE is reported in Table 1. Although both experimental data and predicted content indicate a higher percentage of helical content and lower percentage of unordered structure, the predicted percentages are not significantly (P ≤ 0.5%) different from no addition of TFE. Taken together, these data suggest that SIKE has a minimally stable, helical structure with regions of unordered sequence.
Limited proteolysis and disorder prediction
To identify regions of accessible, dynamic structure, SIKE was subjected to limited tryptic digestion followed by liquid chromatography with tandem mass spectrometry (LC‐MS/MS) to identify cleavage sites. Within the 207 amino acid sequence of SIKE, 16 lysines and 12 arginines are encoded with only K68 (Pro at +1 position) and K207 (C‐terminus) unavailable for cleavage12 [Fig. 2(A), R/K residues]. A heatmap of the five limited proteolysis (LP) experiments summarizes cleavage observed at each site and time point [Fig. 2(B)], providing information on relative accessibility to trypsin cleavage. Observed cleavage for all replicates was the threshold for identifying a site as cleaved. After 1 min exposure to trypsin, four arginines (38, 39, 44, and 194) and one lysine (66) were identified as cleavage sites that clustered near N‐ and C‐termini of the protein [Fig. 2(A), LP 1′]. After 5‐min proteolytic exposure, one additional site was identified corresponding to K129 (Fig. 2(A), LP 5′). At 10‐min exposure to trypsin, residues (R20, K119, and K120) located in regions susceptible to cleavage at earlier time points were identified (Fig. 2(A), LP 10′). These data suggest that residues 130–190 are more protected from proteolytic digest.
Figure 2.
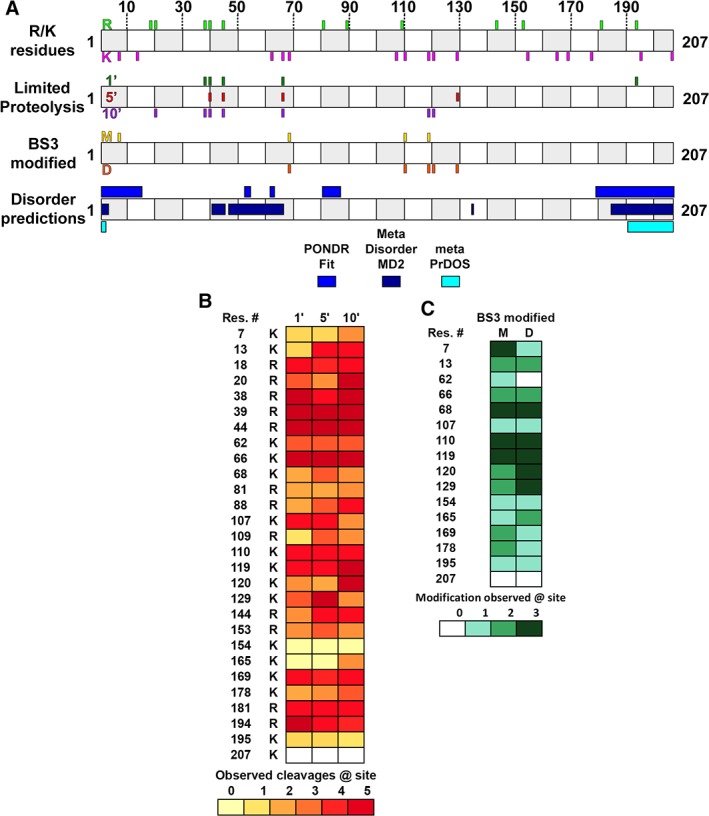
Dynamic regions of SIKE intersect with predicted regions of disorder. (A) Cartoon diagram shows SIKE primary sequence with alternating open and gray boxes demarcating 10 residue lengths. R/K residues: Positions of 12 arginine residues in SIKE are shown above cartoon in green and 16 lysine residues are shown below cartoon in magenta. Experimental data will align with these sites to identify residues. LP: SIKE was subjected to LP with trypsin for 1–10 min and cleavage sites identified by LC–MS/MS. Top shows 1′ cleavage sites in forest green; center shows 5′ cleavage sites in red; bottom shows 10′ cleavage sites in purple. BS3 modified: Lysines monofunctionally modified by BS3 in the excised monomer (M) gel band are shown on top (gold) and in the excised dimer (D) gel band are shown at the bottom (orange). Disordered predictions: Disordered regions were predicted by PONDR FIT, MetaDisorderMD2, and metaPrDOS as indicated. Highlighted regions had residues with predicted disorder parameter ≥0.5 threshold for each program—PONDR FIT: 1–15, 52–54, 61–63, 80–87, 179–207; MetaDisorderMD2: 1–3, 40–44, 46–65, 134–135, 184–207; metaPrDOS: 1–2, 191–207. (B) Heatmap of LP experiments for 1′, 5′, and 10′ data shows the relative cleavage across available K/R residues. Colors range from light yellow (0 cleavages observed) to brick red (cleavage observed in all experiments). K207 is coded white as this is the C‐terminus. (C) Heatmap of BS3 modification experiments for monomer and dimer species shows the relative modification across available K residues. The N‐terminus was not included as no modification was observed at this position. Colors range from light green (0 modifications observed) to deep green (modification observed in all experiments).
Another means to measure accessible residues is via covalent modification. Bis(sulfosuccinimidyl)suberate (BS3) is a homobifunctional crosslinker with a spacer arm of 11.4 Å that reacts with amino groups such as those available at the N‐terminus or lysine R group. If BS3 reacts with only a single amine, this would suggest that the region surrounding the reactive amino group is accessible and >~12 Å from another reactive amino group. SIKE was incubated with BS3 in solution and reactions separated by sodium‐dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE). Gel bands corresponding to a SIKE monomer or SIKE dimer were excised, tryptic digested, extracted, and subjected to LC–MS/MS. Including crosslinker modification as a variable modification within the database search identified monofunctionalized lysines that would represent solvent accessible regions of SIKE. Fully crosslinked BS3 species were excluded from analyses as we could not distinguish if these data represented intramolecular reactions (consistent with solvent accessible regions) or intermolecular reactions related to a homo‐oligomer interface. A heatmap of the three modification experiments summarizes the monofunctionalized sites observed for the monomer and dimer species [Fig. 2(C)]. Observed modification for all replicates was the threshold for classification as modified. For the SIKE monomer, K7, K68, K110, and K119 were modified, whereas, in the SIKE dimer, K68, K110, and K119 as well as K120 and K129 were monofunctionalized (Fig. 2(A), BS3 modified). These data suggest that regions encompassing K7‐K119 are accessible and not involved in the dimer interface as well as being consistent with the LP data that suggested the C‐terminal region of SIKE is less readily accessible.
Residues readily digested or modified suggest that these sites correlate with solvent accessible structure or dynamic/disordered structure. From the native CD spectrum, SIKE secondary structural analyses predicted 11%–19% disordered sequence (Table 1). To identify regions in sequence space that are dynamic or disordered, the SIKE primary sequence was submitted to three meta‐predictors of disordered regions, PONDR FIT,13 metaPrDOS,14 and MetaDisorderMD2.15 These predictors use combinations of individual predictors to significantly improve prediction accuracy compared to individual predictors. PONDR FIT, MetaDisorderMD2, and metaPrDOS predicted 28%, 26%, and 9% disorder within the SIKE sequence, respectively [Fig. 2(A), disorder predictions). Disordered residues are consistently predicted at the N‐ and C‐termini, but PONDR FIT and MetaDisorderMD2 also predict regions of disorder between residues 40 and 90. When predicted disorder regions and LP results at 1 min are compared, these data suggest a correlation. Interestingly, the region unique to the longer LP time points and BS3 modification (residues 110–129) do not contain predicted regions of disorder. These data would suggest that unstructured regions, in addition to accessibility, might contribute to sensitivity to proteolytic digestion.
Model of SIKE structure
To provide a scaffold to assess experimental data, the structure of SIKE was predicted using two publicly available tools, Phyre216 and I‐TASSER.17 Of the top 10 models returned by Phyre2, two models were chosen for analyses based upon Phyre2 confidence value (>0.9) and sequence coverage of the model (>90%) (Fig. 3). I‐TASSER models were assessed based upon their confidence parameter (C score; −5 to 2) that is calculated from the significance of the threading template alignments and the convergence parameters of the structure assembly simulations. The model with the highest C score was chosen for analysis (Fig. 3). This model also had >90% sequence coverage. For each model, the sequence identity between the template structure and SIKE was 9%–11% (Table 2). The Phyre #3 model and I‐TASSER model were nearly identical (RMSD 3.62 Å, 190 Cα) with the I‐TASSER model having less defined secondary structure. All models predicted primarily alpha helical structure, consistent with CD data (Table 1). Residues 38, 39, 44, 66, and 194 identified in the 1 min LP experiments are accessible in all of the models.
Figure 3.
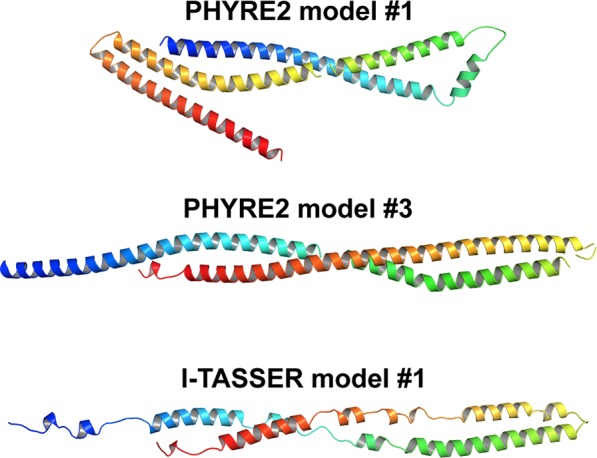
SIKE models predict alpha helical structure. Cartoon diagrams of SIKE models showing C‐α backbone colored from blue to red indicating N‐ to C‐terminus, respectively. PHYRE2 predicted two structures with >90% sequence coverage and confidence (probability [from 0 to 100] that the match between SIKE sequence and template is truly homologous), but sequence identity, a potential measure of model accuracy, between SIKE and template was <15% for each model. The I‐TASSER model with the highest C‐score (−1.86 on a scale of −5, 2; remaining models had values <−2.7) and cluster density (0.1486 whereas remaining four models had values <0.064, indicates structure occurs more often in simulation trajectory suggesting higher quality model) is shown. The TM‐score, scale used to measure structural similarity between two structures, is 0.49 ± 0.15, which falls between a value indicative of correct model topology (≥0.5) and random similarity (<0.17). Both programs predict α helical structure ranging from 61% to 93% that is consistent with CD spectral data.
Table 2.
Summary of SIKE Models
SEC and fluorescence polarization
SIKE has previously been shown to form an oligomer of undefined size through immunoprecipitation.2, 18 Using size exclusion chromatography, the solution molecular weight (MW) of SIKE was determined [Fig. 4(A)]. At ~1 mg/mL (36 μM), SIKE eluted at a volume equivalent to a globular protein of 86.2 kDa (Peak 3) with a smaller peak appearing at an elution volume equivalent to 182.6 kDa (Peak 2) and a small peak of aggregate in the column void volume. As the SIKE concentration was increased twofold to fourfold (1.9–3.6 mg/mL, 71–139 μM), the 182.6 kDa peak grew as did an appreciable peak corresponding to the void volume of the column (Peak 1). When an elution fraction corresponding to Peak 1 was again subjected to SEC, the sample eluted in the void volume of the column, suggesting an irreversible association. With a MW of 25.9 kDa (N‐term 6X His‐tagged SIKE), elution Peaks 2 and 3 would correspond to a globular heptamer and trimer of SIKE, respectively. Using fluorescence polarization, SIKE, prior to SEC separation, behaved as a large multimer in solution even at 3.9 μM [Fig. 4(B)] as compared to three control proteins, glutamate dehydrogenase (GDH, 334 kDa), bovine serum albumin (BSA, 66 kDa), and lysozyme (14.3 kDa). This behavior was unchanged with a twofold to fivefold decrease in concentration. When elution fractions from SEC separation were analyzed, not surprisingly, a pattern similar to the SEC elution was observed [Fig. 4(B)]. These data suggest that even at a low concentration, 3.9 μM, a population of SIKE is trapped in a large multimer/aggregated state.
Figure 4.
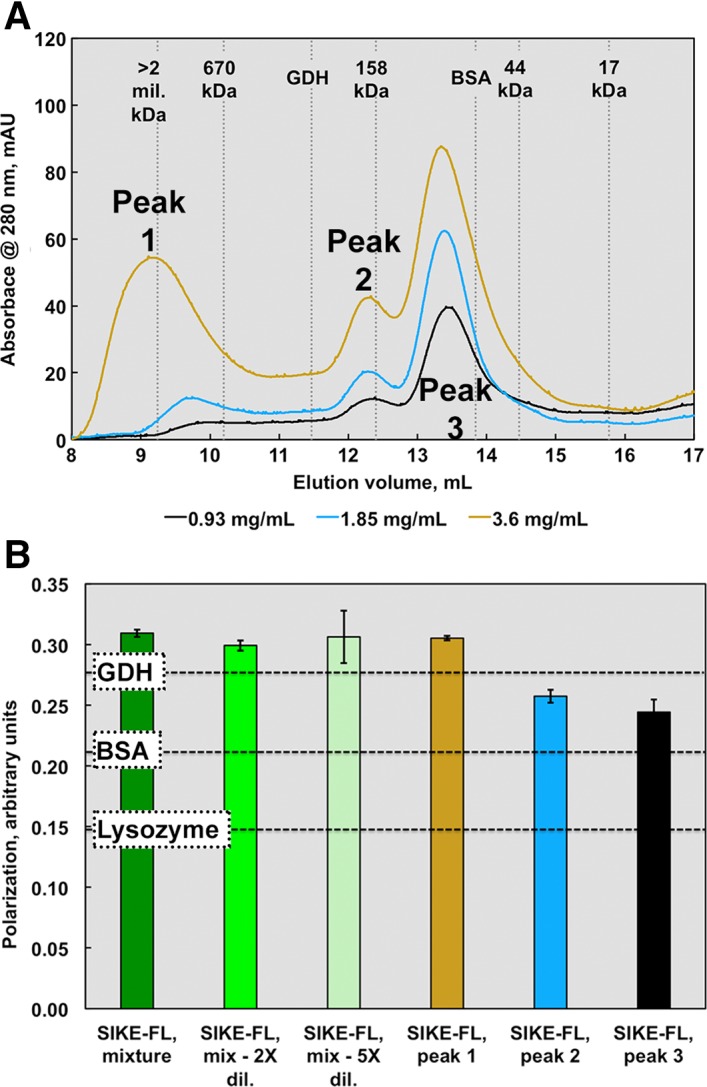
SEC and fluorescence polarization data suggest an elongated or multimeric SIKE structure. SIKE (0.9–3.6 mg/mL or 36–150 μM, colored cyan to dark blue), separated on an ENrich SEC 650 column, showed three distinct species. Peak 1 was assumed an aggregated species with MW corresponding to >1 mil Da. If SIKE behaves as a spherical globular protein in solution similar to the proteins used to derive the standard curve, Peak 2 would correspond to 182 kDa species (7mer based upon a SIKE monomer MW of 25.9 kDa) and Peak 3 a trimer of 86 kDa. The area of Peaks 1 and 2 increased with SIKE concentration. When Peak 1 was subjected to a second SEC separation, the protein retained its initial elution pattern, suggesting aggregation was not reversible (data not shown). (B) Protein fluorescence polarization of SIKE (EX 295 nm; EM 340 nm) at 0.1 mg/mL suggested a species larger than the protein standard GDH (MW 334 kDa) with dilution to 0.05 or 0.02 mg/mL having no effect on overall polarization of sample, consistent with species in an irreversible, aggregated state. Polarization measurements of individual peaks from the 2.5 mg/mL SEC experiment revealed that polarization values from Peaks 2 and 3 were bound by BSA and GDH values, consistent with the SEC data. Both experiments indicate that, in solution, SIKE does not behave as a globular, monomeric 25.9 kDa protein.
Crosslinking
To identify stable oligomers of SIKE, crosslinking experiments utilizing BS3 were completed. As noted above, BS3 containing sulfo‐N‐hydroxysulfosuccinimide esters at each end of an 11.4 Å linker react with primary amine groups. This crosslinking chemistry was chosen as SIKE encodes 16 lysine residues (8% of sequence) in addition to its N‐terminus. Linker length of crosslinker as assessed by BS3 (linker 11.4 Å) versus BS(PEG)5 (linker 21.7 Å) did not affect the pattern of crosslinked species observed (data not shown). As linker length did not alter the gel band pattern observed, BS3 was used for subsequent experiments. SIKE was incubated with a 20‐fold excess of BS3 and the quenched reaction separated by SEC [Fig. 5(A)]. The chromatograph for SIKE and crosslinked SIKE retained the same elution pattern of three major peaks. Peak fractions were subjected to SDS‐PAGE to assess the oligomers present in each peak. The gel band patterns were analyzed with ImageJ gel analysis tools to determine R f values for MW markers and sample lanes.19 From the linear regression of MW marker R f values, MW values for each identified band were derived. Without crosslinking, only the monomeric SIKE species is present [Fig. 5(B)]. The polypeptide MW of the SIKE construct is 25.9 kDa although R f analysis of the SIKE monomer reports a MW of 29.9 kDa. With crosslinking, each of the separated peaks revealed unique homo‐oligomeric states [Fig. 5(C)]. Peak 3 had two gel bands corresponding to the monomer (1×) and dimer (2×; 57–68 kDa). Peak 2 retained a dimer species but also manifested tetramer (4×, 104 kDa) and a dominant hexameric (6×, 139 kDa) species. Peak 1, eluting in the void volume of the column, retained protein in the gel well, suggesting large aggregates were present. Together, the crosslinking data suggest that SIKE forms a stable dimer species that can further associate into a hexameric species.
Figure 5.
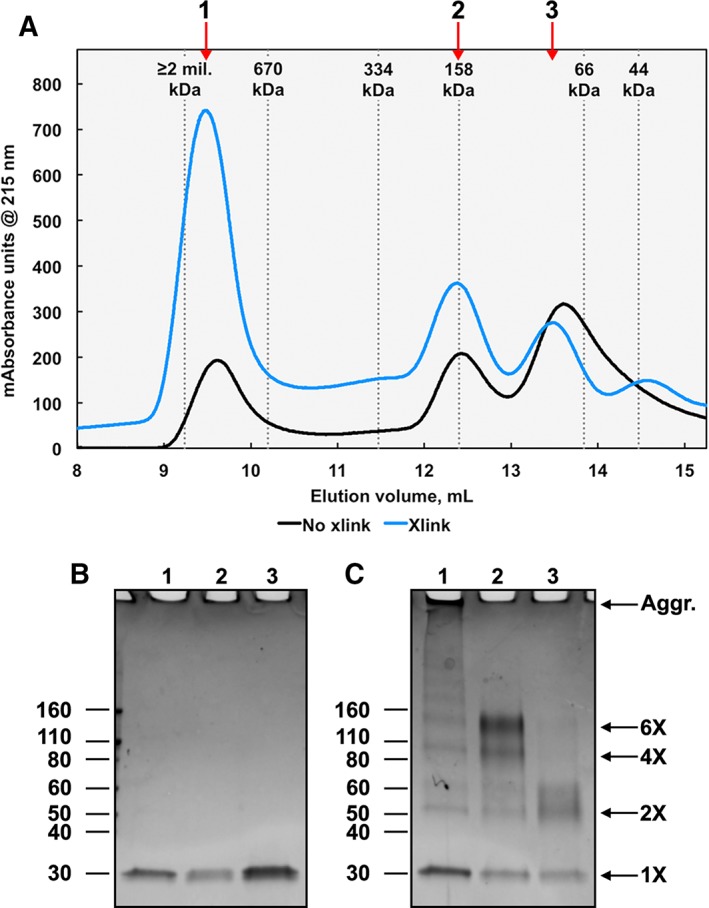
Amine–amine crosslinked SIKE forms dimer species. (A) Recombinant SIKE at 9.5 μM was crosslinked with 20× molar excess of BS3 (11.4 Å linker) at 4°C for 2 h. SIKE (black line) or crosslinked SIKE (blue line) was separated by size exclusion chromatography. Both chromatographs show three major SIKE species (labeled 1–3) similar to Figure 4. Fractions corresponding to each peak for noncrosslinked (B) and crosslinked (C) samples were separated by SDS‐PAGE on a 4%–20% Tris‐glycine gel and stained with SimplyBlue Safestain. (B) Uncrosslinked SIKE separated as a monomer (MW ~29 kDa) for all peaks. (C) Bands corresponding to crosslinked SIKE Peak 1 show monomer, dimer, tetramer, hexamer, and aggregated (Aggr.) species. Peak 2 is composed of primarily the hexamer species, whereas Peak 3 is primarily monomer and dimer species. These results suggest that Peak 1 is an aggregate of undetermined size, Peak 2 is a hexamer, and Peak 3 is a dimer.
Prediction of SIKE oligomer
The SEC, polarization, and crosslinking data supported an oligomeric structure for SIKE. Using the HydroPro software package,20 the radius of gyration (R g) was calculated for GDH, BSA, and the SIKE models (Table 3). GDH and BSA were chosen as reference proteins as they bounded the elution pattern of SIKE in SEC experiments as well as the fluorescence polarization of SIKE Peaks 2 and 3. Phyre2 model #3 and the I‐TASSER model had R g values greater than GDH. This was inconsistent with the experimental data and suggested that these models were incorrect. The Phyre2 model #1 R g value was within the GDH and BSA values. Using ZDOCK21 and ClusPro22 computational docking software, a dimeric structure of SIKE was predicted with the Phyre2 #1 model, but excluding the Lys and Arg residues sensitive to LP at 1 min from the dimeric interface (Res. 38, 39, 44, 66, and 194). Of the top 10 models returned by ZDOCK, four retained R g values consistent with the experimental data. ClusPro returned 30 models from the balanced scoring scheme (balancing van der Waals, electrostatic, and desolvation energy terms) of which 14 retained R g values consistent with the experimental data. To select dimer models consistent with SIKE's dimer structure in solution, the environment of SIKE's single tryptophan was examined. Comparison of N‐acetyl‐l‐tryptophan ethyl ester (NATEE) and SIKE fluorescence emission spectra shows that SIKE's tryptophan emission is blue shifted approximately 15 nm suggesting that the tryptophan is in a nonpolar environment [Fig. 6(E)]. Using these data, 10 of the 14 ClusPro dimer models could be excluded due to solvent exposed Trp R groups or a mixture of exposed and protected Trp R groups, of which neither situation was supported by the emission spectra which suggested a single environment, protected from solvent. The resulting models from ZDOCK and ClusPro were remarkably similar. Figure 6(A–D) depict representative models of the shared dimer orientation. In these dimer predictions, the single tryptophan residue of SIKE is located within the dimer interface. At 295 nm excitation, acrylamide quenching of SIKE's tryptophan fluorescence yielded a Stern‐Volmer constant of 0.026 ± 0.002 compared to NATEE's K SV of 0.042 ± 0.004 consistent with a tryptophan protected from the neutral quencher [Fig. 6(F)]. SIKE also contains three tyrosine residues per polypeptide chain. When excited at 280 nm, tyrosines in close proximity (9–18 Å) to a tryptophan form a Förster's resonance energy transfer pair.23 In a Stern–Volmer plot of tryptophan emission intensities, if tyrosines involved in forster resonance energy transfer (FRET) pairing with a tryptophan are readily quenched, this would result in a larger decrease in measured fluorescence intensity upon addition of quencher compared to quenching of isolated tryptophan fluorescence. In fact, we observed that excitation of tyrosines and tryptophan at 280 nm resulted in a K SV,SIKE280nm of 0.022 ± 0.002, whereas NATEE's K SV,280nm was 0.014 ± 0.0007. These data suggest that a tyrosine residue in SIKE can form a FRET pair with its tryptophan and is readily quenched by acrylamide. For example, in the dimer model of Figure 6(A), Tyr 67 is within 10 Å of Trp 91 [Fig. 6(H,I)] and could act as a FRET partner. The remaining two tyrosines (Y54 and Y108) are ≥20 Å from W91.
Table 3.
Comparison of Radii of Gyration Derived from Experimental Data and Models
| GDH | BSA | Lysozyme | PH2#1 | PH2#3 | IT | |
|---|---|---|---|---|---|---|
| R g (cm)a | 4.5e‐07 | 2.8e‐07 | 1.5e‐07 | 3.6e‐07 | 4.8e‐07 | 4.8e‐07 |
Figure 6.
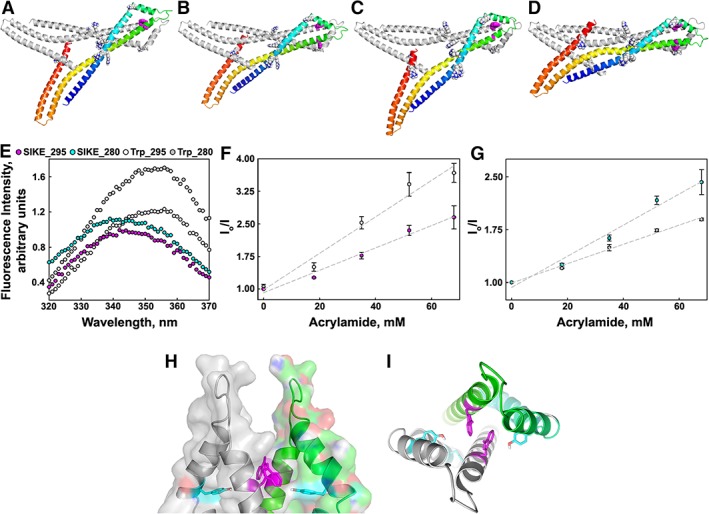
Prediction of SIKE dimer model consistent with Trp environment. SIKE monomers were computationally docked to form dimer species using ZDOCK21 or ClusPro22 with Lys and Arg residues that are susceptible to trypsin cleavage under LP conditions at 1 min (R38, R39, R44, K66, and R194) excluded from the dimer interface. Based upon polarization and SEC data, models with R
g values less than BSA or exceeding GDH were excluded. The remaining dimer models were refined (GalaxyRefineComplex40) to allow interface repacking. Four representative models meeting these criteria are shown in C‐α cartoon diagrams (A,B: ZDOCK; C,D: ClusPro). All models had similar dimer interfaces and protected the Trp residue within the dimer interface. Subunit 1 is colored gray (orientation of model in Fig. 3) and Subunit 2 is colored blue to red indicating N‐ to C‐terminus, respectively. Residues identified as readily accessible by LP experiments are shown in spheres colored by atom. The single tryptophan per polypeptide chain is shown in magenta colored spheres. (E) Fluorescence emission spectra of SIKE (11 μM, dimer species) or NATEE (16.4 μM) excited at 280 or 295 nm show that the emission maxima for SIKE's tryptophan at both excitation wavelengths is blue shifted ~15 nm. Legend:  , SIKE ex. 295;
, SIKE ex. 295;  , SIKE ex. 280; O, Trp ex. 295;
, SIKE ex. 280; O, Trp ex. 295;  , Trp ex. 280. Spectra representative of three independent experiments. Acrylamide quenching of tryptophan fluorescence monitoring only tryptophan (F: excitation 295 nm) or tryptophan fluorescence plus Forster's resonance energy transfer from proximal tyrosine residues (G: excitation 280 nm) showed that SIKE's tryptophan was protected (K
SV,SIKE 295 0.026 ± 0.002; K
SV,Trp 295 0.042 ± 0.004) and that tyrosines contributing to tryptophan fluorescence are readily quenched (K
SV,SIKE 280 0.022 ± 0.002; K
SV,Trp 280 0.014 ± 0.0007). Data are average of three independent experiments. Error is showed as standard deviation. Cartoon diagrams of tryptophan environment in proposed dimer model showing dimer surface highlighting tryptophan pocket (H) and proximity of tyrosine (I). Dimer subunits colored in gray and green, respectively. Tryptophans and nearest tyrosine are shown in magenta and cyan stick, respectively.
, Trp ex. 280. Spectra representative of three independent experiments. Acrylamide quenching of tryptophan fluorescence monitoring only tryptophan (F: excitation 295 nm) or tryptophan fluorescence plus Forster's resonance energy transfer from proximal tyrosine residues (G: excitation 280 nm) showed that SIKE's tryptophan was protected (K
SV,SIKE 295 0.026 ± 0.002; K
SV,Trp 295 0.042 ± 0.004) and that tyrosines contributing to tryptophan fluorescence are readily quenched (K
SV,SIKE 280 0.022 ± 0.002; K
SV,Trp 280 0.014 ± 0.0007). Data are average of three independent experiments. Error is showed as standard deviation. Cartoon diagrams of tryptophan environment in proposed dimer model showing dimer surface highlighting tryptophan pocket (H) and proximity of tyrosine (I). Dimer subunits colored in gray and green, respectively. Tryptophans and nearest tyrosine are shown in magenta and cyan stick, respectively.
Discussion
Structural information provides insight into functional characterization. Our studies were directed toward creating a structural model of SIKE through biochemical and biophysical characterization to probe and interrogate SIKE function. The results reveal a low cooperativity of folding and regions of dynamic and potentially disordered structure that advance the hypothesis that SIKE forms a conformational ensemble of native states that accommodate SIKE's interactions with multiple, distinct protein binding partners.
SIKE is a unique conundrum among proteins. It is ubiquitously expressed implying it may serve a housekeeping role in cells.2 SIKE's primary sequence harbors stretches of predicted helical secondary structure (77% of sequence; PSIPRED24) and coiled coil motifs (residues 71–111 and 164–198; PairCoil225). Fold recognition predictors such as pGenTHREADER26 return high confidence alignments to helical proteins such as α‐ and β‐spectrin,27 tropomyosin,28 and α‐actinin.29 Protein basic local alignment search tool (BLAST)30 and iterative position specific iterated (PSI)‐BLAST31 searches return SIKE orthologs and the protein, fibroblast growth factor receptor 1 oncoprotein 2 (FGFR1OP2, also known as wound inducible transcript 3.0), which shares 50% sequence identity with SIKE. FGFR1OP2 also has no known structure or function beyond an association with accelerated wound healing.32 This association would imply that FGFR1OP2 interacts with cytoskeletal structures, a feature shared with SIKE through its interactions with α‐actinin and tubulin.4 Yet, together, these bioinformatic and sequence analyses do not provide a cohesive picture of SIKE structure to assist in interrogating its function.
Using a complement of biochemical and biophysical approaches, we have built a SIKE structural model consistent with our experimental results that could shed light on its function and properties that could contribute to SIKE's function. Our CD data confirmed that SIKE is a helical structure, consistent with secondary structure, coiled coil, and fold recognition predictions. Not surprisingly, predicted SIKE models were also α‐helical structures. The highest confidence models for SIKE both incorporated coiled coil motifs based on a two‐helices or three‐helices. In the two‐helical coiled coil model, the dimerization domain of the chromatin partitioning protein of Pyrococcus yayanosii's structural maintenance of chromosomes complex served as the template,33 whereas the coiled coil domain of Streptococcus pneumoniae peptidoglycan hydrolase, provided the three‐helix template.34 For each model, the templates shared ≤11% sequence identity with SIKE. The limited stability of SIKE as assessed by thermal denaturation was not unexpected as the recombinant protein is expressed within inclusion bodies and refolded as part of purification.3 Other helical proteins, for example, the spectrin repeats of erythroid spectrin α and β, have thermal stabilities ranging from 21°C to 72°C, which has been attributed in part to the level of hydrophobic residues incorporated into the coiled coil heptad repeat outside the traditional positions “a” and “d” as well as the context of the repeat.35 The thermal melt curve revealed a shallow slope associated with unfolding, reflected in the large cooperativity factor (b value, inversely related to the slope at the transition mid‐point of the thermal melt curve). This shallow slope is indicative of low cooperativity of unfolding, suggesting that local contacts would predominate the 3‐D structure,10 which is consistent with the predicted elongated, helical models. In conjunction with reduced cooperativity of unfolding, an interesting observation was the reversibility of the thermal denaturation, suggesting that SIKE can reversibly undergo large alterations to its structure. The TFE experiments also point to alterations in structure or dynamic structure as increasing TFE concentrations shifted secondary structure to higher helical content, consistent with portions of SIKE that are less structured or are sampling unordered and helical structure depending on environment. These data suggested that the SIKE structure is readily malleable. This raises the possibility as posited by Munoz et al. that structural stability (or instability) could function as a conformation rheostat controlling binding to structurally distinct partners.10 Similarly, regions that are capable of sampling multiple conformations, such as intrinsically disordered regions, provide compact, but robust, conformational sampling that may be exploited in terms of multivalent interactions, as noted for scaffold proteins in signaling cascades36 and more broadly in protein–protein interactions.37 In terms of SIKE function, this pliable structure could contribute to SIKE's ability to interact with two distinct proteins, tubulin and α‐actinin, or provide a means for SIKE's post‐translational modifications, such as serine phosphorylation,3 to alter its structure resulting in preferential binding to tubulin over α‐actinin.
To further investigate the dynamic nature of the SIKE structure and delineate regions accessible for protein interactions, SIKE was subjected to LP and chemical modification. Readily accessible areas were confined to the N‐terminal 2/3 of the protein and the last 13 residues. This left residues 130–190 protected and available for interactions. Fluorescence polarization data of SIKE species in conjunction with protein standards of characterized size and quaternary structure showed that the smallest solution size of the SIKE fell between BSA and the hexameric GDH. To assess the predicted SIKE models for consistency with the experimental data, the hydrodynamic radius (R g) of the SIKE models as well as BSA and GDH were computationally calculated using the HYDROPRO software.38 Comparison of the R g values excluded the two‐helix coiled coil model from further consideration as it exceeded the solution size of GDH, whereas the experimental data showed that SIKE was smaller than GDH. These analyses also show that, although bioinformatics assessments returned helical and coiled coil predictions, SIKE's polypeptide chain cannot fold into a single or two‐helix coiled coil structures as these structures fail to conform to the constraint of SIKE's solution size. SIKE's tertiary structure must be more compact.
A second, necessary component to identify regions accessible for SIKE interactions is defining SIKE's interactions with itself. Our SEC data as well as previous immunoprecipitation data2, 18 suggested that SIKE forms homo‐oligomers that was further refined to a dimer by amine‐based bifunctional crosslinking. By combining LP data to limit residues that could occupy the dimer interface and fluorescence polarization‐based R g to define solution size, a dimer model was predicted. Strikingly, in this orientation, the region consistently protected from LP (residues 130–190) did not serve as the dimerization interface. Instead, residues 44–115 of each subunit formed the primary dimer interface of which only residues 67–129 were protected at the 1 and 5 min LP time points. The C‐terminal 10 residues formed a secondary dimer interface by bridging positions 130 and 190 of the adjacent subunit. As this prediction was unexpected, we probed the dimer model with tryptophan quenching data as SIKE contains a single tryptophan per polypeptide chain. SIKE's tryptophan was not fully solvent exposed as was evident from SIKE's blue shifted emission spectrum and was consistent with the dimer model as the tryptophan from each chain is sandwiched in the dimer interface. Acrylamide quenching restricted to only tryptophan emission maxima (295 nm excitation, emission >320 nm) agreed that the tryptophans were protected from the neutral quencher. Interestingly, when tryptophan quenching by acrylamide was monitored by excitation of tyrosine and tryptophan residues, the Stern–Volmer constant for SIKE exceeded that of free tryptophan. This would imply that the fluorescence intensity quenched in the protein solution was greater than that of free tryptophan even though the two solutions were prepared to have equivalent tryptophan absorbance values. The excited tyrosine residues of SIKE must have contributed the additional intensity by resonance energy transfer to the tryptophans. These data suggest that at least one of the three tyrosines of SIKE is within 9–18 Å of the tryptophan residues. In the current dimer model, Tyr67 of an adjacent subunit meets these distance restraints. Taken together, the LP and quenching data are consistent with the predicted dimer interface centered upon the tryptophan residue at position 91.
The SIKE dimer model provides a platform to develop hypotheses that probe SIKE:cytoskeleton interactions. In particular, SIKE has six phosphorylation sites3 that, when modified, increase the SIKE:tubulin association.4 Within the model, these six serines are clustered at the dimer interface formed by the C‐terminal region of SIKE. These sites are accessible to phosphorylation, more or less so depending upon the splay of the C‐terminal dimer interface. For example, the model of Figure 6(C) is readily accessible, whereas the compactness of Figure 6(D) reduces accessibility to target serines. This suggests that the introduction of negative charges via phosphorylation at a single site may provide a repulsive force to weaken the interface that increases accessibility for additional phosphorylation, which further weakens the dimer interface by increasing the density of clustered, negative charges. This would be consistent with our initial characterization of SIKE phosphorylation by TBK1 in which not all sites were equivalently phosphorylated,3 although explanations other than accessibility (e.g., position of phosphorylation favors/disfavors interaction with TBK1 active site) are also possible. In the context of the current models, SIKE phosphorylation may alter the overall shape of SIKE providing a mechanism for selection of distinct interaction partners. The effect of phosphorylation on SIKE's tertiary and quaternary structure as well as its direct participation in SIKE protein interactions is under active investigation.
In this study, we have characterized the biochemical and biophysical properties of SIKE to probe bioinformatic and modeling predictions. We have shown SIKE to be primarily alpha helical and reversibly denatured, yet minimally stable. SIKE associates to form a dimeric protein that does not conform to polarization values of similarly sized, globular proteins suggesting that SIKE is somewhat nonglobular. The dimer species shields its tryptophans from solvent, allows Tyr → Trp resonance energy transfer, and can further associate into a stable hexamer. Predictive modeling produced a three‐helix coiled coil model that conforms to these parameters. With respect to SIKE function, SIKE has several binding partners ranging from the cytoskeletal proteins, α‐actinin and tubulin, identified by our work4 to TBK12, 3 and STRIPAK complex components18 identified by others. The model provides a starting point for predicting and mapping SIKE surfaces that participate in these protein–protein interactions, which will in turn further improve the model.
Materials and Methods
Materials
All chemicals were purchased from Millipore Sigma (Burlington, MA) unless otherwise stated.
Protein expression and purification
The pET15b SIKE construct was transformed into chemically competent BL21‐CodonPlus (DE3)‐RIPL (Agilent, Santa Clara, CA) following the manufacturer's protocol. A single colony was used to inoculate an overnight culture of Luria broth plus 100 μg/mL ampicillin. The overnight culture was subcultured 1:100 into 1 L LB/amp flasks grown at 37°C until the cell density reached an A600 ≈ 0.6. Protein expression was induced with 1 mM isopropyl‐β‐d‐galactopyranoside and allowed to grow overnight at 20°C. Cells were harvested by centrifugation at 7000g. Cells were resuspended in 50 mM NaH2PO4, pH 8, 300 mM NaCl, 1 mM 2‐mercaptoethanol, 100 μg/mL lysozyme, and 1× Complete ethylenediaminetetraacetic acid (EDTA)‐free protease inhibitor and sonicated to lyse cells (~50 mL). Insoluble material was pelleted by centrifugation (14,000g, 30 min), and the pellet solubilized in guanidine hydrochloride (GudHCl) buffer (6M GudHCl, 50 mM NaH2PO4, pH 8, 300 mM NaCl, and 1 mM 2‐mercaptoethanol). Resuspended insoluble material was clarified by centrifugation at 14,000g, 30 min. The supernatant was mixed with 1.5 mL TALON resin (ClonTech Laboratories, Inc., Mountain View, CA), pre‐equilibrated in GudHCl buffer. The lysate resin mixture was loaded into a column (BioRad, Hercules, CA) and washed by gravity with 50 column volumes (CV) of GudHCl buffer. Bound protein was refolded on the column using a 133‐CV reverse gradient of GudHCl buffer to 50 mM NaH2PO4 (pH 8), 300 mM NaCl, and 1 mM 2‐mercaptoethanol (Buffer 1) and eluted with ~5‐CV of Buffer 1 plus 500 mM imidazole. Protein purity was assessed by SDS‐PAGE analysis and protein concentration quantified by Bradford method (BioRad).
CD and thermal melt studies
CD spectra and thermal melt data were collected on a Jasco J‐810 spectropolarimeter with Peltier and chilled water bath temperature control using a quartz cuvette with 10 mm optical path length (3 mL total volume). SIKE was dialyzed into 5 mM NaH2PO4, pH 8. At a minimum, three independent spectra were collected for each condition at 20°C. Spectral data were submitted to DiChroWeb CONTINLL and SELCON3 software to predict secondary structural content. Thermal melting of SIKE was monitored via MRE at 222 nm from ~10°C to 90°C at a 1°C interval. Three independent melts were collected per condition. Averaged data were fit to a four‐parameter sigmoidal curve (SigmaPlot, San Jose, CA):
where y o is Θ f (MRE of the folded state), a is ΔΘ (Θ f − Θ u; Θ u MRE of unfolded state), x o is the T M, and b is inversely related to the steepness of the curve (cooperativity factor). T M was also derived from first derivative maximum of data (SigmaPlot). Error was calculated as standard deviation.
LP studies
Recombinant SIKE was dialyzed into 10 mM NH4HCO3. SIKE (10 μM) was incubated for 1, 5, or 10 min with trypsin (Trypsin singles, proteomics grade, Millipore Sigma) at 1:200 molar ratio SIKE:trypsin in a 50 μL total volume. Digests were quenched with 0.2% trifluoroacetic acid. Digested peptides were separated from undigested protein by ultrafiltration through a 10 K molecular weight cut‐off (MWCO) filter (Millipore Sigma). Eluant was dried down in a vacuum centrifuge and resuspended in 25 μL 10 mM NH4HCO3 for LC–MS/MS analysis. The LC–MS system consisted of a Thermo Scientific LTQ‐XL Linear Ion Trap mass spectrometer system with an Ion Max API source interfaced to a Dionex Ultimate 3000 UHPLC and a Phenomenex Jupiter 4 μm Proteo 90 Å reversed phase column. Ten microliters of the final solution was injected onto the column, and the peptides were eluted from the column by a 0.1% formic acid (A)/0.1% formic acid–acetonitrile (B) gradient from 5% to 35% Solvent B for 20 min and from 35% to 95% Solvent B for 5 min followed by washing with 95% Solvent B and re‐equilibration with 5% Solvent B (flow rate of 70 μL/min). The API source was operated at 4.5 kV. The digests were analyzed using the double play capability of the instrument acquiring full scan mass spectra (400–2000 m/z) to determine peptide MWs and product ion spectra to determine amino acid sequence in sequential scans. Ions were fragmented by CID using a normalized collision energy of 35. LTQ XL raw data files were analyzed using the MASCOT Server v2.5.1.0. The database was a custom database containing the N‐terminally 6XHis‐tagged SIKE (UniProt Q9BRV8‐1 sequence). The search parameters were set as follows: enzyme Trypsin/P; nine missed cleavages allowed; variable modifications included ammonia loss N‐term, deamidated (NQ), Gln‐ > pyro‐Glu (N‐term Q); Glu‐ > pyro‐Glu (N‐term E); oxidation (M); peptide mass tolerance 1.2 Da; fragment mass tolerance 0.6 Da; and decoy selected. Significance threshold for search results was P < 0.05. Although not valid for the small number of spectra associated with a single protein digest, the decoy search returned a false discovery rate of 0% for all searches. A minimum of three independent experiments was completed for each time point and data reported are representative of each time point. Identified peptides for each experiment are summarized in Supporting Information Tables S1–S3.
Sequence analysis and model building
Disorder predictions
The SIKE sequence (UniProt Q9BRV8‐1 sequence) was submitted to publicly available disorder prediction analyses, PONDR‐FIT,13 MetaPrDOS,14 and metaDisorderMD2,15 to identify regions of SIKE sequence meeting the threshold for disorder. Homology models: Models based on the SIKE sequence were predicted from PHYRE216 and I‐TASSER.17 Secondary structure content of models with ≥90% sequence coverage was determined using STRIDE.39 Radius of gyration was calculated for each model using the HYDROPRO software.20 Dimer complex predictions: Using the select residues feature in ZDOCK21 to exclude R and K residues identified as accessible by LP from a dimer interface, dimer complexes were predicted from the PHYRE2 model #1. Similarly, using the advanced options, repulsion mode to exclude R and K residues identified in the 1 min LP data from the dimer interface, ClusPro22 predicted potential dimer models from the PHYRE2 model #1. R g values were derived for each dimer model using the HYDROPRO software.20 Only complexes with an R g value consistent with experimentally derived range of R g values and consistent with Trp fluorescence and quenching experiments were subjected to refinement of sidechain rotamers, sidechain packing, and overall structure relaxation by molecular dynamic simulation (GalaxyRefineComplex40).
Size exclusion chromatography
Recombinant SIKE was dialyzed into 25 mM NaH2PO4, pH 8, 150 mM NaCl, and 1 mM BME. Dialyzed protein was quantitated by UV absorbance at 280 nm (ε 280nm = 0.37) and concentrated to 36–139 μM using ultrafiltration (Amicon‐Ultra, Millipore Sigma). Five hundred microliters of samples were separated on an Enrich SEC 650 column (BioRad) equilibrated in 25 mM NaH2PO4, pH 8, 150 mM NaCl, and 1 mM BME on an NGC Scout Plus Chromatography system (BioRad). Eluent was monitored at 280 and 215 nm. A standard curve was constructed from the elution profiles of bovine thyroglobulin, bovine g globulin, BSA, chicken ovalbumin, and horse myoglobin as per the manufacturer's protocol (BioRad). Elution of blue dextran marked the void volume of the column. Bovine GDH (0.1 mg/mL 0.3 μM) and hen egg white lysozyme (0.1 mg/mL, 7 μM) were separated under the same conditions.
Fluorescence polarization and fluorescence quenching studies
All fluorescence polarization data were collected on a Photon Technology International QM‐2 spectrofluorimeter equipped with motorized excitation and emission polarizers at room temperature. Fluorescence polarization was measured, setting the excitation at 295 nm and the emission at 340 nm, with bandwidths of 1 nm. Time‐based polarization was calculated using the Felix GX software including the experimental G factor defined as ratio of sensitivities of the detection system for the vertically and horizontally polarized light. Background and G factor data were collected for a 10 s duration; sample data were collected for a 60 s duration. Samples consisted of SIKE at 0.1 mg/mL (39 μM), 0.05 mg/mL (19.5 μM), and 0.02 mg/mL (7.8 μM) in 10 mM NaH2PO4, pH 8 for elution Peaks 1, 2, or 3 of SIKE SEC separation (3.6 and 2.5 mg/mL SEC separations). Fluorescence polarization of bovine GDH (355.5 kDa), BSA (60 kDa), and hen egg white lysozyme (14.3 kDa) at 0.1 mg/mL was measured to provide reference points. A quartz cylindrical microcuvette, 400 μL volume, held in a black anodized holder (Markson) was used for all measurements. Quenching data were collected on a Thermo Spectronic AMINCO‐Bowman Series 2 Luminescence Spectrometer. SEC was used to isolate the dimer species of SIKE (as described previously, but in 1× phosphate buffered saline (PBS)), and the peak fractions corresponding to the dimer species were pooled. SIKE concentration (11 μM) was quantitated by A280 with ε 280nm = 0.37 mL/mg. NATEE was used at 16.4 μM in 1× PBS, a concentration that yielded an equivalent A 295nm to the SIKE sample. Emission spectra of SIKE or NATEE (300 μL) excited at 280 or 295 nm (1 nm width) were collected from 320 to 370 nm as acrylamide (0–100 mM) was titrated into the solution. Three independent experiments were completed for each condition. Error was calculated as standard deviation. Data were analyzed using SigmaPlot software.
Chemical cross‐linking studies
Monofunctional BS3 modification
SIKE was dialyzed into 50 mM NaH2PO4, pH 7.5, and 0.15M NaCl. SIKE (13–18 μM) was crosslinked with BS3 in a molar excess of 20 according to the manufacturer's protocol (ThermoFisher, Waltham, MA). The reactions were quenched after 2 h at 4°C with 50 mM Tris at pH 7.5. Reactions were separated by SDS‐PAGE (4%–20% Tris‐Glycine) and visualized with SimplyBlue SafeStain (ThermoFisher). Gels were fixed with a 1:1 ratio of 5% acetic acid in water:methanol. After fixation, bands corresponding to the SIKE monomer and dimer were excised, and an in‐gel trypsin digest was performed as described.41 Trypsin digestion was performed for 60 h. Digested peptides were extracted from gel using 1:2 (vol/vol) 5% formic acid/acetonitrile solution (50–100 μL; covered gel piece). Digested peptides were dried down in a vacuum centrifuge and resuspended in 25 μL of 10 mM ammonium bicarbonate. LC–MS/MS analyses of samples (10 μL) were as described above except that XLINK: DSS (K) and XLINK DSS (Protein N‐term) were included in the variable modifications. Identified peptides for each experiment are summarized in Supporting Information Table S4. SEC studies: SIKE was dialyzed into 50 mM NaH2PO4, pH 7.5, and 0.15M NaCl. SIKE (9.5 μM) was crosslinked with 190 μM BS3 according to manufacturer's protocol (ThermoFisher). The reactions were quenched after 2 h at 4°C with 50 mM Tris at pH 7.5. Reactions were separated on an ENrich SEC 650 column (10 × 300 mm, BioRad) in 25 mM NaH2PO4, pH 8, 0.15M NaCl, and 1 mM BME. An elution fraction corresponding to each peak in the chromatograph was separated by SDS‐PAGE (4%–20% Tris‐glycine) and visualized with SimplyBlue SafeStain (ThermoFisher). Gels images were captured on a Gel Doc EZ system (BioRad). R f analyses of gel images were completed in ImageJ19 using the Analyze:Gel function (https://imagej.nih.gov/ij/). Approximate MWs of crosslinking reaction bands were calculated from a standard curve based upon MW marker R f values.
Conflict of Interest
The authors declare no conflict of interest in publishing results of this study.
Supporting information
Table S1. Summary of peptides identified by LCMS/MS for LP 1 min timepoint.
Table S2. Summary of peptides identified by LCMS/MS for LP 5 min timepoint.
Table S3. Summary of peptides identified by LCMS/MS for LP 10 min timepoint.
Table S4. Summary of peptides identified by LCMS/MS with BS3 modifications
Acknowledgments
The authors wish to thank Sean W. McKinley for his contributions to refining the SIKE purification protocol, Richard Cruz for his contribution to disorder predictions, and J. Ellis Bell for careful reading of and insightful comments on the manuscript.
Significance Statement: This work describes the biochemical and biophysical characterization of suppressor of IKKepsilon (SIKE) to support an alpha helical, dimeric structural model. The low cooperativity of folding and regions of dynamic and potentially disordered structure advance the hypothesis that SIKE forms a conformational ensemble of native states that accommodate SIKE's interactions with multiple, distinct protein‐binding partners.
References
- 1. Louis C, Burns C, Wicks I (2018) TANK‐binding kinase 1‐dependent responses in health and autoimmunity. Front Immunol 9:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB (2005) SIKE is an IKKepsilon/TBK1‐associated suppressor of TLR3‐ and virus‐triggered IRF‐3 activation pathways. EMBO J 24:4018–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marion JD, Roberts CF, Call RJ, Forbes JL, Nelson KT, Bell JE, Bell JK (2013) Mechanism of endogenous regulation of the type I interferon response by suppressor of IκB kinase ɛ (SIKE), a novel substrate of TANK‐binding kinase 1 (TBK1). J Biol Chem 288:18612–18623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sonnenschein HA, Lawrence KF, Wittenberg KA, Slykas FA, Dohleman EL, Knoublauch JB, Fahey SM, Marshall TM Jr, Marion JD, Bell JK (2018) Suppressor of IKKepsilon forms direct interactions with cytoskeletal proteins, tubulin and α‐actinin, linking innate immunity to the cytoskeleton. FEBS Open Biol 8:1064–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sjoblom B, Salmazo A, Djinovic‐Carugo K (2008) Alpha‐actinin structure and regulation. Cell Mol Life Sci 65:2688–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sreerema N, Woody RW (1993) A self‐consistent method for the analysis of protein secondary structure from circular dichrosim. Anal Biochem 209:32–44. [DOI] [PubMed] [Google Scholar]
- 7. Provencher SW, Glockner J (1981) Estimation of globular protein secondary structure from circular dichrosim. Biochemistry 20:33–37. [DOI] [PubMed] [Google Scholar]
- 8. Van Stokkum IHM, Spoelder HJW, Bloemendal M, Van Grondelle R, Groen FCA (1990) Estimation of protein secondary structure and error analysis from CD spectra. Anal Biochem 191:110–118. [DOI] [PubMed] [Google Scholar]
- 9. Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89:392–400. [DOI] [PubMed] [Google Scholar]
- 10. Munoz V, Campos L, Sadqi M (2016) Limited cooperativity in protein folding. Curr Opin Struct Biol 36:58–66. [DOI] [PubMed] [Google Scholar]
- 11. Abkevich V, Gutin A, Shakhnovich E (1995) Impact of local and non‐local interaction on thermodynamics and kinetics of protein folding. J Mol Biol 252:460–471. [DOI] [PubMed] [Google Scholar]
- 12. Gershon PD (2014) Cleaved and missed sites for trypsin, Lys‐C, and Lys‐N can be predicted with high confidence on the basis of sequence context. J Proteome Res 13:702–709. [DOI] [PubMed] [Google Scholar]
- 13. Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN (2010) PONDR‐FIT: a meta‐predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804:996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishida T, Kinoshita K (2008) Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24:1344–1348. [DOI] [PubMed] [Google Scholar]
- 15. Kozlowski LP, Bujnicki JM (2012) MetaDisorder: a meta‐server for the prediction of intrinsic disorder in proteins. BMC Bioinform 13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I‐TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goudreault M, D'Ambrosio L, Kean M, Mullin M, Larsen B, Sanchez A, Chaudhry S, Chen G, Sicheri F, Nesvizhiskii A, Aebersold R, Raught B, Gingras A‐C (2009) A PP2A phosphatase high density interaction network identifies a novel striatin‐interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics 8:157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ortega A, Amorós D, García de la Torre J (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic‐ and residue‐level models. Biophys J 101:892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pierce B, Wiehe K, Hwang H, Kim B, Vreven T, Weng Z (2014) ZDOCK server: interactive docking prediction of protein‐protein complexes and symmetric multimers. Bioinformatics 30:1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozakov D, Hall D, Xia B, Porter K, Padhorny D, Yueh C, Beglov D, Vajda S (2017) The ClusPro web server for protein‐protein docking. Nat Protoc 12:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lakowicz JR. Principles of fluorescence spectroscopy. 3rd ed. Berline, Germany: Springer, 2006. [Google Scholar]
- 24. Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT (2013) Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res 41:W340–W348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonnell AV, Jiang T, Keating AE, Berger B (2006) Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22:356–358. [DOI] [PubMed] [Google Scholar]
- 26. Lobley A, Sadowski MI, Jones DT (2009) pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25:1761–1767. [DOI] [PubMed] [Google Scholar]
- 27. Yan Y, Winograd E, Viel A, Cronin T, Harrison S, Branton D (1993) Crystal structure of the repetitive segments of spectrin. Science 262:2027–2030. [DOI] [PubMed] [Google Scholar]
- 28. Whitby FG, Phillips GN (2000) Crystal structure of tropomyosin at 7 angstroms resolution. Proteins 38:49–59. [PubMed] [Google Scholar]
- 29. de Almeida Ribeiro E Jr, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Žagrović B, Bordignon E, Pirker KF, Svergun DI, Gautel M, Djinović‐Carugo K (2014) The structure and regulation of human muscle α‐actinin. Cell 159:1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- 31. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin A, Hokugo A, Choi J, Nishimura I (2010) Small cytoskeleton‐assoicated molecule, fibroblast growth factor receptor 1 oncogene partner 2/would inducible transcript‐3.0 (FGFR1OP2/wit3.0), facilitates fibroblast‐driven would closure. Am J Pathol 176:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diebold‐Durand M‐L, Lee H, Ruiz Avila LB, Noh H, Shin H‐C, Im H, Bock FP, Bürmann F, Durand A, Basfeld A, Ham S, Basquin J, Oh B‐H, Gruber S (2017) Structure of full‐length SMC and rearrangements required for chromosome organization. Mol Cell 67:334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartual SG, Straume D, Stamsas GA, Munoz IG, Alfonso C, Martinez‐Ripoll M, Havarstein LW, Hermoso JA (2014) Structural basis of PcsB‐mediated cell separation in Streptococcus pneumoniae . Nat Commun 5:3842. [DOI] [PubMed] [Google Scholar]
- 35. An X, Guo X, Zhang X, Baines AJ, Debnath G, Moyo D, Salomao M, Bhasin N, Johnson C, Discher D, Gratzer WB, Mohandas N (2006) Conformational stabilities of the structural repeats of erythroid spectrin and their functional implications. J Biol Chem 281:10527–10532. [DOI] [PubMed] [Google Scholar]
- 36. Cortese MS, Uversky VN, Dunker AK (2008) Intrinsic disorder in scaffold proteins: getting more from less. Prog Biophys Mol Biol 98:85–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fonin AV, Darling AL, Kuznetsova IM, Turoverov KK, Uversky VN (2018) Intrinsically disordered proteins in crowded milieu: when chaos prevails within the cellular gumbo. Cell Mol Life Sci 75:3907–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de la Torre JG, Huertas ML, Carrasco B (2000) Calculation of hydrodynamic properties of globular proteins from their atomic‐level structure. Biophys J 78:719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heinig M, Frishman D (2004) STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 32:W500–W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heo L, Lee H, Seok C (2016) GalaxyRefineComplex: refinement of protein‐protein complex model structures driven by interface repacking. Sci Rep 6:32153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M (2006) In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1:2856–2860. [DOI] [PubMed] [Google Scholar]
- 42. Phillips GN (1986) Construction of an atomic model for tropomyosin and implications for interactions with actin. J Mol Biol 192:128–131. [DOI] [PubMed] [Google Scholar]
- 43. Ragusa MJ, Stanley RE, Hurley JH (2012) Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 151:1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim JH, Hong SB, Lee JK, Han S, Roh K‐H, Lee K‐E, Kim YK, Choi E‐J, Song HK (2015) Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 11:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Borgnia MJ, Banerjee S, Merk A, Matthies D, Bartesaghi A, Rao P, Pierson J, Earl LA, Falconieri V, Subramaniam S, Milne JLS (2016) Using cryo‐EM to map small ligands on dynamic metabolic enzymes: studies with glutamate dehydrogenase. Mol Pharmacol 89:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bujacz A (2012) Structure of bovine, equine and leporine serum albumin. Acta Cryst D 68:1278–1289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of peptides identified by LCMS/MS for LP 1 min timepoint.
Table S2. Summary of peptides identified by LCMS/MS for LP 5 min timepoint.
Table S3. Summary of peptides identified by LCMS/MS for LP 10 min timepoint.
Table S4. Summary of peptides identified by LCMS/MS with BS3 modifications