

RNA-directed DNA methylation pathway genes act paternally to sensitize endosperm to excess paternal genomes, challenging previous models of dosage sensitivity in endosperm.
Abstract
Seed development is sensitive to parental dosage, with excess maternal or paternal genomes creating reciprocal phenotypes. Paternal genomic excess frequently results in extensive endosperm proliferation without cellularization and seed abortion. We previously showed that loss of the RNA polymerase IV gene NUCLEAR RNA POLYMERASE D1 (NRPD1) in tetraploid fathers represses seed abortion in paternal excess crosses. Here, we show genetically that RNA-directed DNA methylation (RdDM) pathway activity in the paternal parent is sufficient to determine the viability of paternal excess Arabidopsis (Arabidopsis thaliana) seeds. We compared transcriptomes, DNA methylation, and small RNAs from the endosperm of seeds from balanced crosses (diploid × diploid) and lethal (diploid × tetraploid) and viable paternal excess crosses (diploid × tetraploid nrpd1). Endosperms from both lethal and viable paternal excess seeds share widespread transcriptional and DNA methylation changes at genes and transposable elements. Interploidy seed abortion is thus unlikely to be caused by transposable elements or imprinted gene misregulation, and its repression by the loss of paternal RdDM is associated with only modest gene expression changes. Finally, using allele-specific transcription data, we present evidence for a transcriptional buffering system that increases the expression of maternal alleles and represses paternal alleles in response to excess paternal genomic dosage. These findings prompt reconsideration of models for dosage sensitivity in endosperm.
INTRODUCTION
The endosperm of flowering plants is an essential tissue for seed viability. The endosperm, which is most commonly triploid, is formed when the diploid central cell is fertilized by a haploid sperm. In many flowering plants, endosperm proceeds through the early phases of proliferative development as a syncytium before differentiating into three subtypes: micropylar, peripheral, and chalazal endosperm (Li and Berger, 2012). Cellularization is a critical step in endosperm development, after which cell division slows and eventually ceases (Hehenberger et al., 2012; Li and Berger, 2012). In some species, such as Arabidopsis (Arabidopsis thaliana), the endosperm is almost completely degraded at seed maturation as nutrients are assimilated and stored in the embryo (Li and Berger, 2012), whereas in other species, such as grasses, it is persistent and mobilized later during seed germination. The endosperm serves several important functions: it mediates resource transfer from the mother to the growing embryo or germinating seedling (Li and Berger, 2012), signals to the maternal integuments to promote their proliferation and to allow accommodation of the growing offspring (Figueiredo et al., 2016), is required for embryonic development, and influences seed dormancy and germination (Fiume and Fletcher, 2012; Yan et al., 2014; Piskurewicz et al., 2016). Through these activities, endosperm influences seed size.
Balance between the maternal and paternal genomes in endosperm is important for normal endosperm development and, consequently, seed development and viability. Violations of the 2:1 maternal:paternal (m:p) genome ratio in endosperm lead to developmental defects, although species vary in the extent to which they tolerate violations of this ratio (Muntzing, 1936; Håkansson and Ellerström, 1950; Cooper, 1951; Milbocker and Sink, 1969; Esen and Soost, 1973; Scott et al., 1998; Stoute et al., 2012; Povilus et al., 2018). In Arabidopsis, increased maternal genome dosage (maternal excess) leads to premature cellularization of the endosperm and the formation of smaller seeds (Scott et al., 1998). By contrast, increased paternal genome dosage (paternal excess) in crosses between diploid mothers and hexaploid fathers leads to a failure of endosperm cellularization, prolonged cell proliferation, and seed abortion (Scott et al., 1998). However, there is intraspecific variation in the levels of seed abortion observed in paternal excess crosses. For example, whereas tetraploid (4n) Arabidopsis Columbia-0 (Col-0 or Col) induces seed abortion when pollinating diploid mothers, tetraploid C24 and tetraploid Cvi plants do not (Scott et al., 1998; Dilkes et al., 2008; Lu et al., 2012; Piskurewicz et al., 2016).

What determines the threshold between seed lethality and viability under conditions of paternal genomic excess? Mutants that repress seed abortion in interploidy crosses, as well data from incompatible interspecific crosses, which share features with interploidy crosses, have provided some answers to this question. Components in the endosperm, in the gametophytes, or in the parental sporophytes have been proposed to be responsible for endosperm dosage sensitivity in interploid crosses. Some of the elements that determine the critical threshold might be linked to interactions between the endosperm and maternal genotype. In one model, the imbalance is between the paternal dose and the female gametophyte (von Wangenheim and Peterson, 2004; Birchler, 2014). A second model involves interactions between the paternal excess endosperm and the diploid maternal integuments, which develop into the seed coat (Muntzing, 1936). The ability of maternal sporophytic mutations in the flavonoid pathway to repress paternal excess seed abortion is consistent with this model (Dilkes et al., 2008; Doughty et al., 2014). The expression levels of AGAMOUS-LIKE genes (AGLs), the small RNA (sRNA) siRNA854, chromatin regulators, cell wall genes, and defense response genes, all presumed to act in the endosperm, have been linked to paternal excess seed abortion (Walia et al., 2009; Burkart-Waco et al., 2013; Kradolfer et al., 2013; Wolff et al., 2015; Jiang et al., 2017; Wang et al., 2018).
Multiple models suggest that endosperm ploidy incompatibilities are caused by epigenetic abnormalities. The epigenetic mark DNA methylation is established and partially maintained through the RNA-directed DNA methylation (RdDM) pathway. During this process, short noncoding RNAs generated by RNA polymerase IV (RNA Pol IV) are converted into double-stranded RNAs by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2). These RNAs are then subsequently processed by DICER-LIKE 3 (DCL3) into 24-nucleotide sRNAs loaded into an ARGONAUTE complex (usually ARGONAUTE 4 [AGO4] or AGO6), and then interact with a noncoding RNA transcribed by another polymerase, RNA Pol V, to direct DNA methylation by the de novo methyltransferase DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2). In normal triploid endosperm, maternal chromosomes are DNA hypomethylated relative to paternal chromosomes due to the activity of the 5-methylcytosine DNA glycosylase DEMETER (Gehring et al., 2009; Hsieh et al., 2009; Ibarra et al., 2012). DNA methylation represses the expression of some endosperm genes and promotes the expression of others (Satyaki and Gehring, 2017). It has been proposed that a cause of interploid seed abortion is increased expression of transposable elements (TEs) due to epigenetic alterations (Martienssen, 2010). Under this model, a maternally deposited sRNA dose is insufficient to silence the doubled number of paternally inherited TEs. Another model is influenced by the parental conflict theory (Haig and Westboy, 1991). This theory argues that in a polyandrous system in which the mother provisions resources for her offspring by various fathers, genes restricting resource allocation to any one seed and favoring equitable distribution of resources across all the progeny become predominantly maternal in expression (maternally expressed genes [MEGs]). On the other hand, genes promoting resource allocation and larger seed production are predominantly paternally expressed (paternally expressed genes [PEGs]). This type of allele-specific expression is an epigenetic phenomenon referred to as gene imprinting. According to these ideas, an excess dose of paternal chromosomes leads to overexpression of PEGs (genes that promote endosperm proliferation), eventually leading to seed abortion. Consistent with this model, PEGs were reported to be overexpressed in paternal excess seeds (Kradolfer et al., 2013; Schatlowski et al., 2014), and loss-of-function mutations in multiple PEGs repress seed abortion under conditions of paternal genomic excess (Kradolfer et al., 2013; Wolff et al., 2015; Huang et al., 2017; Jiang et al., 2017).
We recently proposed another model based on discoveries about the function of NUCLEAR RNA POLYMERASE D1 (NRPD1), which encodes the largest subunit of RNA Pol IV, in normal triploid endosperm (Erdmann et al., 2017). The expression of 24-nucleotide sRNAs in Arabidopsis endosperm is paternally biased and concentrated in pericentromeric heterochromatin, but a subset of 24-nucleotide sRNAs are expressed predominantly from the maternal alleles of genes (Erdmann et al., 2017). We showed that paternal loss of NRPD1 in 2m:1p triploid endosperm increased the maternal fraction of the transcriptome, suggesting that wild-type NRPD1 represses maternal genome dosage, probably via the production of 24-nucleotide sRNAs. We also discovered that paternally inherited mutations in NRPD1 repress seed abortion caused by excess paternal genomes (i.e. 2n Col × 4n Col), leading to the hypothesis that the increased maternal fraction of the transcriptome in nrpd1 mutant endosperm compensates for increased paternal genomic dosage. In this model, the loss of RNA Pol IV-dependent sRNA production in the endosperm is essential for viability (Erdmann et al., 2017).
Finally, the epigenetically activated small interfering RNA (easiRNA) model argues that NRPD1 and RDR6 act together in a noncanonical pathway in the male gametophyte to produce easiRNAs whose concentration scales with ploidy (Borges et al., 2018; Martinez et al., 2018). easiRNAs are defined as 21- to 22-nucleotide sRNAs produced from the processing of epigenetically activated TE mRNAs (Creasey et al., 2014). It is proposed that easiRNAs are transmitted during fertilization from sperm to the central cell, where they inhibit RdDM in the developing endosperm (Borges et al., 2018; Martinez et al., 2018). In this model, easiRNAs are lost from sperm when the paternal copy of NRPD1 is mutated. The loss of paternal easiRNAs allows for the restoration of a functional RdDM pathway in endosperm using maternal copies of NRPD1. The restored RdDM pathway was proposed to repress excess paternal dosage and restore seed viability.
In this study, to test these models and to further understand the nature of intolerance or tolerance to paternal genomic excess, we investigated the genetic and molecular contributions to interploidy seed abortion and repression in Arabidopsis. We created tetraploid mutants for members of the canonical and noncanonical sRNA pathways to test the genetic requirements for seed abortion in paternal excess crosses. We also profiled sRNAs, DNA methylation, and gene expression in balanced (2n × 2n), lethal paternal excess (2n × 4n), and viable paternal excess (2n × 4n nrpd1) endosperm. We found that genes of the canonical RdDM pathway were necessary only in the male parent for seed abortion induced by paternal genomic excess. Through transcriptomic profiling, we found that there were extensive changes in gene and TE expression associated with lethal paternal genomic excess but that only a small fraction were ameliorated in viable paternal excess endosperm. Our analysis also revealed the signatures of a potential buffering system that attempts to rebalance transcription under conditions of paternal excess by repressing paternal alleles and activating maternal alleles. These observations indicate that endosperm development can tolerate surprisingly large variations in gene expression at most genes, and they suggest that relatively small gene expression changes contribute to the critical dosage that determines seed viability under conditions of paternal genomic excess.
RESULTS
Paternal Loss of Canonical RdDM Pathway Genes Is Sufficient for the Suppression of Interploid Seed Abortion
In crosses between diploid females and tetraploid males, tetraploid nrpd1 mutant fathers repress seed abortion, whereas diploid nrpd1 mothers have no effect (Erdmann et al., 2017; Martinez et al., 2018). We tested if other members of the canonical RdDM pathway behaved genetically in the same manner. Using colchicine-induced tetraploid sawadee homeodomain homolog 1 (shh1), encoding a homeodomain protein required for DNA methylation at some loci, rdr2, dcl3, nrpe1 (encoding the largest subunit of RNA Pol V), and drm2 mutants, we examined seeds obtained from crosses between 2n wild-type (Col) mothers and 4n (Col) fathers that were either wild-type or mutant for one of the RdDM pathway components (Figure 1). Mature seeds were scored as normal, aborted, or abnormal, and multiple independent crosses were analyzed. Typically, the seed abortion percentage in each set of crosses varied by between 20% and 30%, regardless of whether the parents were wild type or mutant (Figure 1A). For example, in wild-type paternal excess crosses (2n Col × 4n Col), the seed abortion ranged from 57.8% to 100%, with a mean percent seed abortion of 81.1%. Therefore, we considered only those mutants capable of substantially enhancing seed viability across multiple crosses as being true repressors of seed abortion.
Figure 1.

Loss of Paternal RdDM Genes but Not RDR6 Represses Seed Abortion in Paternal Excess Crosses.
(A) Each circle represents the seed abortion rate in one cross and represents multiple siliques from a single inflorescence.
(B) Each circle represents the percent of seeds that failed to germinate from each scored collection of seeds. Failure to germinate was defined as the inability to produce either a radicle or a hypocotyl. Bars show median and interquartile range. * at the bottom represents statistically significant difference (P < 0.05) in comparisons between the indicated cross and the cross between wild-type (WT) diploid (2n) Col-0 mothers and wild-type tetraploid (4n) Col-0 fathers. * at the top represents statistically significant differences (P < 0.05) between the indicated crosses. Statistical significance calculated by Wilcoxon test.
All paternally inherited RdDM pathway mutations, except shh1, substantially repressed seed abortion and promoted the production of fully developed seeds capable of germination (Figure 1; Supplemental Figure 1). SHH1 is required for RNA Pol IV recruitment at a subset of its target sites (Law et al., 2013), suggesting that SHH1-independent activity of RNA Pol IV is important for seed abortion. Mutations in nrpe1 and drm2 mirrored nrpd1 in their substantial repression of seed abortion; the mean seed abortion percentage among the examined seeds were 18.9% for nrpd1, 21.4% for nrpe1, and 11.3% for drm2 (Figure 1A; Supplemental Figure 1). rdr2 and dcl3 mutations repressed seed abortion to a lesser extent (Figure 1; Supplemental Figure 1). The previously described redundancy between DCL3 and other DCL paralogs (Gasciolli et al., 2005; Stroud et al., 2013) likely explains the lower repression of seed abortion by the loss of DCL3, but it remains unclear why the loss of RDR2 does not repress seed abortion to the same extent as that observed upon the loss of NRPD1.
For all mutants, the seed abortion ratios were reflected in the percentage of seeds that germinated on plates (Figure 1B; Supplemental Figure 1). Tetraploid drm2 and nrpd1 mutants also suppressed interploidy seed abortion when crossed with wild-type diploid Landsberg erecta (Ler) mothers (Supplemental Figures 1E and 1G; Erdmann et al., 2017), indicating that the effect was not specific to Col mothers. RNA Pol IV has also been proposed to function in partnership with RDR6 in a noncanonical RdDM pathway (Martinez et al., 2018). We therefore tested if tetraploid fathers mutant for rdr6 could suppress paternal excess seed abortion. A paternally inherited rdr6 mutation did not repress seed abortion and did not act additively with rdr2 (Figure 1A; Supplemental Figure 1B). We also tested whether mutations in the CHG methyltransferase gene CMT3 could suppress interploidy seed abortion when inherited through the male—no effect was observed (Supplemental Figures 1D and 1F).
In contrast to the repression of seed abortion by paternally inherited loss-of-function mutations in RdDM pathway genes, most loss-of-function mutations inherited through the diploid mother did not repress seed abortion in paternal excess crosses with wild-type tetraploid fathers (Figure 1; Supplemental Figures 1A and 1C). An exception was the maternal loss of DRM2, which resulted in a statistically significant repression of seed abortion, although the magnitude of the repression was small (mean percentage of aborted seed was 66.9% for drm2 compared with 81.1% for wild type) and did not phenocopy the extensive reduction in seed abortion observed upon the loss of paternal RdDM pathway components (Figure 1; Supplemental Figures 1A and 1C). It was previously reported that the suppression of paternal excess seed abortion also occurs when both parents are mutant for NRPD1 (Erdmann et al., 2017; Martinez et al., 2018). We observed the same effect for all other genes tested: rdr2, dcl3, nrpe1, and drm2 (Figure 1; Supplemental Figures 1A and 1C). In sum, these genetic results indicate (1) that paternal loss of the canonical RdDM pathway is sufficient to suppress seed abortion caused by paternal genomic excess and (2) that a genetically complete canonical RdDM pathway is not required in endosperm itself for the suppression of seed abortion.
Massive, Shared Gene Misregulation in Lethal and Viable Paternal Excess Endosperm
To determine what genes or processes are associated with interploidy seed lethality and its genetic suppression, we examined the transcriptional effects of doubling paternal dosage in the endosperm. We performed mRNA-sequencing (mRNA-seq) to high depth on developing endosperm from three biological replicates (RNA isolated from endosperm of seeds from multiple siliques in separate experiments) of balanced crosses (Ler × Col), lethal paternal excess (Ler × 4n Col), and viable paternal excess (Ler × 4n Col nrpd1; Figure 2; Supplemental Table 1). Surprisingly, comparisons of the three transcriptomes by principal component analysis indicated that lethal and viable paternal excess endosperms were more similar to each other than to balanced endosperm (Figure 2A). Approximately one-third of the transcriptome was significantly misexpressed (2-fold or greater difference at q < 0.05) in lethal paternal-excess endosperm compared with balanced endosperm: 4054 genes were more highly expressed, and 3855 genes exhibited decreased expression (Figure 2B; Supplemental Data Set 1). Gene ontology (GO) analyses performed using PANTHER14.1 (Supplemental Data Set 2) showed that genes with lower transcript abundance in lethal paternal excess relative to balanced endosperm were enriched for those encoding light harvesting proteins, proteins in glucose and starch metabolism, hormone responses, response to abiotic stimuli, and cell wall organization. Whereas the predicted consequences of many of these changes remain unclear, the decreased expression of cell wall genes is consistent with the failure in endosperm cellularization that has been previously described in lethal paternal excess crosses (Wolff et al., 2015). GO analyses of genes with increased expression identified enrichment for those encoding proteins involved in protein deneddylation, ribosome biogenesis, DNA replication, chromosome segregation, and the cell cycle. The increased expression of these genes is consistent with the increased cell proliferation observed in paternal excess endosperm (Scott et al., 1998; Tiwari et al., 2010). Compared to balanced endosperm, viable paternal excess endosperm also showed a similar quantitative change in gene expression: 3150 genes exhibited increased expression and 2845 exhibited decreased expression (Figure 2B; Supplemental Data Set 1). Many of the same genes were misregulated in viable and lethal paternal excess endosperm compared with balanced endosperm. Compared with lethal paternal excess, only 188 genes were downregulated in viable paternal excess endosperm and 426 genes were more highly expressed (Figure 2B; Supplemental Data Set 1). To further test if the viability of paternal excess seeds was predicated on the transcriptome returning to the dosage of balanced endosperm, we examined the extent to which gene expression was “corrected” in viable paternal excess endosperm (Figure 2C). Most genes with increased expression in lethal paternal excess endosperm were not corrected, but a small subset of genes with decreased expression in lethal paternal excess were moderately corrected in viable paternal excess (Figure 2C).
Figure 2.

Lethal and Viable Paternal Excess Endosperms Are Transcriptionally More Similar to Each Other Than to Balanced Endosperm.
(A) PCA plot of read counts for genes from biological replicate mRNA-Seq samples.
(B) Plot of the number of genes differentially expressed in comparisons of balanced endosperm with both lethal (purple) and viable (yellow) ♂ excess endosperm. Only 614 genes were differentially expressed between viable and lethal ♂ excess endosperm (gray).
(C) Correction value in viable paternal excess endosperm for each gene that was called as being significantly differentially expressed in comparisons of balanced and lethal ♂ excess endosperm. The value was calculated as 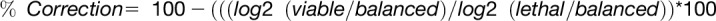 ). A value of 100% indicates that the gene, which was misregulated in lethal ♂ excess, was not differentially expressed in viable ♂ excess relative to balanced endosperm. A value of 0% represents similar misregulation in both lethal and viable ♂ excess relative to balanced endosperm. Fold change values and significance for fold change for (B) and (C) were calculated using Cuffdiff. Boxplot is a Tukey plot.
). A value of 100% indicates that the gene, which was misregulated in lethal ♂ excess, was not differentially expressed in viable ♂ excess relative to balanced endosperm. A value of 0% represents similar misregulation in both lethal and viable ♂ excess relative to balanced endosperm. Fold change values and significance for fold change for (B) and (C) were calculated using Cuffdiff. Boxplot is a Tukey plot.
(D) Lethal paternal excess endosperm was enriched for chalazal endosperm gene expression; viable paternal excess endosperm showed both chalazal and peripheral markers. Tissue enrichment for each biological replicate is shown.
We analyzed the transcriptomic data using a tissue enrichment tool to assess if the abundances of markers enriched in chalazal, peripheral, and micropylar endosperm were altered in paternal excess endosperm (Schon and Nodine, 2017). Paternal excess endosperm adopted a gene expression program characteristic of chalazal endosperm (Figure 2D), which is consistent with the results of phenotypic analysis of lethal paternal excess seeds (Scott et al., 1998; Wolff et al., 2015; Martinez et al., 2018). Viable paternal excess endosperm transcriptomes also showed increased chalazal endosperm marker gene expression, but there was also slightly elevated peripheral endosperm marker gene expression relative to lethal paternal excess endosperm. These results suggest that lethal and viable paternal excess endosperm differ in a similar manner from balanced endosperm in terms of gene expression and subsequent developmental programs.
We also reanalyzed published endosperm transcriptome data from lethal and viable paternal excess endosperm generated using omission of second division1 (osd1) and osd1 nrpd1 mutations (Supplemental Figure 2; Martinez et al., 2018). Consistent with our data, we found that both Ler × Col osd1 (lethal paternal excess) and Ler × Col osd1 nrpd1 (viable paternal excess) showed elevated expression of chalazal markers (Supplemental Figure 2D). Endosperm from Ler × Col osd1 nrpd1 also showed extensive genic misregulation, but the percentage of the transcriptome that was misregulated was lower than in our viable paternal excess endosperm data sets (Figure 2B; Supplemental Figure 2B). Additionally, the extent of gene expression correction in viable paternal excess endosperm was also lower in our data sets compared with theirs (Figure 2C; Supplemental Figure 2C). These discrepancies could stem from multiple differences between our experiments. Our endosperm data had more replicates, higher mappable read depth, and lower levels of seed coat contamination (Supplemental Figure 2; Supplemental Table 1; Martinez et al., 2018). We created paternal excess endosperm using tetraploid fathers while Martinez et al. created paternal excess via the use of the osd1 mutation, which causes omission of the second meiotic division and thus generates diploid pollen. There might be biological differences, as yet unclear, between diploid sperm produced from a diploid parent compared with diploid sperm produced from a tetraploid parent. Also, the osd1 mutation was backcrossed into Col from another accession (d’Erfurth et al., 2009; Martinez et al., 2018), and interploidy seed abortion is sensitive to the genetic background of both parents (Scott et al., 1998; Lu et al., 2012; Piskurewicz et al., 2016).
Misregulation of Imprinted Genes Characterizes Both Lethal and Viable Paternal Excess Endosperm
Imprinted gene misregulation in the endosperm has been suggested to be a culprit for the developmental catastrophe of paternal excess crosses (Haig and Westboy, 1991; Gutierrez-Marcos et al., 2003). We found that imprinted genes are disproportionately more likely to show increased expression than all genes in the genome under conditions of lethal paternal excess (N-1 chi-square test, P = 3 × 10−4 for MEGs and P < 10−4 for PEGs; Figure 3). Of 43 previously identified Col-Ler PEGs and 130 Col-Ler MEGs (Pignatta et al., 2014), 25 PEGs (58%) and 27 MEGs (19%) displayed at least a twofold increase in transcript abundance in lethal paternal excess endosperm (Figure 3A). The abundance of two PEGs and 18 MEGs decreased. The vast majority of these imprinted genes remained misregulated in viable paternal excess endosperm (Figures 3B and 3C). Only two MEGs were differentially expressed in comparisons of viable and lethal paternal excess endosperm— JAGGED LATERAL ORGANS transcript abundance increased 2.6-fold and ARABIDOPSIS GLUTAMINE SYNTHASE1;1 transcript abundance decreased 2.4-fold in viable paternal excess endosperm (Figure 3C; Supplemental Data Set 1). Similarly, only one PEG, AT4G20800, had lower transcript abundance in viable paternal excess endosperm compared with lethal paternal excess endosperm (Figure 3C; Supplemental Data Set 1). These results suggest that misregulation of multiple imprinted genes in endosperm is unlikely to be the cause of endosperm dysfunction and seed lethality in interploidy crosses.
Figure 3.

Imprinted Genes and Transposons Are Misregulated in Both Lethal and Viable Paternal Excess Endosperm.
(A) to (C) Expression of Col-Ler imprinted genes in endosperm. FPKM is normalized expression; statistical significance of difference in abundance was calculated by Cuffdiff. q < 0.05 is represented by black circles. q > 0.05 is represented by gray circles.
(D) to (F) Expression from transposable elements is elevated in lethal and viable ♂ excess endosperm. RNA-Seq reads were mapped to consensus sequences from Repbase. Black circles represent TEs with significant differences in transcript abundances according to DEGseq, q < 0.05. Gray circles represent TEs without significant differences in transcript abundances.
It was previously demonstrated that mutations in a subset of PEGs or their interactors can suppress paternal excess seed abortion when inherited paternally (Kradolfer et al., 2013; Wolff et al., 2015; Huang et al., 2017; Jiang et al., 2017). We found that whereas several of these genes indeed had increased transcript abundance in lethal paternal excess relative to balanced endosperm, their expression levels remained altered in viable paternal excess endosperm (Supplemental Figure 3). This observation suggests that seed viability brought about by the loss of paternal nrpd1 is independent of gene expression normalization in the endosperm for genes whose paternal loss also represses seed abortion.
In summary, these gene expression data indicate that the expression of only a small number of genes distinguishes lethal paternal excess endosperm from viable paternal excess endosperm at this stage of development, and they suggest that correction of gene expression to levels observed in balanced endosperm, even for imprinted genes, is not necessary for paternal excess seed viability.
A Subset of TEs Are Misregulated in Lethal and Viable Paternal Excess Seeds
Increased expression of TEs in paternal excess endosperm has been speculated to be a cause of seed abortion (Martienssen, 2010; Castillo and Moyle, 2012). We compared TE expression levels in endosperm by mapping mRNA-seq reads to 375 consensus TE sequences from REPBASE (Figures 3D–3F; Bao et al., 2015). In lethal paternal excess endosperm, 73 families showed a statistically significant change in transcript abundance, including 40 whose transcript abundance increased by 5-fold or more, including the transpositionally active family ONSEN/ATCOPIA78 (Figure 3D; Supplemental Data Set 3). The TE families ATLINE1-10A and ATGP7 exhibited the largest increases, i.e. at least 52-fold (Figure 3D). Additionally, the transcript abundance of 26 TE families decreased in paternal excess crosses. TAG1, a transpositionally active TE that is present only in the maternally inherited Ler genome (Tsay et al., 1993), was the most repressed (nearly 56-fold; Figure 3D). Like for genes, the expression profiles of TEs in viable paternal excess endosperm were similar to those of lethal paternal excess endosperm (Figures 3E and 3F; Supplemental Data Set 3). Only 14 TE families exhibited decreased transcript abundance in viable paternal excess endosperm relative to lethal endosperm, and 19 TE families exhibited increased transcript abundance (Figure 3F; Supplemental Data Set 3). The observation that lethal and viable paternal excess endosperms exhibit TE misregulation to a similar extent argues that TE misregulation by itself is unlikely to be the cause of seed abortion induced by paternal genomic excess.
The RdDM Pathway Is Attenuated in Paternal Excess Endosperm
A number of proteins that establish or maintain methylation in CG, CHG, and CHH contexts were differentially expressed among balanced, lethal, and viable paternal excess endosperm (Figure 4; Supplemental Data Set 1). The differential expression of genes encoding members of the RdDM pathway is particularly noteworthy. With the exception of NRPD1, the expression of genes encoding subunits of RNA Pol IV and V, along with RDR2, AGO4, and DRM2, was significantly reduced in lethal paternal excess endosperm (Figure 4A; Supplemental Data Set 1). Consistent with these findings, the expression of the 5-methylcytosine DNA glycosylase gene ROS1, whose expression is directly promoted by RdDM (Williams et al., 2015), was also reduced (Figure 4A). The expression of most of these genes appeared to increase slightly in viable paternal excess endosperm compared with lethal endosperm, but only increases in RDR2 and ROS1 were significant (Figure 4A).
Figure 4.

Canonical RdDM Pathway Function in Endosperm Is Affected by Paternal Excess.
(A) Genes encoding RdDM components are downregulated in lethal and viable ♂ excess endosperm. ROS1 expression is a readout of RdDM activity and reflects differential activity of RdDM. * represents statistically significantly different gene expression, q < 0.05.
(B) Small RNA production is impacted in paternal excess endosperm. Nucleotide (nt) size profiles of sRNA reads mapped to the TAIR10 genome for three replicates of balanced endosperm and two replicates each of lethal and viable paternal excess endosperm.
(C) CHH methylation losses in paternal excess endosperm. Venn diagrams show the intersections of CHH DMRs obtained from comparisons of balanced (Bal), lethal, and viable paternal excess endosperm. Lethal and viable paternal excess endosperms share a significant proportion of regions that are hypomethylated relative to balanced endosperm. A smaller subset of DMRs lost more methylation in lethal relative to viable endosperm.
(D) Loss of CHH methylation is associated with the loss of 24-nucleotide (nt) sRNAs. Venn diagrams show the relationship between changes in sRNA abundance and CHH methylation levels. A subset of CHH DMRs is associated with the loss of sRNAs. A smaller subset is associated with gains in 24-nt sRNAs. Significance of overlaps was calculated using the Fisher test option from Bedtools.
We tested if sRNA production was affected in paternal excess endosperm. We sequenced sRNAs from two replicates of lethal and viable paternal excess endosperm (Supplemental Table 1) and compared them to previously published sRNA data from balanced endosperm (Erdmann et al., 2017). Previous evaluations of sRNAs in paternal excess crosses have been performed on whole seeds, not endosperm, but noted reduced 24-nucleotide sRNA levels in lethal paternal excess seeds (Lu et al., 2012; Martinez et al., 2018). We assessed the overall functionality of sRNA production in the endosperm by examining sRNA size profiles. In lethal paternal excess endosperm, 24-nucleotide sRNAs represented the most abundant size class, but their proportion was reduced compared with balanced endosperm (Figure 4B). In viable paternal excess endosperm, the proportion of 24-nucleotide sRNAs was comparable to that of balanced endosperm (Figure 4B). Genic and TE-associated sRNAs in lethal and viable paternal excess showed a pattern similar to that of bulk sRNAs (Supplemental Figures 4A and 4B; Supplemental Data Sets 4 and 5). These observations suggest that the capacity to produce 24-nucleotide sRNAs is relatively normal in viable paternal excess endosperm, presumably due to restored RDR2 expression (Figures 4A and 4B). We also note that the proportion of 21-nucleotide sRNAs increased in both lethal and viable paternal excess endosperm (Figure 4B; Supplemental Figure 4). While the pathways involved in the biogenesis of these 21-nucleotide sRNAs remain unclear, they could represent the activity of posttranscriptional gene silencing (mRNA cleavage) pathways on transcripts that are misregulated in both lethal and viable paternal excess endosperm (Marí-Ordóñez et al., 2013; Borges and Martienssen, 2015).
A hallmark of the activity of the 24-nucleotide sRNA pathway is CHH methylation. We performed whole-genome bisulfite sequencing to profile the methylomes of balanced, lethal, and viable paternal excess endosperm (Supplemental Table 1; Supplemental Data Sets 6–8). To identify regions with altered CHH methylation, we divided the genome into 300-bp windows with 200-bp overlaps and identified those windows with a 10% or greater CHH methylation difference between genotypes (i.e. 20% versus 30% methylation). Windows with significant differences were merged into differentially methylated regions. CHH methylation was slightly higher in viable paternal excess endosperm relative to lethal paternal excess endosperm, but in both genotypes it was drastically reduced relative to balanced endosperm (Figure 4C, Supplemental Data Sets 6–8). Regions with reduced CHH methylation significantly overlapped with regions with lowered sRNA levels in lethal paternal excess endosperm (Figure 4D).
Paternal Excess Differentially Affects the Expression of Maternally and Paternally Inherited Alleles
Total gene expression levels are derived from the contribution of maternally and paternally inherited alleles. Based on the effect of nrpd1 mutations on allele-specific expression in balanced endosperm, we previously proposed that 4n nrpd1 might suppress paternal excess lethality by increasing transcriptional dosage from maternal alleles for many genes (Erdmann et al., 2017). To test this hypothesis, we evaluated allele-specific expression in balanced, lethal, and viable paternal excess endosperm for genes with at least 50 allele-specific reads in both balanced and paternal excess crosses (Figure 5; Supplemental Data Set 9). Balanced endosperm has a 2:1 m:p ratio, and paternal excess endosperm has a 2:2 ratio of m:p genomes. Mirroring the genomic ratios, the median paternal fraction for genes was 33.8% in balanced endosperm, 50.1% in lethal paternal excess endosperm, and 50.6% in viable paternal excess (Figure 5A). To assess potential allelic misregulation of individual genes, we normalized transcriptomic fractions to genomic fractions by calculating the deviation from paternal genomic contributions for each gene (% paternal minus 33% for balanced crosses and % paternalminus 50% for paternal excess). Contrary to our expectation, there was a small but detectable decrease in paternal contribution relative to genomic expectation for specific genes in lethal paternal excess endosperm (two-sided D’Agostino's K2 test, skew = −0.58, P < 2.2e-16; Figure 5B, purple line; Supplemental Data Set 9). A similar bias was observed in viable paternal excess endosperm (two-sided D’Agostino’s K2 test, skew = −0.53, P < 2.2e-16; Figure 5B, yellow line; Supplemental Data Set 9). Thus, in viable relative to lethal paternal excess endosperm, there were few changes in overall allelic bias (two-sided D’Agostino’s K2 test, skew = 0.0443, P = 0.05887; Figure 5B, gray line; Supplemental Data Set 9).
Figure 5.

Allelic Contributions to Expression Differences between Balanced and Lethal Paternal Excess Endosperm.
(A) Paternal fraction in genic transcripts. Boxplot represents all genes with at least 50 allele-specific reads in the indicated genotypes. Number of genes in balanced, 10,388; lethal ♂ excess, 11,709; viable ♂ excess, 11,430. For balanced endosperm, a total of five replicate libraries were analyzed (Erdmann et al., 2017).
(B) Paternal excess endosperm transcriptome is maternally skewed. Frequency distribution plot of % paternal deviation for each gene. Paternal deviation was calculated for genes with at least 50 allele-specific reads in each pair of genotypes being compared. Number of genes in lethal ♂ excess - balanced, 9,550; viable ♂ excess - balanced, 9,639; viable ♂ excess - lethal ♂ excess, 10,919.
(C) Decreased paternal allele contribution at a larger proportion of genes with decreased expression in lethal paternal excess compared with balanced. n = 1910 genes. Impacts of allele-specific changes on gene expression for two examples, WOX8 and AT2G01580, are shown.
(D) Increased maternal allele activation at a larger proportion of genes with increased expression in lethal paternal excess. n = 1608 genes. Impacts of allele-specific changes on gene expression for two examples, SDG21 and HEXO3, are shown. Genes analyzed in (C) and (D) were detected as having significantly different expression levels by Cuffdiff. A gene with an allelic shift of at least 20% was considered to have a modified allelic balance. Boxplots represent median values for paternal deviation from genomic expectation. For gene-specific histograms, the maternal allele is shown in red and the paternal allele is shown in blue.
(E) sRNA populations are increasingly maternally biased in paternal excess endosperm. The paternal fraction of sRNA populations from viable paternal excess endosperm is intermediate between lethal paternal excess and balanced endosperm. Data from three replicates of balanced endosperm and two replicates each of lethal and viable paternal excess endosperm are plotted. sRNA reads mapped to the TAIR10 genome were first split based on size and then into Col and Ler reads using single nucleotide polymorphisms. % of paternal reads and deviation from genomic expectation at each size were calculated.
To explore the contribution of allelic misregulation to genic misregulation, we examined shifts in paternal deviation from genomic expectation for genes with increased or decreased expression in lethal paternal excess endosperm compared with balanced endosperm (Figures 5C and 5D; Supplemental Figure 5). Genes with decreased expression were more heavily influenced by the loss of paternal allele contributions (two-sided D’Agostino's K2 test, skew for down-regulated genes =−0.4506, P = 1.243e-16; Figure 5C). 12.1% of genes with sufficient allele-specific reads showed at least a 20% decrease in paternal bias, indicating increased repression of paternal alleles, whereas 2.3% showed at least a 20% increase in paternal bias, indicating increased repression of maternal alleles (Figure 5C). Genes with increased expression in lethal paternal excess endosperm were more impacted by increases in maternal allele contributions (skew for up-regulated genes = −0.52084, P = 6.61e-16; Figure 5D). 7.4% showed at least a 20% decrease in paternal deviation, indicating increased maternal allele expression, and 4.1% showed at least a 20% increase in paternal bias, indicating increased paternal allele expression (Figure 5D).
A comparison of allelic contributions between viable and lethal endosperm revealed large differences at a very limited number of genes (Supplemental Figures 5B–5D). Among genes with increased transcript abundance in viable compared with lethal paternal excess endosperm, increased paternal allele contributions were observed at nine loci (Supplemental Figures 5B and 5C; Supplemental Data Set 9). Increased maternal allele contributions were observed at another nine loci (Supplemental Figures 5B and 5C; Supplemental Data Set 9). Among genes with decreased transcript abundance, only two genes showed strong evidence for a repression of paternal allele expression, and two genes had evidence for repression of maternal allele expression (Supplemental Figures 5B and 5D).
We also tested if the increased expression of imprinted genes in paternal excess endosperm was related to a breakdown of imprinting (Supplemental Figure 6). In comparisons of balanced endosperm and lethal paternal excess, loss of imprinting was apparent among MEGs. Among 27 MEGs with increased expression in lethal paternal excess endosperm, paternal deviation increased for eight genes, which became biallelically expressed (Supplemental Figure 6). However, strongly maternally biased expression was maintained at 11 MEGs (Supplemental Figure 6). Of the 25 PEGs with increased total expression, 13 had reduced paternal bias (Supplemental Figure 6). However, most of these changes were small, with the exception of AT4G20800, which was biallelically expressed (Supplemental Figure 6). These observations indicate that the loss of imprinting contributes to increases in imprinted gene expression (Figure 3) at only a subset of imprinted genes.
The allele-specific changes in gene expression (Figures 5A–5D) led us to test if sRNA levels change in an allele-specific manner. The sRNA pool in balanced endosperm is overall paternally biased (Figure 5E; Erdmann et al., 2017). For most sRNA size classes, lethal paternal excess endosperm was more maternally biased than expected based on the ratio of maternal and paternal genomes (Figure 5E). For example, in lethal paternal excess, 24-nucleotide sRNAs were 13.8% more maternal than expected given the 2:2 ratio of maternal to paternal genomes. This indicates, counterintuitively, that extra paternal genomes are associated with decreased sRNA levels from paternal genomes (or increased sRNA levels from maternal genomes). sRNAs in viable paternal excess endosperm were slightly more paternal than those of lethal paternal excess endosperm, although to a lesser extent than that observed in balanced endosperm (Figure 5E), which is consistent with the increased functionality of the RdDM pathway in viable paternal excess endosperm (Figure 4A).
In summary, from these analyses, we conclude that increasing paternal genome copy number can affect the expression of only maternal or only paternal alleles for a subset of loci. Thus, maternally and paternally inherited alleles contribute unequally to genome-wide transcriptome misregulation. However, overall, we did not find evidence for our hypothesis that a shift toward maternal allele expression is associated with the repression of paternal excess seed abortion (Erdmann et al., 2017), as both lethal and viable paternal excess endosperm have a higher fraction of maternal transcripts than expected based on parental genomic ratios.
DISCUSSION
Impact on Models for Interploidy Seed Abortion
We have shown that an extra copy of the paternal genome induces regulatory changes at both maternal and paternal alleles and drives massive changes in gene expression in the endosperm, varying little between viable and inviable seeds (Figures 2 to 5). Our data allow us to evaluate a number of proposals regarding the transcriptional changes at genes, TEs, or sRNAs that could result in interploid seed lethality. Inspired by models of transposon-induced dysgenesis in Drosophila leading to atrophied ovaries (reviewed in Kelleher, 2016), it was speculated that an imbalance between sRNAs deposited by a diploid mother and the TE load in a tetraploid father could lead to TE expression, which would then trigger seed abortion (Martienssen, 2010; Castillo and Moyle, 2012). Our finding that TEs are similarly misregulated in lethal and viable paternal excess endosperm (Figure 3) leads us to conclude that TE expression levels are unlikely to be a determinant of interploidy seed viability. While it is perhaps surprising that viable seeds have high TE transcript abundance, it is not unprecedented. Arabidopsis mutants such as methyltransferase1, which have high levels of TE expression (Zilberman et al., 2007; Oberlin et al., 2017), also produce viable seeds (Xiao et al., 2006).
Transcription of imprinted genes is potentially a strong candidate for the critical gene dosage difference separating lethality and viability (Haig and Westboy, 1991; Lafon-Placette and Köhler, 2015; Wolff et al., 2015). However, like TEs, the similar expression levels of imprinted genes between lethal and viable interploid seeds (Figure 3) suggest that their differential expression is not a determinant of viability. We did identify 614 differentially expressed genes between lethal and viable interploidy seeds (Figures 2B and 2C; Supplemental Data Set 1). These 614 differentially expressed genes include several genes encoding proteins involved in phytohormone signaling and developmental regulators—all potentially good candidates for determining seed viability (Supplemental Data Set 1). It is possible that the expression of multiple loci of modest effect or the partial renormalization of expression patterns of multiple loci constitute the dosage threshold between lethal and viable seeds. Based on the present data, we are unable to distinguish if any of the differentially expressed genes or partially corrected genes are causal for the effect on seed viability or whether they represent a consequence of endosperm development associated with seed viability.
Several distinct hypotheses revolving around sRNAs have been postulated to explain how the loss of NRPD1 in tetraploid fathers, but not diploid mothers, represses seed abortion in paternal excess crosses (Erdmann et al., 2017; Martinez et al., 2018). The easiRNA model for interploid seed abortion is based on two key conclusions from the recent data of Martinez et al. (2018). First, a noncanonical RNA Pol IV-RDR6 pathway functions in pollen to produce easiRNAs, which scale with paternal genome dosage. Second, the transfer of increased easiRNAs into the endosperm from diploid pollen inhibits RdDM; it is then the absence of these easiRNAs in nrpd1 mutant pollen that permits the RdDM pathway to function in paternal excess endosperm. Our data do not support this model. We failed to obtain a significant increase in viable paternal excess seeds using 4n rdr6-15 pollen (Figure 1A; Supplemental Figure 1B), suggesting that any sRNAs exclusively dependent on RDR6 are not linked to paternal excess seed abortion. Consistent with this result, paternally produced siRNA854, which is dependent on DCL2/4 and RDR6, is required for the viability of paternal excess seeds (McCue et al., 2012; Wang et al., 2018). By contrast, we found that multiple members of the canonical RdDM pathway restore paternal excess seed viability (Figure 1; Supplemental Figures 1A and 1C). Unlike the reported relatively low levels of viable seeds produced by mutations in non-RdDM genes such as RDR6, DCL2, DCL4, AGO2, and miRNA845 (∼20% to 30% of the seeds are viable; Borges et al., 2018; Martinez et al., 2018), the loss of NRPE1 and DRM2 phenocopies NRPD1 in the large magnitude of the repression of seed abortion (∼80% of the seeds are viable; Figure 1; Supplemental Figure 1). The cause of the discrepancy among the genetic results between our studies remains unclear but may be linked to the use of the osd1 mutation to create conditions of paternal excess. Similar discrepancies have been observed elsewhere; osd1 pickle related2 mutants did not repress paternal excess seed abortion, whereas 4N pickle related2 showed reduced seed abortion (Wolff et al., 2015; Huang et al., 2017). Finally, the easiRNA model also suggests that the RdDM pathway’s recovery in the viable paternal excess endosperm is essential for seed viability. However, the loss of both paternal and maternal copies of NRPD1, RDR2, DCL3, NRPE1, and DRM2 in paternal excess crosses did not abolish the seed viability obtained upon the loss of the paternal copy alone (Figure 1; Supplemental Figure 1; Erdmann et al., 2017; Martinez et al., 2018). This indicates that the activity of a genetically complete canonical RdDM pathway in the endosperm is not necessary for the suppression of paternal excess seed abortion.
Another model posits that RNA Pol IV sRNA levels are determined by maternal dosage and that these sRNAs normally repress AGL expression and trigger endosperm cellularization (Lu et al., 2012). This model argues that in paternal excess endosperm, siRNA levels are disproportionate to target transcript levels of AGL genes, which in turn leads to prolonged endosperm proliferation and cellularization. This model makes a key testable prediction—the requirement for a functional RdDM pathway in the endosperm. However, we found that RdDM in endosperm is not essential for seed viability under conditions of paternal excess (Figure 1; Supplemental Figure 1).
We previously showed that maintaining a 2:1 ratio of maternal to paternal allele transcripts in balanced endosperm is dependent on NRPD1; in its absence, the expression of many genes shifted more maternal (Erdmann et al., 2017). Thus, we suggested that the loss of NRPD1 represses paternal excess seed abortion by raising the maternal fraction of the transcriptome and balancing excess paternal gene dosage (Erdmann et al., 2017). A central tenet of our model was that the reduction in the RNA Pol IV sRNA pathway in endosperm (caused by the loss of paternal NRPD1) is essential for seed viability. However, we found that the RdDM pathway is in fact more functional in viable paternal excess endosperm than in lethal paternal excess endosperm (Figures 4A, 4B, and 5E; Supplemental Figure 4). Additionally, the transcriptome in lethal paternal excess endosperm is already maternally biased relative to genomic expectation. This maternal bias is not increased in viable paternal excess endosperm, as would be predicted under our previous model (Figure 4).
What, then, might be the mechanism by which the suppression of seed lethality occurs? Although our current data cannot address whether mutations in all canonical RdDM genes suppress paternal excess seed lethality in the same manner, given what is known about the RdDM pathway, it is likely that differences in DNA methylation, in either the paternal sporophyte or the male gametophyte (or both), play a key role. It has been shown in diploid plants that the loss of NRPD1 or DRM2 in the father can affect gene expression in the endosperm of balanced crosses (Vu et al., 2013; Erdmann et al., 2017) and that DRM2 establishes methylation patterns at a subset of genomic sites in the male germline (Walker et al., 2018). Some studies suggest that tetraploidization itself induces DNA methylation changes (Mittelsten Scheid et al., 2003; Baubec et al., 2010; Yu et al., 2010; Zhang et al., 2015). In one scenario, DNA methylation changes in tetraploid pollen could ultimately direct changes in gene expression in the endosperm, which would not occur in endosperm fathered by RdDM mutant tetraploids. An alternative possibility is that the loss of RdDM in tetraploids could lead to an altered methylation state not found in either wild-type diploid or tetraploid males. This ectopic paternal methylation state could then dictate a gene expression pattern that represses seed abortion.
Evidence for Buffering Mechanisms in Endosperm
Our results suggest that transcriptional buffering, operational in both lethal and viable paternal excess endosperm, is one feature that could contribute to the ability of the seed to withstand the addition of an entire extra paternal genome. In transcriptional buffering, expression per copy of a gene can be increased or decreased based on the number of copies of the gene relative to that of the rest of the genome (Zhang et al., 2010; Birchler and Veitia, 2012). We observed the signatures of buffering in paternal excess transcriptomes (Figure 5). Decreased paternal transcript levels contributed predominantly to genes with decreased expression in paternal excess, whereas increases in the levels of transcripts from maternal alleles contributed predominantly to genes with increased expression (Figures 5C and 5D). The outcome is that some parts of the transcriptome are more maternal than expected from parental genomic dosage, constituting a push back against excess paternal genomes. While the mechanistic basis of this buffering system remains unclear, it could be linked at least in part to the reduction in the 24-nucleotide sRNA pathway in endosperm (Figure 4). RNA Pol IV function in balanced endosperm has been previously shown to be required for the repression of the maternal transcriptome (Erdmann et al., 2017). The increase in the maternal fraction of the transcriptome for specific genes in paternal excess endosperm (Figure 5B) is therefore consistent with the reduced expression of genes encoding subunits of RNA Pol IV and other downstream members of the RdDM pathway (Figure 4A). It is important to note that the increase in the maternal fraction could be observed in both lethal and viable paternal excess endosperm and is thus unlinked to the repression of seed abortion brought about by paternal loss of RdDM. Other buffering mechanisms might also play important roles and could act through posttranscriptional pathways (Wang et al., 2018), protein degradation pathways, or via signaling pathways that have been shown to influence paternal excess crosses (Doughty et al., 2014; Batista et al., 2019). One striking observation is that even wild-type paternal excess crosses produce ∼20% viable seeds (Figure 1; Supplemental Figure 1), suggesting that seed lethality and viability exist on a continuum. Our data further suggest that the slim margin that separates seed life and death is likely determined by modest changes in the endosperm, which are ultimately instructed by paternal imprints laid down by RdDM.
METHODS
Arabidopsis Strains, Tetraploid Production, and Tissue Collection
The plants used in the experiments were grown at 22°C either in a greenhouse or in a Conviron growth chamber on a 16-h-light/8-h-dark cycle (120 µmol light). The Arabidopsis (Arabidopsis thaliana) mutants used in this study were nrpd1a-4, rdr2-1, dcl3-1, nrpd1b-11, drm2-2, rdr6-15 (CS879578), rdr2-1 rdr6-15 (CS66111), and cmt3-11t (CS16392) and were obtained from the Arabidopsis Biological Resource Center. Tetraploids were generated by applying 0.25% (w/v) colchicine in 0.2% (v/v) Silwet to the apices of 2- to 3-week-old diploid plants. Progeny from treated plants were screened by flow cytometry (BD Sciences, LSRFortessa) to identify tetraploids. For crosses, wild-type diploid Col-0 or Ler buds were emasculated and pollination was performed 2 days later with diploid or tetraploid pollen in the Col-0 background. To assess seed abortion, siliques were harvested after drying and seeds examined under a dissecting microscope. To assess germination rates, seeds were sterilized in 2% (v/v) PPM (Plant Cell Technologies) for 3 days at 4°C and plated on 0.5× Murashige and Skoog/Phytagar medium. Endosperm from ∼100 seeds at 7 days after pollination from at least three siliques was dissected away from embryos and seed coats and pooled for each replicate as previously described (Gehring et al., 2009, 2011). Replicates were collected from different sets of crosses with different individuals as parents. For lethal paternal excess seeds, endosperm was collected only from seeds with arrested embryonic growth. For viable paternal excess seeds, endosperm was collected only from seeds where embryonic growth was progressing normally.
mRNA-Seq Library Preparation and Transcriptome Analyses
Long RNA was isolated as previously described (Erdmann et al., 2017), and mRNA-seq libraries were constructed at the Whitehead Institute Genome Technology Core using a SMARTerUltra-lowPOLYA-V4 kit. Libraries were sequenced on a 40-base, single-read cycle. Sequence data were filtered for quality with “trim_galore -q 25 --phred64 --fastqc --length 20 --stringency 5.” Filtered reads were aligned to the TAIR10 genome using “tophat -i 30 -I 3000 --solexa1.3-quals -p 5 -g 5 --segment-mismatches 1 --segment-length 18 --b2-very-sensitive” (Kim et al., 2013). TopHat version 2.1.1 with Bowtie version 2.3.4 was used. We used Cuffdiff (version 2.2.1) with default settings and the ARAPORT11 genome annotation to calculate changes in gene expression and their statistical significance. Genes with q < 0.05 were considered to be significantly different. We used a custom script (assign_to_allele.py) and single nucleotide polymorphisms between Col-0 and Ler to identify allele-specific reads. Allele-specific reads per gene were assessed using “htseq-count -s no –m union” and the ARAPORT11 annotation. To assess TE transcript levels, reads were aligned to the REPBASE consensus sequence (Jurka et al., 2005) using “bowtie -v 2 -m 3 --best --strata -p 5 --phred64,” and reads mapping to each TE family were summed. META1, ATHILA6A, and ATGP8 were precluded from further analyses because of mapping artifacts. Differential abundance of TE mapping reads were calculated using Fisher’s exact test (P < 0.05) option with multiple-test correction in DEGseq (Wang et al., 2010).
Tissue Enrichment Test
Expression per gene was measured using htseq-count, and analyses were performed using the seed tissue enrichment test (Schon and Nodine, 2017). The time point was set to the bent cotyledon stage.
sRNA-Seq Library Preparation and Analyses
RNA was isolated from manually dissected endosperm using the RNAqueous micro kit (Ambion). sRNA was obtained as previously described (Erdmann et al., 2017). Libraries were built using the NEXTflex sRNA-seq kit v3 (Bioo Scientific). Final library amplification was performed for 24 cycles; the resulting libraries were size selected (135 to 160 bp) with a Pippin Prep (Sage Science). Forty-base single-read sequencing was performed on an Illumina HiSeq 2500. Sequencing reads were trimmed for quality with “fastq_quality_trimmer -v -t 20 -l 25.” Reads were further filtered and adapter containing reads were retained using “cutadapt -a TGGAATTCTCGGGTGCCAAGG --trimmed-only --quality-base 64 -m 24 -M 40 --max-n 0.5.” The reads from libraries produced by the NEXTflex library prep kit include four random nucleotides at both 5′ and 3′ ends of the reads. Taking advantage of these tags, we were able to remove PCR duplicates using prinseq-lite –fastq <infile> -out_format 3 –out_good <outfile> -derep 1. Reads were aligned to TAIR10 with "bowtie -v 1 --best -5 4 -3 4 --sam --phred64-quals or --phred33-quals". We used a custom script (assign_to_allele.py) to identify allele-specific reads. Regions with differential sRNA levels were identified by counting reads in 300-bp windows with 200-bp overlaps. DESeq2 (Love et al., 2014) was used to identify windows with differential abundance of sRNAs (adjusted P value less than or equal to 0.05 and log2 fold change = ±1). Overlapping windows with significant increases or decreases in sRNA were merged to obtain regions with differences in sRNA. To identify genes with differences in 21- and 24-nucleotide sRNAs, mapped reads were separated based on size. Twenty-one- and twenty-four-nucleotide sRNAs mapping to genes were counted using the ARAPORT11 annotation and "htseq-count -s no –m union". To identify TE insertions with differences in 21- and 24-nucleotide sRNAs, we first identified TE insertions that did not overlap with genes and used the "coverageBed -counts" command from Bedtools suite (Quinlan and Hall, 2010) to count reads mapping to TE insertions. We identified genes and TEs with differential abundance in 21- and 24-nucleotide sRNA using DESeq2.
Methylome Library Preparation and Analyses
Genomic DNA was isolated from manually dissected endosperm using the QiaAMP DNA microkit (QIAGEN 56304); dissected tissue was incubated in ATL buffer and proteinase K at 56°C overnight on a shaking incubator. Eighty to one hundred nanograms of DNA obtained from these protocols was subjected to bisulfite treatment using a Methylcode Bisulfite Conversion kit (Invitrogen). Bisulfite libraries were constructed from these materials using Illumina’s Truseq DNA methylation kit. Bisulfite libraries were sequenced on Illumina’s HISeq 2500 in a paired-end configuration. Reads were filtered for quality with "trim_galore --phred64 --fastqc --stringency 5 --length 15 --paired --clip_R1 2 --clip_R2 2". The reads were then aligned to TAIR10 using "bismark -N 1 -L 20". Duplicate reads were removed using Bismark version 0.19. Previously described Bismark methylation extractor and custom scripts (Pignatta et al., 2014, 2015) were used to determine per base methylation. In brief, differences in methylation were calculated genome-wide for 300-bp sliding windows with 200-bp overlaps. To be included, windows had at least three overlapping cytosines in both genotypes with a read depth of at least six reads per cytosine. Windows called as significantly different were at least 10% different between tested genotypes for CHH methylation, 20% for CHG methylation, and 30% for CG methylation. Significance of difference was calculated by Fisher's exact test with a Benjamini-Hochberg correction (P < 0.01). Overlapping windows with significant differences in DNA methylation were merged to obtained differentially methylated regions.
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Information Resource under the following accession numbers: NRPD1, AT1G63020; SHH1, AT1G15215; RDR2, AT4G11130; DCL3, AT3G43920; NRPE1, AT2G40030; DRM2, AT5G14620; RDR6, AT3G49500; CMT3, AT1G69770.
Whole-genome bisulfite sequencing data, sRNA sequencing, and mRNA sequencing data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE126932.
The script assign_to_allele.py can be found at https://github.com/clp90/imprinting_analysis/tree/master/helper_scripts.
Supplemental Data
Supplemental Figure 1. Paternal loss of RdDM genes but not RDR6 and CMT3 represses seed abortion in paternal excess crosses (supports Figure 1)
Supplemental Figure 2. Analysis of gene expression differences in paternal excess endosperm derived using the osd1 mutation (data from Martinez et al., 2018; supports Figure 2)
Supplemental Figure 3. Rescue by loss of NRPD1 does not require normalization of the expression of genes implicated in interploid seed abortion (supports Figure 3)
Supplemental Figure 4. sRNA in balanced, lethal, and viable paternal excess endosperm (supports Figure 4)
Supplemental Figure 5. Allelic contributions to gene expression differences (supports Figure 5)
Supplemental Figure 6. Allelic bias of some imprinted genes is lost in lethal paternal excess endosperm (supports Figure 5)
Supplemental Table 1. Details of sequencing libraries
Supplemental Data Set 1. Cuffdiff output comparing gene expression in balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 2. GO analysis of genes that are differentially expressed between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 3. DEGseq output comparing transposon transcript levels between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 4. Differentially methylated regions in the CHH context in comparisons between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 5. Differentially methylated regions in the CHG context in comparisons between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 6. Differentially methylated regions in the CG context in comparisons between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 7. DESeq output for differential abundance of 21- and 24-nucleotide genic sRNA between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 8. DESeq output for differential abundance of 21- and 24-nucleotide TE sRNA between balanced, lethal, and viable paternal excess endosperm
Supplemental Data Set 9. Allele-specific mRNA read counts and differences in allelic contributions between balanced, lethal, and viable paternal excess endosperm
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We thank Daniela Pignatta for deriving the 4n Col-0 line. This research was supported by the National Science Foundation Division of Molecular and Cellular Biosciences (CAREER award 1453459 to M.G.).
AUTHOR CONTRIBUTIONS
P.R.V.S. and M.G. conceived of and designed the study; P.R.V.S. performed experiments and analyzed data; and P.R.V.S. and M.G. wrote the article.
Footnotes
Articles can be viewed without a subscription.
References
- Bao W., Kojima K.K., Kohany O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista R.A., Figueiredo D.D., Santos-González J., Köhler C. (2019). Auxin regulates endosperm cellularization in Arabidopsis. Genes Dev. 33: 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T., Dinh H.Q., Pecinka A., Rakic B., Rozhon W., Wohlrab B., von Haeseler A., Mittelsten Scheid O. (2010). Cooperation of multiple chromatin modifications can generate unanticipated stability of epigenetic states in Arabidopsis. Plant Cell 22: 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J.A. (2014). Interploidy hybridization barrier of endosperm as a dosage interaction. Front. Plant Sci. 5: 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J.A., Veitia R.A. (2012). Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. USA 109: 14746–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F., Martienssen R.A. (2015). The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 16: 727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F., Parent J.-S., van Ex F., Wolff P., Martínez G., Köhler C., Martienssen R.A. (2018). Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat. Genet. 50: 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart-Waco D., Ngo K., Dilkes B., Josefsson C., Comai L. (2013). Early disruption of maternal-zygotic interaction and activation of defense-like responses in Arabidopsis interspecific crosses. Plant Cell 25: 2037–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo D.M., Moyle L.C. (2012). Evolutionary implications of mechanistic models of TE-mediated hybrid incompatibility. Int. J. Evol. Biol. 2012: 698198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper D.C. (1951). Caryopsis development following matings between diploid and tetraploid strains of Zea mays. Am. J. Bot. 38: 702–708. [Google Scholar]
- Creasey K.M., Zhai J., Borges F., Van Ex F., Regulski M., Meyers B.C., Martienssen R.A. (2014). miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 508: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Erfurth I., Jolivet S., Froger N., Catrice O., Novatchkova M., Mercier R. (2009). Turning meiosis into mitosis. PLoS Biol. 7: e1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilkes B.P., Spielman M., Weizbauer R., Watson B., Burkart-Waco D., Scott R.J., Comai L. (2008). The maternally expressed WRKY transcription factor TTG2 controls lethality in interploidy crosses of Arabidopsis. PLoS Biol. 6: 2707–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty J., Aljabri M., Scott R.J. (2014). Flavonoids and the regulation of seed size in Arabidopsis. Biochem. Soc. Trans. 42: 364–369. [DOI] [PubMed] [Google Scholar]
- Erdmann R.M., Satyaki P.R.V., Klosinska M., Gehring M. (2017). A small RNA pathway mediates allelic dosage in endosperm. Cell Reports 21: 3364–3372. [DOI] [PubMed] [Google Scholar]
- Esen A., Soost R.K. (1973). Seed development in citrus with special reference to 2X x 4X crosses. Am. J. Bot. 60: 448–462. [Google Scholar]
- Figueiredo D.D., Batista R.A., Roszak P.J., Hennig L., Köhler C. (2016). Auxin production in the endosperm drives seed coat development in Arabidopsis. eLife 5: e20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiume E., Fletcher J.C. (2012). Regulation of Arabidopsis embryo and endosperm development by the polypeptide signaling molecule CLE8. Plant Cell 24: 1000–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasciolli V., Mallory A.C., Bartel D.P., Vaucheret H. (2005). Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr. Biol. 15: 1494–1500. [DOI] [PubMed] [Google Scholar]
- Gehring M., Bubb K.L., Henikoff S. (2009). Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324: 1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M., Missirian V., Henikoff S. (2011). Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS One 6: e23687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Marcos J.F., Pennington P.D., Costa L.M., Dickinson H.G. (2003). Imprinting in the endosperm: A possible role in preventing wide hybridization. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D., Westboy M. (1991). Genomic imprinting in endosperm: its effect on seed development in crosses between species, and its implications for the evolution of apomixis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 333: 1–13. [Google Scholar]
- Håkansson A., Ellerström S. (1950). Seed development after reciprocal crosses between diploid and tetraploid rye. Hereditas 36: 256–296. [Google Scholar]
- Hehenberger E., Kradolfer D., Köhler C. (2012). Endosperm cellularization defines an important developmental transition for embryo development. Development 139: 2031–2039. [DOI] [PubMed] [Google Scholar]
- Hsieh T.-F., Ibarra C.A., Silva P., Zemach A., Eshed-Williams L., Fischer R.L., Zilberman D. (2009). Genome-wide demethylation of Arabidopsis endosperm. Science 324: 1451–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F., Zhu Q.-H., Zhu A., Wu X., Xie L., Wu X., Helliwell C., Chaudhury A., Finnegan E.J., Luo M. (2017). Mutants in the imprinted PICKLE RELATED 2 gene suppress seed abortion of fertilization independent seed class mutants and paternal excess interploidy crosses in Arabidopsis. Plant J. 90: 383–395. [DOI] [PubMed] [Google Scholar]
- Ibarra C.A., et al. (2012). Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337: 1360–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Moreno-Romero J., Santos-González J., De Jaeger G., Gevaert K., Van De Slijke E., Köhler C. (2017). Ectopic application of the repressive histone modification H3K9me2 establishes post-zygotic reproductive isolation in Arabidopsis thaliana. Genes Dev. 31: 1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110: 462–467. [DOI] [PubMed] [Google Scholar]
- Kelleher E.S. (2016). Reexamining the P-element invasion of Drosophila melanogaster through the lens of piRNA silencing. Genetics 203: 1513–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kradolfer D., Wolff P., Jiang H., Siretskiy A., Köhler C. (2013). An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev. Cell 26: 525–535. [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C., Köhler C. (2015). Epigenetic mechanisms of postzygotic reproductive isolation in plants. Curr. Opin. Plant Biol. 23: 39–44. [DOI] [PubMed] [Google Scholar]
- Law J.A., Du J., Hale C.J., Feng S., Krajewski K., Palanca A.M.S., Strahl B.D., Patel D.J., Jacobsen S.E. (2013). Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature 498: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Berger F. (2012). Endosperm: Food for humankind and fodder for scientific discoveries. New Phytol. 195: 290–305. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Zhang C., Baulcombe D.C., Chen Z.J. (2012). Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. USA 109: 5529–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marí-Ordóñez A., Marchais A., Etcheverry M., Martin A., Colot V., Voinnet O. (2013). Reconstructing de novo silencing of an active plant retrotransposon. Nat. Genet. 45: 1029–1039. [DOI] [PubMed] [Google Scholar]
- Martienssen R.A. (2010). Heterochromatin, small RNA and post-fertilization dysgenesis in allopolyploid and interploid hybrids of Arabidopsis. New Phytol. 186: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G., Wolff P., Wang Z., Moreno-Romero J., Santos-González J., Conze L.L., DeFraia C., Slotkin R.K., Köhler C. (2018). Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat. Genet. 50: 193–198. [DOI] [PubMed] [Google Scholar]
- McCue A.D., Nuthikattu S., Reeder S.H., Slotkin R.K. (2012). Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8: e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbocker D., Sink K. (1969). Embryology of diploid by diploid and diploid by tetraploid crosses in poinsettia. Genome 11: 598–601. [Google Scholar]
- Mittelsten Scheid O., Afsar K., Paszkowski J. (2003). Formation of stable epialleles and their paramutation-like interaction in tetraploid Arabidopsis thaliana. Nat. Genet. 34: 450–454. [DOI] [PubMed] [Google Scholar]
- Muntzing A. (1936). The evolutionary significance of autopolyploidy. Hereditas 21: 363–378. [Google Scholar]
- Oberlin S., Sarazin A., Chevalier C., Voinnet O., Marí-Ordóñez A. (2017). A genome-wide transcriptome and translatome analysis of Arabidopsis transposons identifies a unique and conserved genome expression strategy for Ty1/Copia retroelements. Genome Res. 27: 1549–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatta D., Erdmann R.M., Scheer E., Picard C.L., Bell G.W., Gehring M. (2014). Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. eLife 3: e03198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatta D., Bell G., Gehring M. (2015). Whole Genome Bisulfite Sequencing and DNA Methylation Analysis from Plant Tissue. Bio Protoc. 5: e1407. [Google Scholar]
- Piskurewicz U., Iwasaki M., Susaki D., Megies C., Kinoshita T., Lopez-Molina L. (2016). Dormancy-specific imprinting underlies maternal inheritance of seed dormancy in Arabidopsis thaliana. eLife 5: e19573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povilus R.A., Diggle P.K., Friedman W.E. (2018). Evidence for parent-of-origin effects and interparental conflict in seeds of an ancient flowering plant lineage. Proc. Biol. Sci. 285: 20172491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyaki P.R.V., Gehring M. (2017). DNA methylation and imprinting in plants: machinery and mechanisms. Crit. Rev. Biochem. Mol. Biol. 52: 163–175. [DOI] [PubMed] [Google Scholar]
- Schatlowski N., Wolff P., Santos-González J., Schoft V., Siretskiy A., Scott R., Tamaru H., Köhler C. (2014). Hypomethylated pollen bypasses the interploidy hybridization barrier in Arabidopsis. Plant Cell 26: 3556–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon M.A., Nodine M.D. (2017). Widespread contamination of Arabidopsis embryo and endosperm transcriptome data sets. Plant Cell 29: 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R.J., Spielman M., Bailey J., Dickinson H.G. (1998). Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 125: 3329–3341. [DOI] [PubMed] [Google Scholar]
- Stoute A.I., Varenko V., King G.J., Scott R.J., Kurup S. (2012). Parental genome imbalance in Brassica oleracea causes asymmetric triploid block. Plant J. 71: 503–516. [DOI] [PubMed] [Google Scholar]
- Stroud H., Greenberg M.V.C., Feng S., Bernatavichute Y.V., Jacobsen S.E. (2013). Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S., Spielman M., Schulz R., Oakey R.J., Kelsey G., Salazar A., Zhang K., Pennell R., Scott R.J. (2010). Transcriptional profiles underlying parent-of-origin effects in seeds of Arabidopsis thaliana. BMC Plant Biol. 10: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay Y.F., Frank M.J., Page T., Dean C., Crawford N.M. (1993). Identification of a mobile endogenous transposon in Arabidopsis thaliana. Science 260: 342–344. [DOI] [PubMed] [Google Scholar]
- von Wangenheim K.-H., Peterson H.-P. (2004). Aberrant endosperm development in interploidy crosses reveals a timer of differentiation. Dev. Biol. 270: 277–289. [DOI] [PubMed] [Google Scholar]
- Vu T.M., Nakamura M., Calarco J.P., Susaki D., Lim P.Q., Kinoshita T., Higashiyama T., Martienssen R.A., Berger F. (2013). RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 140: 2953–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia H., Josefsson C., Dilkes B., Kirkbride R., Harada J., Comai L. (2009). Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr. Biol. 19: 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J., Gao H., Zhang J., Aldridge B., Vickers M., Higgins J.D., Feng X. (2018). Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 50: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Jiang H., Del Toro de León G., Martinez G., Köhler C. (2018). Sequestration of a transposon-derived siRNA by a target mimic imprinted gene induces postzygotic reproductive isolation in Arabidopsis. Dev. Cell 46: 696–705.e4. [DOI] [PubMed] [Google Scholar]
- Wang L., Feng Z., Wang X., Wang X., Zhang X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138. [DOI] [PubMed] [Google Scholar]
- Williams B.P., Pignatta D., Henikoff S., Gehring M. (2015). Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet. 11: e1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff P., Jiang H., Wang G., Santos-González J., Köhler C. (2015). Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. eLife 4: e10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., Brown R.C., Lemmon B.E., Harada J.J., Goldberg R.B., Fischer R.L. (2006). Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 142: 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D., Duermeyer L., Leoveanu C., Nambara E. (2014). The functions of the endosperm during seed germination. Plant Cell Physiol. 55: 1521–1533. [DOI] [PubMed] [Google Scholar]
- Yu Z., Haberer G., Matthes M., Rattei T., Mayer K.F.X., Gierl A., Torres-Ruiz R.A. (2010). Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107: 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Liu Y., Xia E.-H., Yao Q.-Y., Liu X.-D., Gao L.-Z. (2015). Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. USA 112: E7022–E7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Malone J.H., Powell S.K., Periwal V., Spana E., Macalpine D.M., Oliver B. (2010). Expression in aneuploid Drosophila S2 cells. PLoS Biol. 8: e1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D., Gehring M., Tran R.K., Ballinger T., Henikoff S. (2007). Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39: 61–69. [DOI] [PubMed] [Google Scholar]