

Abstract
Introduction:
Cancer is often diagnosed at late stages when the chance of cure is relatively low and although research initiatives in oncology discover many potential cancer biomarkers, few transition to clinical applications. This review addresses the current landscape of cancer biomarker discovery and translation with a focus on proteomics and beyond.
Areas covered:
The review examines proteomic and genomic techniques for cancer biomarker detection and outlines advantages and challenges of integrating multiple omics approaches to achieve optimal sensitivity and address tumor heterogeneity. This discussion is based on a systematic literature review and direct participation in translational studies.
Expert commentary:
Identifying aggressive cancers early on requires improved sensitivity and implementation of biomarkers representative of tumor heterogeneity. During the last decade of genomic and proteomic research, significant advancements have been made in next generation sequencing and mass spectrometry techniques. This in turn has led to a dramatic increase in identification of potential genomic and proteomic cancer biomarkers. However, limited successes have been shown with translation of these discoveries into clinical practice. We believe that the integration of these omics approaches is the most promising molecular tool for comprehensive cancer evaluation, early detection and transition to Precision Medicine in oncology.
Keywords: Cancer biomarker, multi-omics, proteomics, translational research, biomarker discovery
1. Introduction
Advancements in cancer biomarker research parallel the development of technologies. In recent years, great strides in genomic and proteomic analytical methods have uncovered highly complex signaling networks that contribute to disease onset and progression. In turn, the diverse pathophysiology of cancers and the heterogeneous nature of tumors illustrate the compound effect that genetic and protein alterations have on oncogenic signaling [1,2]. Most cancer biomarker studies examine individual genetic variables, transcriptome alterations, and impaired protein function as distinct risk and prognostic factors. These single streamline approaches to biomarker discovery can be attributed to the available analytical methods, which assess genomic (DNA), transcriptome (RNA) or protein composition separately. The introduction of next-generation sequencing (NGS) has tremendously improved the sensitivity and high throughput capabilities of genomic techniques, leading to a disproportionate increase in the identification of genetic variables in cancer [3–5]. NGS allows detection and quantification of low frequency genomic alterations in biopsy samples, generating large datasets of variants observed in cancer populations [6]. These genomic studies further emphasize the high degree of heterogeneity reported within tumors and the compound effect of multiple mutations on disease onset and progression [7]. Despite abundance of genomic data, biomarker discovery is limited by the ability to distinguish driver mutations from variables not contributing to cancer pathophysiology. Integrating genomic data and proteomic analysis, proteogenomics, can generate a comprehensive view of cell signaling that is capable of identifying abnormalities to serve as risk, diagnostic or prognostic markers as well as therapeutic targets.
Deciphering the heterogeneous mechanisms that initiate tumorigenesis requires the integration of omics data stemming from genomic, transcriptomic, epigenetic, and proteomic analysis (Figure 1). The extent of intra and inter-individual variability pertaining to tumor composition and oncogenic signaling can be misinterpreted by a single omic approach. Furthermore, patterns among genetic, transcriptomic, epigenetic, metabolomic, and protein data-sets generated from patient cohorts can be used to identify potential cancer biomarkers [8–13]. Comparative statistical analysis of large multi-omics datasets can lead to the discovery of risk factors that allow early detection in phenotypically healthy individuals and prompt intervention with better outcomes. Patients frequently have unique sets of abnormalities responsible for tumorigenesis, leading to patient-specific disease progression and the need for individualized therapies. The notion that a patient’s unique pathophysiologic profile should guide medical decisions, is part of an ongoing effort in Precision Medicine that aims to tailor risk assessment and treatment on an individual basis [14,15]. This Precision Medicine initiative is central to a wide range of diseases, but it has tremendous potential in oncology with respect to early detection and targeted therapies.
Figure 1.
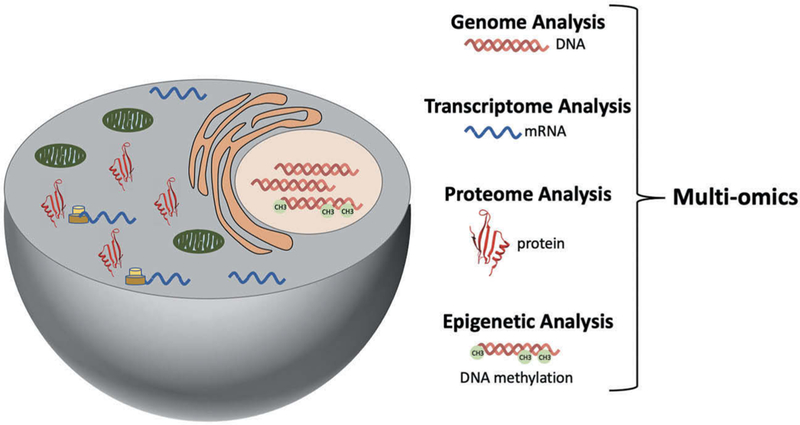
Integration of genomic, epigenetic, transcriptomic, and proteomic data for comprehensive analysis of cell dynamics through multi-omics.
2. Proteomic applications in cancer biomarker discovery
2.1. Proteomic techniques
Mass spectrometry has been invaluable for the growth of proteomics, although detection of very low abundance proteins remains challenging [16]. Techniques such as stable isotope labeling by amino acids in cell culture (SILAC), tandem mass tag (TMT) labeling and isobaric tag for relative and absolute quantification (iTRAQ) have tremendously improved sensitivity and protein detection by mass spectrometry (Figure 2(a,b)) [17–19]. Additionally, these approaches enable simultaneous analysis and peptide quantification for multiple specimens and allow direct comparison across samples. For example, the high throughput capability of TMT LC-MS/MS enables uniform and rapid protein quantification of multiple specimens and comparison among distinct TMT sets when a specific reporter label is dedicated to a reference sample that is included with all LC-MS/MS analysis. The presence of a common reference sample permits direct comparison among specimens from different TMT sets when sample preparation and LC-MS/MS methods are standardized across analytical runs [20]. This ability to analyze and compare large specimen cohorts is integral to harmonization among labs and TMT labeling coupled with LC-MS /MS has become a prominent technique in multi-institution studies aimed at cancer biomarker discovery. Large-scale initiatives, such as the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC), have successfully applied mass spectrometry methods for the identification of cancer-specific proteins and unique protein patterns leading to sub-classification of ovarian, breast and colorectal cancers [21].
Figure 2.
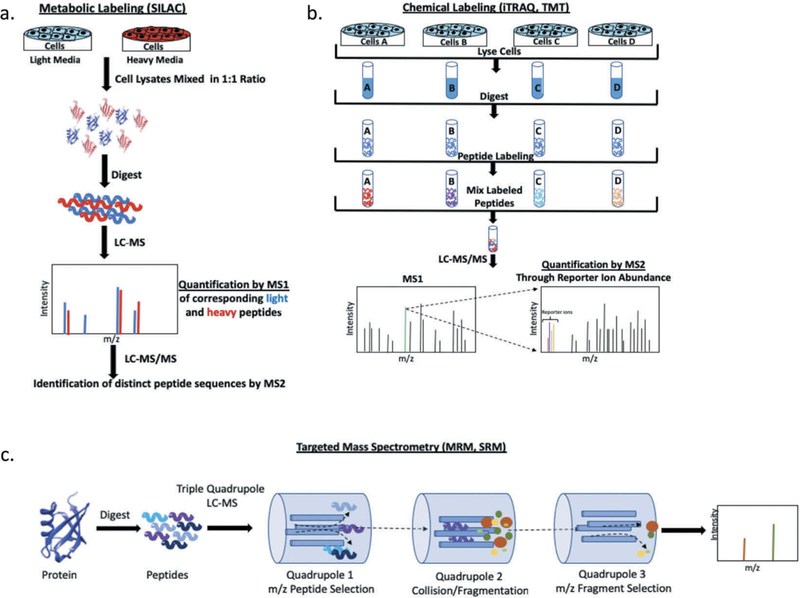
Mass spectrometry techniques for proteomic analysis. (a) Schematic representation of stable isotope labeling by amino acids in cell culture (SILAC). (b). General summary of chemical labeling techniques used for mass spectrometry analysis (TMT, iTRAQ). (c). Overview of targeted mass spectrometry by SRM/MRM using a triple quadrupole instrument.
Protein arrays and antibody-based proteomic methods are targeted approaches for protein detection and quantification with high sensitivity and multiplexing capabilities. These methods are not optimal for biomarker discovery as they use a directed approach, however they have great potential in a clinical setting for monitoring of validated disease biomarkers. Quantitative proteomic analysis is achieved through antibody labels that can be fluorophores, metal isotope tags, or nucleotide sequences. Recent efforts to improve multiplexing capabilities have focused on nucleotide barcode sequences that enable tag amplification and subsequent increase in signal [22]. For example, Ullal et al. demonstrated the utility of antibody barcoding in cancer cells using a panel of 90 antibodies conjugated to photocleavable DNA tags. Multiplexing and quantitative analysis are based on UV light-dependent cleavage of the DNA barcode and subsequent quantification by fluorescent hybridization [23]. Recently Shahi et al. introduced Abseq as a DNA barcoding technique capable of single-cell proteomics [22]. Abseq utilizes microfluid barcoding and DNA sequencing to quantify proteins in more than 10,000 cells in parallel.
Targeted proteomics is not limited to antibody methods. Selected Reaction Monitoring (SRM) and Multiple Reaction Monitoring (MRM) mass spectrometry techniques are emerging as reliable, high throughput assays for cancer biomarkers. Detection of target proteins is achieved with a triple quadrupole mass spectrometer, where specific peptides originating from the protein of interest are selected based on their mass to charge (m/z) ratio and subsequently fragmented into smaller components that are in turn quantified to assess protein abundance (Figure 2(c)) [24,25]. The principle of protein detection is identical for SRM and MRM, with the primary difference that MRM is the application of SRM to multiple peptide fragments. Identifying and quantifying multiple peptides from one or more target proteins simultaneously, makes MRM capable of multiplexing with high sensitivity and the technique of choice for most directed assays [26]. The primary advantage of MRM is the ability to detect multiple isoforms and post-translationally modified species for a given protein with high specificity in a single test run. In an effort to minimize invasive procedures, MRM assays are utilized for the detection of biomarker peptides in serum samples from breast and colorectal cancer patients among others [27,28].
2.2. Post-translational modifications as cancer biomarkers
The proteome is dynamic and regulated by post-translational modifications (PTM), which when aberrant can lead to uncontrolled proliferation and tumorigenesis. Prediction of impaired PTM cannot be achieved through genome and transcriptome analysis, it can only be detected through proteomic approaches that identify abnormal protein activity and/or abundance of the modified variant [29]. Proteins exist in variable PTM states and detection of less abundant forms may be challenging. To improve sensitivity, it has become common practice, when possible, to incorporate an enrichment step in the sample preparation procedure that extracts peptides containing the targeted modification [30,31]. Enriched peptides are subsequently analyzed through mass spectrometry techniques and the PTM of distinct proteins is quantified. Antibody-based techniques can also be used for PTM analysis when the modified epitope is recognized with high specificity and there is no background signal interference from other variants of the protein.
Impaired phosphorylation, usually hyperphosphorylation, is well-established as a cause for dysregulation of cell signaling and carcinogenesis [32]. Therefore, it is not surprising that abnormal kinase activity and overexpression are frequently associated with an oncogenic phenotype. For example, genetic alterations and increased expression of the human epidermal growth factor receptor 2 (HER2) tyrosine kinase are implicated in certain breast and ovarian cancers [33,34]. Although aggressive, HER2 positive breast cancer is susceptible to HER2 targeted therapies that block receptor signaling through antibody binding (trastuzumab and pertuzumab) or by inhibiting kinase activity (lapatinib and neratinib). Unfortunately, these HER2 targeted therapies are not efficacious in HER2 positive ovarian cancer patients, suggesting differences in kinase regulation and signaling [35]. Similarly, genetic alterations in the epidermal growth factor receptor (EGFR) tyrosine kinase are observed in non-small cell lung carcinoma (NSCLC) and while treatment with EGFR-tyrosine kinase inhibitors shows promise in some patients, others develop resistance overtime due to selection for EGFR mutants such asT490M that are not susceptible to kinase inhibition [36,37,38,39].
Altered glycosylation impairs cell signaling, communication, and adhesion and is implicated in the onset and progression of cancer eventually leading to metastasis [40]. Abnormal glycosylation can be due to dysregulation of glycosyltransferase transcription, expression, and activity or alterations in the sequence and conformation of the modified protein that prevents carbohydrate PTM [40]. Glycoproteins represent the majority of clinically utilized serological biomarkers and are routinely used for monitoring disease progression and response to therapeutic intervention. The diagnostic utility of these glycoprotein cancer biomarkers, with the exception of prostate-specific antigen (PSA), is limited by lack of tissue specificity and they are used to monitor disease progression, but are not adequate for screening. For example, SLe tetrasaccharide (CA19–9) is elevated in patients with pancreatic, colorectal and gastric cancer and CA19–9 abundance serves as a prognostic factor as well as a measure of response to therapy [41]. Similarly, carcinoem-bryonic antigen (CEA) is a cell surface glycoprotein, which is normally expressed during fetal development with protein levels decreasing afterbirth [41]. Elevated CEA is observed in patients with colorectal cancer among other carcinomas and fluctuations in CEA blood levels are used to monitor disease recurrence after surgical resection.
Along with phosphorylation and glycosylation, other modifications receiving significant attention in cancer biomarker research include ubiquitination, methylation, and acetylation [42–44]. Impaired ubiquitin-mediated degradation results in constitutive activity of oncoproteins such as transcription factor p53, which is observed across cancer types [45]. Furthermore, mutations in ubiquitin ligase enzymes such as BRAC1 and Mdm2 are established causes of oncogenesis [46–49]. Abnormal epigenetic methylation and acetylation impact transcriptional regulation resulting in impaired protein expression and dysregulated cell signaling [12,50]. Hypermethylation patterns are associated with carcinogenesis and DNA methylation profiles can serve as cancer biomarkers.
2.3. Challenges with proteomic cancer biomarker discovery
The proteome is dynamic and protein composition is continuously modified through highly regulated processes such as transcription, translation, and degradation in response to cell cycle progression and other stimuli. Proteomic assessment at any given time is only representative of the current state of the cells and proteins whose abundance is low at that point can be missed by mass spectrometry-based proteomic analysis. Additionally, proteins with impaired function and/or abnormal conformation can be highly susceptible to degradation, thus rendering them undetectable by proteomic techniques. Another challenge is to distinguish low abundance isoforms of a protein as well as less prevalent post-translationally modified species that represent a small proportion of the total protein. Continuous efforts to overcome these limitations have led to the optimization of sample processing procedures, the introduction of specimen enrichment techniques and the development of innovative labeling methods that improve sensitivity through mass spectrometry detection. Overall, the proteome is the most direct and informative assessment of cell signaling, but whole proteome characterization remains elusive by the currently available methods.
3. Genomic analysis of cancer biomarkers
3.1. NGS and cancer biomarker detection
The development and continuous evolution of NGS techniques have revolutionized genomic as well as transcriptomic analysis [51]. NGS platforms offer deep sequencing, which is capable of detecting very low frequency genetic variants, as well as massively parallel sequencing that can rapidly and comprehensively cover the human genome. Various NGS methods are utilized for whole-genome sequencing, whole-exome sequencing, targeted sequencing and RNA-seq [52–54]. In turn, these applications are used to identify mutations in coding and non-coding genomic regions as well as impaired transcriptome dynamics. These features have propelled NGS to the forefront of cancer biomarker discovery where high sensitivity and massively parallel sequencing allow rapid detection of somatic and germline mutations.
NGS technologies are routinely utilized in clinical research studies, such as The Cancer Genome Atlas project, with the goal of discovering variant patterns that can serve as cancer biomarkers [55]. Consequently, the implementation of NGS in translational research has led to the generation of large, cancer-specific genomic data sets and the identification of genetic variants, including very low frequency mutations, that have diagnostic, prognostic and therapeutic utility. Although cancer onset can be associated with key driver mutations, disease etiology and progression depend on the patient-specific combination of genetic variants. This compound effect of somatic and germline mutations is addressed through Precision Medicine initiatives that utilize genome-wide or targeted NGS sequencing to establish the genomic profile of individual patients [53]. For example, NGS testing performed in NSCLC patients can detect the low frequency EGFR T490M mutant, which is resistant to gefitinib and erlotinib therapy, and can guide medical decisions [38,39].
Clonal evolution and continuous tumor remodeling lead to tumor heterogeneity, which presents a tremendous challenge in oncology with respect to understanding disease etiology, progression and treatment options. Although conventional biopsy specimens are not representative of complete tumor heterogeneity, they often consist of normal cells and multiple clonal cell types that harbor distinct genomic mutations. Distinguishing low frequency mutations from background signal is difficult in these complex samples. However, NGS techniques can successfully detect clonal variants through deep sequencing and identify impaired signaling pathways and potential therapeutic targets [52,53]. A more comprehensive assessment of tumor heterogeneity can be achieved through liquid biopsy specimens, where NGS methods are used to sequence low abundance circulating tumor DNA (ctDNA) that originates from clonal cells throughout the tumor and is representative of primary and metastatic sites. Additionally, liquid biopsy testing is less invasive than conventional biopsies and can be performed routinely to monitor disease progression and response to therapy, rendering it the primary focus of cancer biomarker translational research [56].
3.2. Limitations of genomic cancer biomarkers
Implementation of high sensitivity and ultra-high throughput NGS techniques has generated tremendous amounts of genomic data across cancer types and led to the identification of a myriad of genetic variants that can serve as cancer biomarkers. However, the impact that genetic mutations have on the transcriptome and proteome of cells cannot be inferred directly from sequencing analysis, especially when genomic variables are localized to non-coding regions. Additional challenges with genomic cancer biomarkers stem from lack of standardization pertaining to NGS techniques and difficulties with the validation of rare genetic cancer biomarkers where sufficiently large patient cohorts cannot be obtained. Another limitation of NGS is the cost of performing whole genome or exome sequencing that is necessary for cancer biomarker discovery. However, despite these challenges the FDA recently approved the first NGS clinical test, ClonoSEQ, to be used for detection of minimal residual disease in patients with acute lymphoblastic leukemia and multiple myeloma. In summary, genomic analysis has led to the discovery of many genetic variants that are currently used as cancer biomarkers in lab-developed tests, but in the absence of corresponding proteomic and cell signaling data, these mutations have limited diagnostic, prognostic and therapeutic value.
4. Multi-omics in cancer biomarker discovery
4.1. Compounded contribution of multiple factors to carcinogenesis
Altered protein levels, abnormal structural conformation and impaired function are the most immediate causes of aberrant signaling in tumorigenesis. Protein dynamics are dependent in part on genetic factors, transcriptional regulation and mRNA translation, hence genomic and transcriptome abnormalities are reflected in the proteome [57]. Analyzing the protein composition of tumors has been the focus of immense initiatives such as CPTAC [21]. These studies utilize large cancer and grade-specific cohorts to characterize the proteome of tumor specimens and identify patterns that are indicative of disease onset and progression. Among the challenges with most proteomic analyses are a poor representation of low grade, early stage samples as most patients are diagnosed following the presentation of symptoms that correlate with an advanced disease state. The Early Detection Research Network (EDRN) is another extensive NCI initiative focused on cancer biomarker discovery and validation with the goal of early detection [58]. These multi-institution efforts emphasize the importance of proteomics in understanding disease etiology, but they also recognize the advantage of integrating genomic data thereby establishing proteogenomics as a prominent approach to cancer biomarker discovery [1].
Despite continuous efforts on all fronts, omics areas have advanced at different rates with genomics experiencing a boom in sequencing data acquisition and analysis due to the introduction of NGS. In addition, RNA-seq has propelled transcriptomics and allowed for multiplexing and the detection of low abundance mRNA and regulatory miRNA. Proteomics has also evolved, but to a lesser degree as protein composition is dynamic and low abundance species present a challenge for mass spectrometry methods, while antibody targeted approaches that allow for signal amplification are not optimal for biomarker discovery [59]. Despite the vast amounts of available genomic and transcriptomic data, mapping the signaling networks that initiate carcinogenesis is challenging since protein composition is not in perfect agreement with mRNA abundance and genomic variants in non-coding regions are difficult to assess without knowing if and how they impact protein expression and function.
4.2. Integrating omics data
Integration of ‘omics’ data can identify genomic and transcriptomic alterations that impact the proteome and cell signaling, leading to tumorigenesis. For example, Cohen et al. recently introduced CancerSEEK as a blood test capable of detecting eight different cancer types through the integration of genomic and proteomic analysis [60]. This proteogenomic approach combines targeted genomic analysis of ctDNA using a 61-amplicon panel with a set of eight validated protein cancer biomarkers. CancerSEEK is an excellent example of the superior diagnostic sensitivity and specificity achieved through the integration of two omics approaches [60]. The advantage of integrating omics data is also evident in the work reported by Karagoz et al., where multi-omics analysis were used to identify transcription factors responsible for onset of triple negative breast cancer (TNBC), which lacks expression of estrogen receptor, progesterone receptor and HER2 and presents with aggressive proliferation, large tumor size and rapid progression to metastasis [61]. This study utilized TNBC genomic databases to identify a subset of differentially expressed genes and subsequently analyzed their transcription and protein expression profiles using TNBC transcriptome and protein-interaction databases. Comparing data across omics areas implicated upregulation of JAK-STAT and TNF signaling as key mechanisms contributing to TNBC onset and progression. Furthermore, proteogenomics is utilized by the multi-institution CPTAC initiative in an effort to characterize abnormal signaling across cancer types [62–64]. Colon and rectal tumor samples previously subjected to RNA-seq analysis as part of The Cancer Genome Atlas study are subjected to tandem mass-spectrometry protein detection in an effort to correlate genomic and proteomic profiles [62]. In addition to characterizing the proteome of individual biopsy specimens belonging to the same cancer class, this approach demonstrated that mRNA abundance does not directly predict protein levels [62]. Discrepancy between the transcriptome and proteome emphasizes the impact post-transcriptional regulation has on cell signaling through mechanisms such as miRNA translational repression. This complex and indirect correlation between mRNA and protein levels stresses the need for multi-omics analysis and underscores the potential for misinterpretation when a single omic approach is used.
4.3. Challenges with multi-omics for cancer biomarker discovery
Integrating distinct genomic, transcriptomic, epigenetic, meta-bolomic, and proteomic databases is becoming a prevalent approach to cancer biomarker discovery. However, merging data sets can be challenging if databases use different nomenclatures, when samples show poor correlation due to tumor heterogeneity and if data acquisition and processing are not standardized (different instrumentation, pipeline process). Multi-omics techniques where all data are generated from a single sample can reduce complications due to sample heterogeneity and simplify correlation statistics. However, multi-omics analysis cannot overcome the challenges of merging datasets from different studies with diverse sample preparation and analysis methods. Therefore, it is important that large-scale, multi-institution initiatives such as CPTAC use standardized protocols that are uniformly applied thus enabling data comparison and statistical analysis. Data storage and computational manipulation of multi-omics datasets can be challenging and frequently requires dedicated servers for large-scale studies. Furthermore, in a clinical setting, the resources needed for individual multi-omics data interpretation may be difficult to attain, with integration and storage of data in the patient record being impossible. Additionally, translational studies aimed at cancer biomarker discovery with the goal of identifying genomic, proteomic, or other carcinogenic profiles to be used in a clinical setting, face numerous challenges including method validation, standardization of sample collection and testing procedures, as well as storage and accessibility to patient data.
5. Translating biomarker discovery into clinical applications
5.1. Translational process and challenges
Cancer biomarker discovery most frequently originates in academic research settings, leading to the identification of thousands of protein and genetic markers. However, despite the continuously growing number of potential cancer biomarkers, there have been very few new cancer biomarker-based diagnostics. The discrepancy between the rate of biomarker discovery and implementation of new tests into the clinical lab can be attributed to several challenges associated with the translational process [65].
Transitioning ‘from bench to bedside’ is the foundation of translational research and it requires large-scale validation studies and clinical trials that are difficult for a single academic lab or even institution [66]. Translating biomarker discovery into clinical assays requires a large team, including academic researchers, clinicians, and industry, to define the clinical utility of the test, validation process and study design. Assembling such a team can be a daunting task that requires tremendous resources and poses a major challenge in translational research. The NCI EDRN initiative is a prime example of a successful multi-organization translational research program aimed at the discovery of cancer biomarkers for early detection and risk assessment, where collaboration among academic institutions, industry, and government has been able to address challenges including analytical and clinical validation that require large specimen cohorts and standardized sample preparation [58].
Cancer biomarker discovery conducted in a research setting utilizes complex instrumentation and sample preparation techniques that are not compatible with day to day operations in a clinical lab, which require high throughput, reproducible methods that are straightforward to perform. For example, proteomic-based cancer biomarker discovery relies on mass spectrometers that are highly complex, have low throughput capabilities and require extensive personnel training, making them difficult to integrate in clinical labs with total automation and multiplexing testing platforms. The incompatibility of instruments used in research labs and those utilized for clinical testing, is a major challenge in translational research. Developing a method suitable for clinical testing (high throughput, adequate sensitivity, reproducibility, multiplexing capabilities, specimen compatibility and ease of use among other parameters) can be difficult. In particular, while mass spectrometry might be highly specific, clinical laboratories may want to use an immunoassay platform that can achieve better analytical sensitivity and is easy to incorporate into a clinical setting, but has less specificity for a particular protein variant that serves as the disease biomarker. To further complicate the transition from research to clinical testing, a method must be validated to establish key performance parameters of the assay including a limit of detection, limit of quantitation, precision, accuracy and potential interferences by other substances present in a patient sample [67,68]. Furthermore, this process requires a standardized operating procedure, validated standards, reference materials, and calibrators as well as a large specimen cohort. Interpretation of the results and how they will influence clinical decisions, dictates the performance and analytical requirements of the test such as necessary sensitivity, bias, and coefficient of variation at and near medical decision points.
Analytical and clinical validation studies are essential for an assay to be submitted for FDA clearance or approval [67]. This process can be time-consuming and difficult to achieve due to limited sample availability. Biospecimen repositories and biobanks are key to the translational process as access to patient specimens and detailed clinical history is crucial for the development of assays. The growing need for meticulously categorized specimens with respect to patient status and disease progression has spurred on initiatives to organize large repositories of patient samples and data such as the National Biospecimen Network [69]. Availability of specimens is not enough for proper assay development, standardized sample collection and patient history are necessary for proper validation. Differences in specimen collection, storage and processing can impact results during the analytical validation process posing additional challenges. Patient history and outcome data can be obtained either through prospective trials or retrospective studies, the latter allowing for a shorter validation timeline. Risk assessment biomarker assays further benefit from repositories comprised of specimens from patients and their relatives, such as the Breast Cancer Family Registry, which allows clinical validation through prospective trials that follow individuals with potential risk factors [70]. Such familial repositories are invaluable for the discovery of risk and early diagnostic biomarkers, but unfortunately, they are not readily available for most malignancies.
5.2. Successful translation into clinical testing
Large sample size, detailed patient history and the need to account for sample variability require big data manipulation and superior statistical analysis capabilities. Although this may have been a major challenge in the past, cross-institution efforts such as EDRN have successfully overcome these limitations. The financial backing necessary to take a cancer biomarker from the discovery phase to clinical testing is another limitation that requires a multi-institution effort with ample resources. Overall, the complexity of translational research has presented a bottleneck for the clinical implementation of tests following cancer biomarker discovery, but in recent years tremendous efforts on multiple fronts and increased financial support have been able to address these challenges. Genomic cancer biomarkers have been able to transition from the research to the clinical lab, in part due to the development of NGS and its ability to meet the demands for clinical testing. Primary examples of genomic biomarkers that have successfully transitioned to clinical tests include the Oncotype Dx and Mammaprint assays used for assessment of breast cancer patients whose gene expression profile influences therapy decisions [71–74]. NSCLC patients harboring resistance to tyrosine kinase inhibitor therapy due to the EGFR T790M mutation, have also benefitted from translational research where the presence of EGFR T790M can be detected through clinical tests and used to initiate therapy with osimertinib [75,76]. Certain proteomic cancer biomarkers have also made a successful transition ‘from bench to bedside’, with OVA1 multivariate index assay being a prime example. OVA1 is used for risk assessment of ovarian cancer in patients with pelvic masses who are scheduled to undergo surgery. Risk of malignancy is determined through a combination of markers including CA125, prealbumin, apolipoprotein A1, beta-2-microglobulin, and transferrin [77]. Combining multiple proteins, and even genomic attributes, to serve as a cancer biomarker has tremendous potential and is the current approach to diagnostic and prognostic testing, however, validation studies examining multiple analytes in a single assay can be difficult. One successful example is Cologuard, an FDA approved fecal screening test for colorectal cancer. Cologuard is a clinical assay for tumor-specific DNA changes, including aberrant methylated BMP3 and NDRG4, a mutant form of KRAS, beta-actin, and hemoglobin [78].
Translating research discoveries into clinical assays has been challenging and a very small fraction of genomic and proteomic markers complete the transition to tests routinely offered in the clinical lab. Clinical utility is a major consideration when selecting candidate biomarkers, with patient care, early diagnosis and improved treatment outcome being key to deciding what biomarkers are applicable in a clinical setting [79,80]. Additionally, the ability to develop a method that meets the clinical needs, can be integrated into a clinical lab and is cost-effective also impacts the selection of cancer biomarkers to be implemented in patient care. Increasing multi-institution efforts, collaborations among academia, government and industry and funding support have made translational research the focus of cancer initiatives in an effort to address unmet clinical needs and transition to Precision Medicine.
6. Cancer biomarkers and precision medicine
Genomic analysis has established that numerous, variable mutations exert a compound effect to initiate disease onset and progression. Patients with the same cancer type and stage can respond differently to therapy and exhibit variable rates of disease progression based on the pathophysiological alterations underlying the carcinogenesis. For example, gene expression is assessed in breast cancer patients to guide medical decisions regarding treatment [72]. Patients with estrogen or progesterone receptor positive breast cancer benefit from hormone therapy and those with HER2 positive breast cancer receive HER2 targeted treatments. On the other hand, these therapies are not effective in patients with TNBC, who are likely to benefit from adjuvant chemotherapy. Currently Precision Medicine is primarily utilized in guiding treatment decisions, but nonetheless, it is impacting health-care costs by optimizing therapies, reducing complications, and improving patient outcomes.
Multi-omics initiatives aim to characterize the complex pathophysiology of oncogenesis in an effort to identify risk factors and diagnostic cancer biomarkers. As more patient specimens undergo multi-omics analysis, additional low frequency driver mutations and protein abnormalities will be identified as contributing to oncogenesis. The corresponding increase in the characterization of oncogenic proteogenomic profiles will in turn contribute to the evolution and expansion of Precision Medicine that eventually will enable risk assessment in phenotypically healthy individuals leading to early diagnosis.
7. Conclusion
Cancer biomarker discovery efforts have benefited from technological improvements in instrumentation and the implementation of novel analytical methods. Examples are the development of NGS and mass spectrometry for high throughput genomic and proteomic characterization of biological specimens. However, the intricate cell signaling mechanisms that trigger oncogenesis are not dependent on a single factor, but instead the compound effect of genomic, transcriptomic, epigenetic, metabolomic, and proteomic alterations. Therefore, the comprehensive characterization of various cancer types and stages requires a multi-omics approach. Integrated omics data can serve as the basis for the development of novel multi-omics diagnostic tests. Finally, important lessons learned from the team efforts in moving cancer biomarker discovery through validation to the development of clinical diagnostics, will pave the way for rapid translation into clinical use and transition to Precision Medicine.
8. Expert commentary
The ultimate goal of predicting disease onset and identifying patients during the early stages of cancer, when treatment is most effective, has presented a tremendous challenge in cancer biomarker discovery and translation. A major limitation has been the capability to detect and quantify abnormal biological molecules such as DNA, RNA, proteins, and metabolites, that are present at minute levels early on. Additionally, individual classes of biomarkers have limited specificity and do not provide a comprehensive overview of disease etiology. This is reflective of the currently available clinical tests, which monitor cancer biomarkers such as CA19–9 that lack specificity and are primarily utilized to assess disease progression and response to therapy, but have no diagnostic value. In general, these limitations render an unmet clinical need for cancer biomarkers that offer diagnostic, prognostic, and/or therapeutic utility.
Over the last decade, intense efforts have addressed these challenges through the development of new technologies with improved sensitivity and rapid, large-scale detection capabilities that allowed the identification of many potential cancer biomarkers. For example, in genomics, the introduction of NGS and its application to whole genome and exome sequencing led to a disproportionate increase in the number of genetic mutations associated with cancer. To a lesser degree this has been the case with proteomic mass spectrometry-based techniques such as TMT labeling. Analyzing the dynamic proteome has proven more challenging than an assessment of the static genome, hence ongoing efforts are underway to develop improved proteomic quantification methods. As technological challenges are rapidly overcome, other difficulties involving big data sets are emerging as limiting factors in cancer biomarker translation. These include data storage and the need for complex bioinformatics approaches to manage, process and analyze the data.
Integration of data across omics areas is a promising new approach that gives a comprehensive view of genomic mutations, transcriptional abnormalities and the impact they have on the proteome and cell dynamics. Multi-omics data has become the focus of many large-scale studies that aim to comprehensively characterize genetic, transcriptomic, protein, and other aberrations associated with specific cancer types. The central dogma of biology establishes the relationship between genetic and protein composition, but proteomic dynamics including protein expression, degradation, and PTM cannot be inferred directly from the genome. This emphasizes the need to investigate genomic mutations in the context of epigenetic, transcriptomic, proteomic, and metabolomic abnormalities that may be associated with the disease state. In turn, multi-omics data for a patient provides a detailed overview of the disease etiology and can be used to make prognostic and therapeutic clinical decisions. Examining multi-omics data across cancer-specific patient cohorts generates large datasets that can be used to extrapolate disease-specific patterns in biological molecules that can serve as biomarkers. In addition, this approach provides patient-specific multi-omics profiles that are essential to the transition in clinical practice to Precision Medicine. However, much remains to be improved with multi-omics to enable translation of potential cancer biomarkers into a clinical setting. A major challenge remains the ability to identify key biomarkers that can be readily detected at early stages of disease across patients using methods that are compatible with the automated, high throughput platforms in clinical labs.
The discovery stage of cancer biomarker studies relies on techniques that are not optimal for routine clinical testing due to long analysis time, complex operation procedures and incompatibility with other lab instrumentation. Additionally, data processing and interpretation require expert bioinformatics approaches to extract the necessary information and in the case of multi-omics combine big data sets from different testing platforms. Overcoming the bottleneck of cancer biomarker translation into clinical application will require the development of multiplexing platforms that can rapidly perform multi-omics testing on individual samples along with the necessary data processing that provides a direct result that does not require expert interpretation. To achieve this goal, cancer biomarker translation initiatives are focusing to liquid biopsy testing, which offers analysis of cell-free tumor DNA, circulating tumor cells and exosomes extracted from a single patient blood sample. The ease of sample collection further emphasizes the role liquid biopsies will play in the future of cancer biomarker testing and clinical implementation.
9. Five-year view
Extensive efforts to translate cancer biomarkers into clinical applications will undoubtedly introduce numerous new clinical tests over the next 5 years that utilize NGS platforms and targeted protein detection techniques such as immunoassays and MRM mass spectrometry. However, regulatory requirements and reimbursement issues could hinder the implementation of new biomarkers for clinical laboratories. These tests will most likely be of prognostic and therapeutic value, while reliable diagnostic tests remain further away in the future. Numerous reports of multi-omics tests with improved specificity for early-stage cancers is an indication that in the not too distant future cancer patients will be evaluated using a multitude of biomarkers that generate a patient specific profile indicative of etiology that can be used to guide treatment decisions. A major transition in cancer biomarker testing that will redefine diagnostic and prognostic assessment is liquid biopsy analysis. The less invasive sample collection procedure, coupled with the ability to perform multi-omics analysis on a single specimen are tremendous advantages that will make liquid biopsies the future of diagnostic, prognostic and therapeutic response testing in cancer patients.
Key issues.
Advancements in genomic and proteomic techniques enable detection of very low abundance genetic mutations and protein abnormalities that can serve as disease-specific markers.
Genomic and transcriptomic abnormalities are not directly indicative of protein dynamics, rendering the need for integration of data sets obtained by each omic approach to obtain a comprehensive understanding of disease etiology.
Multi-omics analysis of cancer-specific patient cohorts is the focus of current studies aimed at identifying patterns of biological molecules that can be utilized for early detection and diagnosis.
Management and analysis of multi-omics datasets are challenging due to the lack of standardized specimen processing and data collection procedures, as well as the difficulty of storing and processing big data.
Translating multi-omics cancer biomarkers into clinical testing is currently limited by lack of high throughput, multiplexing instruments that can be easily integrated into the clinical lab as well as big-data storage and analysis.
Liquid biopsy testing is emerging as a promising future for multi-omics clinical testing due to the use of a single sample and the ease of specimen collection.
Acknowledgments
Funding
This paper was supported by the U.S. National Cancer Institute, the Clinical Proteomic Tumor Analysis Consortium (CPTAC, Grant U24CA210985, DWC) and the Early Detection Research Network (EDRN, Grant U24CA115102, DWC).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018. May;19(5):299–310. PubMed PMID: ; PubMed Central PMCID: PMCPMC5990367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018. February;15(2):81–94. PubMed PMID: •Comprehensive overview of the heterogenious nature of tumors and the techniques/approaches to distinguish subpopulations of cells that contibute to the complexity of carcino-geneis and that can be utilized in personalized therapies. [DOI] [PubMed] [Google Scholar]
- 3.Roberts NJ, Vogelstein JT, Parmigiani G, et al. The predictive capacity of personal genome sequencing. Sci Transl Med. 2012. May 9;4 (133):133ra58. PubMed PMID: ; PubMed Central PMCID: PMCPMC3741669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. PubMed PMID: ; PubMed Central PMCID: PMCPMC3656720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kilpivaara O, Aaltonen LA. Diagnostic cancer genome sequencing and the contribution of germline variants. Science. 2013. March 29;339(6127):1559–1562. [DOI] [PubMed] [Google Scholar]
- 6.LeBlanc VG, Marra MA. Next-generation sequencing approaches in cancer: where have they brought us and where will they take us?. Cancers (Basel). 2015. September 23;7(3):1925–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verigos J, Magklara A. Revealing the complexity of breast cancer by next generation sequencing. Cancers (Basel). 2015. November 6;7(4):2183–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sondka Z, Bamford S, Cole CG, et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018. November;18(11):696–705. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borrebaeck CA. Precision diagnostics: moving towards protein biomarker signatures of clinical utility in cancer. Nat Rev Cancer. 2017. March;17(3):199–204. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 10.Xiao L, Xiao T, Wang ZM, et al. Biomarker discovery of nasopharyngeal carcinoma by proteomics. Expert Rev Proteomics. 2014. April;11(2):215–225. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 11.Wang F, Sc W, Chan LW, et al. Multiple regression analysis of mRNA-miRNA associations in colorectal cancer pathway. Biomed Res Int. 2014;2014:676724. PubMed PMID: ; PubMed Central PMCID: PMCPMC4033420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang W, Liu N, Chen XZ, et al. Genome-wide identification of a methylation gene panel as a prognostic biomarker in nasopharyngeal carcinoma. Mol Cancer Ther. 2015. December;14(12):2864–2873. MCT-15–0260. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 13.Armitage EG, Barbas C. Metabolomics in cancer biomarker discovery: current trends and future perspectives. J Pharm Biomed Anal. 2014. January;87:1–11. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 14.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015. February 26;372(9):793–795. PubMed PMID: ; PubMed Central PMCID: PMCPMC5101938.• Introduction to precision medicine NIH initiative and a summary of the importance and future of individualized medicine, in particular pertaining to oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toward precision medicine: building a knowledge network for biomedical research and a new taxonomy of disease. Washington (DC): The National Academies Collection: Reports funded by National Institutes of Health; 2011. [PubMed] [Google Scholar]
- 16.Crutchfield CA, Thomas SN, Sokoll LJ, et al. Advances in mass spectrometry-based clinical biomarker discovery. Clin Proteomics. 2016;13:1 PubMed PMID: ; PubMed Central PMCID: PMCPMC4705754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004. December;3(12):1154–1169. PubMed PMID: • Introduction of multiplex protein quantification in complex mixtures using isobaric labeling of peptides. [DOI] [PubMed] [Google Scholar]
- 18.Thompson A, Schafer J, Kuhn K, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003. April 15;75(8):1895–1904. PubMed PMID: • Development of tandem mass tags (TMT) for mass spectrometry based quantification of complex protein mixtures that allow peptides from different samples to be distinguished and their relative abundance quantified. [DOI] [PubMed] [Google Scholar]
- 19.Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002. May;1(5):376–386. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 20.Clark DJ, Hu Y, Bocik W, et al. Evaluation of NCI-7 cell line panel as a reference material for clinical proteomics. J Proteome Res. 2018. June 1;17(6):2205–2215. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez H, Pennington SR. Revolutionizing precision oncology through collaborative proteogenomics and data sharing. Cell. 2018. April 19;173(3):535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shahi P, Kim SC, Haliburton JR, et al. Abseq: ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci Rep. 2017. March 14;7:44447 PubMed PMID: ; PubMed Central PMCID: PMCPMC5349531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ullal AV, Peterson V, Agasti SS, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med. 2014. January 15;6(219):219ra9. PubMed PMID: ; PubMed Central PMCID: PMCPMC4063286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lange V, Picotti P, Domon B, et al. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222 PubMed PMID: ; PubMed Central PMCID: PMCPMC2583086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006. April 14;312(5771):212–217. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy JJ, Abbatiello SE, Kim K, et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat Methods. 2014. February;11(2):149–155. PubMed PMID: ; PubMed Central PMCID: PMCPMC3922286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HB, Kang UB, Moon HG, et al. Development and validation of a novel plasma protein signature for breast cancer diagnosis by using multiple reaction monitoring-based mass spectrometry. Anticancer Res. 2015. November;35(11):6271–6279. PubMed PMID: . [PubMed] [Google Scholar]
- 28.You J, Kao A, Dillon R, et al. A large-scale and robust dynamic MRM study of colorectal cancer biomarkers. J Proteomics. 2018. June 25 PubMed PMID: DOI: 10.1016/j.jprot.2018.06.013 [DOI] [PubMed] [Google Scholar]
- 29.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003. March;21(3):255–261. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 30.Sun S, Shah P, Eshghi ST, et al. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat Biotechnol. 2016. January;34 (1):84–88. PubMed PMID: ; PubMed Central PMCID: PMCPMC4872599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunn JD, Reid GE, Bruening ML. Techniques for phosphopeptide enrichment prior to analysis by mass spectrometry. Mass Spectrom Rev. 2010. Jan-Feb;29(1):29–54. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 32.Ardito F, Giuliani M, Perrone D, et al. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int J Mol Med. 2017. August;40(2):271–280. PubMed PMID: ; PubMed Central PMCID: PMCPMC5500920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987. January 9;235(4785):177–182. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 34.Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989. May 12;244(4905):707–712. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 35.Teplinsky E, Muggia F. Targeting HER2 in ovarian and uterine cancers: challenges and future directions. Gynecol Oncol. 2014. November;135(2):364–370. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 36.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004. June 4;304(5676):1497–1500. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 37.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004. September 7;101(36):13306–13311. PubMed PMID: ; PubMed Central PMCID: PMCPMC516528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005. March;2(3):e73 PubMed PMID: ; PubMed Central PMCID: PMCPMC549606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005. February 24;352(8):786–792. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 40.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015. September;15(9):540–555. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 41.Kirwan A, Utratna M, O’Dwyer ME, et al. Glycosylation-based serum biomarkers for cancer diagnostics and prognostics. Biomed Res Int. 2015;2015:490531. PubMed PMID: ; PubMed Central PMCID: PMCPMC4609776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kouzarides T Chromatin modifications and their function. Cell. 2007. February 23;128(4):693–705. [DOI] [PubMed] [Google Scholar]
- 43.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006. May;6(5):369–381. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 44.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012. February 1;125(Pt 3):531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science. 1991. July 5;253(5015):49–53. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 46.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by MDM2. Nature. 1997. May 15;387(6630):299–303. [DOI] [PubMed] [Google Scholar]
- 47.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997. December 22;420 (1):25–27. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 48.Hashizume R, Fukuda M, Maeda I, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001. May 4;276 (18):14537–14540. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 49.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002. January 25;108(2):171–182. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 50.Liloglou T, Bediaga NG, Brown BR, et al. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014. January 28;342(2):200–212. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 51.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016. May 17;17(6):333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shyr D, Liu Q. Next generation sequencing in cancer research and clinical application. Biol Proced Online. 2013. February 13;15(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cummings CA, Peters E, Lacroix L, et al. The role of next-generation sequencing in enabling personalized oncology therapy. Clin Transl Sci. 2016. December;9(6):283–292. PubMed PMID: ; PubMed Central PMCID: PMCPMC5351002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018. February;19 (2):93–109. PubMed PMID: 29279605. [DOI] [PubMed] [Google Scholar]
- 55.Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19(1A):A68–77. PubMed PMID: ; PubMed Central PMCID: PMCPMC4322527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heitzer E, Haque IS, Roberts CES, et al. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2018. November 8 PubMed PMID: DOI: 10.1038/s41576-018-0071-5 [DOI] [PubMed] [Google Scholar]
- 57.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012. March 13;13(4):227–232. PubMed PMID: ; PubMed Central PMCID: PMCPMC3654667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner PD, Srivastava S. New paradigms in translational science research in cancer biomarkers. Transl Res. 2012. April;159(4):343–353. PubMed PMID: ; PubMed Central PMCID: PMCPMC3478674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cayer DM, Nazor KL, Schork NJ. Mission critical: the need for proteomics in the era of next-generation sequencing and precision medicine. Hum Mol Genet. 2016. October 1;25(R2):R182–R189. [DOI] [PubMed] [Google Scholar]
- 60.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018. February 23;359(6378):926–930. PubMed PMID: ; PubMed Central PMCID: PMCPMC6080308.• Application of muti-omics liquid biopsy test comprised of pannels of genomic and proteomic markers that detect and differentiate eight different cancer types with high specificity and snesitivity, showing promise for early diagnosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karagoz K, Sinha R, Arga KY. Triple negative breast cancer: a multi-omics network discovery strategy for candidate targets and driving pathways. 2015. February;19(2):115–130. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 62.Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014. September 18;513 (7518):382–387. PubMed PMID: ; PubMed Central PMCID: PMCPMC4249766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016. June 2;534(7605):55–62. PubMed PMID: ; PubMed Central PMCID: PMCPMC5102256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang H, Liu T, Zhang Z, et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016. July 28;166(3):755–765. PubMed PMID: ; PubMed Central PMCID: PMCPMC4967013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maes E, Cho WC, Baggerman G. Translating clinical proteomics: the importance of study design. Expert Rev Proteomics. 2015. June;12 (3):217—219. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 66.Pepe MS, Etzioni R, Feng Z, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001. July 18;93 (14):1054–1061. PubMed PMID: 11459866.•• Proposed guideline for biomarker development and the process of developing and evaluating biomarkers for clinical application prior to implementation. [DOI] [PubMed] [Google Scholar]
- 67.Fuzery AK, Levin J, Chan MM, et al. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013. October 2;10(1):13 PubMed PMID: ; PubMed Central PMCID: PMCPMC3850675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li D, Chan DW. Proteomic cancer biomarkers from discovery to approval: it’s worth the effort. Expert Rev Proteomics. 2014. April;11 (2):135–136. PubMed PMID: ; PubMed Central PMCID: PMCPMC4079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hede K NCI’s National Biospecimen Network: too early or too late?. J Natl Cancer Inst. 2005. February 16;97(4):247–248. [DOI] [PubMed] [Google Scholar]
- 70.John EM, Hopper JL, Beck JC, et al. The breast cancer family registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6(4):R375–89. PubMed PMID: ; PubMed Central PMCID: PMCPMC468645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004. December 30;351(27):2817–2826. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 72.Cronin M, Sangli C, Liu ML, et al. Analytical validation of the Oncotype DX genomic diagnostic test for recurrence prognosis and therapeutic response prediction in node-negative, estrogen receptor-positive breast cancer. Clin Chem. 2007. June;53(6):1084–1091. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 73.van de Vijver MJ, He YD, Van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002. December 19;347(25):1999–2009. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 74.van ‘T Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002. January 31;415(6871):530–536. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 75.Sukari A, Nagasaka M, Wakeling E. EGFR-mutant non-small cell lung cancer in the Era of Precision medicine: importance of germline EGFR T790M testing. J Natl Compr Canc Netw. 2017. October;15 (10):1188–1192. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 76.Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015. April 30;372(18):1689–1699. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z, Bast RC Jr., Yu Y, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004. August 15;64(16):5882–5890. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 78.Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014. April 3;370(14):1287–1297. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 79.Bossuyt PM, Reitsma JB, Linnet K, et al. Beyond diagnostic accuracy: the clinical utility of diagnostic tests. Clin Chem. 2012. December;58 (12):1636–1643. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 80.Moons KG, de Groot JA, Linnet K, et al. Quantifying the added value of a diagnostic test or marker. Clin Chem. 2012. October;58 (10):1408–1417. PubMed PMID: . [DOI] [PubMed] [Google Scholar]