

Abstract
Background
Panniculitis-like T-cell lymphoma is an uncommon type of non-Hodgkin lymphoma, occurring usually in the form of nodules within the subcutaneous fat tissue of the extremities or trunk. In the literature, subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is described as a distinct type of T-cell lymphoma with a variable clinical behavior, depending on molecular phenotype of T-cell receptor (TCR) and on the presence or absence of hemophagocytic syndrome.
Case presentation
We present a bioptic and autoptic case of a 65-years old Caucasian man with panniculitic T-cell lymphoma with morphological and immunohistochemical features of SPTCL, limited to the retroperitoneal and mesenteric mass, i.e. without any cutaneous involvement, and associated with severe hemophagocytic lymphohistiocytosis.
Conclusion
A panniculitic T-cell lymphoma with morphological and molecular features of SPTCL, which is limited to mesentery, i.e. does not involve subcutaneous fat, seems to be exceedingly rare.
Keywords: Panniculitis, T-cell lymphoma, Mesentery, Hemophagocytosis, Lymphohistiocytosis
Background
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a cytotoxic T-cell lymphoma that preferentially infiltrates subcutaneous adipose tissue [1]. It is a rare form of lymphoma, accounting for < 1% of all non-Hodgkin lymphomas. SPTCL can occur at any age, with 20% of patients being < 20 years of age [2]. The diagnosis of SPTCL is challenging. Half of the patients with SPTCL present with non-specific clinical symptoms incl. weight loss, low-grade fever and general malaise, whilst the rest may only have local signs [1, 3]. Locally, there are multiple subcutaneous nodules or plaques ranging from 5 mm to several centimeters in size. They most commonly occur in the subcutaneous tissue of the extremities or trunk. Other locations are rare [4], but may include the mesentery [5, 6]. Lymph node or bone marrow involvement is usually absent [2]. The differential diagnosis includes panniculitis, either unspecific [7] or associated with lupus [3]. Laboratory abnormalities frequently include cytopenia and elevated liver function tests. Panniculitic T-cell lymphoma may be associated with hemophagocytic syndrome in 17–45% cases, depending on molecular phenotype of T-cell receptor (TCR) of tumor cells [3].
Even a biopsy may not lead to a straightforward diagnosis. There are lymphocytic infiltrates involving the fat lobules, but usually sparing the septa. The lymphoma cells vary in size, have irregular and hyperchromatic nuclei. The rimming of the neoplastic cells surrounding individual fat cells is a helpful diagnostic clue. Admixture of reactive histiocytes is usually found in the areas of fat infiltration and destruction [1]. In immunohistochemistry, the neoplastic cells in SPTCL express α/β cytotoxic T-cell phenotype, including CD8 (cluster of differentiation), TIA1 (T-cell intracellular antigen 1), granzyme B and perforin, but not CD56 and CD4 [1].
In this paper, we present case of a patient with panniculitis-like cytotoxic T-lymphoma of the mesentery, with microscopic and immunohistochemical features of SPTCL and both clinical and histopathological signs of severe hemophagocytic syndrome, but without any (sub)cutaneous involvement, which we believe is exceedingly rare.
Case presentation
Clinical history
A 65-years old previously fit and well Caucasian male patient presented to a small district general hospital with a 3 weeks history of recurrent rigors, fevers and night sweats. He was found to have kidney injury and thrombocytopenia. The whole-body CT (computed tomography) scan revealed in the retroperitoneal space a lesion measuring approx. 13x8x8cm (Fig. 1). The patient was referred to our hospital for further evaluation and treatment. At presentation, blood biochemistry showed low platelets (55 × 109/L, reference range (ref.) 150–400), low white blood cells (3.2 × 109/L, ref. 4.0–10.0) with lymphocytopenia (0.47 × 109/l, ref. 0.8–4.0). The liver enzymes were elevated, too (Alanine Aminotransferase 1.16μkat/l, ref. < 0.73; Aspartate Aminotransferase 3.56μkat/l, ref. < 0.67) and albumin was low (21.9 g/l, ref. 35.0–53.0). The patient underwent explorative laparotomy, which revealed a tumor in mesocolon ascendens invading radix mesenterii. A surgical biopsy was performed. Few days later, he developed progressive pancytopenia, coagulopathy (fibrinogen 0,7 g/l) and his serum C-reactive protein concentration increased (140 mg/l). The patient was readmitted to the intensive care unit and treated with platelet transfusions, fibrinogen, prothrombin complex concentrates and broad-spectrum antibiotics. Despite all these measures, he developed multi-organ failure with dominant liver failure (Aspartate aminotransferase 18.64 μkat/l, ref. < 0.67; Bilirubin 158.4 μmol/l, ref. < 21.0) and refractory shock. He died 22 days after the initial presentation. At the time of death, the underlying disease leading to death was not known. An autopsy was performed.
Fig. 1.
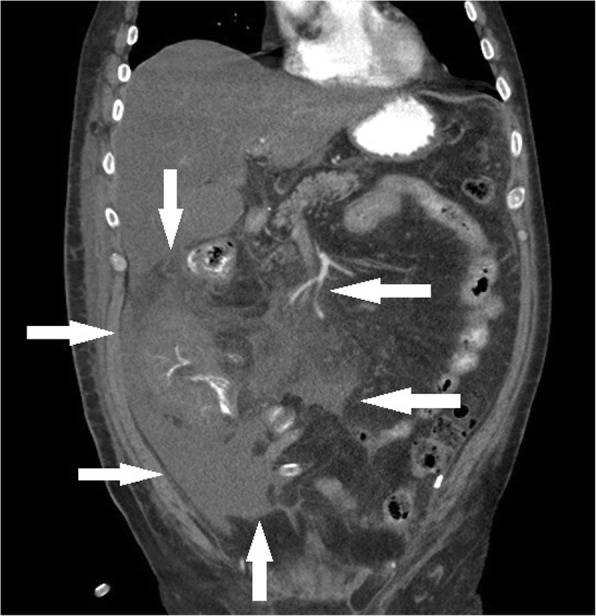
CT scan showing an infiltrate in mesocolon ascendens invading radix mesenterii and the mesenteric vessels
Biopsy findings
Two formalin-fixed adipose tissue fragments with the size of 16x8x4 mm and 12x7x3 mm were sent to the Department of Pathology. Microscopically, we saw adult fat tissue with dense lymphocytic infiltration. The lymphocytic infiltration consisted mostly of small- to medium-sized cells with hyperchromatic irregular nuclei with little rim of pale cytoplasm. There was a pattern of isolated adipocytes surrounded by a dense rim of hyperchromatic lymphocytes (Fig. 2). We also found necrotic adipocytes and reactive macrophages phagocytizing lymphocytes and erythrocytes. Immunohistochemically, the atypical cells rimming the adipocytes stained for CD45(LCA), CD2, CD3, CD5, CD7, CD8, granzyme B, perforin, TIA1 and TCRβF1 and did not stain for CD4, CD20, CD79a, CD56, CD30, EBER, CD1a, S100, myeloperoxidase, cytokeratin CAM5.2 and TCRγ. The proliferation index Ki67 varied between 10 and 50% (Fig. 3).
Fig. 2.

scans of histological slides from the mesenterial fat tissue showing dense lymphocytic infiltration consisting mostly of medium-sized cells with hyperchromatic irregular nuclei. Note a pattern of isolated adipocytes surrounded by a rim of lymphocytes (“rimming”) and large macrophages engulfing lymphocytes (hemophagocytosis). A – HE 60x, B – Giemsa 85x, C – Giemsa 105x, D – HE 145x
Fig. 3.

immunohistochemistry showing neoplastic lymphocytes with positivity of CD3, CD8, perforin, TIA-1 and TCRβF1. CD4 stains rather bystander T-cells and histiocytes, without unequivocal positivity in lymphoma. The neoplastic cells are CD20 negative, note the sparse CD20-positive reactive B-cells. The neoplastic lymphocytes embody proliferation activity (Ki67) about 50%
Genomic deoxyribonucleic acid (DNA) from formalin-fixed paraffin-embedded (FFPE) tissue section was isolated using the QIAamp DNA FFPE Tissue Kit (Qiagen GmbH, Hilden, Germany). The clonality of the TCR rearrangements (TCR β, γ and δ) were tested using standardized multiplex polymerase chain reaction (PCR), as described by the BIOMED2 study group [8]. The clonality of the PCR products was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), with detection of clonal rearrangement of TCR-γ. Tests for clonal rearrangement in TCR-β and TCR-δ were negative.
Based on these morphological, immunohistochemical and molecular findings, we diagnosed panniculitic T-cell lymphoma with morphological and immunohistochemical features of SPTCL.
Autopsy finding
During outer inspection we found yellowish color of entire skin and cutaneous suffusions in the abdominal and genital regions. During the inner inspection, there was a thickened, firm, whitish area approx. 12x10x10 cm in the mesocolon ascendens. Intestine and mesenteric lymph nodes were unremarkable. Spleen was enlarged (470 g) and of soft consistency without focal lesions. Liver (2100 g) had a blunt margin, soft consistency and yellow cut surface. Other organs were unremarkable on gross examination. We collected samples for histopathological examination. We specifically and thoroughly examined subcutaneous adipose tissue and looked for any signs of a tumor. We have only found superficial reddish spots that were also sampled for microscopy.
Histopathological examination of mesenterial fat tissue revealed similar findings to those described above in the biopsy: adult fat tissue with atypical hyperchromatic lymphoid cells rimming the adipocytes, with macrophages engulfing whole lymphocytes and erythrocytes (Fig. 4c). In the spleen there was marked activation of red pulp with abundant histiocytes but no signs of infiltration by lymphoma. The bone marrow had cellularity about 50%, with trilinear hematopoiesis, with admixture of macrophages with signs of hemophagocytosis of lymphocytes and red blood cells (Fig. 4a). The immunohistochemistry of the vertebral bone marrow showed focal infiltration of CD8+ lymphocytes rimming the adipocytes; the finding is highly suspect from presence of lymphoma cells in the bone marrow (Fig. 4b). Liver tissue showed marked steatosis with cholestatic features (Fig. 4d), but no signs of lymphoma (Fig. 4e). Histopathological examination of the other organs, including abdominal skin and subcutaneous fat tissue (Fig. 4f), was unremarkable.
Fig. 4.

scans of histological slides from the autopsy. A – vertebral bone marrow with large macrophages engulfing red cells and lymphocytes (hemophagocytosis). HE, 130x. B – vertebral bone marrow with focal finding of CD8+ lymphocytes rimming the adipocytes; the finding is highly suspect from infiltration by lymphoma, CD8 immunohistochemistry, 55x. C – mesentery with macrophages engulfing lymphocytes, HE, 73x. D – liver tissue showing marked dystrophic changes including steatosis and cholestasis of hepatocytes. HE, 70x. E – liver tissue without apparent lymphoma infiltration. CD3 immunohistochemistry, 12x. F – sample from skin and subcutis tissue without apparent lymphoma infiltration. HE, 2,5x
The autopsy determined the underlying disease as T-cell lymphoma with cytotoxic phenotype, with morphological and immunohistochemical features of SPTCL, associated with signs of hemophagocytic syndrome (clinical findings of fever, bicytopenia and hypofibrinogenemia, autoptic finding of enlarged liver and spleen and histiocytic hemophagocytosis of lymphocytes and red blood cells), probably leading to patients death due to multiorgan failure, in line with reported clinical features.
Discussion
Subcutaneous panniculitis-like T cell lymphoma (SPTCL) was first described by Gonzalez et al. in 1991 [9]. In the largest reported cohort study of SPTCL performed by Willemze et al., 83 cases had been reviewed. None of these had evidence of lymphoma outside the subcutaneous tissue [3], but there were few case reports about mesenterial involvement and simultaneous subcutaneous involvement [5, 6]. To the best of our knowledge, a case of panniculitic T-cell lymphoma with morphological and immunohistochemical features of SPTCL constituting mesenterial mass without involvement of subcutaneous fat, was not yet reported in medical literature.
The histopathological differential diagnosis of SPTCL includes lupus erythematosus panniculitis (LEP, lupus profundus), however, reports of the mesenterial localization of this are exceedingly rare [10]. Clinically, LEP and SPTCL are indistinguishable [11]. According Massone et al., the histopathological features of LEP include presence of B-cell follicles and of plasma cells within the inflammatory infiltrate, septal fat tissue involvement with fibrosis and occasionally presence of eosinophils. In contrast, the most useful features for the diagnosis of SPTCL are the presence of hyperchromatic CD8+ T-lymphocytes and the absence of septal fibrosis, B-cell follicles and plasma cells. Cytotoxic CD8+ T-lymphocytes may be observed in cases of LEP but never constitute the majority of the infiltrate as in SPTCL [11]. In the series of the same authors, all cases of LEP revealed a polyclonal pattern of the TCR-γ gene rearrangement [11]. The PCR analysis may be a feature helpful in the differentiation of LEP from SPTCL. In our case, absence of other lupus signs (i.e. cutaneous involvement), histopathological and molecular findings described above lead us to be confident in our diagnosis of T-cell lymphoma.
Contemporary differential diagnosis of lymphomas is vitally dependent on molecular features. In terms of the present World Health Organization (WHO)-defined categories, our case would best fit into SPTCL because of histology and the presence of major alpha-beta T-cell phenotype (TCRβF1+, CD8+, granzyme B+, perforin +, TIA1+, CD56-, TCR-γ-). However, SPTCL should primarily affect the skin, should have a benign clinical course and unlike gamma-delta phenotypes, only < 20% is associated with hemophagocytic syndrome [3, 12]. Indeed, it can never be ruled out that a subcutaneous lesion was present and missed during autopsy. We consider this very unlikely, because the pathologist performing the autopsy was aware of the biopsy finding and subcutaneous lesions were actively sought for and any suspicious nodules were excised and thoroughly microscopically examined.
According the WHO classification, the term SPTCL is reserved for α/β T-cell phenotype (but not TCRα/β-rearrangement) lymphomas containing CD8+, granzyme B+, perforin+, TIA1+, CD4- and CD56- cells, which is limited to subcutaneous tissue (no dermal and/or epidermal involvement) and bears relatively good prognosis due to a good response to conservative immunosuppressive regimens. SPTCL is distinct from primary cutaneous γ/δ T-lymphomas, which are typically CD4-, CD8-, CD56+, granzyme B+, perforin+, TIA1+, may involve the epidermis and/or dermis [13, 14], may present with panniculitic pattern [15] and invariably have a very poor prognosis [1]. Studies [1, 3, 4, 14–21] showed that TCR α/β lymphomas represent often an indolent disease, whilst γ/δ phenotype harbors a poor prognosis. In the study by Toro et al., median survival was 15 vs. 166 months; 5-years survival 10% vs. 80% in α/β vs. γ/δ T-cell phenotype, respectively [21]. Similar 5-years survival rates were reported by Willemze et al.: 11% vs. 82% [3].
The γ/δ-TCR-rearranged lymphomas are more frequently associated with hemophagocytic syndrome [3], this association was first described by Avionach et al. in 1994 [22]. The hemophagocytic syndrome (hemophagocytic lymphohistiocytosis, HLH) represents a severe hyperinflammatory disease with prolonged fever, cytopenias, hepatosplenomegaly, and hemophagocytosis by activated non-neoplastic macrophages [23]. The diagnosis of HLH is based on presence of at least five of these eight signs: fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, low/absent natural-killer-cell-activity, hyperferritinemia, and high-soluble interleukin-2-receptor levels [24]. Our patient had fever, hepatosplenomegaly, bicytopenia, hypofibrinogenemia and histopathological finding of hemophagocytosis. Plasma triglycerides, ferritin and high-soluble interleukin-2-receptor were not measured.
Frequent non-neoplastic triggers of HLH are infectious agents, mostly viruses of the herpes group, or rheumatic diseases [23]. There are multiple reports of HLH associated with subcutaneous T-cell lymphomas [8, 9, 25–27]. Concerning tumors triggering HLH, the most common are hematological neoplasms (93%), more frequently T-cell than B-cell lymphomas or leukemias, and only rarely solid tumors [28–30]. The pathogenesis of HLH is related to deranged immune response. Dysfunctional cytotoxic CD8+ T lymphocytes (CTLs) and NK cells are unable to initiate appropriate response against malignant or infected cells. Histiocytes proliferate, produce storm of cytokines, invade liver, spleen and lymph nodes, and engulf blood cells and platelets [31]. The immune system is unable to control the hyperinflammatory response, which often leads to multiorgan failure and death.
Conclusion
In conclusion, we described a patient who died of a hemophagocytic syndrome accompanying a mesenteric tumor with morphologic and molecular features of SPTCL without clinical or morphological involvement of subcutaneous tissue.
Acknowledgements
This work was supported by the Charles University research program PROGRES Q 28 (Oncology) and Q37. The fund covers all institutions publication related with oncology. In our article, the fund covered some of used antibodies in immunohistochemistry and publication charges.
Abbreviations
- CD
Cluster of differentiation
- CT
Computer tomography
- CTLs
Cytotoxic T-lymphocytes
- DNA
Deoxyribonucleic acid
- FFPE
Formalin-fixed paraffin-embedded
- HLH
Hemophagocytic lymphohistiocytosis
- INF-γ
Interferon-γ
- LEP
Lupus erythematosus panniculitis
- PCR
Polymerase chain reaction
- Ref.
Reference range
- SPTCL
Sub cutaneous panniculitis-like T-cell lymphoma
- TCR
T-cell receptor
- TIA1
T-cell intracellular antigen 1
Authors’ contributions
“JH analyzed and interpreted the patient’s data, performed the histological examination, took the pictures and was a major contributor in writing the manuscript. VE performed the histological examination and stated a presented diagnosis. JH performed the autopsy and wrote the autopsy description. ZP performed and interpreted some of the immunohistochemistry, analyzed and interpreted the histological examination (2nd opinion), performed and interpreted the molecular examination and was a contributor in writing the manuscript. AR performed and interpreted some of the immunohistochemistry, analyzed and interpreted the histological examination (3rd opinion). FD analyzed and interpreted the patient’s clinical data and contributed to writing the manuscript. All authors reviewed and approved the final version of the manuscript. “.
Availability of data and materials
It is not possible to share research data publicly.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jan Hrudka, Phone: +420 724 579 183, Email: jan.hrudka@lf3.cuni.cz, Email: jan.hrudka@fnkv.cz, Email: jan.hrudka@gmail.com.
Václav Eis, vaclav.eis@email.cz.
Josef Heřman, Email: josef.herman@fnkv.cz.
Zuzana Prouzová, Email: zuzana.prouzova2@fnmotol.cz.
Andreas Rosenwald, Email: rosenwald@uni-wuerzburg.de.
František Duška, Email: frantisek.duska@lf3.cuni.cz.
References
- 1.Jaffe ES, Gaulard P, Cerroni L. Subcutaneous panniculitis-like T-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th edition) Lyon: IARC; 2017. pp. 383–385. [Google Scholar]
- 2.Kumar S, Krenacs L, Medeiros J, Elenitoba-Johnson KS, Greiner TC, Sorbara L, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol. 1998;29(4):397–403. doi: 10.1016/S0046-8177(98)90122-8. [DOI] [PubMed] [Google Scholar]
- 3.Willemze R, Jansen PM, Cerroni L, Berti E, Santucci M, Assaf C, et al. Subcutaneous panniculitis-like T-cell lymphoma: definition, classification, and prognostic factors: an EORTC cutaneous lymphoma group study of 83 cases. Blood. 2008;111(2):838–845. doi: 10.1182/blood-2007-04-087288. [DOI] [PubMed] [Google Scholar]
- 4.Salhany KE, Macon WR, Choi JK, Elenitsas R, Lessin SR, Felgar RE, et al. Subcutaneous panniculitis-like T-cell lymphoma: clinicopathologic, immunophenotypic, and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol. 1998;22(7):881–893. doi: 10.1097/00000478-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Lester L, Ewalt M, Warnke R, Kim J. Systemic panniculitis-like T-cell lymphoma with involvement of mesenteric fat and subcutis. J Cutan Pathol. 2015;42(1):46–49. doi: 10.1111/cup.12436. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Pardee TS, Beaty MW. Subcutaneous panniculitis-like T cell lymphoma with mesenteric involvement. J Hematop. 2013;6(3):155–159. doi: 10.1007/s12308-012-0167-3. [DOI] [Google Scholar]
- 7.Magro CM, Crowson AN, Kovatich AJ, Burns F. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235–247. doi: 10.1034/j.1600-0560.2001.028005235.x. [DOI] [PubMed] [Google Scholar]
- 8.Van Dongen JJ, Langerak AW, Brüggermann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez CL, Medeiros LJ, Braziel RM, Jaffe ES. T-cell lymphoma involving subcutaneous tissue. A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991;15:17–27. doi: 10.1097/00000478-199101000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Dor AM, Kohler JL, Aubrespy P, Scheiner C, Piyyi M, Lebreuil G. Pseudo-tumorous panniculitis of the mesentery. An unusual initial stage of acute lupus erythematosus in a 10-year-old girl. Sem Hop. 1982;58(48):2847–2850. [PubMed] [Google Scholar]
- 11.Massone C, Kodama K, Salmhofer W, Abe R, Shimizu H, Parodi A, Kerl H, Cerroni L. Lupus erythematosus panniculitis (lupus profundus): clinical, histopathological, and molecular analysis of nine cases. J Cutan Pathol. 2005;32(6):396–404. doi: 10.1111/j.0303-6987.2005.00351.x. [DOI] [PubMed] [Google Scholar]
- 12.Oschlies I, Simonitsch-Klupp I, Maldyk J, Konovalov D, Abramov D, Myakova N, et al. Subcutaneous panniculitis-like T-cell lymphoma in children: a detailed clinicopathological description of 11 multifocal cases with a high frequency of haemophagocytic syndrome. Br J Dermatol. 2015;172(3):793–797. doi: 10.1111/bjd.13440. [DOI] [PubMed] [Google Scholar]
- 13.Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 14.Gaulard P, Berti E, Willemze R, Petrella T, Jaffe ES. Primary cutaneous gamma delta T-cell lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoetic and Lymphoid Tissues (Revised 4th edition) Lyon: IARC; 2017. pp. 397–399. [Google Scholar]
- 15.Willemze R. Cutaneous lymphomas with a panniculitic presentation. Semin Diagn Pathol. 2017;34(1):36–43. doi: 10.1053/j.semdp.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Guizzardi M, Hendrickx IAG, Mancini LL, Monti M. Cytotoxic γ/δ subcutaneous panniculitis-like T-cell lymphoma: report of a case with pulmonary involvement unresponsive to therapy. J Eur Acad Dermatol Venerol. 2003;17(2):219–222. doi: 10.1046/j.1468-3083.2003.00600.x. [DOI] [PubMed] [Google Scholar]
- 17.Hoque SR, Child FJ, Whittaker SJ, Ferreira S, Orchard G, Jenner K, et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol. 2003;148(3):516–525. doi: 10.1046/j.1365-2133.2003.05226.x. [DOI] [PubMed] [Google Scholar]
- 18.Massone C, Chott A, Metze D, Kerl K, Citarella L, Vale E, et al. Subcutaneous, Blastic natural killer (NK), NK/T-cell, and other cytotoxic lymphomas of the skin: a morphologic, immunophenotypic, and molecular study of 50 patients. Am J Surg Pathol. 2004;28(6):719–735. doi: 10.1097/01.pas.0000126719.71954.4f. [DOI] [PubMed] [Google Scholar]
- 19.Massone C, Lozzi GP, Egberts F, Fink-Puches R, Cota C, Kerl H, Cerroni L. The protean spectrum of non-Hodgkin lymphomas with prominent involvement of subcutaneous fat. J Cutan Pathol. 2006;33(6):418–425. doi: 10.1111/j.0303-6987.2006.00493.x. [DOI] [PubMed] [Google Scholar]
- 20.Toro JR, Beaty M, Sorbara L, Turner ML, White J, Kingma DW, et al. Gamma delta T-cell lymphoma of the skin: a clinical, microscopic, and molecular study. Arch Dermatol. 2000;136(8):1024–1032. doi: 10.1001/archderm.136.8.1024. [DOI] [PubMed] [Google Scholar]
- 21.Toro JR, Liewehr DJ, Pabby N, Sorbara L, Raffeld M, Steinberg SM, Jaffe ES. Gamma-delta-T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407–3412. doi: 10.1182/blood-2002-05-1597. [DOI] [PubMed] [Google Scholar]
- 22.Avinoach I, Halevy S, Argov S, Sacks M. Gamma/delta T-cell lymphoma involving the subcutaneous tissue and associated with haemophagocytic syndrome. Am J Dermatopathol. 1994;16(4):426–433. doi: 10.1097/00000372-199408000-00014. [DOI] [PubMed] [Google Scholar]
- 23.Janka GE. Haemophagocytic syndromes. Blood Rev. 2007;21(5):245–253. doi: 10.1016/j.blre.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 25.Kao GF, Resh B, McMahon C, Gojo I, Sun CC, Phillips D, Zhao XF. Fatal subcutaneous panniculitis-like T-cell lymphoma gamma/delta subtype (cutaneous gamma/delta T-cell lymphoma): report of a case and review of the literature. Am J Dermatopathol. 2008;30(6):593–599. doi: 10.1097/DAD.0b013e318182c7bf. [DOI] [PubMed] [Google Scholar]
- 26.Rudolph N, Klemke CD, Ziemer M, Simon JC, Treudler R. Haemophagocytic lymphohistiocytosis associated with subcutaneous panniculitis-like T-cell lymphoma. J Dtsch Dermatol Ges. 2016;14(11):1140–1142. doi: 10.1111/ddg.12951. [DOI] [PubMed] [Google Scholar]
- 27. Wang H, Xiong L, Tang W, Zhou Y, Li F. A systematic review of malignancy-associated haemophagocytic lymphohistiocytosis that needs more attentions. Oncotarget. 2017;8(35):59977–85. [DOI] [PMC free article] [PubMed]
- 28.Rivière Sébastien, Galicier Lionel, Coppo Paul, Marzac Christophe, Aumont Cedric, Lambotte Olivier, Fardet Laurence. Reactive Hemophagocytic Syndrome in Adults: A Retrospective Analysis of 162 Patients. The American Journal of Medicine. 2014;127(11):1118–1125. doi: 10.1016/j.amjmed.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 29.Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–1516. doi: 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 30.Lee DE, Martinez-Escala ME, Serrano LM, Zhou XA, Kaplan JB, Pro B, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154(7):828–831. doi: 10.1001/jamadermatol.2018.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arico M, Danesino C, Pende D, Moretta L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol. 2001;114:761–769. doi: 10.1046/j.1365-2141.2001.02936.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
It is not possible to share research data publicly.