

Abstract
The accumulation of misfolded proteins is a common pathological feature of many neurodegenerative disorders, including synucleinopathies such as Parkinson's disease (PD), which is characterized by the presence of α-synuclein (α-syn)-containing Lewy bodies. However, although recent studies have investigated α-syn accumulation and propagation in neurons, the molecular mechanisms underlying α-syn transmission have been largely unexplored. Here, we examined a monogenic form of synucleinopathy caused by loss-of-function mutations in lysosomal ATP13A2/PARK9. These studies revealed that lysosomal exocytosis regulates intracellular levels of α-syn in human neurons. Loss of PARK9 function in patient-derived dopaminergic neurons disrupted lysosomal Ca2+ homeostasis, reduced lysosomal Ca2+ storage, increased cytosolic Ca2+, and impaired lysosomal exocytosis. Importantly, this dysfunction in lysosomal exocytosis impaired α-syn secretion from both axons and soma, promoting α-syn accumulation. However, activation of the lysosomal Ca2+ channel transient receptor potential mucolipin 1 (TRPML1) was sufficient to upregulate lysosomal exocytosis, rescue defective α-syn secretion, and prevent α-syn accumulation. Together, these results suggest that intracellular α-syn levels are regulated by lysosomal exocytosis in human dopaminergic neurons and may represent a potential therapeutic target for PD and other synucleinopathies.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is the second most common neurodegenerative disease linked to the accumulation of α-synuclein (α-syn) in patient neurons. However, it is unclear what the mechanism might be. Here, we demonstrate a novel role for lysosomal exocytosis in clearing intracellular α-syn and show that impairment of this pathway by mutations in the PD-linked gene ATP13A2/PARK9 contributes to α-syn accumulation in human dopaminergic neurons. Importantly, upregulating lysosomal exocytosis by increasing lysosomal Ca2+ levels was sufficient to rescue defective α-syn secretion and accumulation in patient neurons. These studies identify lysosomal exocytosis as a potential therapeutic target in diseases characterized by the accumulation of α-syn, including PD.
Keywords: alpha synuclein, dopaminergic neuron, Kufor–Rakeb syndrome, lysosomal exocytosis, Parkinson's disease, TRPML1
Introduction
Parkinson's disease (PD) is pathologically characterized by the deposition of Lewy bodies and neurites composed of α-synuclein (α-syn), a presynaptic protein implicated in the pathogenesis of sporadic and familial PD (Wong and Krainc, 2017). Increased α-syn expression is toxic to neurons, as duplication of the α-syn SNCA locus causes late-onset PD, whereas triplication leads to early-onset PD, suggesting that α-syn-mediated neurotoxicity is dose dependent (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004). Recent genetic studies have also implicated lysosomal dysfunction as a key player in PD pathogenesis, as several genes encoding lysosomal proteins have been linked to familial forms of PD (McDonald and Krainc, 2017), including ATP13A2/PARK9, whose loss-of-function mutations result in Kufor–Rakeb syndrome (KRS), a rare, hereditary neurodegenerative disorder (Ramirez et al., 2006).
PARK9 encodes a lysosomal type 5 P-type ATPase involved in cation homeostasis (Gitler et al., 2009; Kong et al., 2014; Park et al., 2014; Tsunemi et al., 2014), the loss of function of which leads to lysosomal dysfunction (Usenovic et al., 2012; Dehay et al., 2012; Lopes da Fonseca et al., 2016; Bento et al., 2016). ATP13A2/PARK9 also localizes to multivesicular bodies and contributes to the formation of intraluminal vesicles, and may regulate sorting and trafficking of cargos through inositol phosphate(3,5)P2 binding to the N terminus of PARK9 (Holemans et al., 2015; Demirsoy et al., 2017). Loss of PARK9 function in patient fibroblasts leads to α-syn accumulation (Tsunemi et al., 2014), but whether α-syn also accumulates in patient dopaminergic neurons and if the molecular machinery contributing to α-syn accumulation is amenable to therapeutic interventions remain unknown. Importantly, identifying the cellular pathways and targets that regulate intracellular α-syn levels in human neurons will provide relevant therapeutic strategies for combatting α-syn-mediated neurotoxicity in multiple forms of synucleinopathies.
Using iPSC-derived dopaminergic neurons from KRS (PARK9) patients expressing mutant PARK9, we found that PARK9 plays a critical role in regulating lysosomal exocytosis. Patient neurons exhibit decreased secretion of α-syn from both the axon and the cell body, as well as disrupted lysosomal Ca2+ homeostasis, leading to defective lysosomal exocytosis. Surprisingly, enhancing lysosomal exocytosis using drugs targeting the lysosomal Ca2+ channel transient receptor potential mucolipin 1 (TRPML1) increased lysosomal exocytosis and α-syn secretion and attenuated α-syn intracellular accumulation in patient neurons. Thus, upregulation of neuronal secretion may be a potential key target for developing viable therapies for KRS and other related synucleinopathies such as PD.
Materials and Methods
Cell culture.
Human neuroglioma cell line (H4)-expressing wild-type α-syn under the control of a tetracycline-inducible promoter and primary dermal fibroblasts taken from four normal and two PARK9 patients (1550 C>T; MUT1 and 3176 T>G, 3253 delC; MUT2) were cultured as described previously (Dehay et al., 2012; Grünewald et al., 2012; Usenovic et al., 2012; Tsunemi and Krainc, 2014; Tsunemi et al., 2014; Mazzulli et al., 2016b). Mut1 carries homozygous missense mutations in ATP13A2 alleles (c. 1550C>T/c. 1550C>T) that result in homozygous amino acid substitutions in ATP13A2 proteins (p.T517I/p.T517I). Mut2 carries compound heterozygous mutations: one is a missense mutation that results in an amino acid substitution (c. 3176 T>G, p p.L1059R) and the other is a single nucleotide deletion (3253 delC) that results in many amino acid alternations with an appearance of a premature stop codon. All human induced pluripotent stem cells (iPSCs) were reprogrammed as described previously (Mazzulli et al., 2016a). Briefly, the four distinct factors (OCT4, SOX2, cMYC, and KLF4) were transfected into human skin fibroblasts using retroviral system. Three control iPSCs (Cont 1, Cont 2, and Cont 3) and one mutant (Mut 2) iPSC were characterized previously (Cooper et al., 2012; Mazzulli et al., 2016b). One control (Cont 4) and one mutant (Mut 1) iPSC were characterized for spontaneous differentiation analysis and the expression of pluripotency markers (Fig. 1-1). All iPSCs were cultured on irradiated mouse embryonic fibroblasts in iPSC medium containing DMEM/F12 (STEMCELL Technologies) with 20% knock-out serum replacement (Invitrogen), l-glutamine, nonessential amino acids, 2-mercaptoethanol (Invitrogen), 10 ng/ml FGF-basic (AA1–155) recombinant human protein (Invitrogen), and penicillin/streptomycin at 37°C in 5% CO2. Differentiation toward dopaminergic neurons was conducted following the protocol described previously (Mazzulli et al., 2016a). At 40 d after the initiation of differentiation, we infected lentiviruses depending on the experiments. Immunocytochemical analysis revealed neuralization efficiency in dopaminergic (DA) neurons using neuron-specific β-iii-tubulin and midbrain-specific markers (TH, FOXA2, and LMX1a). The proportion of neurons coexpressing TH and FOXA2 from day 40 and at day 120 was similar among all of the cell lines (Fig. 1-2).
Immunocytochemistry.
Immunocytochemical analysis was conducted as described previously (Tsunemi and Krainc, 2014; Tsunemi et al., 2014). Briefly, after fixation in 4% paraformaldehyde, the cells were permeabilized/blocked in PBS containing 0.1% saponin, 1% BSA, and 5% normal goat serum for 20 min. Specimens were then incubated with primary antibodies overnight, washed in PBS, and then incubated with Alexa Fluor conjugated anti-rabbit or anti-mouse antibodies at 1:400 dilution for 1 h. Confocal imaging was conducted on the Leica TCS SPE confocal system with a Leica DMI 4000B CSQ inverted microscope equipped with an ACS APO 63× [1.3 numerical aperture (NA)] oil-immersion objective. For quantification analysis, 10,000 cells/well were plated in triplication and fluorescence intensity was measured using SpectraMax i3 multimode microplate reader (Molecular Devices). Epifluorescence imaging was performed on a Leica DMI3000 B inverted microscope. Live-cell imaging was conducted on the Zeiss LSM 780 confocal microscope system with the Zeiss AxioObserver. Z1 inverted microscope equipped with an α Plan-Apochromat 100×/1,46 oil differential interference contrast M27 objective. Cells were maintained at 37 C° and 5% CO2 on the temperature-controlled heating stage in a CO2-controlled incubator.
Electrophysiological recordings.
Spontaneous pacemaking activity was recorded on iPSC-derived DA neurons from healthy controls and PARK9 mutant KRS patients (75–85 days after differentiation). Cultures were transferred to a recording chamber on a fixed-stage inverted microscope (Diaphot 200; Nikon). Interleaved recordings from controls and PARK9 patients were performed at 32°C. The recording chamber was perfused (1–2 ml/min) with HEPES-based solution containing the following (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, and 10 sucrose, pH 7.4, osmolarity 300–305 mOsm/L. Blockers of excitatory and inhibitory transmission were included. Patch pipettes (3–5 MΩ) were filled with internal solution containing the following (in mm): 135 K-MeSO4, 5 KCl, 5 HEPES, 0.05 EGTA, 10 phosphocreatine-di(tris), 2 ATP-Mg, and 0.5 GTP-Na, pH 7.25–7.30, osmolarity 285–295 mOsm/L. Recording patch pipettes were prepared with a horizontal puller (model P-97; Sutter Instruments) using borosilicate glass with filament (outer diameter 1.5 mm, inner diameter 0.86 mm). Somatic cell-attached voltage-clamp recordings were obtained with a Multi-Clamp 700B amplifier (Molecular Devices) interfaced to a Pentium-based PC running pClamp 10.6 (Molecular Devices). The signals were filtered at 1 kHz and digitized at 10 kHz with a Digidata 1440A (Molecular Devices). Analyses of instantaneous firing frequency were done in Clampfit version 10.6 (Molecular Devices).
Ca2+ imaging.
Ca2+ imaging was conducted as described previously (Dryanovski et al., 2013). After staining with 1 μm Fura2-AM solution for 30 min, neurons on coverslips were washed once and placed on the imaging chamber mounted on the inverted epifluorescence microscope (IX71; Olympus) with xenon illumination. Neurons were imaged using a CCD camera (I-PentaxMax; Princeton Instruments) operated by a Pentium-based PC running MetaFluor imaging software (Molecular Devices). The imaging chamber was superfused with HEPES-buffered ACSF as the flow rate of 2–3 ml/min. Experiments were conducted at room temperature with a 40×/1.35 NA oil-immersion objective (Olympus). Regions of interests (ROIs) were chosen in the soma and at various distances. The two excitation filters (340 and 380 nm) were mounted on a Lambda 10–2 filter wheel (Sutter Instruments), which allowed for rapid and accurate switch between the two wavelengths. The emission was monitored at 520 nm. Ratiometric images (F340/F380) were taken every 3 s with exposure time of 200 ms. For measuring lysosomal Ca2+ concentration, rhodamine dextran, potassium salt, 10,000 MW, anionic (high-affinity version) and dextran, cascade blue, 10,000 MW were used at the concentrations of 0.25 μg/μl and 0.1 μg/μl, respectively. Cells were incubated for 1 h at 37°C before washing with Ringer's solution containing the following (in mm): 116 NaCl, 2.9 KCl, 1.8 CaCl2, and 5.6 HEPES, pH 7.2.
Cell surface staining and biotinylation assay.
Cell surface staining was conducted as described previously (Samie et al., 2013). Briefly, after PARK9 expression levels were modulated, H4 cells were treated with 200 nm Baf1 for 2 h and incubated with the lysosome-associated membrane protein (LAMP-1) luminal domain antibody (AF 4800; R&D Systems) for 1 h on ice. Cell surface biotinylated proteins were collected as described previously (Tarradas et al., 2013). After PARK9 levels were modulated by transfecting PARK9 shRNA, Scrb shRNA, plasmids containing PARK9 cDNA or empty vectors for 24 h, H4 cells cultured in 6-well dishes were treated with 200 nm Baf1 for the indicated period of time. After washing with ice-cold PBS, cells were incubated in PBS with 300 μm EZ-Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) for 30 min at 4°C. After biotinylated proteins were lysed in RIPA buffer, 10% of cell lysates were put aside for the INPUT samples and the remaining lysates were incubated with NeutrAvidin Agarose (Thermo Fisher Scientific) for 1 h. The agarose beads were washed with PBS and bound proteins were eluted by heating in gel-loading buffer. LAMP-1 levels were analyzed by immunoblotting.
Exosome isolation and nanoparticle tracking analysis.
Exosomes were purified as described previously (Tsunemi et al., 2014). Briefly, exosomes were collected from cell-conditioned media using a basic differential centrifugation method (200 × g for 5 min, 1200 × g for 10 min, and 16,500 × g for 30 min), followed by ultracentrifugation at 110,000 × g for 60 min. After washing in PBS, exosomes were collected by a centrifugation at 110,000 × g for 60 min. Analysis of extracellular vesicles was conducted by the NanoSight LM10 system, configured with a 405 nm laser and a high-sensitivity digital camera system (OrcaFlash2.8, Hamamatsu, and C11440, NanoSight, respectively). Samples were administered and recorded for 1 min under sustained flow controlled by script control system equipped with the NanoSight syringe pump. Videos were analyzed with NTA software version 2.3.
Lysosomal proteolysis in live neurons and lysosomal enzyme activity assays.
Long-lived protein degradation assays were performed by radioactive pulse-chase using tritium-labeled leucine (PerkinElmer, NET460A001MC) as described previously (Kaushik and Cuervo, 2009). Enzyme activity assays were performed using the artificial enzyme substrates 4MU-glucopyranoside (for GCase) and 4MU-sulfate potassium salt (for a-i-2-sulf) as described previously. The β-glucocerebrosidase activities were measured as described previously (Mazzulli et al., 2011). The activities of β-hexosaminidase and acid phosphatase were measured following the manufacture's protocols (Sigma-Aldrich).
α-syn detection.
The α-syn ELISA was conducted as described previously (Tsunemi et al., 2014). α-syn oligomers/fibrils were formed as described previously (Mazzulli et al., 2011). Briefly, after α-syn monomers were incubated at 37°C for 10 d under continuous agitation at 1000 rpm, α-syn oligomers/fibrils were centrifuged at 10,000 × g for 30 min. The pellets were resuspended in PBS and fibril formation was assessed by thioflavin T spectroscopic assay and electron microscopic analysis. Alexa Fluor succinimidyl ester (NHS ester) 555 was conjugated to sonicated α-syn oligomers/fibrils following the manufacturer's instruction (Thermo Fisher Scientific). Briefly, 20 μl of 1 m sodium bicarbonate was added to 200 μl of α-syn oligomers/fibrils (1 mg/ml). Alexa Fluor was mixed to the solution as the degree of labeling (Bliederhaeuser et al., 2016) became three. The reaction mixture was continued at room temperature for 15 min and stopped by adding 22 μl of 1 m Tris, pH 7.4, to the solution. Purification of the proteins from unconjugated dye was performed by Slide-A-Lyzer Dialysis Cassettes (2 K WMCO, 0.5 ml, Thermo Fisher Scientific). Alexa Fluor 555 conjugation was confirmed by mass spectrometry. For the α-syn-Alexa Fluor 555 axonal transport assay, DA neurons were plated on the left chambers in microfluidic devices at day 24 from the start of differentiation (SND450; Xona Microfluidics). At day 60, α-syn-Alexa Fluor 555 was added on the left chambers at a concentration of 1 μm. For live-cell imaging, neurons were prestained with SP-DiOC18(3) (3,3′-dioctadecyl-5,5′-di(4-sulfophenyl)oxacarbocyanine, sodium salt). α-syn fibrils were analyzed by the Zeiss LSM 780 confocal microscope system. After 16 h, media in each chamber were replaced with fresh media. The fluorescence intensities were measured from the media by the microplate reader (Mithwas2 LB 943; Berthold Technologies).
Plasmids.
GCamp3-ML1 was a kind gift from Haoxing Xu. The human ATP13A2 lentivirus was provided by Christopher Rochet. Lentiviruses carrying short hairpin plasmid RNA (shRNA) targeting human SNCA were from Open Biosystems (Dharmacon).
Western blotting.
Immunoblotting was conducted as described previously (Tsunemi and Krainc, 2014; Tsunemi et al., 2014). The antibodies used were anti human LAMP-1 luminal domain (R&D Systems), anti-human LAMP 1 (Santa Cruz Biotechnology), anti-human β-iii tubulin (Covance), human vimentin (BD Biosciences), anti-human GAPDH (Millipore), human ALIX (Santa Cruz Biotechnology), anti-human α synuclein C-20 (Santa Cruz Biotechnology), anti-human Huntingtin (Millipore), anti-human TDP43 (12892–1-AP; Proteintech), anti-human tau (Dako), anti-human TH (Millipore), anti-human Tsg101 (GeneTex), and anti-human CD63 (Developmental Studies Hybridoma Bank).
Mitochondrial respiration analysis.
To measure mitochondrial respiration, we used the Seahorse XF24 extracellular flux analyzer as described previously with minor modifications (Grünewald et al., 2012). We plated 10,000 iPSCs per well at day 24 from the start of differentiation and added 1 μm oligomycin, 1.5 μm carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 1 μm antimycin A for each time point. After analysis, remaining cells were harvested to measure protein levels, which were used for normalizing oxygen consumption rates.
Statistical analysis.
All data were prepared for analysis with standard spreadsheet software (Microsoft Excel). Statistical analysis was performed by one-way ANOVA post hoc Tukey's test or Student's t test. All error bars indicate SEM in the figures.
Results
PARK9 patient DA neurons develop pathogenic phenotypes including lysosomal dysfunction and time-dependent α-syn accumulation
To investigate PARK9 function in neurons, we generated iPSC-derived DA neurons from healthy controls and PARK9 mutant KRS patients (Kriks et al., 2011; Mazzulli et al., 2016a) that were positive for iPSC (Fig. 1-1) and DA neuronal markers (Fig. 1-2). Using time-dependent analysis of pathogenic phenotypes, we found that, by early time points in culture (day 40 after differentiation), patient neurons already demonstrated defective exosome secretion (Fig. 1A) and lysosomal proteolysis (Fig. 1B). Moreover, lysosomal enzyme activity in the medium indicative of impaired lysosomal exocytosis was also significantly decreased in patient neurons (Fig. 1C) (Medina et al., 2011).
Figure 1.

Differentiated from induced pluripotent stem cells (Figure 1-1), PARK9 patient DA neurons (Figure 1-2) develop pathogenic phenotypes including lysosomal dysfunction and time-dependent α-syn accumulation (Figure 1-3). A, Number of exosomes secreted from four control and two PARK9 mutant DA neurons were analyzed at day 40 after the initiation of differentiation. The number of exosomes was normalized by total protein in cell lysates (n = 3, *p = 0.0001). B, Lysosomal proteolysis measured by radioactive pulse chase in four control and two PARK9 mutant DA neurons at day 40 after the initiation of differentiation (n = 3, *p = 0.0001). C, β-glucocerebrosidase (GCase) activity in the secreted media taken from four control and two PARK9 mutant DA neurons at day 40 after the initiation of differentiation. The activity in the media was normalized by the activity in cells and is shown as a percentage of control 1 (n = 3, *p = 0.0001). D, ELISA to quantify α-syn in the media taken from four control and two PARK9 mutant DA neurons at day 40 after the initiation of differentiation (n = 3, *p = 0.0001). E, α-syn ELISA for detecting α-syn proteins in exosomes taken from 4 control and 2 PARK9 mutant DA neurons at day 40 after the initiation of differentiation (n = 3, *p = 0.0001). F, Sequential pathological cascade observed in long-term cultures of iPSC-derived PARK9 DA neurons from day 40 to day 120 after the initiation of differentiation (Figure 1-3). G–I, Immunoblot analysis of α-syn proteins in four control, two PARK9 mutant, and SNCA triplication DA neurons 90 d (G) and 120 d (H) after the initiation of differentiation. After normalization to β-iii-tubulin, the relative α-syn levels are shown as fold changes compared with control 1 (n = 3, *p = 0.0087, **p = 0.0001, ***p = 0.004). Statistical analysis was conducted using one-way ANOVA with Tukey's post hoc test unless otherwise stated.
Characterization of iPSCs (A) Immunocytochemistry for pluripotency markers SSEA4, TRA-1-60, TRA-1-81, OCT4 and NANOG. (B) Images of chromosome G-banding analysis of control (Cont 4) and mutant (Mut 1) iPSCs. (C) Expression of three germ layer-specific markers after spontaneous differentiation. Markers of ectodermal progenitor cells (β-iii-tubulin), endodermal lineage cells (α-fetoprotein) and mesodermal lineage cells (SMA) were used. (D) Direct sequence results of two PARK9 mutant iPSC lines. (E) Gene copy analysis for SNCA in Cont 1, Cont 2, Triplication and Mut 1. Scale bars represent 50 μm for Figure A and 50 μm for Figure C. Download Figure 1-1, TIF file (24.9MB, tif)
Characterization of human iPSC-derived DA neurons (A) DA neurons analyzed by immunocytochemistry using FOXA2 (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage of cells expressing both FOXA2 and TH during differentiation and maturation (n = 30-50). (B) DA neurons analyzed by immunocytochemistry using Lmx1a (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage cells expressing both LMX1a and TH during differentiation and maturation (n = 30-50). (C) DA neurons analyzed by immunocytochemistry using β-iii-tubulin (β-iii-tub) (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage cells expressing both β-iii-tubulin and TH during differentiation and maturation (n = 30-50). Scale bars represent 100 μm. Download Figure 1-2, TIF file (24.9MB, tif)
Pathogenic phenotypes of PARK9 patient DA neurons over time (A) The number of exosomes secreted from four control and two PARK9-mutant DA neurons were analyzed at day 60, 90, 120 after the initiation of differentiation. The number of exosomes was normalized to the total protein level from cell lysates (n = 3, *p = 0.0002, **p = 0.0276). (B) Lysosomal proteolysis measured by radioactive pulse chase in four controls and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation. Data are shown as fold changes normalized to control 1 at day 40 (n = 3, *p = 0.0001, **p = 0.0113, ***p = 0.0155). (C) Released GCase activity in the media taken from four control and two PARK9-mutant DA neurons at Day 60, 90, 120 after the initiation of differentiation. Activity in the media were normalized to the activity in cell lysates, and shown as a percentage of control 1 at day 40 (n = 3, *p = 0.0001, **p = 0.0202, ***p = 0.0250). (D) Enzyme-Linked Immunosorbent Assay (ELISA) to quantify α-syn protein levels in the media taken from four control and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation (n = 3, *p = 0.0001, **p = 0.0001). (E) α-syn ELISA for detecting α-syn protein levels in exosomes taken from four control and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation (n = 3, *p = 0.0052, **p = 0.0001, ***p = 0.0002). (F) Immunoblot analysis of α-syn proteins in four control, two PARK9-mutant and SNCA triplication DA neurons at Day 60 after the initiation of differentiation. After normalization to β-iii-tubulin, the relative α-syn levels are shown as fold changes compared with control 1 (T-x soluble, n = 3, p = 0.0603, Cont1 vs Triplication, SDS soluble, n = 3, p = 0.3726, Cont1 vs Triplication). The statistical analysis was conducted using one-way ANOVA Tukey post hoc test. Download Figure 1-3, TIF file (24.9MB, tif)
Next, we investigated whether this might disrupt α-syn processing and indeed found that patient neurons exhibited decreased secretion of α-syn by day 40 both in the media (Fig. 1D) and in exosomes (Fig. 1E). Importantly, these pathogenic phenotypes persisted until day 120 after differentiation, including defective exosome secretion, lysosomal proteolysis, lysosomal exocytosis, and α-syn secretion (Fig. 1-3). Interestingly, α-syn accumulation occurs later in the pathogenic process, potentially as a result of defective secretion and impaired lysosomal degradation (Fig. 1F). As shown in Fig. 1-3F () and Figure 1, G and H, α-syn did not accumulate intracellularly at early time points, with no observable increase in either soluble or insoluble α-syn levels at day 60 (Fig. 1-3F, ); only by day 90 did α-syn increase in the insoluble fraction (Fig. 1G) and in the soluble fraction by day 120 (Fig. 1H). Thus, PARK9 patient neurons exhibit multiple pathogenic phenotypes including defective α-syn secretion and its increased intracellular accumulation.
Defective α-syn secretion in PARK9 patient DA neurons
To further examine the mechanism of α-syn neuronal secretion, we cultured DA neurons in four-chambered microfluidic devices (Brahic et al., 2016) with top and bottom chambers connected by 450 μm microgroove barriers, allowing for neurons cultured in the top two chambers to extend their axons into the bottom two chambers (Fig. 2A). This system thus enabled us to analyze α-syn release from axons compared with the cell body. DA neurons were cultured in the top chambers 24 d after the start of differentiation (Fig. 2-1, A–C, ), with axons extending through aligned microgrooves into the bottom chambers by day 60 (Fig. 2B). To track α-syn transport in neurons, synthetic α-syn fibrils were generated and labeled with Alexa Fluor 555 (α-syn 555) (Mazzulli et al., 2011). Using a Trypan blue quenching assay, we confirmed the internalization of α-syn 555 (Fig. 2-1D).
Figure 2.
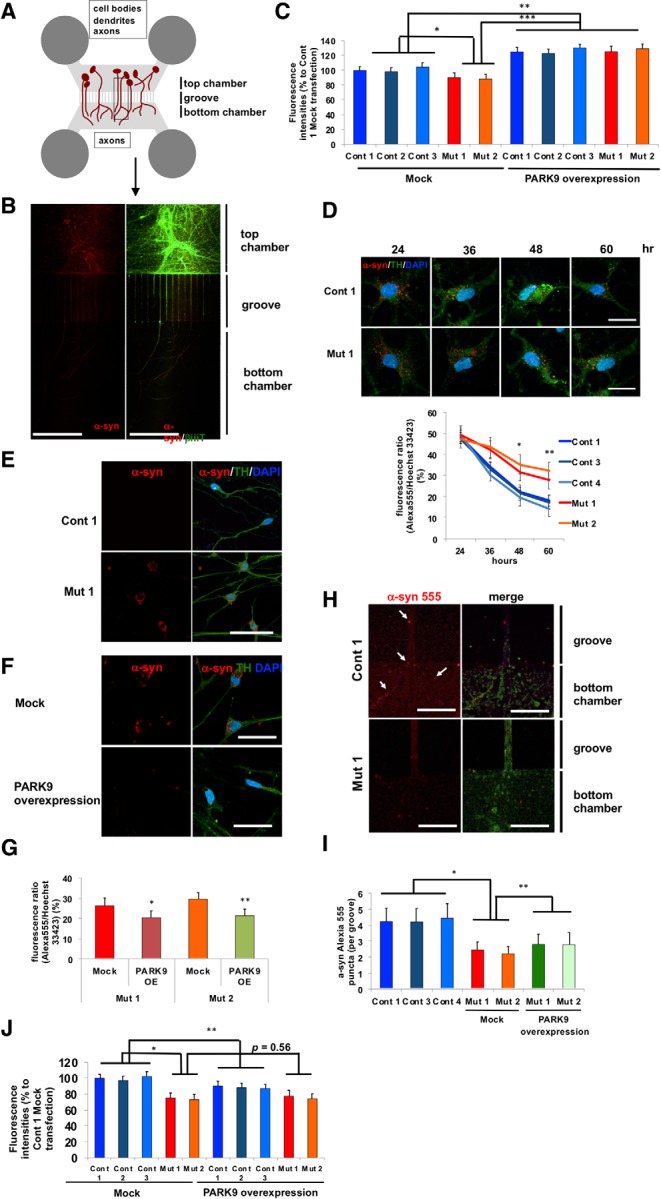
Defective α-syn secretion from both the soma and axons in PARK9 patient DA neurons (Figure 2-1). A, Schematic image of a microfluidic device in which two sets of chambers are connected through 450 μm microgroove groove. Neurons were cultured in the top chambers and extend their axons through grooves into the bottom chamber (Figure 2-1 A–C). B, Representative images of DA neurons cultured with Alexia Fluor-555-labeled α-syn fibrils in microfluidic devices (Figure 2-1 D–E). DA neurons were stained with β-iii-tubulin and visualized as α-syn fibrils conjugated with Alexa Fluor 555. A merged image is shown on the right. C, Fluorescence intensities in the media taken from the top chambers of microfluidic devices. DA neurons were infected with empty lentivirus (left) or lentivirus expressing human PARK9 (right) (Fig. 3-1) (n = 3, *p = 0.0276, **p = 0.0001, ***p = 0.0001). After culturing in media containing α-syn fibrils for 24 h, media was changed to fresh media. After 24 h, the media was collected and fluorescence intensities were analyzed. D, Fluorescence intensities of α-syn fibrils in three control and two PARK9 DA mutant neurons. After culturing in media containing α-syn fibrils for 24 h, the media was replaced with fresh media. The fluorescence intensities were measured at 24, 36, 48, and 60 h (n = 3, *p = 0.0158, **p = 0.0265). E, Representative images of DA neurons (Cont 1 and Mut 1) cultured in media containing Alexa Fluor 555-labeled α-syn fibrils for 24 h and subsequently cultured in fresh media for a week. F, Representative images of PARK9 mutant DA neurons (Mut 1) transfected with empty lentivirus (top) or PARK9 expressing lentivirus (bottom) and subsequently cultured with Alexa Fluor 555-labeled α-syn fibrils. G, Quantification of total α-syn fluorescence intensity in DA neurons (n = 3, *p = 0.0350, **p = 0.0255, Student's t test). H, Representative images of the axons of control (top) and PARK9 mutant (bottom) DA neurons in the bottom chambers of microfluidic devices after adding α-syn fibrils to the top chamber. Arrows show α-syn fibrils. I, Number of α-syn fibrils in each axon of four control and two mutant DA neurons (n = 10–20, *p = 0.0106, **p = 0.0354). J, Fluorescence intensities in the media taken from the bottom chambers of the microfluidic devices. DA neurons were infected with empty lentivirus (left) or lentivirus expressing human PARK9 (right) (n = 3, *p = 0.0016, **p = 0.0354, ***p = 0.0001). After culturing in media containing α-syn fibrils for 24 h, the media was changed to fresh media for another 24 h before the media was collected and fluorescence intensities were analyzed (n = 3, *p < 0.05). Statistical analysis was conducted using one-way ANOVA with Tukey's post hoc test unless otherwise stated. Scale bars: B, 200 μm; D–F, 50 μm; H, 10 μm.
iPSC-derived DA neurons on microfluidics (A) DA neurons analyzed by immunocytochemistry using FOXA2 (red) and tyrosine hydroxylase (TH) (green) at day 60. (B) DA neurons analyzed by immunocytochemistry using LMX1a (red) and tyrosine hydroxylase (TH) (green) at day 60. (C) DA neurons analyzed by immunocytochemistry using β-iii-tubulin (β-iii-tub) (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. (D) After incubation with α-syn 555 for 15 min, DA neurons were fixed and treated with 0.2% trypan blue to quench extracellular α-syn 555. Representative images of control (left) and PARK9-mutated DA neurons (right) in orthogonal projections of confocal z-stacks. Arrows show internalized α-syn 555. (E) The time course of α-syn 555 internalization in control and PARK9-mutated DA neurons. Scale bars represent 100 μm for Figure A to C. White scale bars represent 50 μm and black scale bars represent 7.6 μm for Cont 3 and 13.6 μm for Mut2 in Figure D. Download Figure 2-1, TIF file (24.9MB, tif)
Although the uptake of α-syn 555 was not significantly different between control and PARK9 mutant DA neurons (Fig. 2-1D,E), we found that PARK9 mutant DA neurons showed significantly decreased total α-syn secretion from the cell body (top chamber media) compared with control neurons (Fig. 2C, left). Concomitantly, decreased secretion of α-syn resulted in its gradual accumulation in the soma of PARK9 mutant DA neurons (Fig. 2D), which lasted up to 1 week after fibril addition compared with control neurons, which had cleared α-syn 555 by this time (Fig. 2E). Importantly, increasing PARK9 expression with lentiviral-mediated transduction resulted in increased secretion of α-syn (Fig. 2C, right) and its decreased accumulation in the soma of PARK9 mutant DA neurons (Fig. 2F,G). In contrast, when we examined the axons of DA neurons, we found that the number of α-syn 555 puncta in axonal terminals was significantly reduced in PARK9 mutant neurons (Fig. 2H) and could not be rescued by fully lentiviral-mediated PARK9 overexpression (Fig. 2I). α-syn secretion from the axons of patient neurons was also significantly decreased compared with control neurons (Fig. 2J), potentially due to both decreased trafficking to the axons and defective secretion. Together, these results suggest that defective α-syn secretion both from the cell body and axons contributes to its gradual intracellular accumulation in patient neurons.
PARK9 patient DA neurons exhibit dysfunctional lysosomal Ca2+ homeostasis
As secreted lysosomal enzyme activity was also decreased in PARK9 mutant neurons (Fig. 1C), we next investigated the potential mechanisms contributing to this defect. We first tested the electrophysiological properties of PARK9-deficient DA neurons by conducting cell-attached patch-clamp recordings (Fig. 3A). We found that the rate of spontaneous firing was higher in PARK9 mutant DA neurons compared with controls (Fig. 3B–D). To determine whether the PARK9 mutation affected intracellular Ca2+ levels, the ratiometric fluorescent dye Fura-2 AM was used to measure cytosolic Ca2+ concentration (Shen et al., 2012; Dryanovski et al., 2013). Basal cytosolic Ca2+ levels were significantly higher in both PARK9 mutant fibroblasts (Fig. 3E) and PARK9 mutant DA neurons (Fig. 3F) at day 120. Importantly, lentivirus-mediated PARK9 overexpression (Fig. 3-1A,B) normalized abnormal Ca2+ levels (Fig. 3E,F).
Figure 3.

PARK9 patient DA neurons exhibit dysfunctional lysosomal Ca2+ homeostasis (Figure 3-1). A–D, Spontaneous firing rate of DA neurons taken from control and PARK9 patients. A, Representative images of DA neurons from control individuals (left), PARK9 patients (right) during cell-attached patch-clamp recordings. B, Representative cell-attached recordings from control (top) and PARK9 patient-derived neurons (bottom). C, Box plots showing the distribution of spiking rates in control (n = 10) and PARK9 DA (n = 10) neurons (control median = 4.61 Hz vs PARK9 median = 7.25 Hz; *p = 0.021, unpaired t test with Welch's correction). D, Box plots showing the distribution of coefficient of variation in control (n = 10) and PARK9 DA (n = 10) neurons (control median = 6.1 vs PARK9 median = 4.2; *p = 0.1051, Mann–Whitney test). E, F, Cytosolic Ca2+ levels were measured using Fura-2 AM Ca2+ indicator. The intracellular Ca2+ concentration was measured in two control, two PARK9 mutant, and two PARK9 mutant fibroblasts that were transfected with lentivirus expressing PARK9 (Figure 3-1A) (E) and in iPSC-derived DA neurons (Figure 3-1B) (F) (n = 10, *p = 0.0001). G–I, Ca2+ release from control and PARK9 mutant fibroblasts and DA neurons. G, Change of cytosolic Ca2+ concentration by GPN treatment was monitored by Fura-2 AM fluorescence ratios at 340 nm/380 nm. H, Fura-2 AM fluorescence ratios were shown before and after GPN treatment in two controls and two PARK9 mutant fibroblasts (n = 10, *p = 0.002). I, Ca2+ release from lysosomes was decreased in PARK9 mutant DA neurons. Fura-2 AM ratios were measured before and after GPN treatment (n = 10, *p = 0.0001). J–N, Ca2+ release from lysosomes was analyzed with the lysosome targeted Ca2+ sensor GCamP3-ML1. J, Representative images of GCamp3-ML1 expressing H4 cells labeled with LAMP-1 before and after Baf1 treatment. K, GCamp3-ML1 and LysoTracker intensities were monitored during Baf1 treatment. L, Ratios of green (GCamp3-ML1) to red (LysoTracker red) fluorescence were monitored during Baf1 treatment. M, Ratios of green to red fluorescence under Baf1 treatment were analyzed in H4 cells transfected with Scrb shRNA or the shRNA against human PARK9 (n = 30–50/cells, *p = 0.0005, **p = 0.0422, ***p = 0.0303, ****p = 0.0063). N, Ratios of green to red fluorescence under Baf1 treatment were analyzed in control and PARK9 mutant fibroblasts and two PARK9 mutant fibroblasts transfected with lentivirus expressing PARK9 (Figure 3-1C). (n = 30–50/cells, *p = 0.0001). O, P, Ca2+ levels in lysosomes were measured using the Ca2+ dye rhodamine dextran. O, Representative images of control and mutant fibroblasts labeled with rhodamine dextran and Cascade blue. P, Quantification of rhodamine dextran and Cascade blue fluorescence intensities before and after Baf1 treatment in control and PARK9 mutant fibroblasts (n = 10 cells, *p = 0.0001, Student's t test). Q, Effect of PARK9 deficiency on Ca2+-dependent lysosomal exocytosis. GCase activity was measured in the media before and after 50 μm GPN or 200 nm Baf1 treatment. Treatment with the Ca2+ chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) diminished the effect of GPN and Baf1 (n = 3, *p = 0.0135, **p = 0.0442, ***p = 0.0008, #p = 0.0347, ##p = 0.0023). R, Effect of Ca2+ release from the endoplasmic reticulum (ER) on lysosomal exocytosis was analyzed. GCase activity was measured from the media of two control or two PARK9 mutant fibroblasts before (left) and after 2 μm thapsigargin treatment (right) (n = 3, p = 0.95). Statistical analysis was conducted using one-way ANOVA with Tukey's post hoc test unless otherwise stated. Scale bar, 20 μm in J and O.
Modulation of PARK9 expression levels (A) Real time PCR analysis of human ATP13A2 expressions in Cont1, Cont2, Mut1, Mut2 and Mut1 and Mut2 transfected with lentivirus carrying human ATP13A2 in fibroblasts (n = 3, *p = 0.03, one-way ANOVA Tukey post hoc test). (B) Real time PCR analysis of human ATP13A2 expressions in Cont1, Cont2, Mut1, Mut2 and Mut1 and Mut2 transfected with lentivirus carrying human ATP13A2 in fibroblasts (n = 3, *p = 0.031, **p = 0.0001, one-way ANOVA Tukey post hoc test). (C) Real time PCR analysis of human ATP13A2 expressions in Scrb shRNA, shRNA against human ATP13A2, empty plasmid (Mock) or plasmid containing human ATP13A2 in H4 cells (n = 3, *p = 0.0429, **p = 0.0202, Student t test). Download Figure 3-1, TIF file (24.9MB, tif)
As ATP13A2/PARK9 localizes to endolysosomes (Ramirez et al., 2006; Tsunemi et al., 2014), we investigated whether defective lysosomal Ca2+ handling might contribute to the elevation in cytosolic Ca2+ concentration. First, we measured changes in free cytosolic Ca2+ levels upon GPN (glycyl-l-phenylalanine 2-naphtylamide) treatment, which induces lysosomal Ca2+ release (Fig. 3G–I). Although 50 μm GPN significantly increased cytosolic Ca2+ levels in control fibroblasts, it had no effect in either PARK9 mutant fibroblasts (Fig. 3G,H) or DA neurons (Fig. 3I), suggesting that lysosomal Ca2+ release or Ca2+ sequestration was impaired by PARK9 deficiency. To address this question, we used a genetically encoded lysosomal sensor with a Ca2+-sensitive fluorophore tgat is positioned at the outer surface of the lysosome, allowing it to monitor efflux of Ca2+ (GCaMP3-ML1) (Shen et al., 2012). Bafilomycin A1 (Baf1) (200 nm) treatment (which results in leakage of Ca2+ from lysosomes; Morgan et al., 2015) led to a transient elevation in GCaMP3-ML1 fluorescence (Fig. 3J) even after normalization for changes in lysosomal volume (Usenovic et al., 2012) (Fig. 3K,L). We then depleted PARK9 by shRNA (Tsunemi et al., 2014) (Fig. 3-1) and found that Baf1-induced lysosomal Ca2+ release as measured by GCaMP3-ML1 was significantly reduced compared with control cells (scramble shRNA) (Fig. 3L,M, Fig. 3-1C). Similar results were observed in PARK9-deficient fibroblasts compared with controls and lentivirus-mediated PARK9 overexpression restored the impaired Ca2+ release (Fig. 3H,I,N). We then investigated whether this defect in lysosomal Ca2+ release was caused by impaired lysosomal Ca2+ storage in PARK9-deficient cells. To this end, rhodamine dextran was used to estimate luminal lysosomal Ca2+ levels (Lloyd-Evans et al., 2008). In control fibroblasts, rhodamine dextran fluorescence was robust and rapidly decreased upon Baf1 treatment. In contrast, in PARK9 mutant fibroblasts, rhodamine dextran fluorescence was weak and was not significantly altered after Baf1 treatment (Fig. 3O), consistent with the proposition that PARK9 deficiency leads to decreased lysosomal Ca2+ storage (Fig. 3P).
PARK9 regulates lysosomal exocytosis via modulation of lysosomal Ca2+ homeostasis
Recent studies have shown that high lysosomal Ca2+ concentrations are necessary to trigger trafficking and exocytosis (Xu and Ren, 2015). Lysosomal exocytosis involves lysosomal fusion with the plasma membrane, resulting in the release of contents into the extracellular space (Samie and Xu, 2014). Consistent with what we observed in PARK9 patient neurons (Fig. 1C), lysosomal GCase activity in the media was significantly decreased in PARK9 patient fibroblasts (Fig. 3Q). To determine whether this change in enzymatic activity in the media was driven by lysosomal exocytosis, control cells were treated with 50 μm GPN, which has been shown to drive lysosomal exocytosis, resulting in an increase in GCase activity in the media (Fig. 3Q). Importantly, this was inhibited by the intracellular Ca2+ chelator BAPTA, confirming that lysosomal exocytosis requires a rapid increase in Ca2+ concentration near the lysosomal surface (Xu and Ren, 2015). In contrast, treatment with thapsigargin, an inhibitor of the sarco-/endoplasmic reticulum Ca2+ ATPase which induces Ca2+ release from the ER, did not disrupt GCase activity in the media, further confirming the importance of lysosomal Ca2+ release for activation of lysosomal exocytosis (Raffaello et al., 2016) (Fig. 3R). To further confirm a role for PARK9 in regulating lysosomal exocytosis, we measured the enzymatic activity of several lysosomal enzymes in the media that is normalized to their activity in cell lysate. The media activity of GCase, β-hexosaminidase, and acid phosphatase released from PARK9 mutant fibroblasts were all significantly decreased compared with that from control fibroblasts (Fig. 4A–C), suggesting decreased secretion of lysosomal hydrolases due to PARK9 deficiency. Conversely, increased expression of PARK9 (Fig. 3-1C) led to increased lysosomal enzymatic activity in the media (Fig. 4D–F), indicating an active involvement of PARK9 in this pathway.
Figure 4.

PARK9 regulation of lysosomal exocytosis. A–C, Activities of three lysosomal acid hydrolases in media were measured in PARK9 mutant and control fibroblasts. A, GCase activity in media normalized to activity in cell lysates (n = 3, *p = 0.0103, **p = 0.0093). B, β-hexosaminidase activity in media normalized to activity in cell lysates (n = 3, *p = 0.0417, **p = 0.0462). C, Acid phosphatase activity in media normalized to activity in cell lysates (n = 3, *p = 0.0279, **p = 0.0181). D–F, Activities of lysosomal acid hydrolases released into media were increased in PARK9-overexpressing H4 cells. D, GCase activity in media normalized to activity in cell lysates (n = 3, *p = 0.036, Student's t test). E, β-hexosaminidase activity in media normalized to activity in cell lysates (n = 3, *p = 0.0101, Student's t test). F, Acid phosphatase activity in media normalized to activity in cell lysates (n = 3, *p = 0.0111, Student's t test). G, LAMP-1 surface staining in PARK9-knockdown and PARK9-overexpressing H4 cells. LAMP-1 expression (green fluorescence) on the plasma membrane marked by Dil (red fluorescence) was visualized at steady-state (top) and after 30 min of Baf1 treatment (top middle). LAMP-1 expression on the plasma membrane was also visualized in PARK9-silenced H4 cells with Baf1 treatment (bottom middle) and PARK9-overexpressing H4 cells with Baf1 treatment (bottom). H, Fluorescence intensity ratios (LAMP-1/Dil) are shown (n = 3, *p = 0.0001, **p = 0.0002, Student's t test). I, Cell surface biotinylation assay to analyze the effect of PARK9 expression levels on Baf1-induced cell surface LAMP1. Although overexpression of PARK9 led to increased expression, depletion of PARK9 resulted in reduced LAMP1 expression on the cell surface. J, Quantification of biotinylated LAMP1 proteins against total LAMP-1 proteins (n = 3, *p = 0.0005, **p = 0.0021, ***p = 0.0034, Student's t test). Statistical analysis was conducted using one-way ANOVA with Tukey's post hoc test unless otherwise stated. Values are shown as mean ± SEM. Scale bars, 20 μm in G.
To further assess PARK9's role in lysosomal exocytosis, we conducted LAMP-1 cell surface staining (Fig. 4G,H) with an antibody against the human LAMP-1 topological domain (luminal domain), which is exposed to the cell surface upon lysosomal exocytosis after lysosomes fuse with the plasma membrane. At steady-state conditions (before treatment), LAMP-1 was not present on the cell surface but subsequently translocated to the cell surface upon Baf1 treatment. By contrast, PARK9 silencing inhibited Baf1-induced LAMP-1 translocation to the cell surface, whereas increased PARK9 levels enhanced its translocation (Fig. 4G). In addition, we investigated LAMP-1 translocation biochemically by conducting a cell surface biotinylation assay through which proteins on the plasma membrane are biotinylated and collected by streptavidin beads (Fig. 4I,J). After enhanced or silenced PARK9 expression levels, we treated cells with 200 nm Baf1 for up to 2 h and subjected their lysates to precipitation by streptavidin beads. Both cell lysates and precipitated proteins were subsequently analyzed by immunoblotting with LAMP-1 antibodies (Fig. 4I) and examined for the time course of LAMP-1 translocation to the cell surface after Baf1 treatment (Fig. 4J). Importantly, whereas increased PARK9 expression led to increased LAMP-1 on the cell surface, PARK9 silencing decreased its translocation (Fig. 4I,J), further demonstrating that PARK9 levels regulate LAMP-1 translocation during lysosomal exocytosis.
Upregulation of lysosomal exocytosis with lysosomal Ca2+ channel TRPML1 agonists rescues α-syn secretion defects and intracellular accumulation in PARK9 patient DA neurons
TRPML1 is the main Ca2+ channel responsible for Ca2+ release from lysosomes and the TRPML1 agonist ML-SA1 has been shown to induce lysosomal exocytosis (Shen et al., 2012). We thus investigated whether we could rescue deficient lysosomal exocytosis in PARK9 mutant fibroblasts using three different TRPML1 agonists: ML-SA1, SF-22, and MK6-83. We found that all three agonists increased lysosomal exocytosis, as measured by a significant increase in the extracellular lysosomal enzymatic activity of GCase (normalized to intracellular activity levels) in both control and PARK9-mutated fibroblasts in a dose-dependent manner (Fig. 5A, Fig. 5-1A) (Chen et al., 2014). As a result, intracellular GCase activities were decreased (Fig. 5-1B). This was further confirmed using LAMP-1 cell surface staining, which demonstrated high cell surface staining upon MK6-83 agonist treatment (Fig. 5B, top), but was abolished by PARK9 silencing (Fig. 5B, bottom), further indicating that PARK9 is also involved in TRPML1-induced lysosomal exocytosis. We confirmed the specificity of these TRPML1 agonists by examining the effect of TRPML1 inhibition on TRPML1 agonist-mediated GCase release. Genetic depletion by shRNA of TRPML1 was confirmed by immunoblotting (Fig. 5-1C). Pharmacological inhibition of TRPML1 was achieved by adding either 10 mm adenosine or 100 nm LaCl3 in the culture media 1 h before TRPML1 activation (Dong et al., 2009; Zhong et al., 2017). Importantly, TRPML1 inhibition either by pretreatment with either shRNA-mediated TRPML1 silencing or the TRPML1 inhibitors adenosine and La3+ rendered the cells insensitive to the TRPML1 agonist, demonstrating the direct effect of the TRPML1 agonist on TRPML1 channels (Fig. 5-1D). We further tested whether the Ca2+ storage deficit in PARK9 mutant lysosomes resulted from overactivation of TRPML1, by examining the effect of TRPML1 inhibition on lysosomal Ca2+ release mediated by Baf1, which is the most potent inducer of lysosomal exocytosis (Fig. 3Q). We found that either genetic TRPML1 silencing or chemical inhibition of TRPML1 reduced Ca2+ release from lysosomes in control fibroblasts, suggesting the contribution of TRPML1 on lysosomal Ca2+ release (Fig. 5-1E). In contrast, these treatments did not show any effects on PARK9 mutant cells (Fig. 5-1F), demonstrating that inhibition of TRPML1 does not disrupt impaired Ca2+ release from PARK9 mutant lysosomes. Lysosomal exocytosis was increased either by PARK9 overexpression (Fig. 4) or by TRPML1-agonists, both of which became ineffective when lysosomal Ca2+ was chelated in advance (Fig. 5-1G, Fig. 3Q). Together, these results suggest that decreased lysosomal exocytosis by ATP13A2 deficiency would be largely due to a decrease in lysosomal Ca2+.
Figure 5.

Ca2+-dependent lysosomal exocytosis rescues α-syn secretion in PARK9 patient DA neurons (Figure 5-1). A, Effect of TRPML1 channel agonists on lysosomal exocytosis was analyzed in control and PARK9 mutant fibroblasts. The extracellular and intracellular GCase activities were measured before and after 20 μm ML-SA1, 1 μm SF-22, and 1 μm MK6–83 treatments (Figure 5-1) (n = 3, *p = 0.0103, **p = 0.0045, ***p = 0.0054, ***p = 0.0106, #p = 0.0270, ##p = 0.0444, ###p = 0.0002). B, Effect of the TRPML1 channel agonist ML-SA1 (20 μm) on LAMP1 surface staining in H4 cells transfected with Scrb shRNA (top) or shRNA against PARK9 transfected (bottom). 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (Dil) was used as a plasma membrane marker. C, α-syn levels from the media taken from H4 cells measured by highly sensitive ELISA (n = 3, *p = 0.0348, **p = 0.0011). D, Immunoblot analysis of α-syn levels in H4 cells after treatment with DMSO, ML-SA1, SF-22, or MK6–83 for 24 h. E, Quantification of α-syn levels. After normalization to GAPDH, the relative α-syn levels in treated cells were divided by α-syn levels in cells treated with DMSO (far left) (n = 3, *p = 0.0133, **p = 0.0077, ***p = 0.0046). F, α-syn levels from the media taken from control or PARK9 mutant DA neurons measured by highly sensitive ELISA (n = 3, *p = 0.0001). G, Immunoblot analysis of α-syn levels in DA neurons at day 90 after treatment with DMSO, ML-SA1, SF-22, or MK6–83 for 24 h. H, I, Quantification of α-syn levels. After normalization to β-iii-tubulin (H, Triton-X-soluble fraction) or vimentin (I, SDS-soluble fraction), the relative α-syn levels in the treated cells were normalized to α-syn levels before treatment (green bars) (H, n = 3, *p = 0.0415, **p = 0.0221, ***p = 0.0011, ****p = 0.0001; I, n = 3, *p = 0.0071, **p = 0.0006). J, Mitochondrial respiration analysis using two control, two PARK9 mutant DA neurons and two PARK9 mutant DA neurons pretreated with 1 μm MK6–83 (n = 3, *p = 0.0303). Statistical analysis was conducted using one-way ANOVA with Tukey's post hoc test. Scale bars represent 20 μm for B.
The effect of TRPML1 agonists on lysosomal exocytosis (A) The activity of GCase in the media from Cont1 fibroblasts with treatments of different dose of three TRPML1 agonists, ML-SA1, SF-22 and MK6-83 (n = 3, *p = 0.03, Student t test). (B) The activity of GCase in DA neurons that were treated with three TRPML1 agonists, ML-SA1, SF-22 and MK6-83 (n = 3, *p = 0.03, one-way ANOVA Tukey post hoc test). (C) At 72 hours after transfecting Scrb shRNA or each five different shRNA against human TRPML1 in H4 cells, immunoblotting was conducted to quantify TRPML1 protein levels. (Upper) The representative blotting image. (Bottom) Densiometric analysis of each blots (n = 3, *p = 0.0001, Student t test). (D) Fluorescence of Rhod dextran and Cascade blue was measured before and after 1 μM MK6-83 treatment at 72 hours after transfecting either Scrb shRNA or shRNA against TRPML1 (Left four columns), or at one hour after 10 mM adenosine or 100 uM LaCl3 treatment (Right four columns) (n = 3, *p = 0.03, Student t test). (E, F) (Left four columns) The effect of inhibition of TRPML1 on Baf1-mediated lysosomal exocytosis. (E) At 72 hours after transfecting either Scrb shRNA or shRNA against TRPML1, fluorescence of Rhod dextran and Cascade blue in Cont 1 fibroblast was measured before and at one hour after 200nM Baf1 treatment (n = 3, *p = 0.03, Student t test). (Right six columns) At one hour after 10 mM adenosine or 100 uM LaCl3 treatment, fluorescence of Rhod dextran and Cascade blue was measured before and at one hour after treatment of 200nM Baf1 treatment (n = 3, *p = 0.03, Student t test). (F) The same experiments were conducted using Mut 2 fibroblasts. (G) The activity of GCase released in the media by 1 μM MK6-83 treatment from Cont 1 (left four columns) and Mut 1 fibroblasts (right four columns) was measured after pretreatment(s) of lenti-virus mediated PARK9 overexpression and/or BAPTA-mediated lysosomal Ca2+ depletion (n = 3, *p = 0.0322, **p = 0.0345, ***p = 0.0004, #p = 0.0440, ##p = 0.0174, ###p = 0.0001 , one-way ANOVA Tukey post hoc test). Download Figure 5-1, TIF file (24.9MB, tif)
Next, we investigated whether impaired α-syn secretion could be rescued by upregulating lysosomal exocytosis with TRPML1 agonists. Importantly, treatment with any of the three TRPML1 agonists, ML-SA1, SF-22, or MK6–83, for 24 h was sufficient to increase α-syn secretion (Fig. 5C) and concomitantly reduced intracellular α-syn levels (Fig. 5D,E) from both control and PARK9 mutant DA neurons (Fig. 5F). Moreover, this significantly decreased α-syn intracellular accumulation in both Triton-X-soluble and SDS-soluble fractions in PARK9 mutant DA neurons (Fig. 5G–I). Finally, we examined mitochondrial respiration in PARK9 mutant DA neurons to determine whether TRPML1 agonist treatment could rescue any mitochondrial deficits (Fig. 5J). PARK9 mutant DA neurons exhibited increased in all phases of mitochondrial activities: basal respiration from 0 to 17 min, ATP production from 26 to 43 min, and maximum respiration from 52 to 69 min, which are considered as a response to high energy demand from cellular organelles including lysosomes and consistent with previous studies (Grünewald et al., 2012). Importantly, the TRPML1 agonist MK6-83 (1 μm) normalized this activity back to the level of control neurons. Because TRPML1 agonists are unlikely to have direct effects on mitochondria, our data suggest that this could be the result of improved Ca2+ homeostasis, which enhances lysosomal exocytosis and a-syn secretion. Together, these results highlight a key role for upregulating lysosomal exocytosis as an effective pathway for regulating α-syn levels by increasing its secretion and decreasing its intracellular accumulation in a human DA neuron model of synucleinopathy, and further demonstrate that this pathway is defective in PARK9 patient neurons due to misregulation of lysosomal Ca2+ dynamics.
Discussion
The accumulation of misfolded proteins is a common pathological feature of many neurodegenerative disorders (Eisele et al., 2015). Lewy bodies and neurites are a pathological hallmark of PD, demonstrating a critical role for α-syn accumulation (Lang and Lozano, 1998) and modulation of α-syn levels in Parkinson's pathogenesis (Rubinsztein, 2006). By examining a monogenic form of PD, ATP13A2/PARK9, we found that lysosomal exocytosis is an important pathway that regulates α-syn levels in human neurons. PARK9 deficiency impaired this pathway, resulting in both lysosomal dysfunction and α-syn accumulation. Restoration of lysosomal exocytosis by TRPML1 agonists was able to improve lysosomal exocytosis and reduce α-syn levels in patient DA neurons.
Lysosomal exocytosis is a critical pathway in which lysosomes fuse with the plasma membrane and expel their storage materials outside of the cells (Xu and Ren, 2015). This unconventional exocytotic pathway was initially discovered by studying the protozoan parasite Trypanosoma cruzi (Tardieux et al., 1992); follow-up work revealed that this process exists in many cell types and is required to repair injured plasma membrane (Reddy et al., 2001). We found that PARK9 was able to directly regulate lysosomal exocytosis, which plays an important role in modulating α-syn intracellular levels. In contrast, exosomal secretion only makes a modest contribution to the secretion of α-syn (Tsunemi et al., 2014). Upregulating lysosomal exocytosis may be beneficial, not only for PARK9 DA neurons, but also other disorders including lysosomal storage diseases (Medina et al., 2011; Shen et al., 2012) and Alzheimer's disease (Bae et al., 2014) becauase this pathway can reduce the levels of both soluble and insoluble protein aggregates contributing to these diseases.
α-syn is predominantly found in presynaptic terminals of healthy neurons and has a putative role in synaptic transmission (Burré et al., 2010). However, once its abundance exceeds a certain threshold level, a-syn becomes toxic (Wong and Krainc, 2017). Our results suggest that the exocytotic pathway is indispensable for neurons to reduce α-syn levels. Indeed, α-syn is continuously secreted even under physiological conditions as it is found in the CSF of both PD patient and healthy controls (Borghi et al., 2000). Although neurons can degrade a-syn in several ways, when these mechanisms are overwhelmed, they may use secretory pathways as a last resort to reduce toxic protein accumulation (Rubinsztein, 2006). In this situation, PARK9 deficiency could lead to accumulation of potentially toxic levels of α-syn (Usenovic et al., 2012; Tsunemi et al., 2014).
Using Ca2+ sensors targeted to cytosolic and lysosomal compartments, we found that PARK9 deficiency results in elevated cytosolic and reduced lysosomal Ca2+ concentration. Normally, lysosomes maintain an intraluminal Ca2+ concentration (∼0.5 mm) that is similar to those of the endoplasmic reticulum or mitochondria, both of which have been established as intracellular Ca2+ stores (Raffaello et al., 2016). However, in contrast to these organelles, the mechanisms by which Ca2+ is transported into acidic vesicles is still not well understood (Raffaello et al., 2016). Our results indicate that PARK9 may play a significant role in the sequestration of Ca2+ in lysosomes. Interestingly, deficits in PARK9 function led to an elevation in cytosolic Ca2+ concentration. Previous work has linked elevated cytosolic Ca2+ levels to selective neuronal vulnerability in PD. In particular, Ca2+ entry through Cav1 (L-type) Ca2+ channels during autonomous spiking stimulates mitochondrial respiration and oxidative stress in at-risk neurons, such as the dopaminergic neurons in the substantia nigra, noradrenergic neurons in the locus ceruleus, and cholinergic neurons in the dorsal motor nucleus of the vagus (Surmeier et al., 2017). Deficits in PARK9-mediated sequestration of Ca2+ in lysosomes may further increase the cytosolic Ca2+ loading in these cell types, adding to mitochondrial oxidant stress. It remains to be determined whether the increased spiking rate in PARK9 mutant DA neurons is an attempt to compensate for the deficit in lysosomal Ca2+ storage (by increasing the availability of Ca2+ to be pumped into the lysosome) or is an inadvertent by-product of the failure to adequately sequester Ca2+ in lysosomes. In either case, the PARK-9 mutation adds to the Ca2+ burden on DA neurons and their vulnerability.
In conclusion, we show here that intracellular α-syn levels can be regulated by PARK9-mediated lysosomal exocytosis. Disruption of these pathways evokes a series of cellular dysfunctions observed in PARK9 mutant DA patient neurons, whereas restoring lysosomal exocytosis decreases pathology by decreasing intracellular levels of α-syn via its secretion. Targeting secretory pathway may thus be an important therapeutic strategy for ameliorating α-syn accumulation across multiple synucleinopathies, including PD.
Footnotes
This work was supported by National Institutes of Health (R37 NS096241 to D.K.) and the Japan Society for the Promotion of Science KAKENHI (Grants 16H07185 and 18K07510 to T.T.); the Brain Research Foundation (T.T.); and the Juntendo University Research Institute for Diseases of Old Age and Environmental & Gender-Specific Medicine (T.T.). This work is also supported by Practical Research Project for Rare/Intractable Diseases from AMED (JP17ek0109244 to T.T., W.A., and N.H.) and Strategic Research Foundation Grant-in-Aid for Private Universities (to T.T., Y.I., A.Y., W.A., and N.H.).
The authors declare no competing financial interests.
References
- Bae M, Patel N, Xu H, Lee M, Tominaga-Yamanaka K, Nath A, Geiger J, Gorospe M, Mattson MP, Haughey NJ (2014) Activation of TRPML1 clears intraneuronal abeta in preclinical models of HIV infection. J Neurosci 34:11485–11503. 10.1523/JNEUROSCI.0210-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC (2016) The Parkinson's disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 7:11803. 10.1038/ncomms11803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, Kiechle M, Feiler MS, Freischmidt A, Brenner D, Witting A, Hengerer B, Fändrich M, Ludolph AC, Weishaupt JH, Gillardon F, Danzer KM (2016) Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol 131:379–391. 10.1007/s00401-015-1504-2 [DOI] [PubMed] [Google Scholar]
- Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M (2000) Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett 287:65–67. 10.1016/S0304-3940(00)01153-8 [DOI] [PubMed] [Google Scholar]
- Brahic M, Bousset L, Bieri G, Melki R, Gitler AD (2016) Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol 131:539–548. 10.1007/s00401-016-1538-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. 10.1126/science.1195227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- Chen CC, Keller M, Hess M, Schiffmann R, Urban N, Wolfgardt A, Schaefer M, Bracher F, Biel M, Wahl-Schott C, Grimm C (2014) A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat Commun 5:4681. 10.1038/ncomms5681 [DOI] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, et al. (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson's disease. Sci Transl Med 4:141ra190. 10.1126/scitranslmed.3003985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109:9611–9616. 10.1073/pnas.1112368109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirsoy S, Martin S, Motamedi S, van Veen S, Holemans T, Van den Haute C, Jordanova A, Baekelandt V, Vangheluwe P, Agostinis P (2017) ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum Mol Genet 26:1656–1669. 10.1093/hmg/ddx070 [DOI] [PubMed] [Google Scholar]
- Dong XP, Wang X, Shen D, Chen S, Liu M, Wang Y, Mills E, Cheng X, Delling M, Xu H (2009) Activating mutations of the TRPML1 channel revealed by proline-scanning mutagenesis. J Biol Chem 284:32040–32052. 10.1074/jbc.M109.037184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ (2013) Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci 33:10154–10164. 10.1523/JNEUROSCI.5311-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, Kelly JW (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov 14:759–780. 10.1038/nrd4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S (2009) Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41:308–315. 10.1038/ng.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, Sue CM, Klein C (2012) ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging 33:1843.e1–e7. 10.1016/j.neurobiolaging.2011.12.035 [DOI] [PubMed] [Google Scholar]
- Holemans T, Sørensen DM, van Veen S, Martin S, Hermans D, Kemmer GC, Van den Haute C, Baekelandt V, Günther Pomorski T, Agostinis P, Wuytack F, Palmgren M, Eggermont J, Vangheluwe P (2015) A lipid switch unlocks Parkinson's disease-associated ATP13A2. Proc Natl Acad Sci U S A 112:9040–9045. 10.1073/pnas.1508220112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 364:1169–1171. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM (2009) Methods to monitor chaperone-mediated autophagy. Methods Enzymol 452:297–324. 10.1016/S0076-6879(08)03619-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA (2014) Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-synuclein externalization via exosomes. Hum Mol Genet 23:2816–2833. 10.1093/hmg/ddu099 [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480:547–551. 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Lozano AM (1998) Parkinson's disease. first of two parts. N Engl J Med 339:1044–1053. 10.1056/NEJM199810083391506 [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann-pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14:1247–1255. 10.1038/nm.1876 [DOI] [PubMed] [Google Scholar]
- Lopes da Fonseca T, Pinho R, Outeiro TF (2016) A familial ATP13A2 mutation enhances alpha-synuclein aggregation and promotes cell death. Hum Mol Genet 25:2959–2971. 10.1093/hmg/ddw147 [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. 10.1016/j.cell.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D (2016a) alpha-synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 113:1931–1936. 10.1073/pnas.1520335113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, Patnaik S, Sidransky E, Marugan JJ, Sue CM, Krainc D (2016b) Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J Neurosci 36:7693–7706. 10.1523/JNEUROSCI.0628-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JM, Krainc D (2017) Lysosomal proteins as a therapeutic target in neurodegeneration. Annu Rev Med 68:445–458. 10.1146/annurev-med-050715-104432 [DOI] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 21:421–430. 10.1016/j.devcel.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AJ, Davis LC, Galione A (2015) Imaging approaches to measuring lysosomal calcium. Methods Cell Biol 126:159–195. 10.1016/bs.mcb.2014.10.031 [DOI] [PubMed] [Google Scholar]
- Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM (2014) Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet 23:2802–2815. 10.1093/hmg/ddt623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, Mammucari C, Gherardi G, Rizzuto R (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci 41:1035–1049. 10.1016/j.tibs.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191. 10.1038/ng1884 [DOI] [PubMed] [Google Scholar]
- Reddy A, Caler EV, Andrews NW (2001) Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell 106:157–169. 10.1016/S0092-8674(01)00421-4 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443:780–786. 10.1038/nature05291 [DOI] [PubMed] [Google Scholar]
- Samie MA, Xu H (2014) Lysosomal exocytosis and lipid storage disorders. J Lipid Res 55:995–1009. 10.1194/jlr.R046896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, Gao Q, Slaugenhaupt S, Pickel J, Zolov SN, Weisman LS, Lenk GM, Titus S, Bryant-Genevier M, Southall N, Juan M, et al. (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell 26:511–524. 10.1016/j.devcel.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, Xu H (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3:731. 10.1038/ncomms1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) alpha-synuclein locus triplication causes Parkinson's disease. Science 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113. 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, Andrews NW (1992) Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell 71:1117–1130. 10.1016/S0092-8674(05)80061-3 [DOI] [PubMed] [Google Scholar]
- Tarradas A, Selga E, Beltran-Alvarez P, Pérez-Serra A, Riuró H, Picó F, Iglesias A, Campuzano O, Castro-Urda V, Fernández-Lozano I, Pérez GJ, Scornik FS, Brugada R (2013) A novel missense mutation, I890T, in the pore region of cardiac sodium channel causes Brugada syndrome. PLoS One 8:e53220. 10.1371/journal.pone.0053220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Krainc D (2014) Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet 23:2791–2801. 10.1093/hmg/ddt572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Hamada K, Krainc D (2014) ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J Neurosci 34:15281–15287. 10.1523/JNEUROSCI.1629-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012) Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 32:4240–4246. 10.1523/JNEUROSCI.5575-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2017) alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13. 10.1038/nm.4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77:57–80. 10.1146/annurev-physiol-021014-071649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XZ, Zou Y, Sun X, Dong G, Cao Q, Pandey A, Rainey JK, Zhu X, Dong XP (2017) Inhibition of transient receptor potential channel mucolipin-1 (TRPML1) by lysosomal adenosine involved in severe combined immunodeficiency diseases. J Biol Chem 292:3445–3455. 10.1074/jbc.M116.743963 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of iPSCs (A) Immunocytochemistry for pluripotency markers SSEA4, TRA-1-60, TRA-1-81, OCT4 and NANOG. (B) Images of chromosome G-banding analysis of control (Cont 4) and mutant (Mut 1) iPSCs. (C) Expression of three germ layer-specific markers after spontaneous differentiation. Markers of ectodermal progenitor cells (β-iii-tubulin), endodermal lineage cells (α-fetoprotein) and mesodermal lineage cells (SMA) were used. (D) Direct sequence results of two PARK9 mutant iPSC lines. (E) Gene copy analysis for SNCA in Cont 1, Cont 2, Triplication and Mut 1. Scale bars represent 50 μm for Figure A and 50 μm for Figure C. Download Figure 1-1, TIF file (24.9MB, tif)
Characterization of human iPSC-derived DA neurons (A) DA neurons analyzed by immunocytochemistry using FOXA2 (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage of cells expressing both FOXA2 and TH during differentiation and maturation (n = 30-50). (B) DA neurons analyzed by immunocytochemistry using Lmx1a (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage cells expressing both LMX1a and TH during differentiation and maturation (n = 30-50). (C) DA neurons analyzed by immunocytochemistry using β-iii-tubulin (β-iii-tub) (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. Percentage cells expressing both β-iii-tubulin and TH during differentiation and maturation (n = 30-50). Scale bars represent 100 μm. Download Figure 1-2, TIF file (24.9MB, tif)
Pathogenic phenotypes of PARK9 patient DA neurons over time (A) The number of exosomes secreted from four control and two PARK9-mutant DA neurons were analyzed at day 60, 90, 120 after the initiation of differentiation. The number of exosomes was normalized to the total protein level from cell lysates (n = 3, *p = 0.0002, **p = 0.0276). (B) Lysosomal proteolysis measured by radioactive pulse chase in four controls and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation. Data are shown as fold changes normalized to control 1 at day 40 (n = 3, *p = 0.0001, **p = 0.0113, ***p = 0.0155). (C) Released GCase activity in the media taken from four control and two PARK9-mutant DA neurons at Day 60, 90, 120 after the initiation of differentiation. Activity in the media were normalized to the activity in cell lysates, and shown as a percentage of control 1 at day 40 (n = 3, *p = 0.0001, **p = 0.0202, ***p = 0.0250). (D) Enzyme-Linked Immunosorbent Assay (ELISA) to quantify α-syn protein levels in the media taken from four control and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation (n = 3, *p = 0.0001, **p = 0.0001). (E) α-syn ELISA for detecting α-syn protein levels in exosomes taken from four control and two PARK9-mutant DA neurons at day 60, 90, 120 after the initiation of differentiation (n = 3, *p = 0.0052, **p = 0.0001, ***p = 0.0002). (F) Immunoblot analysis of α-syn proteins in four control, two PARK9-mutant and SNCA triplication DA neurons at Day 60 after the initiation of differentiation. After normalization to β-iii-tubulin, the relative α-syn levels are shown as fold changes compared with control 1 (T-x soluble, n = 3, p = 0.0603, Cont1 vs Triplication, SDS soluble, n = 3, p = 0.3726, Cont1 vs Triplication). The statistical analysis was conducted using one-way ANOVA Tukey post hoc test. Download Figure 1-3, TIF file (24.9MB, tif)
iPSC-derived DA neurons on microfluidics (A) DA neurons analyzed by immunocytochemistry using FOXA2 (red) and tyrosine hydroxylase (TH) (green) at day 60. (B) DA neurons analyzed by immunocytochemistry using LMX1a (red) and tyrosine hydroxylase (TH) (green) at day 60. (C) DA neurons analyzed by immunocytochemistry using β-iii-tubulin (β-iii-tub) (red) and tyrosine hydroxylase (TH) (green) at day 60 after the initiation of differentiation. (D) After incubation with α-syn 555 for 15 min, DA neurons were fixed and treated with 0.2% trypan blue to quench extracellular α-syn 555. Representative images of control (left) and PARK9-mutated DA neurons (right) in orthogonal projections of confocal z-stacks. Arrows show internalized α-syn 555. (E) The time course of α-syn 555 internalization in control and PARK9-mutated DA neurons. Scale bars represent 100 μm for Figure A to C. White scale bars represent 50 μm and black scale bars represent 7.6 μm for Cont 3 and 13.6 μm for Mut2 in Figure D. Download Figure 2-1, TIF file (24.9MB, tif)
Modulation of PARK9 expression levels (A) Real time PCR analysis of human ATP13A2 expressions in Cont1, Cont2, Mut1, Mut2 and Mut1 and Mut2 transfected with lentivirus carrying human ATP13A2 in fibroblasts (n = 3, *p = 0.03, one-way ANOVA Tukey post hoc test). (B) Real time PCR analysis of human ATP13A2 expressions in Cont1, Cont2, Mut1, Mut2 and Mut1 and Mut2 transfected with lentivirus carrying human ATP13A2 in fibroblasts (n = 3, *p = 0.031, **p = 0.0001, one-way ANOVA Tukey post hoc test). (C) Real time PCR analysis of human ATP13A2 expressions in Scrb shRNA, shRNA against human ATP13A2, empty plasmid (Mock) or plasmid containing human ATP13A2 in H4 cells (n = 3, *p = 0.0429, **p = 0.0202, Student t test). Download Figure 3-1, TIF file (24.9MB, tif)
The effect of TRPML1 agonists on lysosomal exocytosis (A) The activity of GCase in the media from Cont1 fibroblasts with treatments of different dose of three TRPML1 agonists, ML-SA1, SF-22 and MK6-83 (n = 3, *p = 0.03, Student t test). (B) The activity of GCase in DA neurons that were treated with three TRPML1 agonists, ML-SA1, SF-22 and MK6-83 (n = 3, *p = 0.03, one-way ANOVA Tukey post hoc test). (C) At 72 hours after transfecting Scrb shRNA or each five different shRNA against human TRPML1 in H4 cells, immunoblotting was conducted to quantify TRPML1 protein levels. (Upper) The representative blotting image. (Bottom) Densiometric analysis of each blots (n = 3, *p = 0.0001, Student t test). (D) Fluorescence of Rhod dextran and Cascade blue was measured before and after 1 μM MK6-83 treatment at 72 hours after transfecting either Scrb shRNA or shRNA against TRPML1 (Left four columns), or at one hour after 10 mM adenosine or 100 uM LaCl3 treatment (Right four columns) (n = 3, *p = 0.03, Student t test). (E, F) (Left four columns) The effect of inhibition of TRPML1 on Baf1-mediated lysosomal exocytosis. (E) At 72 hours after transfecting either Scrb shRNA or shRNA against TRPML1, fluorescence of Rhod dextran and Cascade blue in Cont 1 fibroblast was measured before and at one hour after 200nM Baf1 treatment (n = 3, *p = 0.03, Student t test). (Right six columns) At one hour after 10 mM adenosine or 100 uM LaCl3 treatment, fluorescence of Rhod dextran and Cascade blue was measured before and at one hour after treatment of 200nM Baf1 treatment (n = 3, *p = 0.03, Student t test). (F) The same experiments were conducted using Mut 2 fibroblasts. (G) The activity of GCase released in the media by 1 μM MK6-83 treatment from Cont 1 (left four columns) and Mut 1 fibroblasts (right four columns) was measured after pretreatment(s) of lenti-virus mediated PARK9 overexpression and/or BAPTA-mediated lysosomal Ca2+ depletion (n = 3, *p = 0.0322, **p = 0.0345, ***p = 0.0004, #p = 0.0440, ##p = 0.0174, ###p = 0.0001 , one-way ANOVA Tukey post hoc test). Download Figure 5-1, TIF file (24.9MB, tif)