

Abstract
Background
Autism spectrum disorder (ASD) is a common and etiologically heterogeneous neurodevelopmental disorder. Although many genetic causes have been identified (> 200 ASD-risk genes), no single gene variant accounts for > 1% of all ASD cases. A role for epigenetic mechanisms in ASD etiology is supported by the fact that many ASD-risk genes function as epigenetic regulators and evidence that epigenetic dysregulation can interrupt normal brain development. Gene-specific DNAm profiles have been shown to assist in the interpretation of variants of unknown significance. Therefore, we investigated the epigenome in patients with ASD or two of the most common genomic variants conferring increased risk for ASD. Genome-wide DNA methylation (DNAm) was assessed using the Illumina Infinium HumanMethylation450 and MethylationEPIC arrays in blood from individuals with ASD of heterogeneous, undefined etiology (n = 52), and individuals with 16p11.2 deletions (16p11.2del, n = 9) or pathogenic variants in the chromatin modifier CHD8 (CHD8+/−, n = 7).
Results
DNAm patterns did not clearly distinguish heterogeneous ASD cases from controls. However, the homogeneous genetically-defined 16p11.2del and CHD8+/− subgroups each exhibited unique DNAm signatures that distinguished 16p11.2del or CHD8+/− individuals from each other and from heterogeneous ASD and control groups with high sensitivity and specificity. These signatures also classified additional 16p11.2del (n = 9) and CHD8 (n = 13) variants as pathogenic or benign. Our findings that DNAm alterations in each signature target unique genes in relevant biological pathways including neural development support their functional relevance. Furthermore, genes identified in our CHD8+/− DNAm signature in blood overlapped differentially expressed genes in CHD8+/− human-induced pluripotent cell-derived neurons and cerebral organoids from independent studies.
Conclusions
DNAm signatures can provide clinical utility complementary to next-generation sequencing in the interpretation of variants of unknown significance. Our study constitutes a novel approach for ASD risk-associated molecular classification that elucidates the vital cross-talk between genetics and epigenetics in the etiology of ASD.
Electronic supplementary material
The online version of this article (10.1186/s13148-019-0684-3) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, DNA methylation, Autism spectrum disorder, Genomic variants, Genetic stratification, Heterogeneity
Introduction
Autism spectrum disorder (ASD) comprises a group of complex neurodevelopmental conditions, diagnosed in 1 of 68 (1.4%) individuals in the general population [1]. Significant etiologic heterogeneity is implied by the variable penetrance of a large number of genetic causes involving > 200 ASD-risk loci (i.e., single genes, copy number variants [CNVs]) that have been identified in ~ 25–40% of individuals with ASD [2–6]; no single genetic cause accounts for > 1% of ASD cases [6–9]. Given that ASD is a multifactorial disorder, the expectation is that the combined contribution of multiple etiologic factors will determine an individual’s risk, whereby once a threshold is reached, the clinical features expressed will support a diagnosis of ASD [10]. Other etiologic factors currently implicated in ASD etiology include environmental and epigenetic mechanisms. Many ASD-risk genes are “epigenes” encoding proteins that function as epigenetic regulators, i.e., chromatin remodelers and transcriptional regulators, supporting a role for epigenetic dysregulation in ASD etiology [4, 11–13]. Epigenetic marks such as DNA methylation (DNAm) are precisely programmed spatially and temporally during normal development. DNAm alterations caused by genetic and/or environmental factors could negatively impact biological pathways important for normal brain development.
DNAm is one of the most stable and commonly assessed epigenetic marks. DNAm results from the transfer of a methyl group to cytosine in a CpG dinucleotide, catalyzed by DNA methyltransferase enzymes. These modifications are established and maintained through cell division. We, and others, have shown that there is crosstalk between histone modifications and CpG methylation in DNA; that is, cells from individuals with variants in some epigenes demonstrate functionally relevant genome-wide DNAm alterations that constitute gene-specific signatures. DNAm signatures have been previously identified in peripheral blood for disorders caused by pathogenic variants in epigenes that are associated with neurodevelopmental outcomes including both non-syndromic ASD and syndromic ASD/intellectual disability (ID) (e.g., NSD1 variants associated with Sotos syndrome, CHD7 variants associated with CHARGE syndrome) [14–23]. We have shown that these DNAm signatures represent a promising avenue to improve an increasingly challenging issue in clinical diagnostics. While advancing clinical diagnostics, next-generation sequencing (NGS) has magnified one of the most significant challenges of DNA sequencing-based testing, namely the interpretation of variants of unknown significance (VUS). VUS are increasingly detected as the utilization of diagnostic sequencing expands in both the clinical and research arenas, also true in the context of ASD. Specifically, DNAm signatures are proving to be effective in the functional classification of VUS [14, 15, 18, 19]. A limited number of studies have examined the potential for developing ASD-specific DNAm biomarkers [24–29]. They report inconsistent differences at a variety of genomic sites in peripheral blood [29], brain [26, 27, 30], buccal epithelium [24], and sperm [31]. These studies are limited by small sample sizes and modest DNAm differences between ASD cases and controls likely due not only to inter-tissue and cell type variability, but also more importantly to heterogeneity of DNAm alterations in individuals with ASD with unselected etiologies.
We hypothesized that epigenetic alterations contribute to the molecular etiology of ASD and that the discovery of such epigenetic alterations could be enhanced by genetic stratification of ASD cases. More specifically, we expected that individuals with ASD and variants in specific epigenes would display distinct DNAm signatures, as previously demonstrated for other human disorders caused by variants in epigenes or CNVs overlapping epigenes [14–17, 20, 21, 23]. We also expect that we will be able to use the DNAm signatures to classify additional cases with variants in the specific epigenes or CNVs overlapping epigenes as pathogenic or benign. We therefore undertook several analyses comparing ASD cases and controls. First, we compared genome-wide DNAm in blood from individuals with ASD of undefined etiology (i.e., heterogeneous ASD) to controls. Next, we compared controls to cases with two of the most common genomic variants associated with an increased risk for ASD.
The first genetically homogeneous subgroup comprised individuals with 600 kb deletions in 16p11.2, a CNV known to confer ASD susceptibility (16p11.2del; OMIM# 611913). Individuals with 16p11.2del have a variable phenotype; 20–30% receive a diagnosis of ASD, although the majority of these individuals have behavioral/neurocognitive ASD features [32–34]. The HIRA-interacting protein 3 gene (HIRIP3), located in the 16p11.2del region, is known to be involved in regulating histone metabolism and chromatin function [35], but the full impact of 16p11.2 deletions on epigenetic dysregulation has yet to be elucidated. The second genetically homogeneous subgroup comprised individuals with known pathogenic variants in the chromodomain helicase DNA-binding protein 8 gene (CHD8+/−; OMIM# 615032). More than 85% of individuals with CHD8 variants have ASD, as well as morphological anomalies and other neurodevelopmental features [4, 36, 37]. CHD8 is known to function as an epigenetic regulator, playing a critical role in transcriptional repression through chromatin remodeling [38].
Our results demonstrate that genetically defined subgroups of cases with genomic aberrations at ASD-risk loci involving epigenes have highly sensitive and specific DNAm signatures that distinguish them from heterogeneous ASD cases and controls. We also show that these DNAm signatures identify genomic targets that may contribute to the etiology of specific genetic subgroups of ASD. The differentially methylated gene targets of our DNAm signatures function in biological pathways that are relevant to proposed pathophysiological mechanisms underlying ASD, highlighting the biological significance of our DNAm signatures. Lastly, we show that our DNAm signatures have the potential to be used as a complementary molecular diagnostic tool to help interpret the pathogenicity of VUS, demonstrating their functional utility and potential for clinical application.
Methods and materials
Participants
The ASD group consisted of an etiologically undefined, heterogeneous sample of individuals (heterogeneous ASD; n = 52) not selected for any known genomic features, for whom deep clinical phenotyping and whole genome/exome sequencing data were available through the Province of Ontario Neurodevelopmental Disorders (POND) Network and the Simons Simplex Collection (SSC) (Additional file 3: Table S1). All neurotypical control samples were selected from a collection available in our laboratory (Additional file 3: Table S1). The majority of individuals included in this study are of Caucasian descent; however, ethnicity data were not available for all individuals included. Some individuals in the control group received formal cognitive/behavioral assessments (Dr. Greg Hanna, University of Michigan); others were recruited using physician/parental screening questionnaires. Our 16p11.2del training case group consisted of DNA samples from the blood of individuals with confirmed typical 600 kb deletions in 16p11.2 (16p11.2del; chr16: 29.5–30.1 Mb) that were used for signature derivation (n = 9; 3 with ASD diagnosis) (Table 1). The 16p11.2 test case group consisted of samples from additional individuals with 16p11.2del variants and were used to test the signature (classification) (n = 9; 4 with ASD diagnosis) (Hospital for Sick Children [SickKids], POND; Table 3). Our CHD8+/− training case group consisted of DNA samples from the blood of individuals with confirmed de novo CHD8 pathogenic variants (CHD8+/−; n = 7, all with ASD diagnosis) obtained from SSC (Table 2) for signature derivation. The CHD8+/− test case group consisted of additional CHD8 sequence variants (n = 13; 12 with an ASD diagnosis) for classification. These were obtained from SSC, SickKids, The University of Alberta (Dr. Oana Caluseriu), and the Children’s Hospital of Eastern Ontario (CHEO; Dr. Melissa Carter) (Table 3).
Table 1.
Demographic and deletion information for 16p11.2del training signature cases and age-, sex-matched controls
| Sample ID | Sex | Age (years) | ASD status | 16p11.2del (600 kb region) coordinates | Deletion size (kb) | Inheritance |
|---|---|---|---|---|---|---|
| 435 | M | 12 | Unknown | 29,656,657–30,190,593 | 534 | Unknown |
| 516 | M | 7 | Unknown | 29,656,657–30,190,593 | 534 | Unknown |
| 639 | F | 13 | Unknown | 29,592,751–30,190,593 | 598 | Unknown |
| 797 | M | 7 | Unknown | 29,590,493–30,190,593 | 600 | Unknown |
| 871 | M | 10 | Unknown | 29,590,493–30,190,593 | 600 | Unknown |
| 293 | M | 5 | Unknown | 29,567,295–30,178,406 | 611 | Unknown |
| 3-0269-000 | M | 4 | ASD | 29,567,309–30,177,807 | 610 | Maternal |
| 1-0019-004 | M | 7.5 | ASD | 29,652,799–30,199,507 | 547 | De novo |
| 7-0229-003 | M | 11.5 | ASD | 29,567,295–30,177,916 | 611 | Unknown |
| EP15W | M | 13 | Control | |||
| EP16W | F | 2 | Control | |||
| EP17W | F | 4 | Control | |||
| EP1W | F | 8 | Control | |||
| EP22W | M | 4 | Control | |||
| EP2W | F | 10 | Control | |||
| EP3W | F | 13 | Control | |||
| EP4W | M | 13 | Control | |||
| EP5W | F | 8 | Control | |||
| EP6W | M | 13 | Control | |||
| EP7W | M | 9 | Control | |||
| EP8W | M | 8 | Control | |||
| EP9W | M | 11 | Control | |||
| EP51 | M | 12 | Control | |||
| EP60W | M | 12 | Control | |||
| 460/2048 | M | 11.5 | Control | |||
| EP32W | M | 6 | Control | |||
| EP45W | M | 10 | Control | |||
| EP59W | M | 6 | Control | |||
| EP57W | M | 7 | Control | |||
| CT0011-003 | M | 4 | Control | |||
| MK-311/1601 | M | 10 | Control | |||
| KA-352/1734 | M | 7 | Control |
Mean age 16p11.2del 8.556 ± 3.196
Mean age Control 8.761 ± 3.35
16p11.2.del M, F 8, 1
Control M, F 17, 6
Table 3.
Variant details and respective classification scores for 16p11.2del CNV and CHD8 test cases
| Sample ID | Sex | Age (years) | 16p11.2del coordinates (size in kb) | Diagnostic classification for 16p11.2 typical deletion (pathogenic, benign, variant of unknown significance [VUS]) | Inheritance | ASD status | 16p11.2del score |
|---|---|---|---|---|---|---|---|
| 348 | M | 0.25 | 29,590,493–30,190,593 (600) | Pathogenic | Unknown | Unknown | 0.11505 |
| 228 | M | 0.4 | 29,590,493–30,190,593 (600) | Pathogenic | Unknown | Unknown | 0.12668 |
| 875 | F | 4 | 29,656,657–30,190,593 (534) | Pathogenic | Unknown | Unknown | 0.03073 |
| 172 | F | 6 | 29,590,493–30,190,593 (600) | Pathogenic | Unknown | Unknown | 0.04668 |
| 292 | M | 0.25 | 29,500,252–30,098,094 (598) | Pathogenic | Unknown | Unknown | 0.13978 |
| 2-0088-003 | F | 12 | 29,652,799–30,199,507 (547) | Pathogenic (MOSAIC) | De novo | ASD | -0.00441 |
| 3-0406-000 | M | 1.2 | 29,581,764–30,199,579 (618) | Pathogenic | Unknown | ASD | 0.07735 |
| 6-0196-03 | M | Unknown | 29,500,084–30,098,210 (598) | Pathogenic | Unknown | ASD | 0.04815 |
| 1-0616-003 | M | 7.5 | 28,751,255–28,952,351 (201) | Not determined* | Maternal | ASD | − 0.11936 |
| Sample ID | Sex | Age (years) | CHD8 variant nucleotide change; amino acid change (Exon #) | Diagnostic classification (pathogenic, benign, variant of unknown significance [VUS]) | Inheritance | ASD status | CHD8+/− score |
| 14406.p1 | M | 13 | c.7493_7495del; p.His2498del (37) | VUS | De novo | ASD | − 0.03482 |
| 1-0494-003 | M | 4.75 | c.2219A>G; p.Asn740Ser (9) | Benign | Paternal | ASD | − 0.04451 |
| 2-1228-003 | F | 4.5 | c.6841C>G; p.Pro2281Ala (34) | Benign | Paternal | ASD | − 0.04178 |
| 1-0277-003 | M | Unknown | c.6565G>A; p.Gly2189Arg (33) | Benign | Maternal | ASD | − 0.03065 |
| 1-0507-003 | F | 14 | c.3940G>A; p.Ala1314Thr (19) | Benign | Maternal | ASD | − 0.05507 |
| 7-0167-003 | M | 4 | c.7499A>C; p.His2500Pro (37) | Benign | Unknown | ASD | − 0.05984 |
| 1-0559-003 | M | 7.7 | c.6148dupA; p.Thr2050fs (31) | Pathogenic | De novo | ASD | 0.0437 |
| CHD8-V1 | M | 10 | c.4215G>T; Synonymous (21) | VUS | Paternal | ASD | − 0.04478 |
| CHD8-V2 (father of CHD8-V1) | M | 40 | c.4215G>T; Synonymous (21) | VUS | Not applicable | Unaffected | − 0.04332 |
| CHD8-V3** | M | 6.5 | c.6649C>T, p.Arg2217* (33) | Pathogenic | Paternal | ASD | 0.04616 |
| CHD8-V4 (sibling of CHD8-V3)** | M | 14 | c.6649C>T, p.Arg2217* (33) | Pathogenic | Paternal | ASD | 0.03631 |
| DW0013** | M | 13.5 | c.6947delC, p.Pro2316Leufs*39 (35) | Likely pathogenic | Unknown | ASD | 0.05183 |
| EX0070-W** | M | 6.75 | c.4327C>T, p.Arg1443Cys (21) | VUS | Paternal | ASD | − 0.04131 |
*Deletion encompassing SH2B1 gene, not overlapping 16p11.2del typical deletion region
**Test case data are from EPIC array, classified using 93 probes overlapping 450K and EPIC arrays from CHD8+/− signature
Table 2.
Demographic and variant information for CHD8+/− training signature cases and age-, sex-matched controls
| Sample ID | Sex | Age (years) | ASD status | Nucleotide change | Amino acid change | Exon | Inheritance |
|---|---|---|---|---|---|---|---|
| 11654.p1 | F | 12.5 | ASD | c.3517 A>G | p.Arg1173Gly | 16 | De novo |
| 12714.p1 | M | 5 | ASD | c.185C>G | p.Ser62X | 1 | De novo |
| 12752.p1 | F | 8 | ASD | c.6359_6360del | p.Leu2120ProfsX | 32 | De novo |
| 12991.p1 | M | 16 | ASD | c.6307_6308del | p.Glu2103ArgfsX | 31 | De novo |
| 13844.p1 | M | 5 | ASD | c.3712C>T | p.Gln1238X | 17 | De novo |
| 14016.p1 | M | 14 | ASD | c.4009C>T | p.Arg1337X | 19 | De novo |
| 14233.p1 | M | 10 | ASD | c.7113_7114del | p.Asn2371LysfsX2 | 36 | De novo |
| EP48W | F | 11 | Control | ||||
| EP58W | M | 15 | Control | ||||
| EP41W | M | 16 | Control | ||||
| EH-268 W | F | 6 | Control | ||||
| JS-265 W | M | 16 | Control | ||||
| EP34W | F | 13 | Control | ||||
| TG-278 W | M | 13 | Control | ||||
| TB-342/1706 | M | 16 | Control | ||||
| EP43W | F | 7 | Control | ||||
| EP32W | M | 6 | Control | ||||
| JL-137/1362 | M | 14 | Control | ||||
| EP45W | M | 10 | Control | ||||
| EP59W | M | 6 | Control | ||||
| EP57W | M | 7 | Control | ||||
| CT0009-003 | M | 8 | Control | ||||
| CT0011-003 | M | 4 | Control | ||||
| MK-311/1601 | M | 10 | Control | ||||
| EP39W | F | 13 | Control | ||||
| EP42W | F | 8 | Control | ||||
| KA-352/1734 | M | 7 | Control | ||||
| MM-119/1567 | M | 10 | Control |
Mean age CHD8+/− 10.07 ± 4.325
Mean age Control 10.29 ± 3.836
CHD8+/− M, F 5, 2
Control M, F 15, 6
DNAm analysis
DNA was extracted from whole blood for all cases and controls using standard techniques. Extracted DNA was sodium bisulfite converted using the Qiagen EZ DNA Methylation kit (Qiagen, Valencia, CA), according to the manufacturer’s protocol. All DNA samples were processed according to the manufacturer’s protocol for DNAm analysis using either the Illumina Infinium HumanMethylation450 BeadChip array (450K array) or the Illumina MethylationEPIC (EPIC array; an additional set of CHD8 test case sequence variants only) at The Centre for Applied Genomics (SickKids). The distribution of the samples on the arrays was randomized for all cases and controls and for age and sex variables. The 450K array covers > 485,000 individual CpG sites, whereas the EPIC array covers > 850,000 individual CpG sites genome-wide at single-nucleotide resolution, encompassing > 99% of RefSeq genes and > 96% of annotated CpG islands.
Statistical analysis: DNAm signatures for 16p11.2del and CHD8+/−
Raw genome-wide DNAm data were analyzed using our laboratory’s bioinformatics pipeline employing hierarchical clustering, principal component analysis (PCA) and non-parametric statistical comparisons [15] (Additional file 2: Figure S1). Raw data normalization and pre-processing quality control steps were performed to ensure that only high-quality data were included for group comparisons to identify DNAm differences. Illumina Genome Studio software was used to normalize data (IDAT files) using background subtraction and control normalization to generate methylation values (Beta [β]-values). Before analysis, data for probes located on sex chromosomes, autosomal probes that cross-react with sex chromosome probes, non-specific probes, and probes targeting CpG sites within 5 bp of a SNP with a minor allele frequency > 1% were removed [16] (Additional file 3: Table S2). For signature derivation, batch correction by mean-centering was applied in Qlucore Omics Explorer (QOE, www.qlucore.com) to the comparison of 16p11.2del cases to age- (Mann-Whitney U test, p > 0.05), sex-matched (Chi-square (χ2) test, p > 0.05) controls before performing group comparisons; all other case-control groups were run in single batches. Since there were differences in the composition of age and sex in each group (ASD, 16p11.2del, and CHD8+/−), each training group was compared to a different set of age- and sex-matched controls. The topmost variable probes were used for group comparisons (i.e., least variable CpG sites across the dataset were removed using the QOE 3.2 “filtering by variance” tool), a commonly used prioritization step that increases statistical power while being blinded to group assignment [39]. For example, a variance-filtering parameter “var = 0.02” removes sites with a standard deviation < 2% of the maximal standard deviation. The number of sites retained depended on the specific samples included in each paired group comparison. The derivation of DNAm signatures employed the use of limma regression modeling to account for covariates and the Mann-Whitney U test to account for possible non-parametric effects. Significantly differentially methylated sites between signature training cases and controls were first identified using limma regression, using age, sex, and estimated cell type proportion (minfi) as covariates, with a Benjamini-Hochberg adjusted p value (q-value) < 0.05 and requiring a |Δβ| ≥ 0.05 (at least 5% methylation difference). We then identified significant differentially methylated sites independently using the non-parametric Mann-Whitney U test with a Benjamini-Hochberg adjusted p value (q-value) < 0.05. Optimal q-value and DNAm effect size (Δβ) thresholds were selected using volcano plots (Additional file 2: Figures S2 and S3). To be conservative, the final DNAm signature CpG lists were defined as the overlap among the results of limma regression, the Mann-Whitney U test, and the effect-size requirement.
In addition to incorporating blood cell type proportions as covariates in the derivation of our DNAm signatures (Additional file 1) [40], DNAm data for different blood cell-subtypes [41] were also used to directly ensure that differential methylation at signature sites was not confounded by differences in blood cell-type proportions, as similarly applied in Ref. [14, 15] (Supplementary Information; Additional file 2: Figures S5 and S6).
Comparison of differentially methylated CpG sites across DNAm signatures and differentially methylated regions (DMRs)
To detect consistent patterns of DNAm alterations from our DNAm signatures, we used an additional method to identify significantly differentially methylated regions (DMRs) overlapping and extending from individual CpG sites contained in each respective DNAm signature. We used the bump hunting method [42], which strengthens the detection of regional differences by combining differential methylation patterns across neighboring CpG sites [43]. The analysis detected DMRs that comprise CpGs with |Δβ| ≥ 5% between cases and controls as candidates for the DMRs, with gaps ≤ 500 bp between neighboring CpGs. Statistical significance was established using 1000 randomized bootstrap iterations to account for confounders, as recommended in the Bioconductor bumphunter package documentation. The resulting DMRs were post-filtered to retain only those with p values < 0.05 and average methylation differences |Δβ| ≥ 5% across the DMR. To further enhance robustness and improve stringency, we also required these DMRs to (1) comprise at least three consecutive CpGs and (2) include at least one CpG from the DNAm signatures previously derived for either 16p11.2del or CHD8+/−, as described above (Additional file 3: Tables S3 and S4). DMRs are presented using visualization methods adapted from DMRcate software (Additional file 2: Figure S4) [44].
Testing sensitivity and specificity of 16p11.2del and CHD8+/− DNAm signatures using independent test cases and controls
The 16p11.2del and CHD8+/−-specific DNAm signatures were used to build predictive models to functionally classify independent 16p11.2del CNVs and CHD8 test case variants as well as unrelated controls as pathogenic or benign. These include variants that were reported as pathogenic, benign, or of unknown significance (VUS). To evaluate the predictive models, we used independent, unrelated test cases to determine sensitivity and controls to determine specificity. We evaluated sensitivity on a test cohort of 16p11.2del CNV (n = 9) and CHD8 variants (n = 13), independent of the training cases. Four of the CHD8 variant test cases were run on the EPIC array. These test cases were classified using the 450K-derived CHD8+/− DNAm signature probes that overlapped with those on the EPIC array (92/103 signature sites). To evaluate the specificity of the DNAm classification signatures, we tested the DNAm signatures on a test set of control 450K array data (n = 162, described in further detail below), independent of those used in the signature derivation. To confirm that the two models did not generate overlapping predictions, we applied the 16p11.2del predictive model to the CHD8+/− cohort, and conversely the CHD8+/− predictive model to the 16p11.2del cohort. For each set of training cases and controls used to derive a signature, a reference DNAm profile was generated across the signature-specific CpGs, by computing median DNAm for each CpG site over all samples in the selected cohort (separately for cases and controls) [15]. Classification scores for additional variant test cases and control samples (Table 2; Additional file 3: Table S5) and corresponding classification plots were generated by comparing the correlation of each sample to the two respective reference profiles (corresponding to signature training cases and to controls). For example, blood samples with positive 16p11.2del scores will cluster more closely with the 16p11.2del reference profile, whereas samples with negative 16p11.2del scores will cluster more closely with the control reference profile.
Genomic distribution of DNAm signature CpG sites
The genomic distribution of CpG sites contained in each DNAm signature was compared with that of the respective variance-filtered datasets to identify signature-specific regions of enrichment or depletion using a hypergeometric test (implemented in R).
Additional datasets used for investigating functional significance of DNAm signatures
For additional specificity analysis, we used an independent publicly available set of 450K DNAm data derived from whole blood of control individuals (n = 162) with known ages < 50 years (mean 35.8 years) (Gene Expression Omnibus (GEO) Database; www.ncbi.nlm.nih.gov/geo/) (Additional file 3: Table S5). We did not expect control individuals to have 16p11.2del or CHD8 variants as these genomic variants are present in < 0.04% of the general population [45–47].
To determine the biological relevance of our DNAm signatures, we compared our differentially methylated genes with known ASD-risk genes (SFARI Gene [https://gene.sfari.org/]). To examine cross-tissue and functional relevance of our CHD8+/− signature, we compared the list of differentially methylated genes to those differentially expressed in an independent study using an engineered hemizygous deletion of CHD8 (CHD8+/−; using CRISPR/Cas9) in human induced pluripotent stem cell (iPSC)-derived neuronal precursor cells (NPCs), differentiated neurons [48], and cerebral organoids [49].
We used GREAT 3.0.0 (http://great.stanford.edu; up to 1 Mbp extension from the nearest genes) to identify functional enrichment of biological pathways in the differentially methylated genes of each DNAm signature in the context of wider genomic regions. Enrichment of each set of signature CpGs was defined against the background set of all probes that remained in the data after filtering by variance.
DNAm validation by targeted sodium bisulfite pyrosequencing
Specific differentially methylated sites found in each respective DNAm signature for 16p11.2del or CHD8+/− training signature cases and their age-, sex-matched controls were validated on sodium bisulfite-converted DNA using targeted pyrosequencing assays. These assays were designed using Qiagen Assay Design Software v2.0 to target specific CpGs identified by the microarray experiment as well as adjacent sites (Additional file 1). Selected sites are significantly differentially methylated in both the respective DNAm signatures and DMRs (Additional file 2: Figure S4; Additional file 3: Tables S3 and S4). Pyrosequencing was performed using the PyroMark Q24 system and Pyrosequencing Gold Reagents (Qiagen). Testing for a statistical difference between groups was performed using a Mann-Whitney U test (p < 0.05).
Results
DNAm does not distinguish heterogeneous ASD cases from controls
DNAm profiles of the heterogeneous ASD group (n = 52) were compared with age-, sex-matched controls (n = 30) at 40550 sites following filtering by variance. Neither limma nor Mann-Whitney U analyses identified any significant sites after Benjamini-Hochberg correction for multiple testing (all q > 0.05). Only 22 sites (16 (72.3%) hypomethylated, 6 (27.3%) hypermethylated) passed our criteria of an unadjusted p < 0.001 and |Δβ| > 5%. At this set of sites, no clear distinction between the ASD cases and controls could be made by either hierarchical clustering or principal components analysis (PCA), and thus no DNAm signature was identified.
Given that the genetically homogeneous groups, 16p11.2del and CHD8+/−, were selected for their potential for elucidating epigenetic dysregulation, we assessed whether these groups could be distinguished epigenetically from the heterogeneous ASD group and controls. The 16p11.2del and CHD8+/− cases were therefore classified at the 22 sites where putative ASD differences were detected (p < 0.001, |Δβ| ≥ 5%). No distinct clustering of either group was apparent, i.e., all 16p11.2del and CHD8+/− cases were mixed with both ASD cases and controls (Fig. 1). These data suggested that any potential 16p11.2del- or CHD8+/−-specific DNAm alterations were obscured by the presence of other heterogeneous ASD cases. Therefore, we focused next on the potential discovery of specific DNAm signatures, contingent on direct comparisons of genetically homogeneous ASD-risk groups with controls.
Fig. 1.

Whole blood DNAm comparison between heterogeneous ASD group (n = 52) and age-, sex-matched controls (n = 30). Following filtering by variance (40,550 sites), limma regression and Mann-Whitney U comparison, no CpG sites meet significance criteria of q ≤ 0.05. The 22 sites shown at uncorrected p < 0.001, |Δβ| ≥ 5% do not distinguish clearly between ASD cases and controls. Hierarchical clustering (Euclidian) and principal component analysis (PCA, first 3 principal components labeled) plot show that 16p11.2del (purple) and CHD8+/− (orange) cases are mixed with both heterogeneous ASD cases (gray) and controls (green). Data are normalized for visualization (mean = 0, variance = 1). In heat map, yellow represents high methylation and blue represents low methylation
Identification of a 16p11.2del-specific DNAm signature
Following filtering by variance (24,009 sites remaining), comparison of the DNAm data from the training 16p11.2del group (n = 9; Table 1) to our age-, sex-matched controls (n = 23) showed that our stratification approach allowed cases to be distinguished from controls based on DNAm differences at specific genomic sites. These distinctions are demonstrated by both hierarchical clustering and PCA plot (Fig. 2a). One hundred fifteen sites (77 (67%) hypomethylated, 38 (33%) hypermethylated) distributed across the genome passed our criteria of q < 0.05 and |Δβ| ≥ 5% (|Δβ| range 8–23%). This set of sites constitutes the 16p11.2del DNAm signature (Additional file 2: Figure S2; Additional file 3: Table S6); none of these 115 sites are contained in the 16p11.2del region.
Fig. 2.

a, b DNAm signatures identified in whole blood of individuals with 16p11.2del (600 kb risk locus) or CHD8+/−. a Hierarchical clustering and PCA plot (first 3 principal components labeled) show that 16p11.2del training cases (purple; n = 9) are distinct from age-, sex-matched controls (green; n = 23) at the DNAm signature sites (115 CpG sites; q < 0.05, absolute methylation difference (|Δβ| ≥ 5%). b Hierarchical clustering and PCA plot (first 3 principal components labeled) show that CHD8+/− training cases (orange; n = 7) are distinct from age-, sex-matched controls (green; n = 21) at DNAm signature sites (103 CpG sites; q < 0.01, |Δβ| ≥ 5%) used for classification. Only a single CpG site overlaps between the 16p11.2del and CHD8+/− DNAm signatures: cg25970491 (CLTCL1), which is hypomethylated in both CHD8+/− and 16p11.2del cases relative to controls. All data are normalized for visualization (mean = 0, variance = 1). In heat map, yellow represents high methylation and blue represents low methylation
Identification of a CHD8+/−-specific DNAm signature
We compared DNAm data from a group of individuals with known pathogenic CHD8 variants (n = 7) (Table 2) to age-, sex-matched controls (n = 21). Following filtering by variance (87,662 sites remaining), 422 sites (392 (92.9%) hypomethylated, 30 (7.1%) hypermethylated) distributed across the genome passed our criteria of q < 0.05, |Δβ| ≥ 5% (|Δβ| range 5–54%) (Additional file 3: Table S7) and distinguished all CHD8+/− training cases from controls. This set of sites, which constitutes the CHD8+/− DNAm signature, was used for downstream biological and classification analyses. Importantly, this CHD8+/− DNAm signature (422 sites) also accurately classified all CHD8 test case sequence variants (matching in silico predictions) and classified GEO controls with > 99.3% specificity. This DNAm signature classified all but one heterogeneous ASD case as clustering with controls (data not shown), suggesting the need to increase stringency of the signature for more accurate classification.
Therefore, we investigated whether the use of a more stringent filter of statistical significance (q < 0.01 and |Δβ| ≥ 5%) could be used to derive a more specific DNAm signature. Applying this filter reduced the previously derived DNAm signature to a subset of 103 CpG sites (95 (92.2%) hypomethylated, 8 (7.6%) hypermethylated) with a |Δβ| range of 5–18% (Additional file 2: Figure S3; Additional file 3: Table S7) that was used for classification purposes. Hierarchical clustering and PCA plot demonstrate that at this set of sites, cases are still distinguished clearly from controls (Fig. 2b), and that heterogeneous ASD cases and CHD8 test sequence variants are classified with even greater precision (Fig. 3c) (discussed in greater detail in the “Sensitivity and specificity of the CHD8+/−-specific DNAm signature” section).
Fig. 3.

a–d Evaluating the sensitivity and specificity of the 16p11.2del and CHD8+/− DNAm signatures. The classification models (details in Ref. [15]) are represented by the following classification plots: a The classification model based on the 16p11.2del DNAm signature sites (115 sites; q < 0.05, |Δβ| ≥ 5%) accurately classified CHD8+/− signature cases, and independent test cases consisting of CHD8 sequence variants (orange box with cross) and heterogeneous ASD cases (open gray triangle) as more similar to controls (green C). The model also accurately classified 7 of 9 independent 16p11.2del test cases (solid purple circle) as more similar to 16p11.2del signature cases (open purple circle), demonstrating 100% sensitivity. Two variants (arrows) correctly received negative classification scores: hatched arrow indicates a mosaic 16p11.2del case (2-0088-003), solid arrow indicates a 16p11.2del distal to and not overlapping the 600 kb typical deletion region (1-0616-003). b The 16p11.2del DNAm signature was tested on an independent set of compiled blood control DNAm data (n = 162) extracted from the Gene Expression Omnibus (GEO) Database. All GEO controls (turquoise C) were properly classified with experimental controls (green C), as distinct from 16p11.2del signature training cases (open purple circle), demonstrating 100% specificity. c The classification model based on the CHD8+/− DNAm signature sites for classification (103 sites; q < 0.01, |Δβ| ≥ 5%) accurately classified 16p11.2del signature cases and independent test cases consisting of 16p11.2del variants (purple circle with cross) and heterogeneous ASD cases (gray triangle) as more similar to controls (green C). The model also accurately classified 1 of 9 independent CHD8 test cases consisting of sequence variants (solid orange square) as more similar to CHD8+/− signature training cases (open orange square) (unable to report sensitivity with one positive case, indicated by arrow). Eight variants correctly received negative classification scores (7 missense, 1 with an in-frame deletion in the last exon of CHD8, not predicted pathogenic). Classifications are in agreement with in silico predictions. d The CHD8+/− DNAm signature was tested on the same GEO controls (turquoise C) as in b; all but one GEO control were properly classified with experimental controls (green C), as distinct from CHD8+/− signature cases (open orange square), demonstrating >99.3% specificity
Comparison of DNAm signatures with identified DMRs
Using an alternative analytic approach, we evaluated DMRs to detect regional DNAm differences and to confirm consistent patterns overlapping the significant CpG sites identified in our DNAm signatures. Significant DMRs were defined by p < 0.05, |Δβ| ≥ 5% and at least 3 consecutive CpG sites, of which at least one has already been identified in the 16p11.2del (115 sites) and CHD8+/− (422 sites) DNAm signature CpG sites (Additional file 3: Tables S3 and S4). DMRs found in the case-control comparisons yielded 26 and 70 overlapping CpG sites with the 16p11.2del and CHD8+/− DNAm signatures, respectively. Representative examples of DMR bumps that overlap DNAm signature sites are depicted in Additional file 2: Figure S4.
The 16p11.2del and CHD8+/− DNAm signatures are not confounded by blood cell type
We have taken blood cell type proportion estimates into account as a possible confounding factor by including them as covariates in the derivation of each DNAm signature. To further demonstrate that differences in cell type proportion do not contribute to the differences identified in our 16p11.2del and CHD8+/− DNAm signatures, we show that all normal purified blood cell types from the study by Reinius et al., 2012 [41] (data available from GEO35069) are correctly classified as controls by both signatures (similarly applied in Ref. [14, 15]). None of the methylation profiles for any of the specific blood cell types cluster with or near the signature cases at each respective signature, indicating that cell type-specific methylation profiles are not driving the differences identified at signature sites. Therefore, our DNAm signatures are not confounded by DNAm variation associated with blood cell types (Additional file 1; Additional file 2: Figures S5 and S6).
Sensitivity and specificity of the 16p11.2del-specific DNAm signature
We used the 16p11.2del DNAm signature to classify 9 additional 16p11.2del test case variants. For these cases, 7 of 9 received positive scores. These 7 cases receiving positive scores all carried typical 16p11.2del, clustering closely with signature cases (Fig. 3a; Table 3). Two of 9 cases received negative scores, appearing either on the diagonal border (2-0088-003) or clustering more closely with controls (1-0616-003) (Fig. 3a; Table 3). One of these two intermediate cases is known to be mosaic for the 16p11.2del (referred to as MM0088-003 in Ref. [32]), explaining its borderline position. The second case has a 200 kb deletion distal to the typical deletion region. Thus, our 16p11.2del DNAm signature demonstrates 100% sensitivity, accurately detecting all 7 variants with typical 16p11.2del. The 16p11.2del signature also accurately classified a larger set of independent GEO control subjects (n = 162), clustering them closely with controls, thus demonstrating 100% specificity (Fig. 3b; Additional file 3: Table S5). All CHD8+/− cases were classified as negative using the 16p11.2del DNAm signature.
Sensitivity and specificity of the CHD8+/−-specific DNAm signature
We used the more stringent, 103 CpG site subset of the CHD8+/− DNAm signature to classify additional CHD8 test case sequence variants (n = 13: n = 9 samples using 450K arrays, n = 4 using EPIC arrays) in order to predict pathogenicity. These classifications were compared with results from in silico prediction algorithms: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Assessor (http://mutationassessor.org), SIFT (http://sift-dna.org), Mutation Taster (http://www.mutationtaster.org). One test case (450K) (1-0559-003) received a positive score, classifying it as pathogenic, clustering more closely with the CHD8+/− training signature cases (Fig. 3c; Table 3). This case, which has a de novo heterozygous frameshift nonsense mutation in CHD8, was also classified as pathogenic by Mutation Taster, the only predictor available due to the nature of the variant. All 8 of the other CHD8 test case variants run on the 450K (including VUS) received negative scores, classifying as benign and clustering with controls (Fig. 3c; Table 3). For these 8 CHD8 sequence variants, 5 were missense/synonymous mutations inherited from an unaffected parent (inheritance unknown for 3 samples) and one was an in-frame deletion in the last exon. At least three out of four in silico analyses supported the benign designation of these CHD8 variants. All but one GEO control sample were classified correctly, clustering with controls, thus demonstrating > 99.3% specificity (Fig. 3d; Additional file 3: Table S5). Classification of the 4 CHD8 test cases run on the EPIC array was performed using only the overlapping sites from the 450K DNAm signature (92/103 sites) (Fig. 4). These 92 sites classified 3 test cases as pathogenic, clustering more closely with signature cases, and one VUS as benign, clustering with controls. The 3 test cases receiving positive scores were all known to have pathogenic CHD8+/− variants. The one sample classified as benign was an inherited missense variant, similar to those 450K test cases receiving negative scores. All 16p11.2del cases were classified as negative using the CHD8+/− DNAm signature.
Fig. 4.
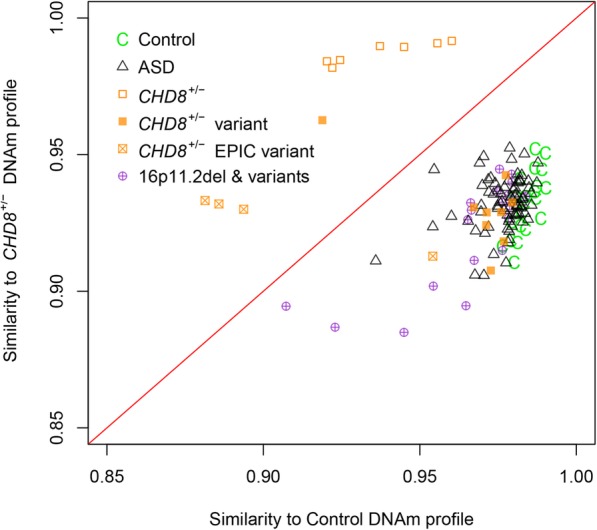
Evaluating the sensitivity of the CHD8+/− DNAm signature using additional CHD8 test cases (EPIC array). Additional independent test cases consisting of CHD8 sequence variants (n = 4; orange squares with cross) were run on the EPIC array. Of the 103 sites in the 450K-derived CHD8+/− DNAm classification signature, 92 sites overlapped those of the EPIC array and were thus used to classify the additional EPIC test cases. One VUS case received a negative classification score. Three of the four additional CHD8 test cases were accurately classified as more similar to CHD8+/− signature training cases (open orange square); these cases were known to have pathogenic CHD8 variants, demonstrating 100% sensitivity. The one sample classified as benign was an inherited missense variant, similar to those 450K test cases receiving negative scores. All other cases and controls (450K) were accurately classified as in Fig. 3
All the heterogeneous ASD cases (n = 52) received negative scores when classified with the 16p11.2del or CHD8+/− DNAm signature used for classification, and all clustered with controls (Fig. 3a, c).
Genomic distribution of DNAm signature CpG sites
CpG sites in the 16p11.2del DNAm signature (115 sites) were found to be significantly enriched in CpG islands, shelves, shores, promoters, and DNase hypersensitivity sites (DHS), and depleted in shelves (p < 0.05) (Additional file 2: Figure S7A). Sites in the 422 probe CHD8+/− DNAm signature were enriched in islands, shores, particularly the “south” shores (S_Shore) directly downstream of CpG islands, differentially methylated regions (DMRs) and DHS, and depleted in shelves and reprogramming-specific DMRs (RDMRs) (p < 0.05); whereas the 103 site CHD8+/− DNAm signature used for classification was enriched in shores, particularly in S_Shore, promoters, and DMRs (Additional file 2: Figure S7B, C).
Biological significance of the 16p11.2del and CHD8+/− signatures
Using GREAT 3.0.0, we identified molecular and biological processes potentially relevant to the phenotype of individuals with 16p11.2del or CHD8+/− using the differentially methylated CpGs from each respective DNAm signature. When we examined the 16p11.2del signature (115 sites), 3 of 5 GO Biological Process terms with significant enrichment (hypergeometric FDR q-value < 0.05) appeared in categories related to immune function (Additional file 3: Table S8). When we examined the CHD8+/− DNAm signature (422 sites), > 20 terms passed with a more stringent hypergeometric FDR q-value < 0.05 (Additional file 3: Table S9). The top 20 terms showed enrichment not only in immune processes, but also in the regulation of inhibitory postsynaptic membrane potential and central nervous system development. Differentially methylated genes specific to 16p11.2del and CHD8+/− signatures overlap with 1 and 10 SFARI Gene ASD-risk genes, respectively (Fig. 5a). The clathrin, heavy chain-like 1 (CLTCL1) gene was hypomethylated in both signatures.
Fig. 5.
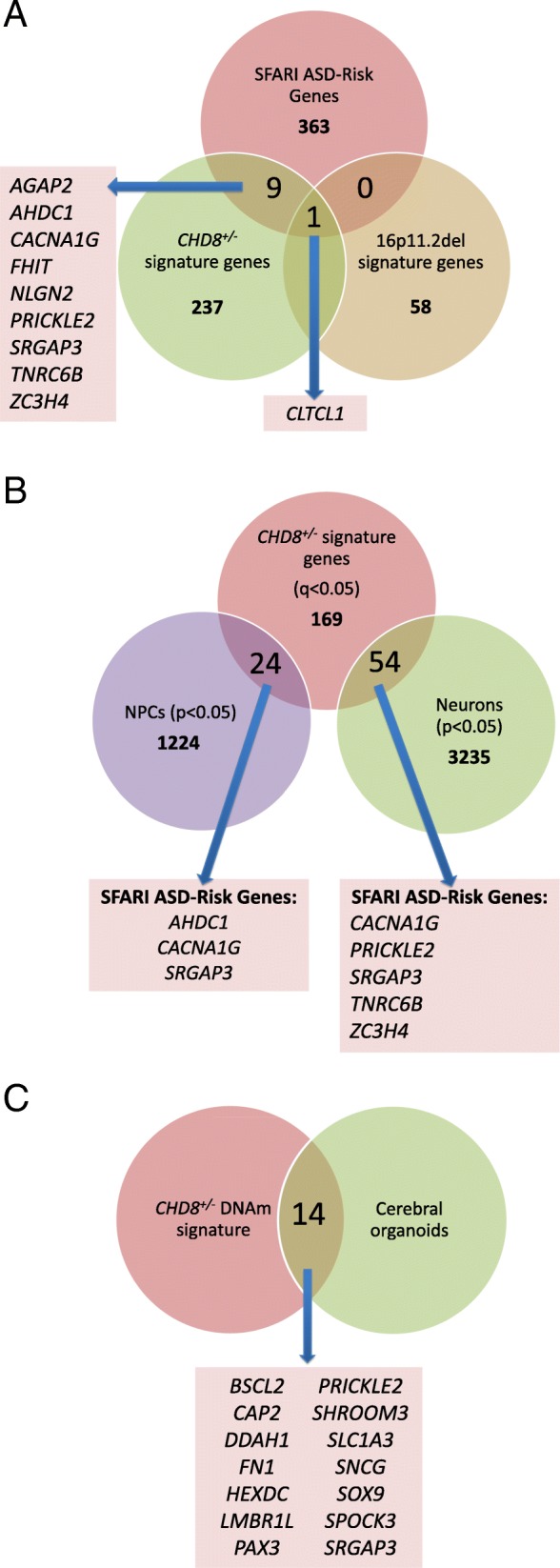
Biological and cross-tissue functional significance of differentially methylated genes. a Differentially methylated genes associated with the 16p11.2del and CHD8+/− DNAm signatures (q < 0.05, |Δβ| ≥ 5%) and the DNAm signature overlap with known ASD-risk genes (SFARI Gene). A single SFARI gene, clathrin heavy chain like 1 (CLTCL1), is significantly hypomethylated in both groups. An independent set of 9 SFARI genes are differentially methylated in the CHD8+/− DNAm signature. b Our findings are further corroborated by an independent study (48) showing that CHD8+/− human iPSC-derived neuronal precursor cells (NPCs) and differentiated neurons result in differentially expressed genes that overlap with some of the differentially methylated genes in CHD8+/− DNAm signature, including known ASD-risk genes (SFARI Gene), and c another study (49) showing that cerebral organoids derived from the iPSCs in b also have differentially expressed genes that overlap our CHD8+/− DNAm signature.
Cross-tissue functional relevance of the CHD8+/− signature
To examine whether DNAm changes downstream of sequence variants in CHD8 in blood are relevant to neuronal function, we compared the overlap between genes in the CHD8+/− DNAm signature to differentially expressed genes (DEGs) in human CHD8+/− iPSC-derived NPCs and neurons (48). Differentially expressed genes in NPCs and neurons overlapped 24 (hypergeometric p value < 0.001) and 54 (hypergeometric p value < 0.001) of our 247 differentially methylated genes in the DNAm signature (q < 0.05, |Δβ| ≥ 5%), respectively (Fig. 5b). Furthermore, 6 of these overlapping genes also overlapped SFARI ASD-risk genes.
More recently, Wang et al. [49] further derived cerebral organoids from their original CHD8+/− iPSCs. These organoids are composed of GABAergic/glutamatergic neurons, and radial glia progenitor cells with gene expression profiles that resemble first-trimester telencephalon [49–52]. The CHD8+/− DNAm signature overlapped 14 DEGs found in the CHD8+/− cerebral organoids (Fig. 5c); 6 of these overlapped DEGs found in both organoids and neurons, and 5 of these overlapped DEGs found in both organoids and NPCs.
Validation of differentially methylated CpG sites with targeted pyrosequencing
Specific CpG sites were selected from the DNAm signatures for validation by targeted pyrosequencing (Additional file 2: Figure S4). These sites overlapped a significant DMR that contained at least two sites within a single gene. For 16p112del cases, the following CpG sites were validated: cg00108944, cg23588049 in GLIPR1L2, cg25983544, and cg06377543 in PSMA8. In the CHD8+/− group, the following CpG sites were validated: cg09819656, cg15089111 in NPAS3, cg27206976, and cg04089788 in PLXNB2. In the 16p11.2del sites, an average gain of methylation (GOM) by 14.5% and 13% at cg00108944 and cg23588049 in GLIPR1L2, respectively, and loss of methylation (LOM) by 7.4% and 10.5% at cg25983544 and cg06377543 in PSMA8, respectively were confirmed by our pyrosequencing analysis (p < 0.005). In the CHD8+/− sites, an average GOM by 17% and 18.8% at cg09819656 and cg15089111 in NPAS3, respectively, and LOM by 11.4% and 18.9% at cg27206976 and cg04089788 in PLXNB2, respectively, were confirmed (p < 0.005). The correlation between methylation β values from the 450K array and % methylation values obtained through pyrosequencing were examined and were found to be highly concordant (r = 0.8524) (Additional file 2: Figure S8).
Discussion
One of the greatest challenges of epigenetic research in ASD is the degree of etiologic and phenotypic heterogeneity. This study uniquely demonstrates that investigating homogeneous subgroups of individuals with genomic alterations at ASD-risk loci involving epigenes enables the identification of distinct functionally and biologically relevant DNAm signatures. Previously, evaluations of DNAm in individuals with ASD have focused on heterogeneous ASD cases in different tissue types using a variety of different methods. These studies of modest sample sizes (n < 50) have identified few, mostly non-overlapping DNAm alterations of small magnitude [24, 26–29, 31].
Heterogeneous ASD cases, when compared with controls, did not identify significant (i.e., corrected for multiple testing) differentially methylated sites, paralleling the aforementioned outcomes of DNAm studies in ASD [24, 26–31]. We expect that the identification of significant epigenetic differences in such a mixed cohort will require large numbers (> 1000) of ASD cases. We therefore undertook a novel alternative approach to identifying etiologically relevant epigenetic alterations in ASD. In contrast to the current literature, we sought to reduce genetic and epigenetic heterogeneity by focusing analyses on groups of individuals with genomic alterations involving epigenes that confer an increased risk for ASD. We show that a relatively small number (< 10) of genetically homogeneous cases can be clearly distinguished from both a heterogeneous ASD group and neurotypical controls based on DNAm. In the future, these findings will need to be replicated using larger cohorts of cases with a wider range of risk-associated variants. Given the expected genomic heterogeneity for ASD, we did not expect that our heterogeneous cohort of ASD patients would contain any individuals with either 16p11.2del or CHD8+/− pathogenic variants as each of these individually account for < 1% of all ASD cases. We detected consistent patterns of DNAm alterations in the 16p11.2del and CHD8+/− blood DNAm signatures by identifying overlapping DMRs. In addition, we validated the top differentially methylated CpG sites in the DNAm signatures by targeted bisulfite pyrosequencing. We demonstrate that our DNAm signatures are robust, overcoming potential confounding factors such as sex, age, blood cell type proportions, and technical variation, illustrated by our classification of a large number of independently assessed GEO control blood DNAm samples. Some additional confounding factors to consider include ethnicity and environmental exposures such as medications, prenatal vitamins, and smoking. Data for these parameters were not collected consistently across our cases or controls, precluding us from incorporating these factors into our models. However, given that our signatures yielded 100% sensitivity and > 99% specificity for both signatures, despite a wide variety of potentially confounding variables, we expect that our signatures are robust enough to overcome these additional confounders. Further, we were able to demonstrate cross-platform applicability of our CHD8+/− DNAm signature derived on the 450K array. Using this signature, we accurately classified 4 CHD8 sequence variant test cases that were run on the EPIC array.
It is important to recognize that DNAm signatures are dynamic; their stability and robustness depend on the size and composition of the group of signature cases used to derive the signature. DNAm signatures can be optimized in various ways depending on the specific purposes for which they are to be utilized. For example, one signature may provide more information for the examination of underlying biological pathways of disease, whereas classification of sequence variants in a clinical diagnostic setting may require a more refined signature to optimize sensitivity and specificity. DNAm signatures can be further optimized to reduce type I error inflation, which may confound the performance of a signature in a classification model (see Additional file 1). Although we were only able to test our signatures on a small group of test cases, we were able to demonstrate 100% sensitivity. Refining a signature for clinical diagnostic application will require a larger discovery cohort with a wider range of variant types and genomic locations (e.g., alterations across different functional domains) to improve classification performance and to better understand their impact on pathogenicity and functional consequences.
The identification of the 16p11.2del and CHD8+/− DNAm signatures highlights both the scientific relevance and the potential for clinical utility. Highly sensitive and specific genome-wide DNAm signatures have the potential to be used to identify individuals with pathogenic variants among groups of heterogeneous ASD cases, an approach currently in development for molecular diagnostics. As well, our DNAm signatures are able to facilitate the interpretation of VUS of the respective CNV or epigene variants detected using sequencing/array-based diagnostics. This particular application underscores the potential clinical utility of this molecular classification system beyond simply genotyping using NGS technologies. Given that there are already gene-specific panels available to test for variants in ASD-risk populations, we propose that in the future, a parallel panel could be used as a complementary second-tier diagnostic test that would employ various ASD risk-specific DNAm signatures. Finally, differentially methylated genes that constitute the downstream genomic targets of ASD-associated CNVs and sequence variants in epigenes elucidate the biological and molecular pathophysiology of ASD and identify potential therapeutic targets. Expanding this work in the future could also lead to the identification of biomarkers for earlier prediction of neurodevelopmental outcomes, which would be of tremendous value given the variable penetrance of ASD with each ASD-risk locus. Although our current study does not have sufficient statistical power to accomplish this, the future study of large numbers of cases with 16p11.2del and CHD8 sequence variants in conjunction with deep clinical phenotyping may enable the identification of more refined epigenotype-phenotype correlations. Further investigation will also be required to better understand the impact of altered methylation on downstream targets for each DNAm signature in a cell type-, tissue-, and developmental time-specific manner.
Genes and pathways relevant to the clinical phenotypes of individuals with 16p11.2del or CHD8+/− are elucidated by their respective DNAm signatures. Interestingly, the single gene, CLTCL1, found to be similarly hypomethylated in both the 16p11.2del and CHD8+/− signatures, is found in the 22q11.2 deletion syndrome (22qDS; OMIM# 611867) deletion region. 22qDS is associated with a range of clinical phenotypes, but notably, several neurodevelopmental (ASD, ADHD), and neuropsychiatric (schizophrenia) outcomes [53]. In the 16p11.2del signature, multiple sites in GLI pathogenesis-related 1 like 2 (GLIPR1L2; 8 hypermethylated sites, |Δβ| > 10%) and proteasome subunit alpha 8 (PSMA8; 3 hypomethylated sites, |Δβ| > 8%) were found to be significantly differentially methylated. GLIPR1L2 and PSMA8 have not previously been associated with ASD, but encode proteins that function in immunity and histone degradation, respectively. The CHD8 gene is known to negatively regulate beta catenin (CTNNB1) and WNT pathways, both important for development and growth, through the direct interaction between CHD8 and CTNNB1. Our study extends the current knowledge of downstream targets regulated by CHD8 through its role as a chromatin remodeler. The genes with the greatest differential DNAm in our CHD8+/− signature highlight pathways that are relevant to the pathophysiology of ASD. These include Plexin B2 (PLXNB2; 4 hypomethylated sites, |Δβ| > 12%) and neuronal PAS domain protein 3 (NPAS3; 2 hypermethylated sites, |Δβ| > 15%), both of which are expressed in the brain during embryonic development [54, 55]. PLXNB2 encodes a transmembrane receptor protein that participates in axon guidance and is required for normal embryonic development/differentiation and migration of neuronal cells during brain corticogenesis [54, 56]. PLXNB2 is deleted in the Phelan-McDermid, 22q13.3 deletion syndrome region, which is associated with dysmorphic features, neurologic deficits and ASD. This overlap could suggest that epigenetic dysregulation of shared biological pathways across disorders is reflected in overlapping clinical features. This has been demonstrated in the case of CHARGE and Kabuki syndromes, two clinically distinct syndromes sharing a subset of phenotypes [14]; each syndrome has a distinct DNAm signature, each of which also includes some overlapping sites affecting genes involved in pathways relevant to specific concordant syndromic features. Mouse models with Plxnb2 deletions are perinatal lethal and have abnormal cerebellar development, reflecting the critical role of the gene in neurodevelopment [56]. NPAS3, expressed by GABAergic interneurons, has been shown to be an important transcription factor during neurogenesis; chromosomal aberrations encompassing this gene are associated with schizophrenia and mental retardation [55, 57]. The finding that immune-related genes are enriched in both the 16p11.2del and CHD8+/− signatures supports recent data highlighting contributions of altered immune function to ASD pathophysiology [58–60]. Individuals with 16p11.2del or CHD8+/− do not demonstrate consistent immune phenotypes; however, the dysregulation of immune genes could mediate changes in early embryogenesis that may affect neurogenesis and subsequently ASD risk. Immune dysregulation likely only contributes to the pathophysiology of disease for a subset of individuals with ASD, rather than all individuals. Importantly, the identification of these pathways could help to identify targets for potential drug therapies and facilitate more individualized interventions.
We searched for overlap between differentially methylated sites in our 16p11.2del and CHD8+/− blood DNAm signatures and previous DNAm studies of ASD. Two differentially methylated genes (DNPEP, RAD50) in the CHD8+/− signature overlapped with the single 450K array study of DNAm in the blood of individuals with ASD [29]. Differentially methylated genes identified in the brain regions Brodmann areas (BA) 10 and 24 of the cerebral cortex (27) overlap a number of differentially methylated DNAm signatures genes in our study: 16 genes in the 16p11.2del signature, 51 genes in the CHD8+/− signature in BA10 and 22 genes in the 16p11.2del signature, and 82 genes in the CHD8+/− signature in BA24. A more recent study also examining DNAm in the postmortem brain (prefrontal and temporal cortices, cerebellum and cross-cortical analysis) in individuals with idiopathic ASD identified region-specific significant differentially methylated sites [30]. Although none of our signature sites overlapped those identified in Wong et al. [30], both studies identified pathway enrichment in immune and neuronal processes, which were found using two different analytical pipelines (GREAT in our study, weighted gene co-methylation network analysis in Ref. [30]). These data suggest that certain DNAm alterations found in blood are likely to be reflected in the brain or in genes of related biological pathways, which is of particular relevance for research in neurodevelopmental disorders (NDDs). Although ASD is an NDD with heterogeneous etiology(ies) and phenotypes, where the brain is undoubtedly an important organ to study, the pathophysiology of ASD is still unclear, affecting multiple systems (e.g., gastrointestinal) and pathways (e.g., metabolic, immune). DNAm in blood also reflects environmental exposures (e.g., gestational alcohol exposure). It is important to distinguish between the different potential utilities of DNAm signatures. From a biomarker perspective, whether genes identified by a DNAm signature in blood can be causally linked to the disease phenotype is less important than identifying DNAm marks that predict the phenotype in a stable, sensitive, and specific manner. From a biological perspective, an increasing number of studies do in fact demonstrate an intersection between DNAm patterns in the blood and brain using similar genome-wide assessment platforms [61–63], which may help to elucidate underlying pathophysiological mechanisms. Our findings in blood are further corroborated by an independent study [48, 49] showing that CRISPR/Cas9-mediated creation of CHD8+/− human iPSC-derived NPCs, differentiated neurons, and cerebral organoids results in differentially expressed genes that overlap with some of the differentially methylated genes in our CHD8+/− DNAm signature (Fig. 5b, c), including known ASD-risk genes (SFARI Gene).
It is possible that epigenetic marks other than DNAm at CpG sites, such as 5-hydroxymethylcytosine (5-hmC), may also demonstrate differences between individuals with ASD and neurotypical controls. 5-hmC is abundant in the brain and not in the blood [64, 65] and is yet to be explored with respect to ASD in humans, although mouse models of ASD have demonstrated genome-wide differences [66]. A potential role for altered histone acetylation in ASD has been demonstrated in a recent human brain histone acetylome study [67]. Animal studies have more extensively investigated the effects of in utero exposure to a histone deacetylase inhibitor, valproic acid, on autistic-like behavioral and cognitive outcomes in offspring [68–70]. Further research exploring downstream molecular functional studies such as gene and protein expression are also necessary to fully understand the functional consequences of either the ASD-associated CNV or gene variants of interest in our study. Nevertheless, our DNAm signatures represent novel DNA biomarkers in an easily accessible tissue (blood) that are more stable than histone marks and present novel clinical diagnostic applications. Our DNAm signatures include alterations in genes that may be important for understanding the underlying pathophysiology of ASD and demonstrate parallel relevance in gene expression profiles within cell types representative of the brain.
Conclusions
In summary, we present a novel approach that enhances the detection of epigenetic dysregulation in ASD etiology. Our successful identification of highly sensitive and specific CNV/gene mutation-specific DNAm signatures provides significant opportunities for clinical impact, offering more precise ASD subgroup molecular classification with the potential for developing diagnostic and predictive biomarkers. Our investigations provide a more comprehensive understanding of the molecular landscape underlying genetically homogeneous groups of ASD, illustrating the convergence of epigenetic and genetic mechanisms. These findings will also help to further elucidate the molecular pathophysiology of ASD and facilitate the identification of novel therapeutic targets to support precision therapies for individuals with ASD.
Additional files
Supplementary information. (DOCX 18 kb)
Figure S1. Data analysis flowchart. Outline of the training cohorts and analysis pipeline used to derive DNAm signatures and test cohorts (independent samples), on which the DNAm signatures were tested. Figure S2. Identification of differentially methylated CpG sites when comparing 16p11.2del signature cases with age-, sex-matched controls. Significant sites for the DNAm signature for 16p11.2del represented by the overlap from limma regression and Mann Whitney U analysis are shown. Each volcano plot represents the distribution of significant sites at various statistical parameters (y-axis is negative log q-value, Benjamini-Hochberg corrected) and Δβ differences (x-axis is the average difference in DNAm between 16p11.2del and controls). Red horizontal line represents q < 0.05, red vertical lines represent |Δβ| ≥ 5%. Figure S3. Identification of differentially methylated CpG sites when comparing CHD8+/− signature cases with age-, sex-matched controls. Significant sites for the DNAm signature for CHD8+/− represented by the overlap from limma regression and Mann Whitney U analysis are shown. Each volcano plot represents the distribution of significant sites at various statistical parameters (y-axis is negative log q-value, Benjamini-Hochberg corrected) and Δβ differences (x-axis is the average difference in DNAm between CHD8+/− and controls). Red horizontal line represents q < 0.05, red vertical lines represent |Δβ| ≥ 5%. Figure S4. Targeted sodium bisulfite pyrosequencing of selected 16p11.2del and CHD8+/− DNAm signature CpG sites overlapping differentially methylated regions (DMRs). Specific CpG sites from our DNAm signatures that overlapped DMRs found using bump hunting were selected for targeted sodium bisulfite pyrosequencing validation in signature cases and age-, sex-matched controls. In the 16p11.2del group, the following CpG sites were validated: A) cg00108944 and cg23588049 in GLIPR1L2 showing a gain of methylation, B) cg25983544 and cg06377543 in PSMA8 showing a loss of methylation. In the CHD8+/− group, the following CpG sites were validated: C) cg09819656 and cg15089111 in NPAS3 showing a gain of methylation, D) cg27206976 and cg04089788 in PLXNB2 showing a loss of methylation. Upper graphs in each panel depict mean methylation (y-axis) over each DMR (x-axis, position of CpG sites along the chromosome) for the 16p11.2del (purple), CHD8+/− (orange) and control (green) groups. Box plots represent median methylation at the specific CpG sites indicated with min/max whiskers. Figure S5. Correlation of pyrosequencing and array data. We demonstrate that there is high correlation (r = 0.8524) between the bisulfite pyrosequencing validation % methylation and array methylation beta (β) values. Data include all samples run at all 8 of the specific CpG sites assayed for validation. Figure S6. Comparison of the 16p11.2del DNAm signature with blood cell type composition. Blood cell type DNAm data was extracted from Reinius et al. [41] (GEO series GSE35069) representing whole blood, peripheral blood mononuclear cells, granulocytes and isolated cell populations (CD4+ T cells, CD8+ T cells, CD56+ NK cells, CD19+ B cells, CD14+ monocytes) from 6 different control individuals. Blood cell type data was combined with our signature cases and control data using only the CpGs from the 16p11.2del-specific DNAm signature. The PCA plot demonstrates that the 16p11.2del signature is independent of blood cell type composition where signature cases remain separated from controls cases (whole blood and purified blood cell types). Abbreviations: PBMC peripheral blood mononuclear cells; WB whole blood. Figure S7. Comparison of the CHD8+/− DNAm signature with blood cell type composition. Blood cell type DNAm data was extracted from Reinius et al. (41) (GEO series GSE35069) representing whole blood, peripheral blood mononuclear cells, granulocytes and isolated cell populations (CD4+ T cells, CD8+ T cells, CD56+ NK cells, CD19+ B cells, CD14+ monocytes) from 6 different control individuals. Blood cell type data was combined with our signature cases and control data using only the CpGs from the CHD8+/−-specific DNAm signature. The PCA plot demonstrates that the CHD8+/− signature is independent of blood cell type composition where signature cases remain separated from controls cases (whole blood and purified blood cell types). Abbreviations: PBMC peripheral blood mononuclear cells; WB whole blood. Figure S8. Genomic distribution of DNAm signature CpG sites. Bar charts representing the genomic distribution of CpG sites in the 16p11.2del (A) and CHD8+/−-specific DNAm signatures (B, C [classification signature]), according to Illumina 450K array annotations. In mammalian cells, DNAm occurs predominantly on cytosines located in CpG dinucleotides. CpG islands refer to regions that are especially CpG-rich. Specific locations of CpGs relative to the CpG island can be denoted as “shores” and “shelves”. A shore is 0–2 kb from a CpG island, a shelf is 2–4 kb from a CpG island. N refers to regions upstream (5′) and S refers to regions downstream (3′) of a CpG island. We compared the distribution of the CpGs between each respective variance filtered dataset with each signature training case group for the regulatory feature group, relation to CpG island and other functional categories such as overlapping enhancer region, DNase hypersensitive sites (DHS) and type of differentially methylated regions (Reprogrammed DMR (RDMR), cancer-specific DMR (cDMR), other DMR) in addition to relation to RefSeq group annotation. N refers to north and S refers to south. Asterisk denotes significant enrichment or depletion by hypergeometric test comparison (p < 0.05). Figure S9. The quantile-quantile plot showing the log-transformed distributions of limma regression p-values associated with the DNAm differences in CHD8+/− cases compared to matching controls. The results are shown before (left) and after (right) correction by the BACON method, with inflation patterns evident in both cases. Figure S10. The quantile-quantile plot showing the log-transformed distributions of limma regression p-values associated with the DNAm differences in 16p11.2del cases compared to matching controls. The results are shown before (left) and after (right) correction by the BACON method, with inflation patterns evident in both cases. (PPTX 2477 kb)
Table S1. Demographic information for heterogeneous ASD cases and age- and sex-matched neurotypical controls. Table S2. Number of probes removed and remaining for analysis following quality control. Table S3. List of overlapping DMRs for 16p11.2del. Table S4. List of overlapping DMRs for CHD8+/−. Table S5. List of samples extracted from GEO database and respective 16p11.2del and CHD8+/− signature scores for specificity analysis. All sample data are from blood and from individuals with known ages < 50 years old. Table S6. List of significant (q < 0.05, |Δβ| ≥ 5%) sites in 16p11.2del DNAm signature. Table S7. List of significant (q < 0.05, |Δβ| ≥ 5%) sites in CHD8+/− DNAm signature. Table S8. GO Biological Process enrichment terms following GREAT enrichment analysis of differentially methylated genes in 16p11.2del DNAm signature. Table S9. GO Biological Process enrichment terms following GREAT enrichment analysis of differentially methylated genes in CHD8+/− DNAm signature. (XLSX 156 kb)
Acknowledgements
We would like to thank all of the patients and families for participating in our research studies and the physicians, genetic counselors, and clinical staff for their assistance with patient recruitment. We would also like to thank Khadine Wiltshire for her valuable contributions to this work. We thank Dr. Greg Hanna (University of Michigan) for contributing blood DNA samples from neurotypical control individuals who had received cognitive/behavioral assessments. These samples had previously been obtained as part of an independent study as described in Ref. [71].
Abbreviations
- 16p11.2del
16p11.2 deletion
- ASD
Autism spectrum disorder
- BA
Brodmann area
- cDMR
Cancer-specific DMRs
- CHARGE
Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and development, and Ear abnormalities and deafness syndrome
- CHD7
Chromodomain helicase DNA-binding protein 7 gene
- CHD8
Chromodomain helicase DNA-binding protein 8 gene
- CLTCL1
Clathrin, heavy chain-like 1 gene
- CNV
Copy number variant
- CTNNB1
Beta catenin
- DEG
Differentially expressed gene
- DHS
DNase hypersensitivity site
- DMR
Differentially methylated region
- DNAm
DNA methylation
- GEO
Gene Expression Omnibus database
- GLIPR1L2
GLI pathogenesis-related 1 like 2 gene
- HIRIP3
HIRA-interacting protein 3 gene
- iPSC
Induced pluripotent stem cell
- NPAS3
Neuronal PAS domain protein 3 gene
- NPC
Neuronal precursor cells
- NSD1
Nuclear receptor binding SET domain protein 1 gene
- PCA
Principal component analysis
- PLXNB2
Plexin B2 gene
- PSMA8
Proteasome subunit alpha 8 gene
- QOE
Qlucore Omics Explorer
- RDMR
Reprogramming-specific DMR
- VUS
Variant of unknown significance
- WB
Whole blood
- Δβ
Effect size of differential DNA methylation
Authors’ contributions
MTS and ALT analyzed the data and drafted the manuscript. DTB, SC, and MB helped with project design, data analysis, and manuscript revision. CC provided clinical input and helped with manuscript revisions. DJS, SW, and SWS collected patient samples and performed genome-wide sequencing to identify variants. YL performed bisulfite conversion and targeted bisulfite pyrosequencing analysis. RN, SG, PS, and EA comprise the executive committee of the POND Network, which provided patient cohorts and phenotype data. RW is the principal investigator responsible for project design, data analysis, and manuscript revision. All authors read and approved the final manuscript.
Funding
This research was funded by the Canadian Institutes of Health Research, The Rare Diseases Foundation, the Serruya Family Endowment Fund, and the Ontario Brain Institute (OBI). OBI is an independent non-profit corporation, funded partially by the Ontario government. The opinions, results, and conclusions are those of the authors, and no endorsement by the Ontario Brain Institute is intended or should be inferred. Bioinformatic analyses were supported in part by the Canadian Centre for Computational Genomics (C3G), part of the Genome Technology Platform (GTP), funded by Genome Canada through Genome Quebec and Ontario Genomics (ALT, MB), and Genome Canada through Ontario Genomics (ALT, SC, MB, and RW). We would like to acknowledge the support of the Ontario Research Fund, and the Centre for Applied Genomics (SickKids). Dr. Stephen W. Scherer holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and SickKids.
Availability of data and materials
The datasets used for the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All participants were enrolled and consented in studies approved by the Research Ethics Boards of the respective institutions (University of Michigan, SickKids, Holland Bloorview Kids Rehabilitation Hospital, Western University, McMaster University).
Competing interests
The authors declare they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.CDC . Centers for Disease Control and Prevention. 2014. [Google Scholar]
- 2.Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine. 2015;21(2):185–191. doi: 10.1038/nm.3792. [DOI] [PubMed] [Google Scholar]
- 3.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nature reviews Genetics. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. American journal of human genetics. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CY RK, Merico D, Bookman M, LH J, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20(4):602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter MT, Scherer SW. Autism spectrum disorder in the genetics clinic: a review. Clin Genet. 2013;83(5):399–407. doi: 10.1111/cge.12101. [DOI] [PubMed] [Google Scholar]
- 8.Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- 9.Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903. doi: 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 10.Hoang Ny, Cytrynbaum Cheryl, Scherer Stephen W. Communicating complex genomic information: A counselling approach derived from research experience with Autism Spectrum Disorder. Patient Education and Counseling. 2018;101(2):352–361. doi: 10.1016/j.pec.2017.07.029. [DOI] [PubMed] [Google Scholar]
- 11.Lasalle JM. Autism genes keep turning up chromatin. OA Autism. 2013;1(2):14. doi: 10.13172/2052-7810-1-2-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, et al. CHARGE and kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. American journal of human genetics. 2017;100(5):773–788. doi: 10.1016/j.ajhg.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, Chen YA, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207. doi: 10.1038/ncomms10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grafodatskaya D, Chung BH, Butcher DT, Turinsky AL, Goodman SJ, Choufani S, et al. Multilocus loss of DNA methylation in individuals with mutations in the histone H3 lysine 4 demethylase KDM5C. BMC Med Genomics. 2013;6:1. [DOI] [PMC free article] [PubMed]
- 17.Strong E, Butcher DT, Singhania R, Mervis CB, Morris CA, De Carvalho D, et al. Symmetrical dose-dependent dna-methylation profiles in children with deletion or duplication of 7q11.23. American journal of human genetics. 2015;97(2):216–227. doi: 10.1016/j.ajhg.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aref-Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, et al. Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. American journal of human genetics. 2018;102(1):156–174. doi: 10.1016/j.ajhg.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aref-Eshghi E, Schenkel LC, Lin H, Skinner C, Ainsworth P, Pare G, et al. The defining DNA methylation signature of kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics. 2017;12(11):923–933. doi: 10.1080/15592294.2017.1381807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hood RL, Schenkel LC, Nikkel SM, Ainsworth PJ, Pare G, Boycott KM, et al. The defining DNA methylation signature of Floating-Harbor Syndrome. Scientific reports. 2016;6:38803. doi: 10.1038/srep38803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kernohan KD, Cigana Schenkel L, Huang L, Smith A, Pare G, Ainsworth P, et al. Identification of a methylation profile for DNMT1-associated autosomal dominant cerebellar ataxia, deafness, and narcolepsy. Clinical epigenetics. 2016;8:91. doi: 10.1186/s13148-016-0254-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schenkel LC, Aref-Eshghi E, Skinner C, Ainsworth P, Lin H, Pare G, et al. Peripheral blood epi-signature of Claes-Jensen syndrome enables sensitive and specific identification of patients and healthy carriers with pathogenic mutations in KDM5C. Clinical epigenetics. 2018;10:21. doi: 10.1186/s13148-018-0453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schenkel LC, Kernohan KD, McBride A, Reina D, Hodge A, Ainsworth PJ, et al. Identification of epigenetic signature associated with alpha thalassemia/mental retardation X-linked syndrome. Epigenetics Chromatin. 2017;10:10. doi: 10.1186/s13072-017-0118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berko ER, Suzuki M, Beren F, Lemetre C, Alaimo CM, Calder RB, et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS genetics. 2014;10(5):e1004402. doi: 10.1371/journal.pgen.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain transcriptional and epigenetic associations with autism. PloS one. 2012;7(9):e44736. doi: 10.1371/journal.pone.0044736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Molecular psychiatry. 2014;19(8):862–871. doi: 10.1038/mp.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Translational psychiatry. 2014;4:e433. doi: 10.1038/tp.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24(8):3036–3051. doi: 10.1096/fj.10-154484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Molecular psychiatry. 2014;19(4):495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong Chloe C Y, Smith Rebecca G, Hannon Eilis, Ramaswami Gokul, Parikshak Neelroop N, Assary Elham, Troakes Claire, Poschmann Jeremie, Schalkwyk Leonard C, Sun Wenjie, Prabhakar Shyam, Geschwind Daniel H, Mill Jonathan. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Human Molecular Genetics. 2019;28(13):2201–2211. doi: 10.1093/hmg/ddz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feinberg JI, Bakulski KM, Jaffe AE, Tryggvadottir R, Brown SC, Goldman LR, et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. International journal of epidemiology. 2015;44(4):1199–1210. doi: 10.1093/ije/dyv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. Journal of medical genetics. 2010;47(3):195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 33.Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biological psychiatry. 2015;77(9):785–793. doi: 10.1016/j.biopsych.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. Journal of medical genetics. 2012;49(10):660–668. doi: 10.1136/jmedgenet-2012-101203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorain S, Quivy JP, Monier-Gavelle F, Scamps C, Lecluse Y, Almouzni G, et al. Core histones and HIRIP3, a novel histone-binding protein, directly interact with WD repeat protein HIRA. Mol Cell Biol. 1998;18(9):5546–5556. doi: 10.1128/mcb.18.9.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Merner N, Forgeot d’Arc B, Bell SC, Maussion G, Peng H, Gauthier J, et al. A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): a case report and literature review. Am J Med Genet A. 2016;170A(5):1225–1235. doi: 10.1002/ajmg.a.37566. [DOI] [PubMed] [Google Scholar]
- 38.Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol Cell Biol. 2008;28(12):3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourgon R, Gentleman R, Huber W. Independent filtering increases detection power for high-throughput experiments. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(21):9546–9551. doi: 10.1073/pnas.0914005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PloS one. 2012;7(7):e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. International journal of epidemiology. 2012;41(1):200–209. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li D, Xie Z, Pape ML, Dye T. An evaluation of statistical methods for DNA methylation microarray data analysis. BMC bioinformatics. 2015;16:217. doi: 10.1186/s12859-015-0641-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, R VL, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009;52(2-3):77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 46.Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 47.Barnard RA, Pomaville MB, O’Roak BJ. Mutations and modeling of the chromatin remodeler chd8 define an emerging autism etiology. Front Neurosci. 2015;9:477. doi: 10.3389/fnins.2015.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang P, Lin M, Pedrosa E, Hrabovsky A, Zhang Z, Guo W, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Molecular autism. 2015;6:55. doi: 10.1186/s13229-015-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Molecular autism. 2017;8:11. doi: 10.1186/s13229-017-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(51):15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin M, Zhao D, Hrabovsky A, Pedrosa E, Zheng D, Lachman HM. Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. PloS one. 2014;9(4):e94968. doi: 10.1371/journal.pone.0094968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(31):12770–12775. doi: 10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jonas RK, Montojo CA, Bearden CE. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biological psychiatry. 2014;75(5):351–360. doi: 10.1016/j.biopsych.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konopka G, Friedrich T, Davis-Turak J, Winden K, Oldham MC, Gao F, et al. Human-specific transcriptional networks in the brain. Neuron. 2012;75(4):601–617. doi: 10.1016/j.neuron.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pickard BS, Malloy MP, Porteous DJ, Blackwood DH, Muir WJ. Disruption of a brain transcription factor, NPAS3, is associated with schizophrenia and learning disability. Am J Med Genet B Neuropsychiatr Genet. 2005;136B(1):26–32. doi: 10.1002/ajmg.b.30204. [DOI] [PubMed] [Google Scholar]
- 56.Deng S, Hirschberg A, Worzfeld T, Penachioni JY, Korostylev A, Swiercz JM, et al. Plexin-B2, but not Plexin-B1, critically modulates neuronal migration and patterning of the developing nervous system in vivo. J Neurosci. 2007;27(23):6333–6347. doi: 10.1523/JNEUROSCI.5381-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brunskill EW, Ehrman LA, Williams MT, Klanke J, Hammer D, Schaefer TL, et al. Abnormal neurodevelopment, neurosignaling and behaviour in Npas3-deficient mice. Eur J Neurosci. 2005;22(6):1265–1276. doi: 10.1111/j.1460-9568.2005.04291.x. [DOI] [PubMed] [Google Scholar]
- 58.Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Molecular psychiatry. 2012;17(4):389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noriega DB, Savelkoul HF. Immune dysregulation in autism spectrum disorder. Eur J Pediatr. 2014;173(1):33–43. doi: 10.1007/s00431-013-2183-4. [DOI] [PubMed] [Google Scholar]
- 60.Voineagu I, Eapen V. Converging pathways in autism spectrum disorders: interplay between synaptic dysfunction and immune responses. Front Hum Neurosci. 2013;7:738. doi: 10.3389/fnhum.2013.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nikolova YS, Swartz JR, Hariri AR. Can we identify meaningful epigenetic effects on human brain function and related risk for mental illness? Epigenomics. 2016;8(10):1307–10. doi: 10.2217/epi-2016-0099. [DOI] [PubMed] [Google Scholar]
- 62.Smith AK, Kilaru V, Klengel T, Mercer KB, Bradley B, Conneely KN, et al. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(1):36–44. doi: 10.1002/ajmg.b.32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tylee DS, Kawaguchi DM, Glatt SJ. On the outside, looking in: a review and evaluation of the comparability of blood and brain “-omes”. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(7):595–603. doi: 10.1002/ajmg.b.32150. [DOI] [PubMed] [Google Scholar]
- 64.Li W, Liu M. Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids. 2011;2011:870726. doi: 10.4061/2011/870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22(3):467–477. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Papale LA, Zhang Q, Li S, Chen K, Keles S, Alisch RS. Genome-wide disruption of 5-hydroxymethylcytosine in a mouse model of autism. Hum Mol Genet. 2015;24(24):7121–7131. doi: 10.1093/hmg/ddv411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun W, Poschmann J, Cruz-Herrera Del Rosario R, Parikshak NN, Hajan HS, Kumar V, et al. Histone acetylome-wide association study of autism spectrum disorder. Cell. 2016;167(5):1385–97 e11. doi: 10.1016/j.cell.2016.10.031. [DOI] [PubMed] [Google Scholar]
- 68.Hara Y, Ago Y, Taruta A, Katashiba K, Hasebe S, Takano E, et al. Improvement by methylphenidate and atomoxetine of social interaction deficits and recognition memory impairment in a mouse model of valproic acid-induced autism. Autism Res. 2016;9(9):926–939. doi: 10.1002/aur.1596. [DOI] [PubMed] [Google Scholar]
- 69.Roullet FI, Lai JK, Foster JA. In utero exposure to valproic acid and autism--a current review of clinical and animal studies. Neurotoxicol Teratol. 2013;36:47–56. doi: 10.1016/j.ntt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 70.Yamaguchi H, Hara Y, Ago Y, Takano E, Hasebe S, Nakazawa T, et al. Environmental enrichment attenuates behavioral abnormalities in valproic acid-exposed autism model mice. Behav Brain Res. 2017;333:67–73. doi: 10.1016/j.bbr.2017.06.035. [DOI] [PubMed] [Google Scholar]
- 71.Hanna GL, Liu Y, Isaacs YE, Ayoub AM, Torres JJ, O’Hara NB, et al. Withdrawn/depressed behaviors and error-related brain activity in youth with obsessive-compulsive disorder. J Am Acad Child Adolesc Psychiatry. 2016;55(10):906–13 e2. doi: 10.1016/j.jaac.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information. (DOCX 18 kb)
Figure S1. Data analysis flowchart. Outline of the training cohorts and analysis pipeline used to derive DNAm signatures and test cohorts (independent samples), on which the DNAm signatures were tested. Figure S2. Identification of differentially methylated CpG sites when comparing 16p11.2del signature cases with age-, sex-matched controls. Significant sites for the DNAm signature for 16p11.2del represented by the overlap from limma regression and Mann Whitney U analysis are shown. Each volcano plot represents the distribution of significant sites at various statistical parameters (y-axis is negative log q-value, Benjamini-Hochberg corrected) and Δβ differences (x-axis is the average difference in DNAm between 16p11.2del and controls). Red horizontal line represents q < 0.05, red vertical lines represent |Δβ| ≥ 5%. Figure S3. Identification of differentially methylated CpG sites when comparing CHD8+/− signature cases with age-, sex-matched controls. Significant sites for the DNAm signature for CHD8+/− represented by the overlap from limma regression and Mann Whitney U analysis are shown. Each volcano plot represents the distribution of significant sites at various statistical parameters (y-axis is negative log q-value, Benjamini-Hochberg corrected) and Δβ differences (x-axis is the average difference in DNAm between CHD8+/− and controls). Red horizontal line represents q < 0.05, red vertical lines represent |Δβ| ≥ 5%. Figure S4. Targeted sodium bisulfite pyrosequencing of selected 16p11.2del and CHD8+/− DNAm signature CpG sites overlapping differentially methylated regions (DMRs). Specific CpG sites from our DNAm signatures that overlapped DMRs found using bump hunting were selected for targeted sodium bisulfite pyrosequencing validation in signature cases and age-, sex-matched controls. In the 16p11.2del group, the following CpG sites were validated: A) cg00108944 and cg23588049 in GLIPR1L2 showing a gain of methylation, B) cg25983544 and cg06377543 in PSMA8 showing a loss of methylation. In the CHD8+/− group, the following CpG sites were validated: C) cg09819656 and cg15089111 in NPAS3 showing a gain of methylation, D) cg27206976 and cg04089788 in PLXNB2 showing a loss of methylation. Upper graphs in each panel depict mean methylation (y-axis) over each DMR (x-axis, position of CpG sites along the chromosome) for the 16p11.2del (purple), CHD8+/− (orange) and control (green) groups. Box plots represent median methylation at the specific CpG sites indicated with min/max whiskers. Figure S5. Correlation of pyrosequencing and array data. We demonstrate that there is high correlation (r = 0.8524) between the bisulfite pyrosequencing validation % methylation and array methylation beta (β) values. Data include all samples run at all 8 of the specific CpG sites assayed for validation. Figure S6. Comparison of the 16p11.2del DNAm signature with blood cell type composition. Blood cell type DNAm data was extracted from Reinius et al. [41] (GEO series GSE35069) representing whole blood, peripheral blood mononuclear cells, granulocytes and isolated cell populations (CD4+ T cells, CD8+ T cells, CD56+ NK cells, CD19+ B cells, CD14+ monocytes) from 6 different control individuals. Blood cell type data was combined with our signature cases and control data using only the CpGs from the 16p11.2del-specific DNAm signature. The PCA plot demonstrates that the 16p11.2del signature is independent of blood cell type composition where signature cases remain separated from controls cases (whole blood and purified blood cell types). Abbreviations: PBMC peripheral blood mononuclear cells; WB whole blood. Figure S7. Comparison of the CHD8+/− DNAm signature with blood cell type composition. Blood cell type DNAm data was extracted from Reinius et al. (41) (GEO series GSE35069) representing whole blood, peripheral blood mononuclear cells, granulocytes and isolated cell populations (CD4+ T cells, CD8+ T cells, CD56+ NK cells, CD19+ B cells, CD14+ monocytes) from 6 different control individuals. Blood cell type data was combined with our signature cases and control data using only the CpGs from the CHD8+/−-specific DNAm signature. The PCA plot demonstrates that the CHD8+/− signature is independent of blood cell type composition where signature cases remain separated from controls cases (whole blood and purified blood cell types). Abbreviations: PBMC peripheral blood mononuclear cells; WB whole blood. Figure S8. Genomic distribution of DNAm signature CpG sites. Bar charts representing the genomic distribution of CpG sites in the 16p11.2del (A) and CHD8+/−-specific DNAm signatures (B, C [classification signature]), according to Illumina 450K array annotations. In mammalian cells, DNAm occurs predominantly on cytosines located in CpG dinucleotides. CpG islands refer to regions that are especially CpG-rich. Specific locations of CpGs relative to the CpG island can be denoted as “shores” and “shelves”. A shore is 0–2 kb from a CpG island, a shelf is 2–4 kb from a CpG island. N refers to regions upstream (5′) and S refers to regions downstream (3′) of a CpG island. We compared the distribution of the CpGs between each respective variance filtered dataset with each signature training case group for the regulatory feature group, relation to CpG island and other functional categories such as overlapping enhancer region, DNase hypersensitive sites (DHS) and type of differentially methylated regions (Reprogrammed DMR (RDMR), cancer-specific DMR (cDMR), other DMR) in addition to relation to RefSeq group annotation. N refers to north and S refers to south. Asterisk denotes significant enrichment or depletion by hypergeometric test comparison (p < 0.05). Figure S9. The quantile-quantile plot showing the log-transformed distributions of limma regression p-values associated with the DNAm differences in CHD8+/− cases compared to matching controls. The results are shown before (left) and after (right) correction by the BACON method, with inflation patterns evident in both cases. Figure S10. The quantile-quantile plot showing the log-transformed distributions of limma regression p-values associated with the DNAm differences in 16p11.2del cases compared to matching controls. The results are shown before (left) and after (right) correction by the BACON method, with inflation patterns evident in both cases. (PPTX 2477 kb)
Table S1. Demographic information for heterogeneous ASD cases and age- and sex-matched neurotypical controls. Table S2. Number of probes removed and remaining for analysis following quality control. Table S3. List of overlapping DMRs for 16p11.2del. Table S4. List of overlapping DMRs for CHD8+/−. Table S5. List of samples extracted from GEO database and respective 16p11.2del and CHD8+/− signature scores for specificity analysis. All sample data are from blood and from individuals with known ages < 50 years old. Table S6. List of significant (q < 0.05, |Δβ| ≥ 5%) sites in 16p11.2del DNAm signature. Table S7. List of significant (q < 0.05, |Δβ| ≥ 5%) sites in CHD8+/− DNAm signature. Table S8. GO Biological Process enrichment terms following GREAT enrichment analysis of differentially methylated genes in 16p11.2del DNAm signature. Table S9. GO Biological Process enrichment terms following GREAT enrichment analysis of differentially methylated genes in CHD8+/− DNAm signature. (XLSX 156 kb)
Data Availability Statement
The datasets used for the current study are available from the corresponding author on reasonable request.