

Abstract
New insights into the biological mechanisms involved in modulating periodontal inflammation and alveolar bone loss are paving the way for novel therapeutic strategies for periodontitis. The neutrophil adhesion cascade for transmigration in response to infection or inflammation is a key paradigm in immunity. Developmental endothelial locus-1 (Del-1) is one of several newly identified endogenous inhibitors of the leukocyte adhesion cascade. Del-1 competes with intercellular adhesion mole-cule-1 (ICAM-1) on endothelial cells for binding to the LFA-1 integrin on neutrophils, thereby regulating neutrophil recruitment and local inflammation. In animal periodontitis models, Del-1 deficiency resulted in severe inflammation and alveolar bone loss, but local treatment with recombinant Del-1 prevented neutrophil infiltration and bone loss. The expression of Del-1 is inhibited by the pro-inflammatory cytokine IL-17. Nucleic-acid-receptor-mediated inflammatory responses may be important in periodontal disease pathogenesis. Bacterial nucleic acids released during inflammation are detected by host microbial DNA sensors, e.g., Toll-like receptor-9 (TLR-9), leading to the activation of pro- and/or anti-inflammatory signaling pathways. DNA from periodontitis-associated bacteria induced pro-inflammatory cytokine production in human macrophage-like cells through the TLR-9 and NF-κB signaling pathways, but had less effect on human osteoblasts. Inhibition of TLR-9 signaling in human macrophages reduced cytokine production in response to P. gingivalis DNA. Differential expression of a polymorphic site in the TLR-9 gene promoter region and increased TLR-9 gene and protein expression were reported in chronic periodontitis. Further research to confirm that periodontal bacterial DNA contributes to destructive inflammation in vivo could provide alternative therapeutic targets to control periodontitis.
Keywords: neutrophils, Del-1, periodontal disease, Toll-like receptor, AIM-2, DAI
Introduction
Recent research has produced new insights into the biological mechanisms that may be important in modulating the pathological processes involved in destructive periodontitis, including localized persistent inflammation and alveolar bone loss.
The influx of neutrophils from the circulation into affected tissues is a key feature of the inflammatory response to infection or injury. The process by which neutrophils are recruited to inflamed tissues is complex, involving their adhesion to endothelial cells, crawling to detect locations for transmigration, extravasation into the tissues, and migration to the site of injury. Many of the molecular interactions involved in neutrophil adhesion are well understood, but a picture is now emerging of the locally produced endogenous molecules that inhibit adhesion and suppress the recruitment of neutrophils, thereby promoting the regulation or resolution of inflammation. Data relating to one such molecule, developmental endothelial locus-1 (Del-1), and its relevance to periodontal disease are outlined in the following.
The role of the periodontal microbiota in triggering the destructive inflammatory changes observed in chronic peri-odontitis is well established. However, the detection of bacterial DNA originating from periodontitis-associated bacteria by specific host receptors is now being explored as a further contributory factor in periodontal inflammation. Genetic variations in DNA receptor expression are also of interest, and analysis of data suggests that they may be important in periodontitis.
Del-1, An Endogenous Regulator of Neutrophil Recruitment to the Periodontium
The neutrophil adhesion cascade for transendothelial migration in response to infection or inflammation is one of the major paradigms in immunity (Fig. 1) (Ley et al., 2007; Nourshargh et al., 2010). This process involves a tightly regulated cascade of low- and high-affinity adhesive interactions between the neutro-phils and the endothelium lining the blood vessels of the infected or inflamed tissue. The first step involves transient rolling interactions, mediated by the binding of endothelial cell-surface molecules (P- or E-selectin) to their glycoprotein ligands on neutrophils. This rolling-dependent deceleration of neutrophils is followed by their firm adhesion to and subsequent crawling on the endothelium, during which neutrophils seek an appropriate site for transmigration. Firm adhesion and crawling are primarily mediated by heterodimeric β2-integrins, each with a distinct CD11 subunit and a common CD18 subunit. The lymphocyte-function-associated antigen 1 (LFA-1) integrin (CD11a/CD18) plays a crucial role in firm adhesion, which involves interactions with endothelial counter-receptors, such as the intercellular adhesion molecule-1 (ICAM-1). LFA-1-dependent firm adhesion to the endothelium is required for subsequent extravasation and migration of neutrophils to peripheral tissues (Ley et al., 2007; Phillipson and Kubes, 2011; Hajishengallis et al., 2012). The various steps of neutrophil extravasation can be modulated by cytokines or chemokines. Tissue-derived cytokines can up-regulate endothelial adhesion molecule expression, whereas tissue-derived chemokines decorating the apical endothelial cell surface can trigger conformational changes on neutrophil integrins, leading to the activation of their high-affinity binding state (Shamri et al., 2005; Ley et al., 2007; Vestweber, 2007).
Figure 1.
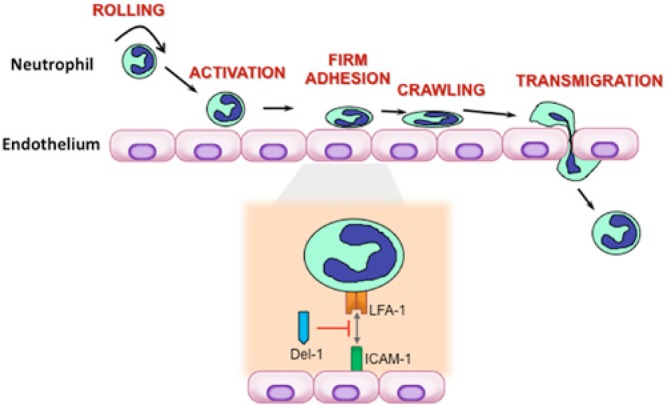
Leukocyte adhesion cascade and mode of action of the endogenous inhibitory regulator Del-1. The leukocyte adhesion cascade represents a sequence of adhesion and activation events culminating with the transmigration of the cells to inflamed or infected tissue. Shown are the major steps in the cascade, namely rolling, activation, firm adhesion, crawling, and transmigration. The interactions between the LFA-1 integrin on neutrophils and the intercellular adhesion molecule (ICAM)-1 on endothelial cells are critical for firm adhesion of neutrophils to the endothelium and the subsequent transmigration process. Del-1 binds LFA-1 and blocks its interaction with ICAM-1, thereby suppressing LFA-1-dependent neutrophil transmigration and recruitment to sites of inflammation.
Local Inhibition of Neutrophil Extravasation
In contrast to the plethora of adhesion molecules promoting the different steps of neutrophil extravasation that have been described over the past 3 decades, very little was known until recently about locally produced inhibitory regulators of this inflammatory process. The discovery of several such regulators, including developmental endothelial locus-1 (Del-1), pentraxin 3, and growth-differentiation factor 15, has evolved into a new field of endogenous inhibitors of the leukocyte adhesion cascade (Hajishengallis et al., 2012).
One of these regulators, Del-1 (also known as EDIL3 for EGF-like repeats and discoidin I-like domains 3) was recently shown to mediate a local homeostatic mechanism in the periodontium for protection against the destructive potential of recruited inflammatory cells (Eskan et al., 2012). Del-1 is an endothelial-cell-secreted 52-kDa glycoprotein originally described for its role in embryonic vascular development (Hidai et al., 1998). More recently, Del-1 was identified as a novel ligand for the LFA-1 integrin (Choi et al., 2008). As outlined earlier, the adhesive interactions between LFA-1 on neutrophils and ICAM-1 on endothe-lial cells are crucial for firm adhesion of neutrophils onto the vascular endothelium and the ensuing extravasation (Ley et al., 2007; Hajishengallis and Chavakis, 2013). However, unlike ICAM-1, which promotes transmigration, Del-1 antagonizes the LFA-1-dependent transmigration of neutrophils (Choi et al., 2008; Hajishengallis and Chavakis, 2013) (Fig. 1). In fact, at equimolar amounts, Del-1 outcompetes ICAM-1 for binding to LFA-1 (Choi et al., 2008). These findings suggested that Del-1 could act homeostatically to regulate local inflammation.
This concept was demonstrated by studies in animal models of periodontitis (Eskan et al., 2012), a chronic inflammatory disease that leads to inflammatory destruction of tooth-supporting tissues (Pihlstrom et al., 2005) and, in its severe form, adversely affects systemic health, thereby increasing the risk for atherosclerosis and diabetes (Genco and Van Dyke, 2010; Kebschull et al., 2010; Lalla and Papapanou, 2011). Specifically, Del-1-deficient mice were shown to spontaneously develop excessive neutrophil infiltration in the periodontium, leading to severe inflammation and alveolar bone loss (Eskan et al., 2012). In contrast, excessive neutrophil recruitment and bone loss were prevented in mice with dual deficiency in Del-1 and LFA-1. Intriguingly, Del-1 expression was diminished in the gingival tissue of old mice, correlating with excessive neutrophil recruitment and inflammatory bone loss (Eskan et al., 2012) (Fig. 2). Importantly, both pathologic features (i.e., neutrophil infiltration and bone loss) were blocked by local treatment with recombinant Del-1 (Eskan et al., 2012).
Figure 2.
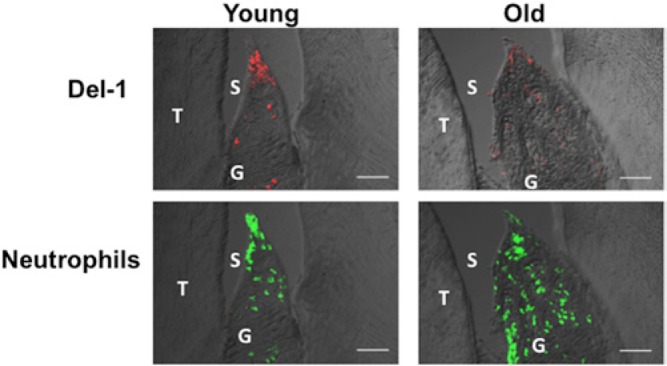
Lower expression of Del-1 in old mice causes increased infiltration of neutrophils. Overlays of differential interference contrast and fluorescent confocal images of sagittal sections of interdental gingiva stained for Del-1 or the neutrophil-specific marker Ly6G. The images show that old mice (18 months of age) have lower Del-1 expression but higher recruitment of neutrophils to the periodontium compared with young mice (8 to 10 weeks of age). T, tooth; G, gingiva; S, sulcus. Scale bars, 50 μm. From Eskan et al., 2012.
Del-1 vs. Interleukin-17 Expression in the Inflammatory Response
Interleukin (IL)-17 (also known as IL-17A) is a pro-inflammatory cytokine that promotes granulopoiesis and orchestrates the recruitment, activation, and survival of neutrophils (Ye et al., 2001; von Vietinghoff and Ley, 2008). Interestingly, IL-17 inhibits the expression of Del-1 (Eskan et al., 2012). Thus, down-regulation of this endogenous inhibitory regulator of neu-trophil transmigration may be a novel mechanism by which IL-17 facilitates neutrophil recruitment to inflamed or infected tissues. IL-17-mediated down-regulation of Del-1 expression may be beneficial in the acute defense response against pathogen infection. However, chronic IL-17 signaling can lead to persistent recruitment of neutrophils, thereby contributing to chronic inflammatory conditions (Hajishengallis, 2014).
IL-17 and Del-1 Interactions in Animal Models of Periodontitis
IL-17 is the most profoundly up-regulated inflammatory mediator in the gingiva of periodontitis-susceptible Del-1-deficient mice (Eskan et al., 2012). Furthermore, IL-17 can mediate the destruction of connective tissue and bone resorption by inducing the expression of matrix metalloproteases and the osteoclastogenic factor RANK ligand (Eskan et al., 2012; Miossec and Kolls, 2012). It was therefore suspected that IL-17 could be the driving force behind the pathologic loss of alveolar bone in Del-1-deficient mice. However, since IL-17 also promotes antimicrobial immunity (Dubin and Kolls, 2008), it was also possible that the up-regulation of IL-17 in the gingiva of Del-1-deficient mice represented a compensatory mechanism to fight periodontal infection, which developed into a dysbiotic state in these mice (Eskan et al., 2012). This question was addressed by the generation of mice with combined deficiency in Del-1 and the IL-17 receptor. These doubly deficient mice were protected from inflammation and bone loss (Eskan et al., 2012), indicating that IL-17 mediates destructive inflammation in this model of periodontitis.
Detailed analysis has shown that Del-1 and IL-17 are reciprocally cross-regulated: Whereas Del-1 inhibits LFA-1-dependent neutrophil recruitment and IL-17 production, IL-17 down-regulates Del-1 expression in endothelial cells and thereby promotes neutrophil recruitment (Eskan et al., 2012). An inverse expression of Del-1 and IL-17 was also demonstrated in human gingival biopsy samples, with Del-1 dominating in healthy gingiva and IL-17 in inflamed gingiva (Eskan et al., 2012). Moreover, it was demonstrated that human Del-1 inhibits LFA-1-dependent transendothelial migration of human neutrophils (Eskan et al., 2012), thus acting as a gatekeeper for normal neutrophil recruitment.
It should be noted that IL-17 receptor signaling can also stimulate antimicrobial immunity and was associated with protection in a mouse model of periodontitis induced by implantation of a human periodontal pathogen (Porphyromonas gingivalis) (Yu et al., 2007). This apparent difference from the study described above (Eskan et al., 2012) might reflect the different nature of the two models (naturally occurring chronic bone loss vs. the relatively acute P. gingivalis-induced periodontitis model). In this regard, chronic IL-17 receptor signaling can potentially turn an acute inflammatory response into chronic immunopathology, as in rheumatoid arthritis (Gaffen and Hajishengallis, 2008; Lubberts, 2008).
Microbial DNA Sensing in Periodontal Inflammation
The recognition of invading micro-organisms is paramount to the survival of the host, and the disruption of tissue homeostasis leads to the pathology associated with various infectious and inflammatory conditions, including periodontitis. Host-pathogen interactions are mainly initiated through host recognition of conserved molecular structures known as pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs). During infection and inflammation, nucleic acids from bacteria, viruses, and host cells can be released and detected by specific host receptors, activating inflammatory signaling cascades. Toll-like receptor-9 (TLR-9) is considered the main sensor for microbial DNA through detection of unmethylated/ hypomethylated CpG (cytosine-phosphate-guanosine) motifs. It not only activates nuclear factor kappa B (NF-κB), the activator protein-1 (AP-1), and mitogen-activated protein (MAP) kinases signaling pathways, which stimulate pro-inflammatory activities, but also triggers the interferon regulatory factor pathway, which can induce type I interferon and anti-inflammatory activities (Akira, 2009). Besides TLR-9, there are other cyto-plasmic DNA sensors, which include “absent in melanoma-2” (AIM-2) and DNA-dependent activator of IFN-regulatory factors (DAI) (Thompson et al., 2011). Hence, recognition of DNA by the host immune system can be accomplished through distinct receptors and signaling pathways, contributing to the pathogenesis of several infectious and inflammatory conditions, including autoimmune diseases, gastrointestinal and pulmonary infections, preterm labor, cardiovascular diseases, and cancer (Hirsch et al., 2010).
Periodontal structures are exposed to constant microbial challenge, and periodontal tissue homeostasis is tightly regulated by the interaction of host cells that form different components of the periodontium with this continuous microbial insult. Continual cell turnover leads to the accumulation of extracellular (e-DNA) and intracellular DNA in periodontal tissues and in the dental biofilm, which can contribute to periodontal inflammation through activation of DNA receptors and inflammatory signaling pathways. Therefore, a study of the microbial DNA interactions with different cells of the periodontium is of particular importance to a full elucidation of the immune responses initiated by periodontal bacterial DNA.
In vitro Studies
Although it has been well-studied in relation to other diseases, the role of microbial DNA sensing in the context of periodontal inflammation has received attention only in the last few years. It has now been shown that bacterial DNA of periodontitis-associated bacteria including Porphyromonas gingivalis (P. gingivalis), Tannerella forsythia (T. forsythia), and Fusobacterium nucleatum can induce pro-inflammatory cytokine production in human macrophage-like cells through the TLR-9 and NF-κB signaling pathways (Sahingur et al., 2010, 2012). Similar responses were reported for gingival fibroblasts and mouse macrophages (Nonnenmacher et al., 2003). Overall, current evidence suggests that DNA from periodontitis-associated bacteria has the ability to trigger inflammatory signaling. Yet, not all oral bacterial DNA has the same capacity to activate immune responses, possibly due to differences in composition such as the abundance of CpG content or methylation pattern. For example, unlike DNA from periodon-tal pathogens, DNA from Streptococcus sanguinis is unable to induce cytokine production from human macrophages (Sahingur et al., 2012) and gingival epithelial cells (Wara-Aswapati et al., 2013). Such variations in the activation of inflammatory responses were reported related to DNA from other bacterial species as well (Dalpke et al., 2006).
The multi-microbial nature of periodontitis and various cells forming the periodontal structures creates an environment for the simultaneous interaction of distinct PRRs and periodontal PAMPs, leading to periodontal inflammation. Of the PRRs, TLR-2 and TLR-4 are the most widely studied extracellular innate receptors that recognize various PAMPs and are likely to play a role in the pathogenesis of periodontitis (Hajishengallis and Lambris, 2011). In a living organism, physiological responses require interaction of multiple host cells and receptors with various microbial products. In parallel with this notion, cross-talk among TLR-2, TLR-4, and TLR-9 has been reported in other disease models (De Nardo et al., 2009; Bhan et al., 2010). Similarly, it has also been shown that inhibition of TLR-9 signaling in human macrophages reduces cytokine production, in response to not only P. gingivalis DNA but also to LPS, implying that TLR-9 signaling can contribute to the pathogenesis of periodontitis, either alone or through communication with other signaling pathways (Sahingur et al., 2012). Overall, these results warrant further investigation to determine how variations in DNA structure among different species and cross-talk among different host factors affect biological responses in periodontitis.
Recent research has revealed that periodontal bacterial DNA can stimulate inflammatory mediators through TLR-9 from both myeloid and non-myeloid cells forming the periodontal structures. Osteoblasts are mainly involved in periodontal tissue homeostasis through bone formation, but also respond to pathogenic insult by producing inflammatory mediators (Gruber, 2010). P. gingivalis can invade osteoblasts, raising the question of whether TLR-9 activation through P. gingivalis DNA released during cell lysis contributes to periodontal inflammation (Zhang et al., 2013). This possibility was investigated, and it was found that bacterial DNA is less effective in initiating pro-inflammatory responses in human osteoblasts than in macrophages (Sahingur et al., unpublished observations). P. gingivalis DNA stimulated increased IL-8 production but failed to induce IL-1β and TNF-α production in MG-63 human osteoblastic cells. Depending on the pathology and the cell type being investigated, TLR-9 signaling can elicit either a protective or a destructive immune response, and analysis of available data further implies that cells with different effector functions involved in various stages of periodontal pathology can respond to bacterial DNA in different ways (Hotte et al., 2012; Tuvim et al., 2012; Bhan et al., 2013; Xu et al., 2013). Hence, future investigations in in vivo periodontitis models will fully characterize the extent of involvement of microbial DNA sensing in periodontal inflammation.
Clinical Studies in Periodontitis Patients
It is accepted that genetic background affects susceptibility to periodontitis (Kinane et al., 2005). Recently, two clinical studies compared the presence of single-nucleotide polymorphisms in the TLR-9 gene in individuals with chronic periodontitis vs. healthy individuals and revealed differential expression of a specific polymorphic site in the TLR-9 gene (Holla et al., 2010; Sahingur et al., 2011). As determined by in silico analyses, these polymorphisms are located in the promoter region of the TLR-9 gene, corresponding to a possible transcriptional activator binding site (NF-κB and Sp-1), presumably having a functional role in TLR-9 expression (Hamann et al., 2006; Ng et al., 2010). Studies are under way to determine whether the presence of these polymorphisms has any effect on the extent of inflammatory responses in periodontitis. Another clinical study reported increased TLR-9 and DAI mRNA expression in periodontitis sites (Sahingur et al., 2013). Further immunohistochemical analyses revealed constitutive expression of these sensors, even in healthy tissues (Fig. 3). The receptor expression, however, was up-regulated prominently at the basal epithelial layers and connective tissues in the diseased sites. The same study also revealed significantly increased mRNA expression of TLR-8 in the diseased tissues (Sahingur et al., 2013). TLR-8 is another intracellular innate receptor that recognizes viral and bacterial RNA (Akira, 2009; Cervantes et al., 2012). Interestingly, TLR-9 and DAI can also respond to viral DNA (Thompson et al., 2011). While the bacterial etiology of periodontitis is well-accepted, the contribution of viruses in periodontal disease pathology has also been supported by several studies (Slots, 2005). In addition, the association between bacteria and viruses has been proposed in various conditions, suggesting that these interactions create a favorable environment for pathogen survival and persistence, as well as an enhanced inflammatory response (Bakaletz, 1995; Grande et al., 2011). Hence, the interactions of bacteria and viruses with the intracellular nucleic acid sensors within periodontal tissues and the effects of such interactions on overall periodontal health need to be determined in future studies. In summary, combined with the results of in vitro studies, analysis of the data obtained from clinical studies further substantiates a role for microbial DNA sensing in periodontitis.
Figure 3.

Immunohistochemical detection of TLR-9 and DAI expression. Tissue sections from healthy subjects (H) and periodontitis patients (P) were immunostained with the specific antibodies (upper panels); lower panels: negative controls. Original magnification, x100. From Sahingur et al., 2013.
Conclusion
Continued research is providing many advances in our understanding of the interrelated biological systems involved in promoting disease and/or maintaining health. These new findings, particularly those relating to inflammation, the immune response, and bone turnover, could ultimately lead to new therapeutic approaches in the management of periodontal disease. The inhibitory regulator Del-1 provides a mechanism whereby the periodontal tissue can locally self-regulate persistent inflammation associated with chronic recruitment of neutrophils, a concept that can potentially be exploited therapeutically to treat human periodontitis. It should be noted that both unrestrained and, conversely, diminished neutrophil recruitment to the peri-odontium can cause inflammatory bone loss. Indeed, patients with leukocyte adhesion deficiency, a group of inherited disorders that inhibit the extravasation of circulating neutrophils to sites of infection or inflammation, develop aggressive periodontitis affecting both the primary and permanent dentition (Deas et al., 2003; Hajishengallis and Hajishengallis, 2014). Therefore, any deviation from the normal recruitment of neutrophils to the periodontium can potentially cause disruption of tissue homeo-stasis and lead to periodontitis.
Recent studies have highlighted the importance of nucleic-acid-receptor-mediated inflammatory responses as alternative, and as yet not fully explored, pathways contributing to periodontal disease pathogenesis. Furthermore, given that oral bacterial DNA can be identified at sites distant from the oral cavity, such as atheromatous plaques (Gaetti-Jardim et al., 2009) and synovial fluid of arthritis patients (Reichert et al., 2013), often without the presence of live bacteria, these observations also raise another interesting question – that of whether the engagement of host receptors by periodontal bacterial DNA contributes to systemic inflammation. Investigators working in this field believe that further research on microbial DNA and its sensors will lead not only to identification of alternative therapeutic targets to control periodontal inflammation but also to a possible link between oral and systemic diseases.
Acknowledgements
Work performed in the laboratory of author GH was supported by National Institutes of Health (NIH) grants DE015254, DE017138, DE021580, and DE021685. SES was supported by NIH grants DE022836 and KL2TR000057-03; by A.D. Williams Research Funds, VCU, Richmond, VA; by Thomas F. and Kate Miller Jeffress Memorial Trust Funds, Richmond, VA; and by Presidential Research Quest Funds, VCU, Richmond, VA. The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Akira S. (2009). Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev 227: 5–8. [DOI] [PubMed] [Google Scholar]
- Bakaletz LO. (1995). Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol 3: 110–114. [DOI] [PubMed] [Google Scholar]
- Bhan U, Ballinger MN, Zeng X, Newstead MJ, Cornicelli MD, Standiford TJ. (2010). Cooperative interactions between TLR4 and TLR9 regulate interleukin 23 and 17 production in a murine model of Gram negative bacterial pneumonia. PLoS One 5: e9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhan U, Newstead MJ, Zeng X, Podsaid A, Goswami M, Ballinger MN, et al. (2013). TLR9-dependent IL-23/IL-17 is required for the generation of Stachybotrys chartarum-induced hypersensitivity pneumonitis. J Immunol 190: 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes JL, Weinerman B, Basole C, Salazar JC. (2012). TLR8: the forgotten relative revindicated. Cell Mol Immunol 9: 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. (2008). Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 322: 1101–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke A, Frank J, Peter M, Heeg K. (2006). Activation of Toll-like receptor 9 by DNA from different bacterial species. Infect Immun 74: 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D, De Nardo CM, Nguyen T, Hamilton JA, Scholz GM. (2009). Signaling crosstalk during sequential TLR4 and TLR9 activation amplifies the inflammatory response of mouse macrophages. J Immunol 183: 8110–8118. [DOI] [PubMed] [Google Scholar]
- Deas DE, Mackey SA, McDonnell HT. (2003). Systemic disease and peri-odontitis: manifestations of neutrophil dysfunction. Periodontol 2000 32: 82–104. [DOI] [PubMed] [Google Scholar]
- Dubin PJ, Kolls JK. (2008). Th17 cytokines and mucosal immunity. Immunol Rev 226: 160–171. [DOI] [PubMed] [Google Scholar]
- Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. (2012). The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol 13: 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetti-Jardim E, Jr, Marcelino SL, Feitosa AC, Romito GA, Avila-Campos MJ. (2009). Quantitative detection of periodontopathic bacteria in atherosclerotic plaques from coronary arteries. J Med Microbiol 58(Pt 12): 1568–1575. [DOI] [PubMed] [Google Scholar]
- Gaffen SL, Hajishengallis G. (2008). A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J Dent Res 87: 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genco RJ, Van Dyke TE. (2010). Prevention: reducing the risk of CVD in patients with periodontitis. Nat Rev Cardiol 7: 479–480. [DOI] [PubMed] [Google Scholar]
- Grande SR, Imbronito AV, Okuda OS, Pannuti CM, Nunes FD, Lima LA. (2011). Relationship between herpesviruses and periodontopathogens in patients with HIV and periodontitis. J Periodontol 82: 1442–1452. [DOI] [PubMed] [Google Scholar]
- Gruber R. (2010). Cell biology of osteoimmunology. Wien Med Wochenschr 160: 438–445. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. (2014). Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 35: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Chavakis T. (2013). Endogenous modulators of inflammatory cell recruitment. Trends Immunol 34: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis E, Hajishengallis G. (2014). Neutrophil homeostasis and its impact on periodontal health in children and adults. J Dent Res 93: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Lambris JD. (2011). Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol 11: 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Darveau RP, Curtis MA. (2012). The keystone-pathogen hypothesis. Nat Rev Microbiol 10: 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann L, Glaeser C, Hamprecht A, Gross M, Gomma A, Schumann RR. (2006). Toll-like receptor (TLR)-9 promotor polymorphisms and atherosclerosis. Clin Chim Acta 364: 303–307. [DOI] [PubMed] [Google Scholar]
- Hidai C, Zupancic T, Penta K, Mikhail A, Kawana M, Quertermous EE, et al. (1998). Cloning and characterization of developmental endothe-lial locus-1: an embryonic endothelial cell protein that binds the alphav-beta3 integrin receptor. Genes Dev 12: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch I, Caux C, Hasan U, Bendriss-Vermare N, Olive D. (2010). Impaired Toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol 31: 391–397. [DOI] [PubMed] [Google Scholar]
- Holla LI, Vokurka J, Hrdlickova B, Augustin P, Fassmann A. (2010). Association of Toll-like receptor 9 haplotypes with chronic periodontitis in Czech population. J Clin Periodontol 37: 152–159. [DOI] [PubMed] [Google Scholar]
- Hotte NS, Salim SY, Tso RH, Albert EJ, Bach P, Walker J, et al. (2012). Patients with inflammatory bowel disease exhibit dysregulated responses to microbial DNA. PLoS One 7: e37932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull M, Demmer RT, Papapanou PN. (2010). “Gum bug, leave my heart alone!”-Epidemiologic and mechanistic evidence linking peri-odontal infections and atherosclerosis. J Dent Res 89: 879–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinane DF, Shiba H, Hart TC. (2005). The genetic basis of periodontitis. Periodontol 2000 39: 91–117. [DOI] [PubMed] [Google Scholar]
- Lalla E, Papapanou PN. (2011). Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol 7: 738–748. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7: 678–689. [DOI] [PubMed] [Google Scholar]
- Lubberts E. (2008). IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine 41:84–91. [DOI] [PubMed] [Google Scholar]
- Miossec P, Kolls JK. (2012). Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Disc 11: 763–776. [DOI] [PubMed] [Google Scholar]
- Ng MT, Van't Hof R, Crockett JC, Hope ME, Berry S, Thomson J, et al. (2010). Increase in NF-κB binding affinity of the variant C allele of the Toll-like receptor 9 -1237T/C polymorphism is associated with Helicobacter pylori-induced gastric disease. Infect Immun 78: 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonnenmacher C, Dalpke A, Zimmermann S, Flores-De-Jacoby L, Mutters R, Heeg K. (2003). DNA from periodontopathogenic bacteria is immu-nostimulatory for mouse and human immune cells. Infect Immun 71: 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourshargh S, Hordijk PL, Sixt M. (2010). Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol 11: 366–378. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Kubes P. (2011). The neutrophil in vascular inflammation. Nat Med 17: 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrom BL, Michalowicz BS, Johnson NW. (2005). Periodontal diseases. Lancet 366: 1809–1820. [DOI] [PubMed] [Google Scholar]
- Reichert S, Haffner M, Keysser G, Schafer C, Stein JM, Schaller HG, et al. (2013). Detection of oral bacterial DNA in synovial fluid. J Clin Periodontol 40: 591–598. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Alamgir S, Honma K, Sharma A, Schenkein HA. (2010). DNA from Porphyromonas gingivalis and Tannerella forsythia induce cytokine production in human monocytic cell lines. Mol Oral Microbiol 25: 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Gunsolley J, Schenkein HA, Genco RJ, De Nardin E. (2011). Single nucleotide polymorphisms of pattern recognition receptors and chronic periodontitis. J Periodontal Res 46: 184–192. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Schifferle RE. (2012). Oral bacterial DNA differ in their ability to induce inflammatory responses in human monocytic cell lines. J Periodontol 83: 1069–1077. [DOI] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Voth SC, Yeudall WA, Gunsolley JC. (2013). Increased nucleic acid receptor expression in chronic periodontitis. J Periodontol 84: e48–e57. [DOI] [PubMed] [Google Scholar]
- Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, et al. (2005). Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemo-kines. Nat Immunol 6: 497–506. [DOI] [PubMed] [Google Scholar]
- Slots J. (2005). Herpesviruses in periodontal diseases. Periodontol 2000 38: 33–62. [DOI] [PubMed] [Google Scholar]
- Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. (2011). Pattern recognition receptors and the innate immune response to viral infection. Viruses 3: 920–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvim MJ, Gilbert BE, Dickey BF, Evans SE. (2012). Synergistic TLR2/6 and TLR9 activation protects mice against lethal influenza pneumonia. PLoS One 7: e30596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D. (2007). Adhesion and signaling molecules controlling the transmigration of leukocytes through endothelium. Immunol Rev 218: 178–196. [DOI] [PubMed] [Google Scholar]
- von Vietinghoff S, Ley K. (2008). Homeostatic regulation of blood neutro-phil counts. J Immunol 181: 5183–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wara-Aswapati N, Chayasadom A, Surarit R, Pitiphat W, Boch JA, Nagasawa T, et al. (2013). Induction of Toll-like receptor expression by Porphyromonas gingivalis. J Periodontol 84: 1010–1018. [DOI] [PubMed] [Google Scholar]
- Xu L, Wen Z, Zhou Y, Liu Z, Li Q, Fei G, et al. (2013). MicroRNA-7-regulated TLR9 signaling-enhanced growth and metastatic potential of human lung cancer cells by altering the phosphoinositide-3-kinase, regulatory subunit 3/Akt pathway. Mol Biol Cell 24: 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. (2001). Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194: 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. (2007). An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109: 3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ju J, Rigney T, Tribble G. (2013). Integrin alpha5beta1-fimbriae binding and actin rearrangement are essential for Porphyromonas gingivalis invasion of osteoblasts and subsequent activation of the JNK pathway. BMC Microbiol 13: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]