

Abstract
Bidirectional signaling by cell-adhesion molecules is thought to mediate synapse formation, but the mechanisms involved remain elusive. Here we found that the adhesion-G-protein-coupled receptors latrophilin-2 and latrophilin-3 selectively directed formation of perforant-path and Schaffer-collateral synapses, respectively, to hippocampal CA1-region neurons. Latrophilin-3 binds to two trans-cellular ligands, fibronectin leucine-rich-repeat transmembrane proteins (FLRTs) and teneurins. In vivo, both binding activities were required for input-specific synapse formation, suggesting that coincident binding of both ligands is necessary for synapse formation. In vitro, teneurin or FLRT alone did not induce excitatory synapse formation, whereas together they potently did so. Thus, postsynaptic latrophilins promote excitatory synapse formation by simultaneous binding of two unrelated presynaptic ligands, which is required for formation of synaptic inputs at specific dendritic localizations.
INTRODUCTION:
In brain, synaptic connections form neuronal communication networks, thereby constructing neural circuits. Synaptic connections are exquisitely specific and dynamic, but the underlying molecular mechanisms remain largely unexplored. In the hippocampus, Schaffer-collateral axons from the CA3 region form synapses on CA1 region pyramidal neurons exclusively on dendritic domains in the S. oriens and S. radiatum of these neurons. In contrast, perforant-path axons from the entorhinal cortex form synapses on CA1 region pyramidal neurons exclusively on dendritic domains in the S. lacunosum-moleculare. How this synaptic input specificity is achieved, however, and what signaling mechanisms maintain the two classes of synapses, is unknown.
RATIONALE:
Synapse formation is thought to involve bidirectional signaling by trans-synaptic cell-adhesion molecules. Building on recent observations that the adhesion G-protein coupled receptor (GPCR) latrophilin-2 is essential for synapses in the S. lacunosum-moleculare of the CA1 region, we asked whether distinct latrophilins are localized to different dendritic domains of CA1 region neurons. Moreover, latrophilins are known to form trans-cellular interactions with two classes of cell-adhesion molecules, teneurins and fibronectin leucine-rich-repeat transmembrane proteins (FLRTs). Thus we hypothesized that latrophilins may act in synapse formation via trans-synaptic interactions with these adhesion molecules as ligands, and that such interactions may contribute to the specificity of synapse formation.
RESULTS:
We produced genetic manipulations that allow monitoring the localization of endogenous latrophilin-2 and latrophilin-3 in vivo and that enable their conditional deletion. Using these manipulations, we found that latrophilin-2 and latrophilin-3 were specifically localized to postsynaptic spines in non-overlapping dendritic domains of CA1 region pyramidal neurons. Latrophilin-2 was targeted only to excitatory synapses in the S. lacunosum-moleculare, whereas latrophilin-3 was targeted only to excitatory synapses in the S. oriens and S. radiatum, corresponding to distinct presynaptic inputs onto CA1 region pyramidal neurons. Deletion of latrophilin-3 selectively decreased Schaffer-collateral synapses in the S. radiatum and S. oriens, whereas deletion of latrophilin-2 selectively decreased entorhinal cortex-derived synapses in the S. lacunosum-moleculare of CA1 neurons. In vivo rescue experiments with latrophilin-3 mutants that selectively lack binding to only FLRTs or only teneurins revealed that both binding activities were required for input-specific synapse formation, as monitored by electrophysiology and retrograde rabies tracing. Thus, coincident binding of both latrophilin-3 ligands was necessary for synapse formation. Moreover, in in vitro synapse formation assays teneurin-2 or FLRT3 alone were unable to induce excitatory synapse formation, whereas together they potently did so. However, even in combination FLRT3 and teneurin-2 only induced excitatory synapses when teneurin-2 was expressed as a splice variant that is competent to interact with latrophilins, indicating that simultaneous binding of both FLRT3 and teneurin-2 to latrophilins was necessary to induce synapse formation.
CONCLUSION:
We suggest that latrophilin-2 and latrophilin-3 are postsynaptic adhesion GPCRs that are targeted in CA1 pyramidal neurons to non-overlapping dendritic domains, where they promote excitatory synapse formation by specific and distinct presynaptic inputs. The function of latrophilin-3 in synapse formation required simultaneous binding of two unrelated presynaptic ligands, FLRTs and teneurins, suggesting a coincidence signaling mechanism that could account for the specificity of synaptic connections.
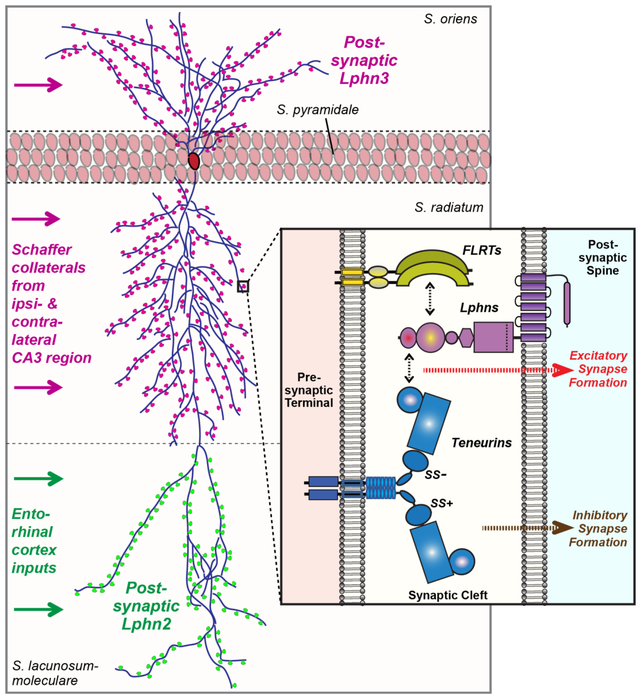
Graphical Abstract
Postsynaptic latrophilin-2 and latrophilin-3 mediate synapse specificity by simultaneous binding to presynaptic FLRTs and teneurins. Latrophilin-2 and latrophilin-3 are exclusively localized to dendritic domains of CA1 pyramidal neurons in the S. lacunosum-moleculare or the S. oriens and S. radiatum, respectively. In these locations, latrophilins are essential for synapse formation by simultaneously interacting with two different presynaptic cell-adhesion molecules, FLRTs and teneurins.
One Sentence Summary:
Postsynaptic latrophilin-3 mediates hippocampal synaptogenesis by coincidence signaling via presynaptic FLRTs and teneurins
Most synapses form during postnatal development, but many synapses continue to turn over throughout life. Although intensely studied, it is unclear how synapses form. Synapses are shaped by trans-synaptic cell-adhesion molecules. Many such molecules have been described, but for most such molecules it is unclear whether they are physiologically important – even the question of whether a particular candidate synaptic cell-adhesion molecule is pre- or postsynaptic is often unanswered (1-3).
Latrophilins are adhesion G-protein-coupled receptors (GPCRs) containing extensive N-terminal extracellular sequences followed by a classical 7-transmembrane region GPCR architecture (Figure 1A). The extracellular domains of latrophilins bind to at least two trans-cellular ligands, FLRTs (4) and teneurins (5-7). Like all adhesion GPCRs, latrophilins contain a characteristic extracellular GAIN-domain (for “GPCR-Autoproteolysis INducing domain”) that catalyzes autoproteolysis at a site just N-terminal to the first transmembrane region (8). Although an invariant feature of adhesion-GPCRs, the physiological significance of latrophilin autoproteolysis remains unclear. One well-supported idea suggests that GAIN domain-mediated adhesion cleavage exposes in an adhesion-GPCR a new N-terminal sequence, the so-called “Stachel”, which then serves as an intrinsic ligand for the GPCR (9-12). An alternative hypothesis is that ligand-binding activates the adhesion GPCR directly via a stachel-independent mechanism (13-15). One latrophilin, Lphn2, is essential for synapse formation in hippocampal CA1 neurons in a specific dendritic domain, the S. lacunosum-moleculare (16). Lphn2 may thus be postsynaptic, consistent with previously reported interaction between the intracellular C-terminus of latrophilins and postsynaptic Shank scaffolding proteins (17,18). However, other studies have indicated a presynaptic location of latrophilins (4,5), illustrating the difficulty of assigning functions and locations to synaptic molecules. Collectively, these studies raise questions about how latrophilins contribute to synapse formation. Do latrophilins act as pre- or postsynaptic molecules, do different latrophilins perform complementary or overlapping roles, and are their autoproteolytic cleavage or their ligand-binding activities required for synapse formation?
Figure 1: Latrophilin-3 (Lphn3) is essential for formation of a subset of excitatory synapses in cultured hippocampal neurons.

A. Schematic of the Lphn3 domain structure with location of the HA-tag in Lphn3 cKO mice.
B. Lphn3 is postsynaptic as revealed by sparse expression of Cre-recombinase in transfected hippocampal neurons that were cultured from Lphn3 cKO mice.
C-E. Postsynaptic Lphn3 deletion in cultured hippocampal neurons decreases the density of dendritic spines and the number of excitatory but not of inhibitory synapses.
F-I. Postsynaptic Lphn3 deletion suppresses the frequency of spontaneous mEPSCs but not mIPSCs monitored in the presence of tetrodotoxin.
J & K. Postsynaptic deletion of Lphn3 decreases the amplitudes of evoked EPSC but not IPSCs.
Data are means ± SEM (numbers of cells/experiments are indicated in bars). Statistical significance was assessed by two-tailed t-tests (** denotes p<0.01; * denotes p<0.05).
Lphn3 is a postsynaptic GPCR essential for excitatory synapse formation.
We generated conditional (cKO) and constitutive knockout (KO) mice for Lphn3 (Figure 1A, S1A [19]). Both were viable and fertile, but constitutive Lphn3 KO mice exhibited a decreased body weight (Figure S1B, S1C). In the cKO mice, we tagged Lphn3 with N-terminal HA-epitopes, allowing us to monitor Lphn3 protein in the absence of suitable antibodies (Fig. 1A). Developmentally, Lphn3 was expressed in a pattern paralleling synaptogenesis, with peak expression at approximately postnatal day 12 (P12; Figure S1D). Lphn3 was robustly expressed in dissociated cultures containing neurons, but not in cultures composed only of glia (Figure S1E). In hippocampal neurons, Lphn3 was localized to excitatory synapses (Figure S2A). Sparse postsynaptic expression of Cre-recombinase in a small percentage of neurons in culture abolished synaptic Lphn3 expression within transfected neurons, whereas expression of mutant inactive Cre-recombinase (ΔCre, as a control) had no effect (Figure 1B, S2B). Similarly, global Cre-expression abolished all Lphn3 expression in the cKO neurons (Figure S2C, S2D). Thus, Lphn3 is a postsynaptic adhesion-GPCR in cultured neurons.
To examine the function of Lphn3, we sparsely transfected cultured hippocampal neurons with ゔCre or Cre. We analyzed their spine density after filling individual neurons with biocytin via a patch pipette, and their synapse numbers by staining the neurons for pre- and postsynaptic marker proteins. Postsynaptic Cre-recombinase-mediated deletion of Lphn3 produced a ~50% loss of dendritic spines and a ~40% loss of excitatory synapses, but no change in inhibitory synapses (Figure 1C-1E, S3A-S3C). We then performed electrophysiological recordings from sparsely transfected Lphn3 cKO neurons expressing Cre or ΔCre. These neurons exhibited identical passive electrical properties, but displayed a ~50% decrease in the frequency of spontaneous miniature EPSCs (mEPSCs) without a change in miniature IPSCs (mIPSCs). Moreover, these neurons displayed a ~40% decrease in evoked EPSCs but not in evoked IPSCs (Figure 1F-1K, S3D-S3J). Thus, postsynaptic Lphn3 in cultured hippocampal neurons is essential for the selective formation and/or maintenance of approximately half of excitatory but not inhibitory synapses.
Lphn3 function in synapse formation requires FLRT- and teneurin-binding but not autoproteolysis.
Although adhesion-GPCRs represent the second most numerous family of human GPCRs, their mechanism of action has remained unclear (11,14). Because all adhesion-GPCRs contain an autoproteolytic GAIN-domain (8), ligand binding to an adhesion-GPCRs may activate its GPCR moiety by pulling off its extracellular sequences, thereby exposing a short N-terminal stub that serves as an intrinsic ligand for the GPCR similar to the activation mechanism of thrombin receptors (20,21). A complicating factor is that for most adhesion-GPCRs, no ligands have been identified, rendering a test of the hypothesis difficult. However, for latrophilins two high-affinity ligands are known that bind with nanomolar affinity – FLRTs and teneurins (4-7). Is binding of both of these ligands physiologically relevant, and do these ligands –separately or together– activate latrophilins by pulling off their extracellular domains, thereby exposing a tethered agonist in the new free N-terminal sequence?
To address these questions, we generated three Lphn3 mutants (Figure 2A). The “4A” mutation changes four amino acids in the olfactomedin-like domain and selectively blocks FLRT binding (22), the “ΔLEC” mutation deletes the lectin-like domain of Lphn3 and abolishes teneurin binding (6), and the “T869G” mutation with the eponymous amino acid substitution blocks autoproteolysis by the GAIN domain without impairing its folding (8). All three Lphn3 mutants were well expressed at the expected size (Figure S4A). The 4A mutation blocked FLRT- but not teneurin-mediated aggregation, while the ΔLEC mutation blocked teneurin- but not FLRT-mediated aggregation, and the T869G mutation had no effect on either teneurin- or FLRT-mediated aggregation (Figure 2B, S4B). Thus, the mutants acted as designed, were transported to the cell surface, and were properly folded. Moreover, when expressed in neurons all mutants were also efficiently localized to the cell surface (Figure S4C-S4E).
Figure 2: Lphn3-dependent synapse formation in cultured hippocampal neurons requires both teneurin- and FLRT-binding by Lphn3 but not Lphn3 autoproteolysis.

A. Domain structures of Lphn3 mutants that selectively block binding of Lphn3 to FLRTs (Lphn3-4A) or teneurins (Lphn3-ΔLEC), or that abolish Lphn3 autoproteolysis (Lphn3-T869G).
B. Lphn3 mutants are efficiently transported to the cell surface and mediate cell adhesion via the Lphn3 binding partners whose binding site was not mutated (for quantifications, see Figure S2).
C & D. Wild-type Lphn3 and Lphn3-T869G lacking autoproteolysis activity efficiently rescue the loss of synapse density (C) or the decrease in mEPSC frequency (D) in Lphn3-deficient neurons, whereas the Lphn3 mutants that do not bind to FLRTs or to teneurins do not rescue.
Data are means ± SEM (numbers of cells/experiments are indicated in bars). Statistical significance was assessed by one-way ANOVA (** denotes p<0.01; * denotes p<0.05).
We then tested the ability of these mutants to rescue the decrease in excitatory synapse density and mEPSC frequency induced by the Lphn3 deletion in cultured hippocampal neurons (Figure 2C, 2D). Both the 4A mutant that selectively blocked FLRT binding and the ΔLEC mutant that selectively blocked teneurin binding abolished rescue. In contrast, the T869G mutation that abolished Lphn3 autoproteolysis did not impair rescue. Thus, surprisingly, Lphn3 function in synapse formation requires binding of both FLRT and teneurin but does not depend on autoproteolysis.
Lphn3 function in CA1 pyramidal neurons is restricted to Schaffer collateral synapses.
In the CA1 region of the hippocampus, Lphn2 is selectively enriched in spine synapses in the S. lacunosum-moleculare (16). These synapses are formed by perforant-path axons originating from the entorhinal cortex. To analyze the localization of Lphn3 compared to Lphn2, we generated mice co-expressing mVenus-tagged Lphn2 and HA-tagged Lphn3 (Figure 3A). Using these mice, we confirmed that Lphn2 was highly concentrated in the S. lacunosum-moleculare (Figure 3A, 3B). Lphn3, however, was enriched in the S. oriens and S. radiatum, corresponding to the other dendritic domains of CA1 pyramidal neurons, and was absent from the S. lacunosum-moleculare (Figure 3A, 3B). Higher magnification images of sections that were double-labeled for Lphn2 and Lphn3 revealed that Lphn3 was undetectable in the S. lacunosum-moleculare, whereas Lphn2 was excluded from the S. radiatum (Figure 3C, S5). Similar to cultured neurons, Lphn3 was observed only in excitatory but not inhibitory synapses in the CA1 region (Figure 3D, 3E). Thus, Lphn2 and Lphn3 exhibit complementary, non-overlapping distributions in excitatory synapses of pyramidal CA1 neurons. Interestingly, the spatial segregation of Lphn2 and Lphn3 into different dendritic domains was lost in cultured hippocampal neurons, although in cultured neurons Lphn2 and Lphn3 were still largely localized to distinct synapses in neurons (Figure S6).
Figure 3: Postsynaptic Lphn2 and Lphn3 are targeted to distinct non-overlapping dendritic domains of pyramidal CA1 neurons.

A & B. Lphn3 is broadly distributed in the hippocampal formation except for the S. lacunosum-moleculare of the CA1 region, whereas Lphn2 is present only in the S. lacunosum-moleculare. Overview images (A) depict the entire hippocampal formation (left) or the CA1 region and dentate gyrus (right) viewed in a cryosection from knockin mice expressing mVenus-tagged Lphn2 (16) and HA-tagged Lphn3. The section was stained for Lphn2-mVenus, HA-tagged Lphn3, and nuclei (DAPI). Quantifications of relative Lphn2 and Lphn3 levels (B) show that Lphn2 is specifically targeted in the CA1 region to the S. lacunosum-moleculare, while Lphn3 is present in the S. oriens and S. radiatum but not the S. lacunosum-moleculare (means ± SEM; n=5 mice/experimental group; controls are wild-type mice).
C. High-magnification images showing that the S. radiatum of the CA1 region and the dentate gyrus contain only Lphn2-positive puncta but not Lphn3-positive puncta, whereas the S. moleculare-lacunosum contains only Lphn2-positive puncta but not Lphn3-positive puncta. Images were taken from a cryosection obtained and stained as described for panel A.
D. Low- and high-magnification images showing that Lphn3-positive puncta in the S. radiatum overlap with excitatory vGluT1-positive synapses (left, overview of the S. pyramidale and S. radiatum; right, higher magnification images).
E. Low- and high-magnification images showing that Lphn3-positive puncta in the S. radiatum do not overlap with inhibitory vGAT-positive synapses (left, overview of the S. pyramidale and S. radiatum; right, higher magnification images). Note that most inhibitory synapses are perisomatic in the S. pyramidale.
F. AAV-mediated Cre-recombination deletes Lphn3 in a large area of the CA1 region of HA-Lphn3 cKO mice. Image shows hippocampal section of a mouse that had been stereotactically injected with AAVs encoding eGFP-tagged Cre-recombinase. Section was labeled for Lphn3 and eGFP.
G. High-resolution images of the S. radiatum and the subiculum from control mice and mice after AAV-mediated deletion of Lphn3 in CA1 region (see F). Note that the deletion of Lphn3 abolishes all Lphn3-positive puncta in the CA1 region, excluding the presence or Lphn3-positive presynaptic inputs, but the CA1 region deletion has no effect on Lphn3-positive puncta in the subiculum, ruling out a presence of Lphn3 on CA1-derived presynaptic outputs.
We next asked whether Lphn3 was pre- or postsynaptic in CA1 region neurons. Deletion of Lphn3 in large areas of the CA1 region by stereotactic infection with adeno-associated viruses (AAVs) expressing an eGFP-fusion protein of Cre-recombinase caused a complete loss of Lphn3 from the infected area, but had no effect on Lphn3 levels in the subiculum (Figure 3F, 3G, S7). Since the CA1 region deletion of Lphn3 would not affect Lphn3 expression in the CA3 region or the entorhinal cortex, the complete ablation of Lphn3 by the deletion in the CA1 region means that the presynaptic inputs into the CA1 do not contain Lphn3. Moreover, since the major target area for presynaptic projections from the CA1 region is the subiculum, the absence of a change in Lphn3 staining in the subiculum suggests that the Lphn3 signal here is not derived from CA1 region neurons. Thus, Lphn3 is primarily a postsynaptic protein in the hippocampus.
To examine the function of Lphn3 in vivo, we sparsely expressed eGFP-tagged Cre in CA1 pyramidal neurons of newborn Lphn3 cKO mice using unilateral stereotactic injections of lentiviruses (Figure 4A). We analyzed infected CA1 pyramidal neurons (identified by nuclear eGFP expression) and uninfected control neurons in the opposite hemisphere of the same mouse three weeks later by imaging and slice physiology. We filled individual neurons expressing Cre vs. controls with biocytin for visualization of the entire dendritic arbor (Figure 4B), and measured their spine density in the three major synaptic layers (S. oriens, S. radiatum, and S. lacunosum-moleculare). Deletion of Lphn3 in vivo caused a significant loss of spines in the S. oriens and S. radiatum but not in the S. lacunosum-moleculare, consistent with the selective localization of Lphn3 to these dendritic domains (Figure 4C-4E).
Figure 4: Lphn3 is selectively essential for Schaffer-collateral synapse formation in the CA1 region.

A. Unilateral infection of the neonatal hippocampal CA1 region with lentiviruses expressing EGFP-tagged Cre-recombinase causes sparse deletion of Lphn3 in Lphn3 cKO mice. Image shows a hippocampal section stained for DAPI; infected neurons are identified via their EGFP fluorescence.
B. Filling of individual neurons with biocytin via a patch pipette enables analysis of dendritic morphology. Image shows representative pyramidal neuron filled with biocytin; Cre-recombined and uninfected control neurons were examined in opposite hemispheres of the same mice.
C-E. Quantifications of biocytin-filled, patched CA1 neurons show that postsynaptic Lphn3 deletion decreases dendritic spine densities in the S. oriens and S. radiatum but not the S. lacunosum-moleculare.
F. Diagram of the electrophysiological recording configuration in acute slices with sparse lentiviral expression of Cre-recombinase.
G-I. Postsynaptic Lphn3 deletion in CA1 region neurons decreases the mEPSC but not mIPSC frequency (G, representative mEPSC and mIPSC traces; H & I, summary graphs of the mean mEPSC (H) and mIPSC frequency (I)).
J. Postsynaptic Lphn3 deletion in CA1 neurons decreases the Schaffer-collateral synaptic strength (left, representative traces; right, summary graph of EPSC amplitude). Input-output curves were used to control for differences in stimulus strength.
K. Postsynaptic Lphn3 deletion in CA1 neurons had no effect on the synaptic strength of entorhinal cortex inputs (left, representative traces; right, summary graph of EPSC amplitude).
L. Measurements of paired-pulse ratios of Schaffer-collateral EPSCs as a function of the interstimulus interval show that the Lphn3 deletion has no effect on release probability (left, representative traces; right, summary plot).
M. Same as J, but measured in acute slices from mice that were sparsely infected with Cre-expressing lentiviruses at P21, and analyzed at P40.
Numerical data are means ± SEM (numbers of cells/mice are indicated in bars). Statistical significance was assessed by two-tailed t-tests (** denotes p<0.01; * denotes p<0.05).
We then examined infected cells electrophysiologically (Figure 4F, S8). We observed no change in capacitance or input resistance, but detected a ~50% decrease in the mEPSC frequency (Figure 4G-4I, S8A-S8C). The mEPSC amplitude was unchanged, as were the mIPSC frequency and amplitude. Thus, the Lphn3 deletion caused a selective loss of spines and of excitatory synaptic inputs specifically in the more proximal dendritic domains of CA1 pyramidal neurons innervated by Schaffer collaterals.
To confirm that the decrease in mEPSC frequency was caused by a selective decrease in Schaffer-collateral but not perforant-path synapses, we separately monitored EPSCs evoked by Schaffer-collateral or perforant-path stimulation in the same patched CA1 pyramidal neuron, using input/output curves to control for the variability in stimulus strength (Figure 4F, S8D). The postsynaptic deletion of Lphn3 decreased Schaffer-collateral EPSCs ~50%, but had no effect on perforant-path EPSCs (Figure 4J, 4K). The decrease in Schaffer-collateral synapse strength was not due to a decrease in release probability because the paired-pulse ratio and the co-efficient of variation of EPSCs were unchanged by the Lphn3 deletion (Figure 4L, S8E). These manipulations were performed using sparse postsynaptic deletions of Lphn3 in newborn mice, but deletions by stereotactic Cre-recombinase expression in juvenile mice produced the same phenotype (Figure 4M). In these measurements, EPSCs monitor almost exclusively AMPA-receptor-mediated responses, but parallel experiments assessing NMDA-receptor-mediated responses also showed a decrease in synaptic strength of Schaffer-collateral but not perforant-path synapses (Figure S8F-S8H). NMDA-receptor mediated responses, however, were decreased less strongly in Lphn3-deficient neurons than AMPA-receptor-mediated responses, suggesting that the remaining synapses may be relatively less mature (Figure S8H). Thus, the postsynaptic Lphn3 deletion in CA1 pyramidal neurons selectively impaired synapses formed by Schaffer collaterals onto CA1 pyramidal neurons in the S. oriens and S. radiatum, but had no effect on synapses formed by perforant path axons in the S. lacunosum-moleculare.
These results suggest two major conclusions: Lphn2 and Lphn3 perform similar postsynaptic functions in distinct dendritic domains of CA1 pyramidal neurons, and at least in cultured neurons, the Lphn3 function requires Lphn3 binding of two different ligands that interact with distinct Lphn3 domains. To further test the validity of these conclusions, we performed in vivo rescue experiments. We sparsely infected CA1 neurons of newborn Lphn3 cKO mice with lentiviruses that express Cre alone, or co-express Cre with either Lphn2 or Lphn3 (Figure 5A, S4C). We then patched infected or control neurons in acute slices, and measured the input/output relations of EPSCs elicited by Schaffer-collateral or perforant-path axon stimulation.
Figure 5: Neither wild-type Lphn2 nor mutant Lphn3 lacking FLRT- or teneurin-binding activity rescue the decrease in Schaffer-collateral synaptic strength induced by Lphn3 deletion.

Experiments were performed by whole-cell patch-clamp recordings from CA1 neurons in acute slices from Lphn3 cKO mice that were lentivirally-infected at P0 as described in Figure 4A and 4F.
A & B. Wild-type Lphn3 but not wild-type Lphn2 fully rescues the decrease in Schaffer-collateral synaptic strength induced by loss of Lphn3 (A), while none of the manipulations affects entorhinal cortex-derived synapses (B).
C. Although wild-type Lphn3 fully rescues the decrease in Schaffer-collateral synaptic strength induced by Lphn3 deletion, mutant Lphn3 lacking FLRT-binding (4A) or teneurin-binding (ΔLEC) was unable to rescue.
D. Wild-type Lphn3 also fully rescues the decrease in mEPSC frequency induced by Lphn3 deletion, but again mutant Lphn3 lacking FLRT-binding (4A) or teneurin-binding (ΔLEC) was unable to rescue, with none of the manipulations having any effect on mEPSC amplitude.
Numerical data are means ± SEM (numbers of cells/experiments are indicated in bars). Statistical significance was assessed by one-way ANOVA (* denotes p<0.05).
Wild-type Lphn3 fully rescued the loss of Schaffer-collateral synapse strength in Lphn3-deficient neurons, whereas wild-type Lphn2 had no effect (Figure 3A). None of the manipulations changed synaptic responses induced by perforant-path axon stimulation (Figure 3B). Thus, Lphn2 and Lphn3 mediate similar postsynaptic functions in synapse formation in CA1 pyramidal neurons, but perform these functions in different nonoverlapping dendritic domains that form excitatory synapses with distinct classes of axons. Even overexpression of Lphn2, which is inherent in rescue experiments, did not rescue the deletion of Lphn3, suggesting a tight regulation of the functional differences of Lphn2 and Lphn3.
Blocking teneurin or FLRT3 binding independently ablates Lphn3 function in vivo.
The rescue experiments in cultured neurons suggested that surprisingly, Lphn3 may require simultaneous binding of both FLRTs and teneurins for function. To investigate this, we examined rescue of Schaffer-collateral synaptic strength in Lphn3-deficient CA1 neurons by two Lphn3 mutants, Lphn3-4A (deletes FLRT binding) and Lphn3-ΔLEC (deletes teneurin binding)(Figure 2A). We used rescue with wild-type Lphn3 as a positive control. Strikingly, neither the FLRT-binding nor the teneurin-binding mutant of Lphn3 rescued the loss of synaptic strength, whereas wild-type Lphn3 reversed the phenotype (Figure 5C). Furthermore, measurements of spontaneous mEPSCs in the presence of tetrodotoxin also demonstrated that the two ligand-binding mutants of Lphn3 were unable to rescue the large decrease in mEPSC frequency observed in Lphn3-deficient neurons, whereas wild-type Lphn3 again fully compensated for the Lphn3 deletion (Figure 5D).
To independently validate the electrophysiological conclusions, we used retrograde transmission of recombinant rabies viruses as a tool to measure synaptic connectivity (Figure 6A; 23). In these experiments, we first unilaterally stereotactically infected the hippocampal CA1 region of newborn Lphn3 cKO mice with lentiviruses encoding Cre alone or Cre co-expressed with Lphn3 rescue proteins, as performed for the electrophysiological analyses. We then re-infected the hippocampal CA1 region at P21 with AAVs encoding the Cre-inducible receptor and packaging proteins for replication-defective pseudotyped rabies virus, and finally administered, again stereotactically, the pseudotyped replication-defective rabies viruses at P35 to mediate retrograde tracing of presynaptic inputs onto the CA1 neurons (Figure 6A). We measured three such inputs: Synaptic inputs from the ipsilateral CA3 region, from the contralateral CA3 region, and from the ipsilateral entorhinal cortex, and quantified connectivity by image analysis (Figure 6B).
Figure 6: Rabies virus-mediated retrograde tracing demonstrates selective functions and ligand-dependence of Lphn2 and Lphn3 in hippocampal synaptic connectivity.

A. Experimental approach. The CA1 region of newborn Lphn3 cKO or control mice was sparsely infected unilaterally with lentiviruses encoding Cre-recombinase without or with co-expression of Lphn3 rescue constructs. At P21, the same CA1 region was infected with AAVs encoding the mCherry-tagged receptor and packaging proteins for pseudotyped rabies viruses. At P35, the same CA1 region was infected with the pseudotyped rabies virus encoding eGFP, and at P40, the expression of eGFP transcribed from trans-synaptically transferred rabies virus was analyzed in the ipsi- and contralateral hippocampal CA3 region and the ipsilateral entorhinal cortex.
B. Exemplary images of synaptic inputs mapped by pseudotyped rabies virus administered into the hippocampal CA1 region. Red mCherry-TVA expression marks neurons (starter cells) in which the rabies virus was originally introduced, while green eGFP expression marks neurons with synaptic inputs onto the starter cells.
C. Exemplary higher-magnification images of starter neurons (top row) and neurons providing synaptic inputs onto starter neurons (middle and bottom rows) after Lphn3 manipulations.
D. Quantifications of presynaptic inputs onto postsynaptic CA1 neurons as a function of Lphn3 manipulations, determined by rabies virus tracing and normalized for starter cell numbers.
E & F. Representative images (E) and summary graphs of the presynaptic inputs onto postsynaptic CA1 neurons (F) as a function of Lphn2 deletions. Experiments were performed analogous to those of C and D. Note that the Lphn2 deletion selectively impairs entorhinal cortex inputs into the CA1 region.
Data in D and F are means ± SEM (number of mice analyzed are indicated in bars). Statistical significance was assessed by one-way ANOVA (** denotes p<0.01; * denotes p<0.05).
Lphn3 deletion selectively impaired inputs to CA1 region pyramidal neurons from both the ipsilateral and the contralateral CA3 region but not from the entorhinal cortex (Figure 6C). Whereas wild-type Lphn3 was able to rescue this phenotype fully, neither the FLRT-binding defective nor the teneurin-binding defective Lphn3 mutant produced any rescue. Quantifications revealed that the Lphn3 deletion blocked ~70% of Schaffer-collateral connectivity (Figure 6D). To ensure the specificity of these measurements, we also analyzed the effect of the Lphn2 deletion in the same experimental paradigm (Figure 6E). In contrast to the Lphn3 deletion, the Lphn2 deletion had no effect on Schaffer-collateral synaptic connectivity, but decreased the synaptic connections to the entorhinal cortex by ~60% (Figure 6E, 6F). Thus, deletion of postsynaptic Lphn2 and Lphn3 in the CA1 region selectively decreases the synaptic connectivity of entorhinal cortex or Schaffer-collateral afferents, and the function of Lphn3 in maintaining Schaffer-collateral synapse formation again requires binding sites for both FLRTs and teneurins.
Teneurin-2 and FLRT3 elicit synapse formation only when co-expressed.
To further examine the role of teneurin and FLRT ligand binding to Lphn3 in synapse formation, we turned to an in vitro synapse-formation paradigm (24,25). In this assay paradigm, expression of a candidate synaptic adhesion molecule in a non-neuronal cell, such as a HEK293T cell or COS cell, elicits formation of synaptic contacts with co-cultured neurons that elaborate pre- or postsynaptic specialization at the point of contact (24,25). This assay broadly suggests a role for an adhesion molecule that is active in this assay as a pre- or postsynaptic signaling molecule, depending on what type of specialization it elicits (reviewed in 3).
When we tested Lphn3 in this assay, we observed potent induction of presynaptic excitatory specializations, consistent with a function of a postsynaptic adhesion molecule (Figure S9A-S9C). However, FLRT3 on its own was unable to induce pre- or postsynaptic specializations. Moreover, although the splice variant of teneurin-2 that does not bind to latrophilins actively induced inhibitory postsynaptic but not presynaptic specializations (see also 7), the splice variant of teneurin-2 that binds to Lphn3 was unable to induce pre- or postsynaptic specializations (Figure 7, S9D). However, when FLRT3 was co-expressed in HEK293T cells with the teneurin-2 splice variant that binds to Lphn3, the two molecules together potently induced excitatory but not inhibitory postsynaptic specializations (Figure 7). In this combination, the teneurin-2 splice variant that does not bind to Lphn3 again was inactive, even if combined with FLRT3. Thus, teneurins and FLRTs exclusively act as presynaptic adhesion molecules, and are only able to stimulate excitatory synapse formation in combination but not separately.
Figure 7: Teneurin-2 and FLRT3 expressed in HEK293T cells induce postsynaptic specializations in co-cultured neurons only when FLRT3 is co-expressed with the latrophilin-binding splice variant of teneurin-2.

A & B. Representative images of in vitro synapse-formation assays in which the indicated cell-adhesion molecules are co-expressed with eGFP in HEK293T cells that are then co-cultured with cortical neurons, and subsequently immunostained for the postsynaptic excitatory synapse marker PSD95 (A) or inhibitory synapse marker GABAAα2-receptor (B). For other in vitro synapse formation immunostaining experiments, see Figure S6.
C. Summary graphs showing that only combined expression of Teneurin-2SS− that binds to latrophilins and of FLRT3 that also binds to latrophilins induced excitatory postsynaptic specializations. In contrast, expression of Teneurin-2SS+ alone, but not of Teneurin-2SS−, induced inhibitory postsynaptic specializations. In these experiments, Nrxn1β is used as a positive control that equally induced excitatory and inhibitory postsynaptic specializations (Graf et al., 2004).
D. Summary graphs showing that FLRT3 and teneurin-2 splice variants, alone or in combination, do not induce presynaptic excitatory or inhibitory specializations, whereas Nlgn1, used as a positive control, potently does (n.d. = non-detectable).
Data in C & D are means ± SEM (numbers of cells/experiments are indicated in bars). Statistical significance was assessed by one-way ANOVA (*** denotes p<0.001; ** denotes p<0.01; * denotes p<0.05).
SUMMARY.
How synapses form are maintained, and what molecular processes establish specificity in synaptic connections remains a fundamental unanswered question in neuroscience (1-3). Here, we provide three findings that reveal mechanisms involved in input-specific synapse formation in the brain and suggest an explanation for synapse specificity (Figure S10).
First, we show that Lphn3 is specifically targeted to the dendritic domains of the S. oriens and S. radiatum of hippocampal CA1 pyramidal neurons, whereas the highly homologous Lphn2 is specifically targeted to the S. lacunosum-moleculare in the same neurons. Both Lphn2 and Lphn3 are essential for subsets of excitatory synapses on the dendritic domain to which they are targeted, suggesting that different isoforms of the same postsynaptic protein family differentially function in distinct synapses. These findings thus provide an explanation for the evolution of homologous adhesion-GPCRs and their co-expression in the same neuron, and reveal that different isoforms of a postsynaptic cell-recognition molecule can be targeted to distinct dendritic domains.
Second, we show that autoproteolysis mediated by the GAIN domain (11), a canonical feature of adhesion GPCRs, is not required for Lphn3 function, suggesting that their activation does not involve the exposure of an intrinsic tethered agonist that is rendered competent for receptor binding by removal of the extracellular domains of Lphn3.
Third, we show that individual inactivation of FLRT- or teneurin-binding to Lphn3 blocked its function in synapse formation, and that in the in vitro synapse formation paradigm, teneurin-2 and FLRT3 only induced excitatory synapse formation when they were co-expressed. Even when FLRT3 and teneurin-2 were co-expressed, only the teneurin-2 splice variant capable of binding to latrophilins was active in synapse formation. These results suggest that the requirement for two simultaneous ligands enables a higher specificity in synapse formation. More generally, the co-incidence signaling by multiple ligands we describe here contributes to the emerging realization that signal integration and coincidence detection are a key feature in synaptic plasticity and neural circuits (26,27). Our results suggest that input-specific synapse formation requires integration of multiple trans-synaptic signals acting on latrophilin adhesion GPCRs.
Our results are at odds with several previous results. It has been proposed that latrophilins are presynaptic and FLRTs are postsynaptic (4), but this conclusion was largely based on the notion that latrophilins as α-latrotoxin receptors should be presynaptic. Moreover, a recent study arguing for a postsynaptic localization of FLRT2 is confounded by the use of an antibody targeting the FLRT2 extracellular region for localization analysis, which is presumably localized in the synaptic cleft, and the use of shRNA-mediated knockdowns, which are difficult to control (28). It was also proposed that teneurins act in synapse formation not as heterophilic but as homophilic cell-adhesion molecules (29, 30), but in our assays teneurins do not engage as homophilic cell-adhesion and teneurins act exclusively as presynaptic cell-adhesion molecules.
Our results raise multiple questions. For example, what is the nature of the postsynaptic signal that is activated by latrophilins during synapse formation? How is the specificity of Lphn2 and Lphn3 for different dendritic domains in the same pyramidal neuron determined – is this due to intrinsic sequence determinants or to differential ligand-binding affinities? What postsynaptic ligands mediate teneurin action in inhibitory synapse formation? FLRT3 can simultaneously bind to Lphn3 and to Unc5 in a trans-configuration (22), suggesting that the trans-synaptic teneurin-latrophilin-FLRT complex may be even larger and include postsynaptic Unc5, which in turn could possibly bind to yet another presynaptic adhesion molecule. These large, multi-protein trans-synaptic complexes may be modular and differ in distinct synapse subtypes to increase specificity and generate functional diversity. Thus, the overall portrait of synapse formation emerging from these data is that different latrophilin isoforms are targeted to defined postsynaptic dendritic domains, where they mediate specific excitatory synapse formation by binding to presynaptic FLRTs and teneurins on incoming axons.
METHODS SUMMARY
All procedures conformed to National Institutes of Health Guidelines for the Care and Use of Laboratory Mice and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Generation of Lphn3 cKO mice.
Mutant Lphn3 conditional mice containing an N-terminal double HA tag were generated by introducing the tag into the coding region of the 3’ exon following the exon with the start codon and the sequence encoding for the signal peptide. A frt-site flanked neomycin selection cassette was introduced 5’ to the coding exon as well as a single loxP site. A second loxP-site is flanking the coding exon 3’. The presence of frt-sites allows for Flp-mediated deletion of the selection cassette and the the loxP-sites allow for the Cre-mediated deletion of the HA-epitope bearing exon to generate a Lphn3 null allele. Lphn2-mVenus cKO mice were previously described (16).
Neuronal cultures.
Primary hippocampal cultures were generated by dissecting hippocampi from P0 neonatal mice, dissociating cells by papain digestion, filtering through a 70 μm cell strainer, and plating on Matrigel-coated 0 thickness glass coverslips in 24-well plates. Primary hippocampal neurons were infected with lentiviruses at 3-5 DIV and analyzed at 14-16 days in vitro (DIV). Sparse transfections of cultured neurons were performed 7 days after plating using a calcium phosphate method, and neurons were also analyzed at 14-16 DIV. Whole-cell voltage-clamp recordings in primary hippocampal neurons were conducted at 14-16 DIV as previously described (32).
Stereotactic injections and electrophysiological and imaging analyses in acute slices.
Stereotactic injections into newborn mice and mice at P21 were performed under anesthesia as described previously (16,33). Whole-cell voltage clamp recordings in acute transverse hippocampal slices were performed at P21-25 following P0 neonatal viral injections or at P40-45 following P21 injections as described (16,33). Cells were filled with biocytin for morphological analyses, and images were acquired using a Nikon A1 Eclipse Ti confocal microscope with a 10x, 20x, 60x and 100x objective, operated by NIS-Elements AR acquisition software.
Monosynaptic retrograde rabies viral tracing experiments were conducted by injecting lentiviral Cre or Cre co-expressed with rescue cDNAs via a P2A sequence at P0, followed by injection of rabies complementing AAVs containing CAG-FLEX-TCB-mCherry and CAG-FLEX-RG at P21, subsequent injection of replication-deficient, pseudotyped rabies virus expressing GFP at P35, and analysis at P40.
In vitro synapse formation assays were performed by transfecting HEK293T cells with indicated plasmids and subsequently plating them on 16 DIV cortical neurons 24-hrs post-transfection. Co-cultures were immunolabeled for the indicated synaptic markers 24-hrs later, and analyzed by confocal microscopy.
Detailed Materials and Methods, including a description of the all reagents and of standard procedures, are located in the Supplemental Materials.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. A. Olsen (Stanford Neuroscience Microscopy Facility) for assistance with microscopy, Dr. K. Ritola (Janelia Farm Viral Core facility) for assistance with rabies virus tracing, and Dr. D. Arac (University of Chicago) for sharing plasmids.
Funding: This study was supported by grants from the NIH (F32-MH108230 to RS), K99-MH117235 to RS, NS069375 to A. Olsen, and R37-MH052804 to TCS).
Footnotes
Competing interests: The authors declare no conflict of interest.
Data and materials availability: All data are available in the manuscript or supplementary material. For reagents, please contact T.C.S. at tcs1@stanford.edu.
REFERENCES and NOTES
- 1.Sanes JR, Yamagata M, Many paths to synaptic specificity. Annu. Rev. Cell Dev. Biol. 25, 161–195 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Nusser Z, Creating diverse synapses from the same molecules. Curr. Opin. Neurobiol. 51, 8–15 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Südhof TC, Towards an understanding of synapse formation. Neuron, in press (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Sullivan ML, de Wit J, Savas JN, Comoletti D, Otto-Hitt S, Yates JR 3rd, Ghosh A, FLRT proteins are endogenous latrophilin ligands and regulate excitatory synapse development. Neuron. 73, 903–910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silva JP, Lelianova VG, Ermolyuk YS, Vysokov N, Hitchen PG, Berninghausen O, Rahman MA, Zangrandi A, Fidalgo S, Tonevitsky AG, Dell A, Volynski KE, Ushkaryov YA, Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. USA. 108, 12113–12118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucard AA, Maxeiner S, Südhof TC, Latrophilins function as heterophilic cell-adhesion molecules by binding to teneurins: regulation by alternative splicing. J. Biol. Chem. 289, 387–402 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Shalev-Benami M, Sando R, Jiang X, Kibrom A, Wang J, Leon K, Katanski C, Nazarko O, Lu YC, Südhof TC, Skiniotis G, Arac D, Structural basis for teneurin function in circuit-wiring: A toxin motif at the synapse. Cell. 173, 735–748 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arac D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC, Brunger AT, A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langenhan T, Aust G, Hamann J, Sticky signaling--adhesion class G protein-coupled receptors take the stage. Sci. Signal. 6, re3 10.1126/scisignal.2003825 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Promel S, Frickenhaus M, Hughes S, Mestek L, Staunton D, Woollard A, Vakonakis I, Schoneberg T, Schnabel R, Russ AP, Langenhan T, The GPS motif is a molecular switch for bimodal activities of adhesion class G protein-coupled receptors. Cell Rep. 2, 321–331 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liebscher I, Schöneberg T, Tethered Agonism: A Common Activation Mechanism of Adhesion GPCRs. Handb. Exp. Pharmacol. 234, 111–125 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Muller A, Winkler J, Fiedler F, Sastradihardja T, Binder C, Schnabel R, Kungel J, Rothemund S, Hennig C, Schoneberg T, Promel S, Oriented cell division in the C. elegans embryo is coordinated by G-protein signaling-dependent on the adhesion GPCR LAT-1. PLoS Genetics. 11, e1005624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paavola KJ, Hall RA, Adhesion G protein-coupled receptors: signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 82, 777–783 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purcell RH, Hall RA, Adhesion G Protein-Coupled Receptors as Drug Targets. Annu. Rev. Pharmacol. Toxicol. 58, 429–449 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salzman GS, Zhang S, Gupta A, Koide A, Koide S, Araç D, Stachel-independent modulation of GPR56/ADGRG1 signaling by synthetic ligands directed to its extracellular region. Proc. Natl. Acad. Sci. U S A. 114, 10095–10100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson GR, Maxeiner S, Sando R, Tsetsenis T, Malenka RC, Südhof TC, Postsynaptic adhesion GPCR latrophilin-2 mediates target recognition in entorhinal-hippocampal synapse assembly. J. Cell Biol. 216, 3831–3846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tobaben S, Südhof TC, Stahl B, The G protein-coupled receptor CL1 interacts directly with proteins of the Shank family. J. Biol. Chem. 275, 36204–36210 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Kreienkamp HJ, Soltau M, Richter D, Bockers T, Interaction of G-protein-coupled receptors with synaptic scaffolding proteins. Biochem. Soc. Trans. 30, 464–468 (2002). [DOI] [PubMed] [Google Scholar]
- 19.For methods and supplementary figures, see Supplementary Online Materials.
- 20.Liebscher I, Schön J, Petersen SC, Fischer L, Auerbach N, Demberg LM, Mogha A, Cöster M, Simon KU, Rothemund S, Monk KR, Schöneberg T, A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 9, 2018–2026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vu TK, Hung DT, Wheaton VI, Coughlin SR, Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 (1991). [DOI] [PubMed] [Google Scholar]
- 22.Lu YC, Nazarko OV, Sando R, Salzman GS, Li NS, Südhof TC, Arac D, Structural basis of latrophilin-FLRT-UNC5 interaction in cell adhesion. Structure. 23, 1678–1691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Callaway EM, Transneuronal circuit tracing with neurotropic viruses. Curr. Opin .Neurobiol. 18, 617–623 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T, Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101, 657–669 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM, Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicoll RA, A Brief History of Long-Term Potentiation. Neuron 93, 281–290 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Sakmann B, From single cells and single columns to cortical networks: dendritic excitability, coincidence detection and synaptic transmission in brain slices and brains. Exp. Physiol. 102, 489–521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder A, Vanderlinden J, Vints K, Ribeiro LF, Vennekens KM, Gounko NV, Wierda KD, de Wit J, A modular organization of LRR protein-mediated synaptic adhesion defines synapse identity. Neuron 99, 329–344 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Hong W, Mosca TJ, Luo L, Teneurins instruct synaptic partner matching in an olfactory map. Nature 484, 201–207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berns DS, DeNardo LA, Pederick DT, Luo L, Teneurin-3 controls topographic circuit assembly in the hippocampus. Nature 554, 328–333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC, RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell 2, 282–295 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maximov A, Pang ZP, Tervo DG, Südhof TC, Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci. Methods 161, 75–87 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.