

Abstract
The gene SCN5A encodes the main cardiac sodium channel NaV1.5. This channel predominates the cardiac sodium current, INa, which underlies the fast upstroke of the cardiac action potential. As such, it plays a crucial role in cardiac electrophysiology. Over the last 60 years a tremendous amount of knowledge regarding its function at the electrophysiological and molecular level has been acquired. Furthermore, genetic studies have shown that mutations in SCN5A are associated with multiple cardiac diseases (e.g. Brugada Syndrome, Long QT syndrome, conduction disease and cardiomyopathy), while genetic variation in the general population has been associated with differences in cardiac conduction and risk of arrhythmia through genome wide association studies. In this review we aim to give an overview of the current knowledge (and the gaps therein) on SCN5A and NaV1.5.
Introduction
Voltage-gated sodium channels are responsible for the inward sodium current (INa) in excitable cells. As such, they induce a fast depolarization, thereby initiating an action potential(Weidmann 1955). In the heart, INa is crucial for fast impulse propagation through the tissue(Buchanan et al. 1985). The main protein generating the cardiac sodium current is the pore-forming alpha subunit NaV1.5, encoded by the gene SCN5A (Gellens et al. 1992; Abriel 2010). Since the cloning of this gene in 1992(Gellens et al. 1992), a substantial amount of genetic and molecular biological information has been obtained regarding the role of this gene and its corresponding protein in health and disease. Mutations have been identified in families with inherited cardiac arrhythmia syndromes and genetic variation in the general population at the SCN5A-locus has been associated with electrocardiographic differences (Remme, 2013). We here provide an overview of the current knowledge concerning the regulation and function of the gene SCN5A and its corresponding protein NaV1.5 and discuss the association of gene mutations and common variants in relation to inherited cardiac diseases. For some of the pertinent topics of this review excellent recent and highly detailed reviews are available (e.g. sodium channel interaction partners(Abriel 2010), posttranslational modifications(Marionneau & Abriel 2015)), human genetics and splice regulation (Schroeter et al. 2010). In these cases we here provide only a short summary and refer the reader to these reviews for further reading.
Gene SCN5A
Gene structure
SCN5A, i.e. sodium channel, voltage gated, type V alpha subunit, NaV1.5, (ENSG00000183873, HGNC:10593, NCBI Gene ID: 6331), is part of a family of 10 genes encoding sodium channel alpha subunits. Of these, NaV1.1, NaV1.2, NaV1.3 NaV1.6 and Nav2.1 (Nax) are the main sodium channels in the central nervous system, NaV1.7, NaV1.8 and NaV1.9 in the peripheral nervous system, NaV1.4 in skeletal muscle and NaV1.5 which is encoded by SCN5A is the main sodium channel in the heart (reviewed in Catterall, 2014). SCN5A is a large, highly conserved gene that is present from platypus to birds and human (Catterall 2014). The gene SCN5A spans more than 100kb on human chromosome 3p22 and consists of 28 exons, of which exon 1 and in part exon 2 forms the 5’ untranslated region (5’ UTR) and exon 28 the 3’ untranslated region (3’ UTR) of the RNA.
Expression pattern
SCN5A transcripts are mainly found in the heart, however transcript levels have been demonstrated in smooth muscle cells of the intestines(Holm et al. 2002) and in macrophages(Black et al. 2013). Also, the “neonatal” splice isoform of SCN5A (see below) is expressed in the central nervous system and in certain types of cancer (Catterall 2014). The functional role of SCN5A in these non-cardiac tissues is only slowly beginning to emerge. In the heart, SCN5A transcripts are highly abundant in working myocardium and conductive tissue, whereas the expression in the sinoatrial and atrioventricular nodes is relatively low (Remme et al. 2009). Within the sinoatrial node, the central part is devoid of Nav1.5 expression, while expression has been demonstrated in the periphery(Lei et al. 2004). Accross the ventricular wall, a transmural expression gradient exists, as reflected by higher expression of SCN5A in the subendocardial layer as compared to the subepicardium(Remme et al. 2009).
Splice variants
More than 10 different splice isoforms have been described and are predicted based on sequence and cDNA. Many of these splice isoforms are differentially expressed in the heart and other tissues and are known to have different electrophysiological properties. However, not all splice isoforms have been studied in detail and discrepancies in the nomenclature between studies hamper easy comparison of the results. In Table 1 we provide an overview of the annotated splice variants with the corresponding reference IDs in different databases and papers. A clear comprehensive overview of the functional differences between the different splice isoforms is given in a recent review (Schroeter et al. 2010).
Table 1, SCN5A transcript variants.
Overview of the described splice variants in SCN5A. As different sources refer to the different variants with different numbers and letters, we here aim to provide an unambiguous link between the different annotations. Ensembl transcript number and ID refer to identifications of the Ensembl consortium (www.ensembl.org); CCDS refers to the consensus cDNA (http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi) number. Uniprot ID refers to the reference protein ID (http://www.uniprot.org/); RefSeq ID and cDNA name refers to the reference transcript and protein IDs and the isoform names of NCBI respectively (http://www.ncbi.nlm.nih.gov). Characteristics provides a short summary of the essential differences between the isoforms.
| Ensembl transcript nr | Ensembl ID | length of cDNA | length of protein | CCDS | UniPROT ID | RefSeq IDs (transcript, protein) | NCBI names (transcript, protein) | cDNA name (Schroeter et al) | characteristics |
|---|---|---|---|---|---|---|---|---|---|
| SCN5A-014 | ENST00000333535 | 8456 | 2016 | CCDS46796 | Q14524 | NM_198056, NP_932173 | transcript variant 1, isoform a | NaV1.5c | “adult” exon 6b, inclusion of Q1077 |
| SCN5A-003 | ENST00000423572 | 8362 | 2015 | CCDS46797 | Q14524 | NM_000335, NP_000326 | transcript variant 2, isoform b | NaV1.5 | alternative 5’UTR, ΔQ1077, “adult” exon 6b |
| SCN5A-001 | ENST00000413689 | 8504 | 2016 | CCDS46799 | H9KVD2 | NM_001099404, NP_001092874 | transcript variant 3, isoform c | Nav1.5e | “neonatal” exon 6a, inclusion of Q1077 |
| SCN5A-002 | ENST00000455624 | 7170 | 1983 | CCDS54570 | E9PHB6 | NM_001160160, NP_001153632 | transcript variant 5, isoform e | Nav1.5e | alternative 5’UTR, ΔQ1077, “neonatal” exon 6a |
| SCN5A-004 | ENST00000414099 | 8303 | 1998 | CCDS46798 | E9PG18 | - | - | Nav1.5f | alternative 5’UTR, Q1077, lacking exon 24 |
| SCN5A-008 | ENST00000450102 | 6284 | 1962 | CCDS54569 | K4DIA1 | - | - | n.d. | “neonatal” exon 6a, lacking |
| exon 18, alternative 3’UTR | |||||||||
| SCN5A-010 | ENST00000449557 | 5898 | 1962 | - | A0A0A0MT39 | - | - | NaV1.5a | “adult” exon 6b, lacking exon 18, alternative 3’UTR |
| SCN5A-201 | ENST00000425664 | 8450 | 1998 | CCDS46798 | E9PG18 | NM_001099405, NP_001092875 | transcript variant 4, isoform d | Nav1.5f | predicted transcript, Q1077, lacking exon 24 |
| SCN5A-202 | ENST00000451551 | 8343 | 1962 | CCDS54569 | K4DIA1 | NM_001160161, | transcript variant 6, | n.d. | predicted transcript, lacking |
| NP_001153633 | isoform f | exon 18, “neonatal” exon 6a | |||||||
| SCN5A-203 | ENST00000612060 | 714 | 223 | - | Q86V90 | - | - | n.d. | incomplete |
| SCN5A-006 | ENST00000327956 | 335 | 65 | - | A3EY21 | - | - | n.d. | incomplete |
| SCN5A-005 | ENST00000491944 | 1382 | - | - | - | - | - | n.d. | non-coding |
| SCN5A-013 | ENST00000476683 | 774 | - | - | - | - | - | n.d. | non-coding |
| SCN5A-011 | ENST00000464652 | 591 | - | - | - | - | - | n.d. | non-coding |
There are four major different coding SCN5A isoforms, of which SCN5A-003 (NM_000335) is the most abundant transcript in murine and human heart (Onkal et al. 2008; Schroeter et al. 2010) (figure 1). This transcript, also referred to as the “adult” isoform, replaces the “neonatal” SCN5A-001 (NM_001099404, NaV1.5e) within a few days after birth in mice. The two isoforms differ in exon 6 (exon 6b in the adult and exon 6a in the neonatal isoform), resulting in a difference of 7 amino-acids. (Onkal et al. 2008; Schroeter et al. 2010). SCN5A-001 is also abundantly expressed in neonatal murine brain and at low levels in adult brain (Chioni et al. 2005). Functionally these two isoforms differ significantly: compared to the adult isoform, the neonatal splice isoform exhibits slower rate of activation and inactivation and a depolarized shift in voltage dependence of activation(Onkal et al. 2008). Apart from these two isoforms, SCN5A-010 (no refseq ID) & SCN5A-014 (NM_198056) have been studied in some detail. SCN5A-010, which lacks exon 18, is expressed in the rat brain, heart and hippocampal progenitor cells, howeverthis splice isoform has not been found in human tissue as yet (Schroeter et al. 2010). SCN5A-014 includes an additional CAG at the exon 17–18 splice boundary resulting in an additional glutamine at position 1077 (1077Q). This transcript is expressed in the human heart albeit less abundantly than SCN5A-003 (ratio approximately 1:2) (Makielski et al. 2003; Schroeter et al. 2010). The transcript including 1077Q encodes channels that exhibit less INa(Makielski et al. 2003). Remarkably, certain polymorphisms demonstrate more pronounced effects in either the SCN5A-014 or the SCN5A-003 splice variant. (Tan et al. 2005). In addition to the coding splice variants described above, several different exon 1 variants have been described leading to alternative 5’UTRs. It is likely that these differences in 5’UTR play a role in transcriptional regulation.
Figure 1: Illustrative scheme of splice variants of SCN5A.
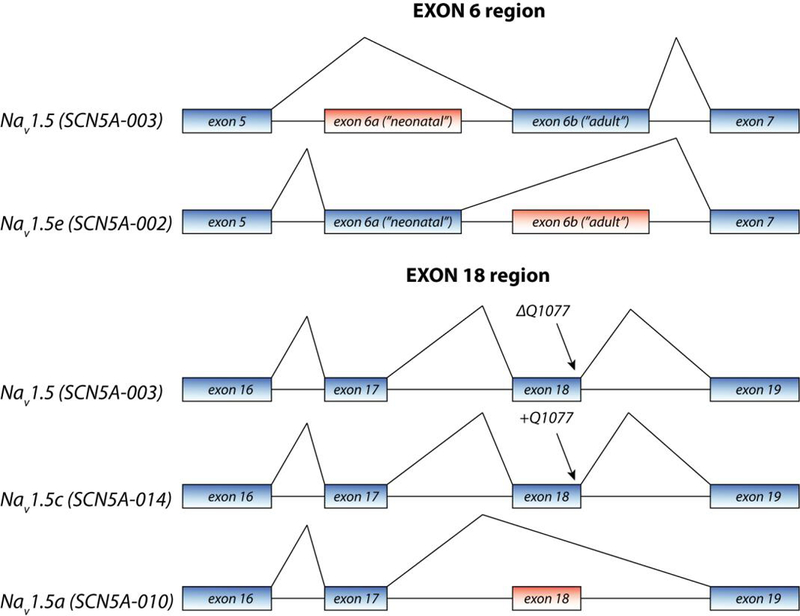
Annotations are according to Schroeter et al (2010), with in parenthesis the ID as can be found in the Ensembl database (see Table 1). Alternative splicing mainly occurs at the region of exon 6 and exon 18.
Interestingly, the different exons 1 are partially species specific as they are different between the murine and human genome (van Stuijvenberg et al. 2010; Shang & Dudley 2005). Functional consequences of these exon 1 splice variations remain to be elucidated.
Transcriptional regulation
Currently, three distinct promoter regions have been identified for SCN5A, corresponding to 4 different transcripts with alternative 5’ UTRs (Yang et al. 2004; van Stuijvenberg et al. 2010). The original identified promoter consists of 2.8 kilobases and exceeds exon1 and partially intron 1 (Yang et al. 2004). Several transcription factors that influence gene expression have been identified to date, including Forkhead Box O1 (Foxo1)(Cai et al. 2014; Mao et al. 2012), nuclear factor-κ Β (NF-KappaB) (Shang et al. 2008) and TBX5 (Arnolds et al. 2012; Van Den Boogaard et al. 2012). Foxo1 and NF-KappaB are both involved in gene regulation upon oxidative stress, present for example during myocardial infarction or upon hypertrophic stimuli. Production of reactive oxygen species (ROS) leads to the nuclear translocation of these transcription factors and consequently inhibits transcription of SCN5A by directly binding the promoter region. TBX5, which plays a fundamental role during cardiac development, stimulates SCN5A expression in the adult cardiac conduction system by binding gene enhancer elements (Arnolds et al. 2012; Van Den Boogaard et al. 2012). Apart from regulation through direct transcriptional control, regulation of SCN5A at the posttranscriptional level through microRNAs has been demonstrated, showing increased transcript and protein levels upon expression of miR-219 (Daimi et al. 2015).
Structure and function of NaV1.5
Protein structure and function
NaV1.5 is a large transmembrane protein with 4 repetitive transmembrane domains (DI-DIV) with 6 transmembrane spanning sections each (S1–S6). The four S5–S6 and intervening loop domains together are the central pore forming region of the α-subunit of the sodium channel, while the remaining S domains act as voltage sensors, in which the positively charged S4 domains play a crucial role (Figure 2) (Chen-Izu et al. 2015; Catterall 2014). As other voltage-gated sodium channels, NaV1.5 exhibits different biophysical properties regarding its voltage and time-dependent conformational state (termed ‘gating’), which determines whether the channel is opened (i.e. able to conduct Na+-ions) or closed. During diastole, when the transmembrane electrical potential is around −85 mV, NaV1.5-channels are in a closed state. As the membrane depolarizes upon a stimulus and a certain threshold is reached, channels become activated within 1ms. This is mediated through a simultaneous outward movement of the S4 segments of all 4 transmembrane domains, resulting in the opening of the channel pore and, due to the electrochemical gradient, inward conductance of Na+-ions. Consequently, a fast depolarization of the membrane is realized, reflecting phase 0 of the cardiac action potential (Figure 3A). Immediately upon depolarization, NaV1.5 channels are closed through a process called ‘fast inactivation’. Again the S4 segments, especially those from domain III and IV, are moved outward, while the intracellular loop between domains III and IV functions as a ‘lid’ to close the channel pore. In the latter event, the amino acid sequence IFM (located at position 1488–1490) plays a key role (West et al. 1992; Kellenberger et al. 1996). Activation and inactivation of NaV1.5 channels is voltage-dependent, as depicted in Figure 3B. In physiological conditions, when inactivated, channels remain in closed state until the cell membrane is repolarized, allowing them to recover from inactivation and becoming available for activation again. While the membrane is still depolarized, NaV1.5 channels undergo more conformational changes, reaching different states of inactivation, i.e. the ‘intermediate-’ and ‘slow-inactivation’ state. During the cardiac action potential, Na+-channels never reach the full slow-inactivated state, as this happens only after a time frame of >60 seconds(O’Reilly et al. 1999). The intermediate-inactivated state however, can be reached during the action potential, albeit only by a small fraction of channels (Nuss et al. 1995; Veldkamp et al. 2003). All these states require different times to recover during the repolarization phase: while recovery from fast-inactivation happens within 10 ms, Na-channels that reside in intermediate and slow-inactivation states require ~50 ms and >5 s, respectively, until they are available for activation again. The exact biophysical processes underlying the different state of inactivation are not completely understood; here, important roles for different structural parts of the Na-channel (e.g. the pore, intracellular loops and voltage-sensor) have been suggested, pointing out different responsible mechanisms (Groome 2014).
Figure 2. NaV1.5 protein structure.
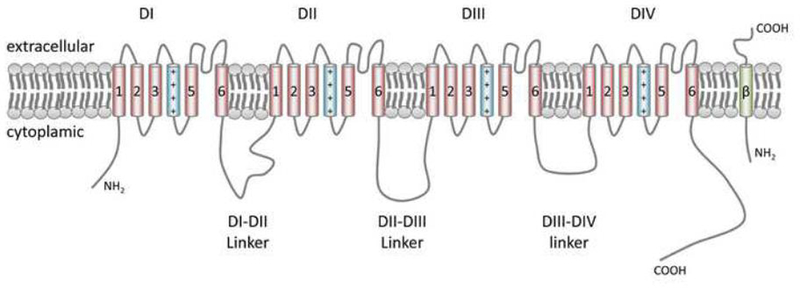
The transmembrane segments S1–S6 are indicated by numbered cylinders; the fourth positively charged S segment, important in voltage sensing is depicted in blue. The transmembrane segment depicted in green resembles one of the beta-subunits.
Figure 3. Nav1.5-driven INa in normal conditions.
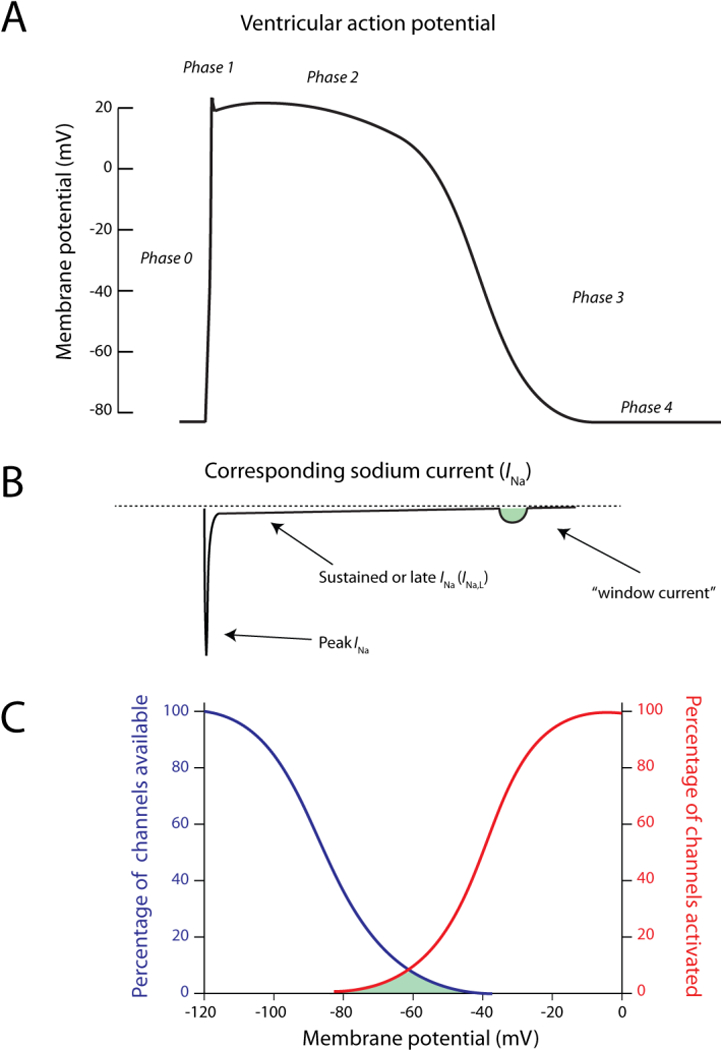
A) The ventricular action potential as a function of time and B) the corresponding INa in the physiological situation. The window current is depicted in green C) Illustration of the percentage of available (blue) and activated (red) channels as a function of the membrane potential. The window current (indicated in green) is formed at potentials in which inactivation and activation are overlapping.
During the action potential, a very small fraction of sodium current persists and does not inactivate completely. This current is called ‘sustained current’, ‘late current’ or ‘INa,L’ (Sakmann et al. 2000; Maltsev et al. 1998). Finally, some channels may reactivate during the repolarizing phase of the action potential at a range of potentials where inactivation is not complete and shows overlap with activation, generating the so-called “window current” (Figure 3A and 3B) (Attwell et al. 1979). Both the window current and the sustained current can play important roles in genetic and acquired cardiac diseases, as discussed below.
Sub-units and protein interaction partners
To date, a wide scale of interacting proteins regulating function or membrane expression of NaV1.5 have been identified (extensively reviewed in Abriel, 2010). NaV1.5 is part of a macromolecular complex in which different proteins interact and modify the trafficking, function, or structure of the channel. An important group of interacting proteins is formed by the 4 beta-subunits, transmembrane proteins encoded by the genes SCN1B to SCN4B that consist of only one transmembrane segment. While different voltage gated sodium channels do not exhibit a response to each beta-subunit, for the cardiac sodium channel NaV1.5 a role has been assigned for each one of them. Although results on the direct exact physiological effects of beta-subunits on NaV1.5-driven Na+-current are conflicting (Meadows & Isom 2005; Abriel 2010), in general, beta-subunits increase currents by increase in trafficking of the channel to the cell surface or change in the intrinsic properties of the sodium channel, such as voltage dependence of (in)activation. Moreover, the interaction of NaV1.5 with some other important interacting proteins is dependent on the presence of beta-subunits (Meadows & Isom 2005). Finally, the crucial role of beta-subunits is highlighted by the fact that mutations in the genes SCN1B to SCN4B are implicated in different cardiac arrhythmia syndromes (Watanabe et al. 2009; Watanabe et al. 2008; Hu et al. 2009; Medeiros-Domingo et al. 2007). For example, SCN1B, which encodes the β1-subunit, has been associated with Brugada syndrome (Watanabe et al. 2008), conduction disease and atrial fibrillation.
Apart from the beta-subunits, other proteins interacting and modulating function of NaV1.5 have been identified, such as calmodulin, calmodulin kinase II δc, ankyrin-G and plakophilin-2, of which some have also been linked to genetic and acquired cardiac diseases (Cerrone et al. 2014; Herren et al. 2013) These interactions and postranslation modifications of the cardiac sodium channel have been reviewed in detail (see Abriel, 2010).
Genetics
SCN5A mutations: association with disease
Mutations in SCN5A can disrupt proper function of NaV1.5 and as such lead to different, mainly cardiac, diseases. Both loss- and gain-of-function mutations are described, while occasionally a mutation results in functional channels with aspects of both, leading to a disease with overlapping phenotypes (Remme et al. 2006). Most commonly, pathogenic SCN5A mutations show an autosomal dominant inheritance pattern, with incomplete penetrance, but also recessive forms with homozygous or compound heterozygous mutations are described (Lupoglazoff et al. 2002; Neu et al. 2010; Lopez et al. 2011; Frigo et al. 2007; Bezzina et al. 2003; Baskar et al. 2014). Interestingly, the observed phenotypes in SCN5A mutation carriers are highly diverse. This diversity could in part be explained by the fact that different biophysical aspects of the channel (e.g. voltage dependence of (in)-activation, conductivity, INa,L) could be affected by the mutation, leading to both loss- and gain-of-function (Figure 4 and 5, respectively). However, even within families, different clinical phenotypes can be observed, suggesting important roles for environmental and other (common) genetic factors (Nakajima et al. 2013; Bezzina et al. 1999; Remme et al. 2006).
Figure 4. Reduced function of INa as a consequence of loss-of-function mutation in SCN5A.
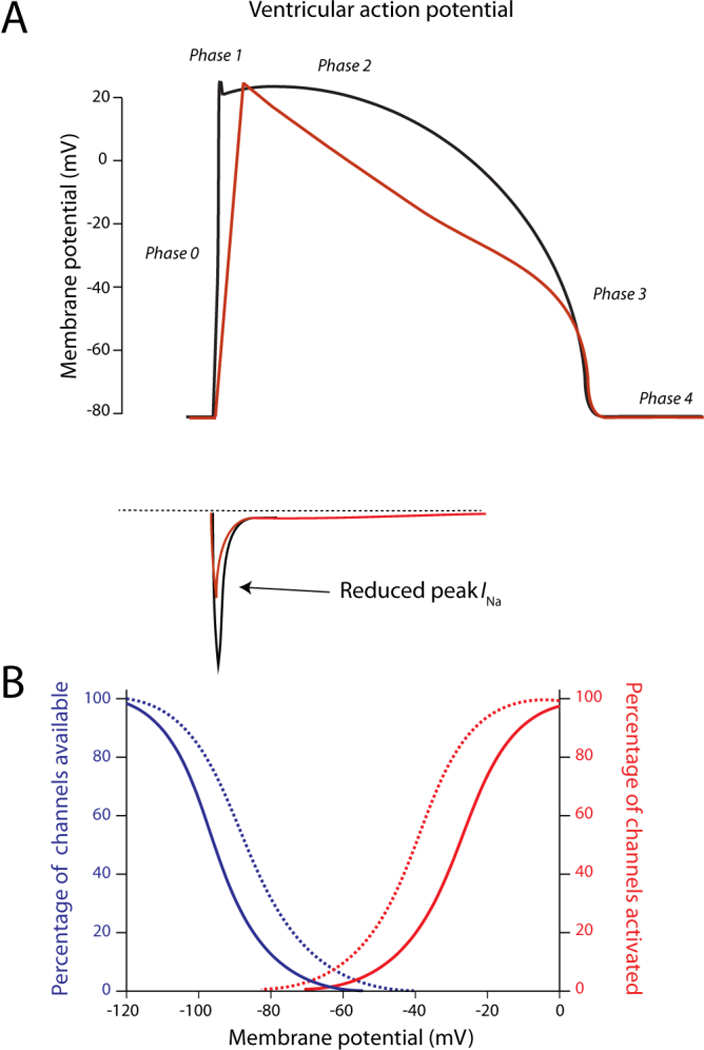
A) Decreased peak INa lowers the upstroke velocity of the action potential (red trace). B) Shifts in voltage dependence of (in)activation (dashed lines) that result in loss-of-function of INa. Apart from mutations that lower the amount of channels at the membrane or reduce conductivity of the channel, this phenomenon forms an alternative mechanism for reduced functionality.
Figure 5: Functional effects of gain-of-function mutations in SCN5A.
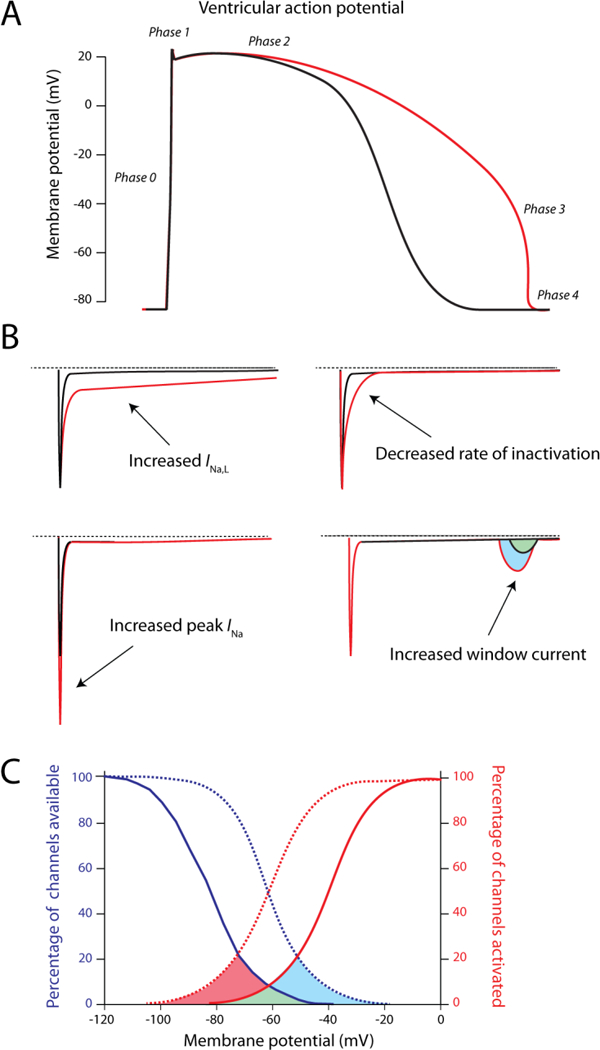
A) Consequences of gain-of-function on the ventricular action potential. Due to an increased net influx of Na+-ions, action potential duration is increased (red trace), which can evoke arrhythmic events. B) Different mechanisms that can be responsible for the gain-of-function in INa, i.e. increased late current (INa,L), increased peak INa, decreased rate of inactivation and increased window current. Most commonly, an increase in sustained current (INa,L) is observed. C) Shifts in voltage dependence of (in)activation (dashed lines) that lead to an increased window current, as illustrated by the red and blue areas under the curve.
To date, loss-of-function mutations have been associated with Brugada syndrome (BrS) (Chen et al. 1998; Bezzina et al. 1999; Remme et al. 2006) progressive cardiac conduction disease (Lev-Lenègre disease) (Schott et al. 1999; Tan et al. 2001), dilated cardiomyopathy (DCM) (McNair et al. 2004; Olson et al. 2005; Laurent et al. 2012; Bezzina et al. 2003), sick sinus syndrome (Benson et al. 2003; Smits et al. 2005) and atrial fibrillation (Makiyama et al. 2008). Mutations resulting in a gain-of-function are causal for Long QT syndrome type 3 (Wang et al. 1995; Remme et al. 2006) and are also more recently implicated in Multifocal ectopic Purkinje-related premature contractions (MEPPC)(Mann et al. 2012; Swan et al. 2014; Laurent et al. 2012). Some gain-of-function mutations are also associated with AF and DCM (Olson et al. 2005).
Brugada syndrome
BrS is a familial arrhythmia syndrome characterized by ST-elevations in the right precordial leads on the ECG. Patients with Brugada syndrome are at substantial risk for development of tachyarrhythmia and sudden cardiac death. While originally described as a disease in which no cardiac structural defects are present, different reports have pointed out subtle microscopic structural abnormalities in hearts from BrS patients, specifically fibrosis (Coronel et al. 2005). Also, in many cases BrS patient present with cardiac conduction slowing. The exact mechanism underlying the ECG abnormalities and the arrhythmia are still controversial (Hoogendijk et al. 2011). However, given that ~25% of BrS patients possess a loss-of-function mutation in SCN5A and that the typical ECG features can be evoked by challenge with sodium channel blockers (e.g. ajmaline), the cardiac sodium channel plays an important role in this disease (Lippi et al. 2012). However, apart from incomplete penetrance of mutations, presence of the disease in absence of the familial SCN5A mutation is not uncommon, indicating the genetic complexity of the disease (Probst et al. 2009; Marsman et al. 2013).
Progressive cardiac conduction disease (Lev-Lenègre disease)
While the mechanism of loss-of-function mutations underlying BrS is still heavily debated, the role of SCN5A mutations in progressive cardiac conduction disease, or Lev-Lenègre disease, is more clear, given the important role of NaV1.5 in the specialized cardiac conduction system (Remme et al. 2009). Patients with Lev-Lenègre disease present usually at more advanced age with widened QRS complexes on the ECG, in combination with a left- or right-bundle-branch block that can eventually culminate into complete atrioventricular block. Although it is a very common cardiac condition, Lev-Lenègre disease shows a familial inheritance only sporadically. Mutations in SCN5A as a cause of Lev-Lenègre disease were first described in 1999 (Schott et al. 1999), after which more reports followed (Tan et al. 2001).
Sick sinus syndrome (SSS)
Sick sinus syndrome (SSS) is a disease characterized by malfunction of the sinus node in which patients exhibit sinus bradycardia, sinus arrest and reduced chronotropic response (Abe et al. 2014; Butters et al. 2010; Benson et al. 2003). Although the disease usually manifests at later age, mostly due to structural defects related to fibrosis or ischemia, families in which SSS manifests at younger age and inherits according to Mendelian patterns, are described. One of the genes that has been linked to SSS is SCN5A (Makita et al. 2005; Benson et al. 2003; Smits et al. 2005; Veldkamp et al. 2003; Makiyama et al. 2008). Since expression of SCN5A is low in central sinus nodal cells and sodium channels are mostly inactivated during the relatively positive diastolic potential, mutations in SCN5A have only small impact on individual primary pacemaker cells. However, in the periphery of the sinus node, in which SCN5A is expressed(Lei et al. 2004), loss-of-function mutations can affect the amount of ‘window’ current during the diastolic phase, thereby lowering the speed of diastolic depolarization at the single cell level (Abe et al. 2014; Butters et al. 2010). The role of Nav1.5 in sinus node cells has been highlighted in mouse models, demonstrating sinus node dysfunction in Scn5a knockout mice(Lei et al. 2005) and slowed sinus node conduction upon Nav1.5 blockade(Lei et al. 2004). Considering the connections between sinus node and atrium, there is less inward current in the sinus node counteracting the more negative diastolic potential of atrial cells, resulting in hyperpolarization of the central sinus node cells that leads to slowing of the firing rate. Moreover, reduced excitability of atrial cells due to the mutation can lead to exit block, a common feature in SSS (Butters et al. 2010).
Atrial fibrillation
Atrial fibrillation (AF) is the most common arrhythmia syndrome in the Western World (Zoni-Berisso et al. 2014). During AF, atria exhibit involuntary contractions rather than the simultaneous single contraction normally occurring after depolarization of the atria, which is the result of a continuous chaotic and unorganized electrical activity. In general, the disease occurs in the context of structural heart disease. In the absence of any structural abnormalities, especially when it arises in relatively young people, and when a familial pattern is observed, a genetic cause is suspected, however the genetic causes of AF remain largely elusive thus far (Napolitano 2013). Emerging evidence suggests a possible link between mutations in SCN5A and familial AF: (i) a high prevalence of SCN5A mutations, that co-segregated among family members was noted in AF patients (Ellinor et al. 2008; Darbar et al. 2008); (ii) there is a high degree of overlap between atrial fibrillation and other diseases associated with SCN5A mutations, i.e. BrS, LQT3 and conduction disease (Laitinen-Forsblom et al. 2006; Rossenbacker et al. 2004; Darbar et al. 2008); (iii) the common polymorphism NaV1.5-H558R, that results in a slight reduction of function, is more prevalent among AF patients than in controls (Smith et al. 2009; Nikulina et al. 2015). As both gain- and loss-of-function mutations have been described, the possible underlying pathogenic mechanism remains unclear.
Long QT syndrome type III
The first mutation described for SCN5A was ΔKPQ and was linked to congenital Long QT syndrome (LQT type III). Patients with LQT syndrome exhibit prolonged QT-intervals at the ECG, which is reflected as increased action potential duration at the cellular level (Figure 5A). Moreover, patients are at increased risk for the development of polymorphic ventricular tachycardia, specifically torsade de pointes (Wang et al. 1995). From all successfully genotyped LQTS patients, 5–10% carry a mutation in SCN5A, whereas 90% of the genotyped patients possess a mutation in the potassium channel encoding genes KCNQ1 and KCNH2 (manifesting as LQT1 and LQT2, respectively)(Mizusawa et al. 2014). Gain-of-function mutations in SCN5A that result in LQT3 usually impact on the inactivation characteristics of the sodium channel, which is either slowed or incomplete (Figure 5B). Due to failure to inactivate completely, the late component of the sodium current is increased, leading to a persistent inward current during the plateau phase of the action potential and subsequently a prolongation. An alternative mechanism is a shift in voltage dependence of inactivation, resulting in an increase in window current (Figure 5C4B). Finally, an increased rate of recovery from inactivation can result in an increased rate of channel reopening during the repolarization phase, as illustrated for the mutation I1768V (Clancy et al. 2003)
Multifocal Ectopic Purkinje-Related Premature Contractions
Recently, the mutation R222Q in SCN5A was identified in different unrelated families in which mutation carriers exhibited frequent premature ventricular complexes arising from the Purkinje system, leading to ventricular tachycardia and sudden death in some cases (Nair et al. 2012; Laurent et al. 2012; Mann et al. 2012). Moreover, this arrhythmia syndrome, annotated as Multifocal Ectopic Purkinje-Related Premature Contractions (MEPPC), was associated with dilated cardiomyopathy, probably secondary to the arrhythmias. Functional studies revealed that the R222Q mutation exhibits a negative shift in both voltage dependence of activation and inactivation, indicating both a gain- and loss-of-function, however the net effect gives rises to an increase and shift in window current. Modeling studies performed in that study showed that the mutation affects the action potential of Purkinje cells, i.e. repolarization is delayed, while ventricular cells are not affected by the mutation, in alignment with the origin of the premature contractions. Also, the simulated effects were more profound at rest, a phenomenon also observed in the mutation carriers.
Apart from R222Q, a different study identified another mutation located in the same region, R225P, which generates a similar phenotype with similar biophysical changes. While premature ventricular complexes are mainly present at rest in MEPPC cases, a recent study has linked a variant in SCN5A to a family with patients experiencing a high frequency of exercise-induced premature beats (Swan et al. 2014). Also this variant (I141V) exhibited an increased window current, similar to the R222Q mutation.
Dilated cardiomyopathy (DCM)
The association of SCN5A mutations and dilated cardiomyopathy (DCM) (Olson et al. 2005; Laurent et al. 2012; Bezzina et al. 2003; McNair et al. 2004), a structural heart disease characterized by dilated chambers, pump failure and a high incidence of arrhythmia, is possibly the most intriguing and surprising one. To date, the mechanism underlying the disease in case of SCN5A mutations is mainly speculative. This has been complicated by the fact that the identified mutations show a high degree of functional divergence (both gain- and loss-of-functions, different types of biophysical changes), and the disease is highly heterogeneous (Nguyen et al. 2008). Several pathophysiological mechanisms have been proposed. First, DCM could develop secondary to frequent arrhythmia or sinus node dysfunction, as is observed with MEPPC. (Alves et al. 1985; Luchsinger & Steinberg 1998). In case of sinus node dysfunction, which is described in several patients with DCM and SCN5A-mutations, low heart rate can lead to remodeling and hypertrophy, as described in the dog model of chronic atrioventricular block (de Groot et al. 2000). In the second hypothesis, it is proposed that increased window current or persistent current causes disturbance in Na+ homeostasis, which in turn leads to alterations in intracellular Ca2+ and pH, through the Na+/Ca2+- and Na+/H+-exchanger, respectively. It should be noted however that in LQT3 patients persistent Na+-influx does not lead to such a pronounced structural phenotype. Recent studies demonstrated the presence of a proton-based leak current (Moreau et al. 2015; Gosselin-Badaroudine et al. 2012) caused by the mutations R225W, R222Q and R219H, which could alternatively induce cellular acidification and secondary to that cellular remodeling. Interestingly, all these mutations are located in the same domain while generating diverging biophysical effects (Gosselin-Badaroudine et al. 2012). Finally, the mechanism by which SCN5A-mutation cause DCM could be solely non-electrical. At the intercalated disks of cardiomyocytes, NaV1.5 is part of a macromolecular complex (Delmar 2004) that includes structural proteins, and it is conceivable that disruption of these interactions can cause downstream structural problems.
SCN5A variations in the general population
Genetic variations in SCN5A, i.e. single nucleotide polymorphisms (SNPs), which are present at relatively high frequencies within the general population have been described in both coding and non-coding regions of the gene. Regarding non-coding region variants, several have been identified in the promoter region and are known to alter transcriptional activity of SCN5A (Yang et al. 2004; Van Den Boogaard et al. 2012). As such, these SNPs affect conduction in healthy and diseased patients (Syrris et al. 2006) and arrhythmia susceptibility in SCN5A mutation carriers (Clancy et al. 2003). As for variants in the coding region, several have been linked to arrhythmia syndromes and are studied functionally. For example H558R, present at a allele frequency of 20–30%, is known to aggravate or attenuate the effects of disease-causing mutations (Viswanathan et al. 2003; Gui et al. 2010), while presence in a wildtype channel reduces current, depending on the presence of the splice variant that includes Q1077 (Makielski et al. 2003). Other studied polymorphisms include S1102Y (Splawski et al. 2002) (10% in Blacks) and R1193Q (8% in Asians) (Wang et al. 2004), which have been linked to BrS and LQT3, respectively.
Rare variants: evidence of pathogenicity
Most phenotypes associated with SCN5A mutations have been identified through candidate gene studies rather than unbiased, genome wide studies such as linkage analysis in large pedigrees (see Table 2 for details). This is mainly a result of the lack of large enough families to perform such studies. The evidence of involvement of SCN5A mutations in the phenotypes described above thus depends on the large enrichment of these kind of mutations in the patient cohorts (5–10% in LQTS3, 25% in BrS) and on segregation testing within families in addition to functional characterization of the identified mutations. However, ascribing pathogenicity to a rare variant of unknown significance (VUS) identified in SCN5A in a patient is not straight forward (Kapplinger et al. 2015). With the advent of exome sequencing, large panels of individuals have now been sequenced for SCN5A, the results of which are publicly available through online databases such as the Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP) n.d.) and ExAC (Exome Aggregation Consortium 2015). Inspection of these databases shows that between 2–7% of the individuals screened in these cohorts carry rare (population frequency <1%) protein altering variants in SCN5A (Table 3). The majority of these are missense mutations, which could in theory lead to gain- and loss–of-function phenotypes. Given the low prevalence of SCN5A mutation associated disorders it is highly improbable that all these VUSs significantly contribute to disease, although subtle effects of variations could modify disease susceptibility. In clinical practice this means that ascribing pathogenicity to SCN5A missense VUS identified in individual patients is not straightforward, especially when no affected relatives are available for segregation testing (Lodder & Wilde 2012; Kapa et al. 2009). In contrast, variants that dramatically alter protein structure (splice, stop gain and frameshift) are very rare in these cohorts (Table 3), indicating that these variants are most likely pathogenic when encountered in patients with a loss-of-function phenotype. In addition to the distinction between dramatic change and missense change, information from in silico prediction tools and the affected protein domain can be informative. A combination of all the available information helps to distinguish pathogenic variants from background genetic noise. Unfortunately, even then many variants remain in the limbo as VUS (Kapplinger et al. 2015).
Table 2, Diseases and phenotypes associated with SCN5A genetic variation.
Overview of the diseases and phenotypes associated with genetic variation in SCN5A, with the level of evidence, Online Mendelian Inheritance in Man (OMIM) database numbers and pertinent references.
| OMIM | disease/phenotype | evidence | references |
|---|---|---|---|
| 614022 | Familial Atrial Fibrillation | Candidate gene approach | (Laitinen-Forsblom et al. 2006; Ellinor et al. 2008; Darbar et al. 2008) |
| 601144 | Brugada syndrome | Candidate gene approach, cosegregation, functional studies, confirmed in many patients | (Crotti et al. 2012; Bezzina et al. 1999; Chen et al. 1998; Remme et al. 2006) |
| 601154 | Dilated Cardiomyopathy | Linkage analysis, contradicting results | (Olson et al. 2005; Laurent et al. 2012; Bezzina et al. 2003; McNair et al. 2004) |
| 113900 | Cardiac Conduction Disease | Linkage analysis and functional studies | (Schott et al. 1999; Tan et al. 2001) |
| 603830 | Long QT syndrome | Linkage analysis and functional studies | (Tester et al. 2005; Wang et al. 1995; Bezzina et al. 1999; Veldkamp et al. 2003; Remme et al. 2006) |
| 608567 | Sick sinus syndrome | Candidate gene approach | (Benson et al. 2003) |
| 603829 | Familial ventricular fibrillation | Candidate gene approach | (Akai et al. 2000) |
| 272120 | Sudden infant death syndrome, susceptibility to | Candidate gene approach | (Schwartz et al. 2000; Ackerman et al. 2001; Plant et al. 2006) |
| 613601 | Early Repolarisation syndrome | Candidate gene approach | (Watanabe et al. 2013; Watanabe et al. 2011) |
| 108980 | QRS interval duration | GWAS signal replicated in many independent studies | (Ritchie et al. 2013; Jeff et al. 2013; Smith et al. 2009) |
| 610141 | QTc interval duration | GWAS signal replicated in many independent studies | (Smith et al. 2011; Holm et al. 2010; Noseworthy et al. 2011; Sotoodehnia et al. 2010; Gaunt et al. 2012; Chambers et al. 2010) |
| 108980 | PR interval duration | GWAS signal replicated in many independent studies | (Jeff et al. 2011; Smith et al. 2011; Holm et al. 2010; Chambers et al. 2010; Sotoodehnia et al. 2010; Gaunt et al. 2012) |
| - | Multifocal ectopic Purkinje-related premature contractions (MEPPC) | Candidate gene approach | (Laurent et al. 2012; Mann et al. 2012; Nair et al. 2012; Swan et al. 2014) |
Table 3: SCN5A genetic variation in the general population.
Frequencies of different types of variants with a minor allele frequency of <1% in the general population as can be found in the databases of Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/) and Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/)
| Type of mutation | frequency in EVS (n>6500) | Frequency in ExAC (n>120,000) |
|---|---|---|
| Missense | 1.89% | 7.40% |
| Splice | 0.02% | 0.01% |
| Stopgain | - | 0.01% |
| Frameshift | - | 0.01% |
| In frame deletion | - | 0.02% |
| Total | 1.91% | 7.45% |
Common variants: Genome Wide Association Studies
Genome Wide Association Studies (GWAS) have employed common genetic variation to identify genetic loci associated with variability in phenotypic traits. In the cardiovascular field this powerful technique has been used to detect genomic loci involved in variation in electrocardiographic parameters (i.e. PR-, QR- and QTc-interval duration) in the general population (reviewed in Lodder and Bezzina, 2013). The rationale behind this technique is that common genetic variation present in the general population can influence cardiac conduction in non-diseased individuals. These studies have been tremendously successful in identifying novel loci that impact on cardiac conduction. Interestingly, these studies consistently identified the SCN5A andSCN10A genomic region on chromosome 3 to be associated with variation in QTc-interval, QRS duration and PR-interval (see Table 3 and (Lodder & Bezzina 2013) for a recent overview). These results are consistent with the notion that common variants at loci implicated in “Mendelian” disease can confer smaller effects within the general population on related or intermediate phenotypes such as ECG parameters. Furthermore, the SCN5A locus has been implicated in the risk of sudden cardiac death in a candidate gene study in the general population (Albert et al. 2008). Whether the effects of the independent GWAS signals in SCN5A and SCN10A (the genes are juxtaposed at 60 kb from each other at chr3p22) are mediated through SCN5A alone (Van Den Boogaard et al. 2012) or partially to the Nav1.8 encoding gene SCN10A as well (Verkerk et al. 2012; Chambers et al. 2010) is still a matter of debate. Of note, transcript levels of SCN10A did not exceed the detection limit of RNA sequencing in ventricular tissues(Van Den Boogaard et al. 2014), suggesting a limited role of this gene in cardiomyocytes.
NaV1.5 as a pharmacological target
The cardiac sodium channel NaV1.5 has since long been a common target in the pharmacological treatment of arrhythmic events. Classically, sodium channel blockers that block the peak sodium current are classified as Class I anti-arrhythmic agents and further subdivided in class IA, IB and IC, depending on their ability to change the length of the cardiac action potential (Milne et al. 1984). The mode of action of these blockers may depend on the biophysical state of the Na+-channel: while some blockers bind channels in activated state (open-state block), others block when the channels are in inactivated state (closed-state block). This paradigm is described in the “modulated receptor hypothesis” (Balser 2001; Courtney 1981). While the blockade of sodium channels can stop reentrant wavefronts by reducing excitability of cardiomyocytes and increasing refractory period, the same mechanism may actually exert opposing effects and can evoke arrhythmia in specific situations. This however depends on the intrinsic parameters of the drug (use-dependency, dissociation and binding rates, concomitant block of K+ and Ca2+ channels) and on the condition during which the drug is applied (e.g. ischemia). Use of sodium channel blockers is among others indicated in patients with ventricular reentrant tachyarrhythmia in the setting of cardiac ischemia and in patients with atrial fibrillation in absence of structural heart disease (Balser 2001).
Apart from acting on peak sodium current, sodium channel blockers also impact on the late component of the sodium current (INa,L) to different extents. Drugs that inhibit mainly INa,L are of potential clinical interest as an increase in INa,L is involved in different conditions. Ranolazine, the most selective INa,L that is currently in clinical use, is approved by the FDA for the treatment of angina pectoris. During ischemia, where INa,L is enhanced, ranolazine reduces intracellular Ca2+ concentration indirectly through the Na+/Ca2+-exchanger, and thereby cardiac workload. As an antiarrhythmic agent, ranolazine is especially of interest in the treatment of LQT3, where INa,L directly prolongs action potential duration and causes QT prolongation, as proven by several preclinical and clinical studies (Huang et al. 2011; Moss et al. 2008). Furthermore, different studies suggest that ranolazine is a potential candidate in treating atrial fibrillation (Murdock et al. 2012; De Ferrari et al. 2015; Gupta et al. 2015). Given that ranolazine can exert unwanted effects by blocking repolarizing potassium currents (Zaza et al. 2008), possibly evoking drug-induced Long QT-syndrome, more selective drugs are currently under development (Remme & Wilde 2014).
Non-canonical roles of NaV1.5
Apart from the role of voltage-gated sodium channels in excitable cells, there is emerging evidence for a function of these channels in non-excitable cells, in particular in different type of cancer cells. This also holds for NaV1.5 of which the “neonatal” isoform is expressed and functional in amongst others breast cancer cells, colon cancer and brain astrocytoma (Brisson et al. 2011; Pittman et al. 2010; Xing et al. 2014; Baptista-Hon et al. 2014). In these cells NaV1.5 function is associated with increased invasiveness and the development of metastases (Besson et al. 2015). In breast cancer cells, NaV1.5 co-localizes with the Na+/H+-exhanger (NHE-1) (Brisson et al. 2011), where it affects activity of NHE-1 allosterically, leading to local extracellular acidification. This acidification results in an increased activity of proteolytic enzymes (cathepsins) that are pH-sensitive and responsible for breakdown of extracellular matrix. Influx of Na+-ions is also of importance, as blocking the channels in cancer cells directly with sodium current blockers affects the degree of invasion(Xing et al. 2014; Baptista-Hon et al. 2014).
Recent studies have identified NaV1.5 in endosomes of macrophages present within lesions of the neurological disease multiple sclerosis (Black et al. 2013). Here the channels contribute to phagocytosis and pH-regulation within the endosome. It is suggested that also for this disease, targeting NaV1.5 would form a putative therapeutic approach.
Summary
SCN5A and its gene product NaV1.5 are essential for proper cardiac function. The NaV1.5 mediated Na+-current INa underlies the fast depolarization phase of the cardiac action potential. Mutations in SCN5A are implicated in several inherited cardiac arrhythmia syndromes and dilated cardiomyopathy as described in detail above. Genetic variation in SCN5A in the general population is associated with variation in ECG-parameters. While a tremendous amount has been learned about the channel and this knowledge has lead to improved clinical care, much remains to be discovered. The underlying mechanisms of some of the SCN5A-associated arrhythmia syndromes (i.e. Brugada syndrome) and the genetic variability in the general population remain to be solved. Furthermore, the non-canonical roles of SCN5A are interesting new avenues to explore.
Acknowledgments
Although we have tried to refer as much as possible to the original research papers, it is impossible to cite all of the more than 1500 papers on SCN5A listed in Pubmed today. We therefore apologize to all researchers whose original articles were not directly cited. This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series--a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM089820and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. We acknowledge the support from the “Netherlands CardioVascular Research Initiative”: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development and the Royal Netherlands Academy of Sciences (PREDICT project).
Abbreviation list, Veerman et al:
- AF
atrial fibrillation
- BrS
Brugada syndrome
- DCM
dilated cardiomyopathy
- ECG
electrocardiogram
- Foxo1
Forkhead Box O1
- GWAS
Genome Wide Association Study
- INa
sodium current
- INa,L
late component of the sodium current
- Kb
kilobases
- LQTS
Long QT syndrome
- MEPPC
Multifocal ectopic Purkinje-related premature contractions
- miR
microRNA
- NaV1.5
voltage-gated sodium channel type v alpha subunit
- NF-KappaB
nuclear factor-κ Β
- NHE-1
Na+/H+-exhanger
- Ros
reactive oxygen species
- SNPs
single nucleotide polymorphisms
- Tbx5
T-Box 5
- UTR
untranslated region
- VUS
variant of unknown significance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The corresponding Gene Wiki entry for this review can be found here: http://en.wikipedia.org/wiki/Nav1.5 “
References
- Abe K et al. , 2014. Sodium channelopathy underlying familial sick sinus syndrome with early onset and predominantly male characteristics. Circulation: Arrhythmia and Electrophysiology, 7(3), pp.511–517. [DOI] [PubMed] [Google Scholar]
- Abriel H, 2010. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. Journal of Molecular and Cellular Cardiology, 48(1), pp.2–11. [DOI] [PubMed] [Google Scholar]
- Ackerman MJ et al. , 2001. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA : the journal of the American Medical Association, 286(18), pp.2264–2269. [DOI] [PubMed] [Google Scholar]
- Akai J et al. , 2000. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Letters, 479(1–2), pp.29–34. [DOI] [PubMed] [Google Scholar]
- Albert CM et al. , 2008. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation, 117(1), pp.16–23. [DOI] [PubMed] [Google Scholar]
- Alves LE, Buser JW & Rose EP, 1985. Cardiomyopathy due to chronic tachycardias. JAMA : the journal of the American Medical Association, 253(21), p.3092. [PubMed] [Google Scholar]
- Arnolds DE et al. , 2012. TBX5 drives Scn5a expression to regulate cardiac conduction system function. The Journal of clinical investigation, 122(7), pp.2509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D et al. , 1979. The steady state TTX-sensitive (“window”) sodium current in cardiac Purkinje fibres. Pflügers Archiv European Journal of Physiology, 379(2), pp.137–142. [DOI] [PubMed] [Google Scholar]
- Balser JR, 2001. The cardiac sodium channel: gating function and molecular pharmacology. Journal of molecular and cellular cardiology, 33(4), pp.599–613. [DOI] [PubMed] [Google Scholar]
- Baptista-Hon DT et al. , 2014. Potent inhibition by ropivacaine of metastatic colon cancer SW620 cell invasion and NaV1.5 channel function. British journal of anaesthesia, 113 Suppl , pp.i39–i48. [DOI] [PubMed] [Google Scholar]
- Baskar S et al. , 2014. Compound Heterozygous Mutations in the SCN5A-Encoded NaV1.5 cardiac sodium channel resulting in atrial standstill and His-Purkinje system disease. The Journal of Pediatrics, 165(5), pp.1050–1052. [DOI] [PubMed] [Google Scholar]
- Benson DW et al. , 2003. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). Journal of Clinical Investigation, 112(7), pp.1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson P et al. , 2015. How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochimica et Biophysica Acta (BBA) - Biomembranes [DOI] [PubMed]
- Bezzina C et al. , 1999. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circulation research, 85(12), pp.1206–1213. [DOI] [PubMed] [Google Scholar]
- Bezzina CR et al. , 2003. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circulation research, 92(2), pp.159–168. [DOI] [PubMed] [Google Scholar]
- Black JA, Newcombe J & Waxman SG, 2013. Nav1.5 sodium channels in macrophages in multiple sclerosis lesions. Multiple sclerosis (Houndmills, Basingstoke, England), 19(5), pp.532–42. [DOI] [PubMed] [Google Scholar]
- Van Den Boogaard M et al. , 2014. A common genetic variant within SCN10A modulates cardiac SCN5A expression. Journal of Clinical Investigation, 124(4), pp.1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Boogaard M et al. , 2012. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. Journal of Clinical Investigation, 122(7), pp.2519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson L et al. , 2011. NaV1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene, 30(17), pp.2070–2076. [DOI] [PubMed] [Google Scholar]
- Buchanan JW, Saito T & Gettes LS, 1985. The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circulation research, 56(5), pp.696–703. [DOI] [PubMed] [Google Scholar]
- Butters TD et al. , 2010. Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circulation Research, 107(1), pp.126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B et al. , 2014. Deletion of FoxO1 leads to shortening of QRS by increasing Na+ channel activity through enhanced expression of both cardiac NaV1.5 and β3 subunit. Journal of molecular and cellular cardiology, 74, pp.297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. a, 2014. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annual review of pharmacology and toxicology, 54, pp.317–38. [DOI] [PubMed] [Google Scholar]
- Cerrone M et al. , 2014. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation, 129(10), pp.1092–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JC et al. , 2010. Genetic variation in SCN10A influences cardiac conduction. Nature genetics, 42(2), pp.149–52. [DOI] [PubMed] [Google Scholar]
- Chen Q et al. , 1998. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature, 392(6673), pp.293–296. [DOI] [PubMed] [Google Scholar]
- Chen-Izu Y et al. , 2015. Na+ channel function, regulation, structure, trafficking and sequestration. The Journal of Physiology, 593(6), pp.1347–1360. Available at: http://doi.wiley.com/10.1113/jphysiol.2014.281428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chioni AM et al. , 2005. A novel polyclonal antibody specific for the NaV1.5 voltage-gated Na+ channel “neonatal” splice form. Journal of Neuroscience Methods, 147(2), pp.88–98. [DOI] [PubMed] [Google Scholar]
- Clancy CE et al. , 2003. Non-equilibrium gating in cardiac Na+ channels: An original mechanism of arrhythmia. Circulation, 107(17), pp.2233–2237. [DOI] [PubMed] [Google Scholar]
- Coronel R et al. , 2005. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation, 112(18), pp.2769–77. [DOI] [PubMed] [Google Scholar]
- Courtney KR, 1981. Slow sodium channel inactivation and the modulated receptor hypothesis Application to phenobarbital. Biochimica et Biophysica Acta (BBA) - Biomembranes, 642(2), pp.433–437. [DOI] [PubMed] [Google Scholar]
- Crotti L et al. , 2012. Spectrum and prevalence of mutations involving BrS1-Through BrS12-susceptibility genes in a cohort of unrelated patients referred for brugada syndrome genetic testing: Implications for genetic testing. Journal of the American College of Cardiology, 60(15), pp.1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daimi H et al. , 2015. Regulation of SCN5A by microRNAs: miR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart rhythm : the official journal of the Heart Rhythm Society, 12(6), pp.1333–1342. [DOI] [PubMed] [Google Scholar]
- Darbar D et al. , 2008. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation, 117(15), pp.1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmar M, 2004. The intercalated disk as a single functional unit. Heart Rhythm, 1(1), pp.12–13. [DOI] [PubMed] [Google Scholar]
- Ellinor PT et al. , 2008. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm, 5(1), pp.99–105. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium, 2015. ExAC database Cambridge, MA ExAC, p. http://exac.broadinstitute.org. [Google Scholar]
- De Ferrari GM et al. , 2015. Ranolazine in the treatment of atrial fibrillation: Results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following An ELectricaL CardiOversion) study. Heart rhythm : the official journal of the Heart Rhythm Society, 12(5), pp.872–8. [DOI] [PubMed] [Google Scholar]
- Frigo G et al. , 2007. Homozygous SCN5A mutation in Brugada syndrome with monomorphic ventricular tachycardia and structural heart abnormalities. Europace, 9(6), pp.391–397. [DOI] [PubMed] [Google Scholar]
- Gaunt TR et al. , 2012. Integration of genetics into a systems model of electrocardiographic traits using HumanCVD BeadChip. Circulation. Cardiovascular genetics, 5(6), pp.630–8. [DOI] [PubMed] [Google Scholar]
- Gellens ME et al. , 1992. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proceedings of the National Academy of Sciences of the United States of America, 89(2), pp.554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin-Badaroudine P et al. , 2012. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS ONE, 7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groome JR, 2014. Voltage Gated Sodium Channels. Handbook of experimental pharmacology, 221, pp.7–32. [DOI] [PubMed] [Google Scholar]
- De Groot SH et al. , 2000. Contractile adaptations preserving cardiac output predispose the hypertrophied canine heart to delayed afterdepolarization-dependent ventricular arrhythmias. Circulation, 102(17), pp.2145–2151. [DOI] [PubMed] [Google Scholar]
- Gui J et al. , 2010. Mutation-specific effects of polymorphism H558R in SCN5A-related sick sinus syndrome. Journal of cardiovascular electrophysiology, 21(5), pp.564–73. [DOI] [PubMed] [Google Scholar]
- Gupta T et al. , 2015. Antiarrhythmic properties of ranolazine: A review of the current evidence. International journal of cardiology, 187, pp.66–74. [DOI] [PubMed] [Google Scholar]
- Herren AW, Bers DM & Grandi E, 2013. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. American journal of physiology. Heart and circulatory physiology, 305(4), pp.H431–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm AN et al. , 2002. Sodium current in human jejunal circular smooth muscle cells. Gastroenterology, 122(1), pp.178–187. [DOI] [PubMed] [Google Scholar]
- Holm H et al. , 2010. Several common variants modulate heart rate, PR interval and QRS duration. Nature genetics, 42(2), pp.117–22. [DOI] [PubMed] [Google Scholar]
- Hoogendijk MG et al. , 2011. ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart rhythm : the official journal of the Heart Rhythm Society, 8(1), pp.111–8. [DOI] [PubMed] [Google Scholar]
- Hu D et al. , 2009. A mutation in the beta-3 subunit of the cardiac sodium channel associated with brugada ECG phenotype. Circulation: Cardiovascular Genetics, 2(3), pp.270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H et al. , 2011. Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of NaV1.5 channels. American journal of physiology. Heart and circulatory physiology, 300(1), pp.H288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeff JM et al. , 2013. Generalization of Variants Identified by Genome-Wide Association Studies for Electrocardiographic Traits in African Americans. Annals of human genetics, 77(4), pp.321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeff JM et al. , 2011. SCN5A variation is associated with electrocardiographic traits in the Jackson Heart Study. Circulation. Cardiovascular genetics, 4(2), pp.139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapa S et al. , 2009. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation, 120(18), pp.1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD et al. , 2015. Enhanced Classification of Brugada Syndrome- and Long QT Syndrome-Associated Genetic Variants in the SCN5A-Encoded NaV1.5 Cardiac Sodium Channel. Circulation: Cardiovascular Genetics [DOI] [PMC free article] [PubMed]
- Kellenberger S, Scheuer T & Catterall W. a., 1996. Movement of the Na+ channel inactivation gate during inactivation. Journal of Biological Chemistry, 271(48), pp.30971–30979. [DOI] [PubMed] [Google Scholar]
- Laitinen-Forsblom PJ et al. , 2006. SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. Journal of Cardiovascular Electrophysiology, 17(5), pp.480–485. [DOI] [PubMed] [Google Scholar]
- Laurent G et al. , 2012. Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol, 60(2), pp.144–156. [DOI] [PubMed] [Google Scholar]
- Lei M et al. , 2004. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. The Journal of physiology, 559(Pt 3), pp.835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M et al. , 2005. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. The Journal of physiology, 567(Pt 2), pp.387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippi G et al. , 2012. Genetic and clinical aspects of Brugada syndrome: an update. Advances in clinical chemistry, 56, pp.197–208. [DOI] [PubMed] [Google Scholar]
- Lodder EM & Bezzina CR, 2013. Genomics of cardiac electrical function. Briefings in functional genomics [DOI] [PubMed]
- Lodder EM & Wilde AAM, 2012. Clinical assessment of the pathogenicity of unknown variants in long-QT syndrome: does the pendulum swing back? Journal of cardiovascular electrophysiology, 23(6), pp.643–4. [DOI] [PubMed] [Google Scholar]
- Lopez KN et al. , 2011. Homozygous mutation in SCN5A associated with atrial quiescence, recalcitrant arrhythmias, and poor capture thresholds. Heart Rhythm, 8(3), pp.471–473. [DOI] [PubMed] [Google Scholar]
- Luchsinger J. a. & Steinberg JS, 1998. Resolution of cardiomyopathy after ablation of atrial flutter. Journal of the American College of Cardiology, 32(1), pp.205–210. [DOI] [PubMed] [Google Scholar]
- Lupoglazoff JM et al. , 2002. Homozygotous mutation of the SCN5A gene responsible for congenital long QT syndrome with 2/1 atrioventricular block, [PubMed]
- Makielski JC et al. , 2003. A Ubiquitous Splice Variant and a Common Polymorphism Affect Heterologous Expression of Recombinant Human SCN5A Heart Sodium Channels. Circulation Research, 93(9), pp.821–828. [DOI] [PubMed] [Google Scholar]
- Makita N et al. , 2005. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm, 2(10), pp.1128–1134. [DOI] [PubMed] [Google Scholar]
- Makiyama T et al. , 2008. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. Journal of the American College of Cardiology, 52(16), pp.1326–34. [DOI] [PubMed] [Google Scholar]
- Maltsev V. a et al. , 1998. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation, 98(23), pp.2545–2552. [DOI] [PubMed] [Google Scholar]
- Mann SA et al. , 2012. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. Journal of the American College of Cardiology, 60(16), pp.1566–1573. [DOI] [PubMed] [Google Scholar]
- Mao W et al. , 2012. Reactive oxygen species suppress cardiac NaV1.5 expression through Foxo1. PloS one, 7(2), p.e32738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C & Abriel H, 2015. Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. Journal of Molecular and Cellular Cardiology, 82, pp.36–47. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0022282815000589. [DOI] [PubMed] [Google Scholar]
- Marsman RF, Tan HL & Bezzina CR, 2013. Genetics of sudden cardiac death caused by ventricular arrhythmias. Nature reviews. Cardiology, 11(2), pp.96–111. [DOI] [PubMed] [Google Scholar]
- McNair WP et al. , 2004. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation, 110(15), pp.2163–2167. [DOI] [PubMed] [Google Scholar]
- Meadows LS & Isom LL, 2005. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res, 67(3), pp.448–458. [DOI] [PubMed] [Google Scholar]
- Medeiros-Domingo A et al. , 2007. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation, 116(2), pp.134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JR et al. , 1984. Class 1 antiarrhythmic drugs--characteristic electrocardiographic differences when assessed by atrial and ventricular pacing. European heart journal, 5(2), pp.99–107. [DOI] [PubMed] [Google Scholar]
- Mizusawa Y, Horie M & Wilde AA, 2014. Genetic and Clinical Advances in Congenital Long QT Syndrome. Circulation Journal, 78(12), pp.2827–2833. [DOI] [PubMed] [Google Scholar]
- Moreau A et al. , 2015. Gating pore currents are defects in common with two NaV1.5 mutations in patients with mixed arrhythmias and dilated cardiomyopathy. The Journal of general physiology, 145(2), pp.93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ et al. , 2008. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. Journal of cardiovascular electrophysiology, 19(12), pp.1289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdock DK, Kaliebe J & Larrain G, 2012. The use of ranolazine to facilitate electrical cardioversion in cardioversion-resistant patients: a case series. Pacing and clinical electrophysiology : PACE, 35(3), pp.302–7. [DOI] [PubMed] [Google Scholar]
- Nair K et al. , 2012. Escape capture bigeminy: Phenotypic marker of cardiac sodium channel voltage sensor mutation R222Q. Heart Rhythm, 9(10), pp.1681–1688. [DOI] [PubMed] [Google Scholar]
- Nakajima S et al. , 2013. A novel SCN5A mutation demonstrating a variety of clinical phenotypes in familial sick sinus syndrome. Internal medicine (Tokyo, Japan), 52(16), pp.1805–8. [DOI] [PubMed] [Google Scholar]
- Napolitano C, 2013. The Contradictory Genetics of Atrial Fibrillation: The Growing Gap between Knowledge and Clinical Implications. Journal of cardiovascular electrophysiology, pp.1–3. [DOI] [PubMed]
- Neu A et al. , 2010. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Human Mutation, 31(8), pp.1609–1621. [DOI] [PubMed] [Google Scholar]
- Nguyen TP et al. , 2008. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circulation Research, 102(3), pp.364–371. [DOI] [PubMed] [Google Scholar]
- NHLBI GO Exome Sequencing Project (ESP), Exome Variant Server, p.http://evs.gs.washington.edu/EVS/.
- Nikulina SY et al. , 2015. An Investigation of the Association of the H558R Polymorphism of the SCN5A Gene with Idiopathic Cardiac Conduction Disorders. Genetic Testing and Molecular Biomarkers, 19(6), p.150414120233001. [DOI] [PubMed] [Google Scholar]
- Noseworthy P a et al. , 2011. Common genetic variants, QT interval, and sudden cardiac death in a Finnish population-based study. Circulation. Cardiovascular genetics, 4(3), pp.305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss HB et al. , 1995. Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu 1) sodium channel alpha subunits expressed in Xenopus oocytes. The Journal of general physiology, 106(6), pp.1171–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly JP et al. , 1999. Comparison of slow inactivation in human heart and rat skeletal muscle Na+ channel chimaeras. The Journal of physiology, 515 ( Pt 1, pp.61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson TM et al. , 2005. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA : the journal of the American Medical Association, 293(4), pp.447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onkal R et al. , 2008. Alternative splicing of NaV1.5: An electrophysiological comparison of “neonatal” and “adult” isoforms and critical involvement of a lysine residue. Journal of Cellular Physiology, 216(3), pp.716–726. [DOI] [PubMed] [Google Scholar]
- Pittman AM et al. , 2010. Allelic variation at the 8q23.3 colorectal cancer risk locus functions as a cis-acting regulator of EIF3H. PLoS genetics, 6(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant LD et al. , 2006. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. Journal of Clinical Investigation, 116(2), pp.430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst V et al. , 2009. SCN5A Mutations and the role of genetic background in the pathophysiology of brugada syndrome. Circulation: Cardiovascular Genetics, 2(6), pp.552–557. [DOI] [PubMed] [Google Scholar]
- Remme CA, 2013. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. The Journal of physiology, 591(Pt 17), pp.4099–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme CA et al. , 2006. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation, 114(24), pp.2584–2594. [DOI] [PubMed] [Google Scholar]
- Remme CA et al. , 2009. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol, 104(5), pp.511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme CA & Wilde AAM, 2014. Targeting sodium channels in cardiac arrhythmia. Current opinion in pharmacology, 15, pp.53–60. [DOI] [PubMed] [Google Scholar]
- Ritchie MD et al. , 2013. Genome- and Phenome-Wide Analysis of Cardiac Conduction Identifies Markers of Arrhythmia Risk. Circulation, 127(13), pp.1377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossenbacker T et al. , 2004. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm, 1(5), pp.610–615. [DOI] [PubMed] [Google Scholar]
- Sakmann BF et al. , 2000. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circulation research, 87(10), pp.910–914. [DOI] [PubMed] [Google Scholar]
- Schott JJ et al. , 1999. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet, 23(1), pp.20–21. [DOI] [PubMed] [Google Scholar]
- Schroeter A et al. , 2010. Structure and function of splice variants of the cardiac voltage-gated sodium channel NaV1.5. Journal of Molecular and Cellular Cardiology, 49(1), pp.16–24. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ et al. , 2000. A molecular link between the sudden infant death syndrome and the long-QT syndrome. The New England Journal of Medicine, 343(4), pp.262–267. [DOI] [PubMed] [Google Scholar]
- Shang LL et al. , 2008. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. American journal of physiology-Cell physiology, 294(1), pp.C372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang LL & Dudley SC, 2005. Tandem promoters and developmentally regulated 5’- and 3’-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. Journal of Biological Chemistry, 280(2), pp.933–940. [DOI] [PubMed] [Google Scholar]
- Smith JG et al. , 2011. Genome-wide association studies of the PR interval in African Americans. PLoS Genet, 7(2), p.e1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JG et al. , 2009. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm, 6(5), pp.634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits JPP et al. , 2005. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. Journal of molecular and cellular cardiology, 38(6), pp.969–81. [DOI] [PubMed] [Google Scholar]
- Sotoodehnia N et al. , 2010. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nature genetics, 42(12), pp.1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I et al. , 2002. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science (New York, N.Y.), 297(5585), pp.1333–6. [DOI] [PubMed] [Google Scholar]
- Van Stuijvenberg L et al. , 2010. Alternative promoter usage and splicing of the human SCN5A gene contribute to transcript heterogeneity. DNA and cell biology, 29(10), pp.577–87. [DOI] [PubMed] [Google Scholar]
- Swan H et al. , 2014. Gain-of-Function Mutation of the SCN5A Gene causes exercise-induced polymorphic ventricular arrhythmias. Circulation: Cardiovascular Genetics, 7(6), pp.771–781. [DOI] [PubMed] [Google Scholar]
- Syrris P et al. , 2006. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation, 113(3), pp.356–364. [DOI] [PubMed] [Google Scholar]
- Tan BH et al. , 2005. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm, 2(7), pp.741–747. [DOI] [PubMed] [Google Scholar]
- Tan HL et al. , 2001. A sodium-channel mutation causes isolated cardiac conduction disease. Nature, 409(6823), pp.1043–1047. [DOI] [PubMed] [Google Scholar]
- Tester DJ et al. , 2005. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm, 2(5), pp.507–517. [DOI] [PubMed] [Google Scholar]
- Veldkamp MW et al. , 2003. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circulation Research, 92(9), pp.976–983. [DOI] [PubMed] [Google Scholar]
- Verkerk AO et al. , 2012. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circulation research, 111(3), pp.333–43. [DOI] [PubMed] [Google Scholar]
- Viswanathan PC, Benson DW & Balser JR, 2003. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. The Journal of clinical investigation, 111(3), pp.341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q et al. , 1995. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell, 80(5), pp.805–811. [DOI] [PubMed] [Google Scholar]
- Wang Q et al. , 2004. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. Journal of medical genetics, 41(5), p.e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H et al. , 2011. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circulation. Arrhythmia and electrophysiology, 4(6), pp.874–81. [DOI] [PubMed] [Google Scholar]
- Watanabe H et al. , 2009. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med, 15(4), pp.380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H et al. , 2013. SCN5A mutation associated with ventricular fibrillation, early repolarization, and concealed myocardial abnormalities. International journal of cardiology, 165(2), pp.e21–3. [DOI] [PubMed] [Google Scholar]
- Watanabe H et al. , 2008. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest, 118(6), pp.2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann S, 1955. The effect of the cardiac membrane potential on the rapid availability of the sodium-carrying system. The Journal of physiology, 127(1), pp.213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW et al. , 1992. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proceedings of the National Academy of Sciences of the United States of America, 89(22), pp.10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D et al. , 2014. Expression of neonatal NaV1.5 in human brain astrocytoma and its effect on proliferation, invasion and apoptosis of astrocytoma cells. Oncology reports, 31(6), pp.2692–700. [DOI] [PubMed] [Google Scholar]
- Yang P, Kupershmidt S & Roden DM, 2004. Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovascular research, 61(1), pp.56–65. [DOI] [PubMed] [Google Scholar]
- Zaza A, Belardinelli L & Shryock JC, 2008. Pathophysiology and pharmacology of the cardiac “late sodium current.”. Pharmacology & therapeutics, 119(3), pp.326–39. [DOI] [PubMed] [Google Scholar]
- Zoni-Berisso M et al. , 2014. Epidemiology of atrial fibrillation: European perspective. Clinical epidemiology, 6, pp.213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]