

Abstract
Background: The farnesoid X receptor (FXR) regulates bile acid (BA) metabolism and possesses tumor suppressor functions. FXR expression is reduced in colorectal tumors of subjects carrying inactivated adenomatous polyposis coli (APC). Identifying the mechanisms responsible for this reduction may offer new molecular targets for colon cancer prevention.
Objective: We investigated how APC inactivation influences the regulation of FXR expression in colonic mucosal cells. We hypothesized that APC inactivation would epigenetically repress nuclear receptor subfamily 1, group H, member 4 (FXR gene name) expression through increased CpG methylation.
Methods: Normal proximal colonic mucosa and normal-appearing adjacent colonic mucosa and colon tumors were collected from wild-type C57BL/6J and Apc-deficient (ApcMin/+) male mice, respectively. The expression of Fxr, ileal bile acid-binding protein (Ibabp), small heterodimer partner (Shp), and cyclooxygenase-2 (Cox-2) were determined by real-time polymerase chain reaction. In both normal and adjacent colonic mucosa and colon tumors, we measured CpG methylation of Fxr in bisulfonated genomic DNA. In vitro, we measured the impact of APC inactivation and deoxycholic acid (DCA) treatment on FXR expression in human colon cancer HCT-116 cells transfected with silencing RNA for APC and HT-29 cells carrying inactivated APC.
Results: In ApcMin/+ mice, constitutive CpG methylation of the Fxrα3/4 promoter was linked to reduced (60–90%) baseline Fxr, Ibabp, and Shp and increased Cox-2 expression in apparently normal adjacent mucosa and colon tumors. Apc knockdown in HCT-116 cells increased cellular myelocytomatosis (c-MYC) and lowered (∼50%) FXR expression, which was further reduced (∼80%) by DCA. In human HCT-116 but not HT-29 colon cancer cells, DCA induced FXR expression and lowered CpG methylation of FXR.
Conclusions: We conclude that the loss of APC function favors the silencing of FXR expression through CpG hypermethylation in mouse colonic mucosa and human colon cells, leading to reduced expression of downstream targets (SHP, IBABP) involved in BA homeostasis while increasing the expression of factors (COX-2, c-MYC) that contribute to inflammation and colon cancer.
Keywords: farnesoid X receptor, bile acid metabolism, adenomatous polyposis coli, deoxycholic acid, epigenetics, CpG methylation, inflammation, colon cancer
Introduction
The farnesoid X receptor (FXR)7 regulates bile acid (BA) homeostasis through the enterohepatic circulation. In the intestine, FXR activates the expression of ileal bile acid-binding protein (IBABP) and small heterodimer partner (SHP). In turn, SHP represses the intestinal expression of the sodium-dependent BA transporter. In the liver, FXR induces SHP expression, which then inhibits the expression of the cytochrome P450 A1 enzyme that catalyzes the de novo synthesis of BA from cholesterol (1). In human colorectal neoplasms, FXR expression becomes repressed at the late adenoma stage during the transition to carcinoma (2) and is inversely correlated with the grade of malignancy and poor clinical outcome (3). In Fxr−/−knockout mice, the loss of FXR function increases susceptibility to chemically induced colon tumorigenesis (4). Conversely, Fxr transgene overexpression in intestinal cells reduces tumor growth (5). Thus, conditions that interfere with normal intestinal cell FXR expression and signaling may compromise normal BA homeostasis, leading to the increased production of a tumor-promoting secondary BA such as deoxycholic acid (DCA) (6–10).
Intestinal expression of FXR is substantially reduced in patients with autosomal dominantly inherited familial adenomatous polyposis (FAP) (5), which is caused by germline mutations in the adenomatous polyposis coli (APC) gene. Somatic APC mutations occur early in colorectal tumorigenesis (11). Patients with FAP develop numerous colorectal adenomas in their first 2 decades of life that inevitably progress to colorectal cancer unless prophylactic panproctocolectomy is performed (12). Fxr expression is reduced in the ApcMin/+ mouse (4), which carries an inactivating mutation in the Apc gene. The APC protein sequesters β-catenin in the cytosol, thereby preventing the activation of the protumorigenic wingless-type mouse mammary tumor virus integration site family (Wnt) signaling pathway (13, 14). When APC expression is reduced, β-catenin has been shown to translocate to the nucleus and induce members of the transcription factor/lymphoid enhancer-binding factor to form transcription complexes, which in turn increase the expression of protumorigenic cyclin D1 (15, 16) and cellular myelocytomatosis (c-MYC) (17). Although these downstream effects are understood, the initial mechanisms that link APC inactivation to reduced FXR expression remain largely unknown.
Our first objective in this study was to investigate the mechanisms that link the inactivation of Apc to reduced nuclear receptor subfamily 1, group H, member 4 (Fxr gene name) expression in colon tumors. We used the ApcMin/+ mouse because it carries mutated Apc (18) and is regarded as a good model of multistage colon carcinogenesis (19). We extended these studies to in vitro experiments with human colon cancer cells (HCT-116) carrying wild-type APC (20) and transfected with silencing RNA for APC (siAPC) or human colon cancer cells (HT-29) harboring inactivated APC (20) and treated with DCA. We selected DCA as a prototype secondary BA because its fecal excretion increases with high-fat diet (HFD) consumption (21, 22), and the accumulation of DCA has been linked to an increased risk of polyps and colorectal tumors (6, 7).
Methods
Mice models.
Control C57/BL6J male mice were purchased from Jackson Laboratories. Mice were killed at 12 wk of age, and colonic tissue was isolated for further analyses as described previously (23). Briefly, the large bowel was cut open longitudinally along the main axis and washed with ice-cold PBS. Proximal colonic mucosa was scraped, and colonic cells were separated by centrifugation. Fxrα−/− mice in pure C57BL/6J background were gifts from Frank Gonzalez (Laboratory of Metabolism, National Cancer Institute). C57BL/6J-ApcMin/+ mice were purchased from Jackson Laboratories. Female Fxrα−/− mice were crossed with male ApcMin/+ to produce Fxrα+/− ApcMin/+ mice. Female Fxrα+/− were then crossed with male Fxrα+/− ApcMin/+ mice to produce Fxrα−/− ApcMin/+, Fxrα+/− ApcMin/+, and ApcMin/+ genotypes. All genotypes were viable and fertile. All animal procedures with Fxr and Apc genotypes were approved by the Institutional Animal Care and Use Committee of the Burnham Institute for Medical Research and the University of Arizona. FXRα genotyping of tail DNA was performed using PCR as described previously (24). ApcMin/+ genotyping was conducted following the protocol from Jackson Laboratories. Mice were housed in conventional cages under a 12-h light/dark cycle with free access to Teklad global rodent diet (Harlan Laboratories) and tap water. The mice were killed by CO2 asphyxiation at 12 wk of age. Colon tumors and adjacent (1 cm away from the tumor site), normal-appearing colon tissue were collected, rinsed with PBS, and stored at −80°C until further analysis.
Tissue culture and reagents.
Human HCT-116 and HT-29 colon cancer cells were obtained from American Type Culture Collection and maintained in DMEM from Sigma-Aldrich supplemented with 10% FBS (Hyclone Laboratories) as described previously (25). At the end of the treatment periods, cells were washed with PBS, harvested, and stored at −80°C until further analysis. DCA was purchased from Sigma-Aldrich.
qRT-PCR.
Total RNA was extracted from mucosa scraped from the proximal colon according to the protocol described previously (26) and purified using the Quick-RNA MiniPrep kit according to the manufacturer's instructions (Zymo Research). The concentration and quality of RNA were verified using the Thermo Scientific NanoDrop1000 spectrophotometer. Equal amounts of total RNA (500 ng) were transcribed into cDNA using qScript cDNA SuperMix (Quanta Biosciences). PCR products were amplified from the cDNA fragments using PerfeCTa SYBR Green FastMix, ROX (Quanta Biosciences). Briefly, reactions were run at a final volume of 10 μL consisting of the following master mix: 5 μL of SYBR Green FastMix, 1 μL each of forward and reverse primers (10 nmol/L), 2 μL of nuclease-free water, and 1 μL of cDNA. Amplification of Gapdh (GAPDH for HCT-116 and HT-29) was used for normalizing mRNA expression. The mouse and human primers (Sigma-Aldrich) used for qRT-PCR are shown in Supplemental Table 1.
Western blotting and silencing RNA experiments.
Immunodetection by Western blotting was performed using antibodies obtained from Santa Cruz Biotechnology (H-130 FXR, β-ACTIN) and EMD Millipore (c-MYC anti-phospho Thr58/Sr62). Silencing RNA experiments were carried out according to the manufacturer's instructions (Dharmacon) as described previously (27). Briefly, 5 × 105 HCT-116 cells were plated in 6-well plates and transfected using the DharmaFECT 2 transfection reagent with nontargeting pool and smart pool human siAPC for 48 h. Cells were then cultured in control DMEM or DMEM supplemented with 50 μmol/L DCA for an additional 72 h. At the end of the incubation period, cells were harvested for qRT-PCR and Western blotting analyses.
CpG methylation.
Promoter methylation was analyzed as described previously (28). Briefly, genomic DNA was isolated from 10–15 mg of proximal colon mucosa using the DNeasy Blood & Tissue Kit (Qiagen). Genomic DNA (1 μg) was subjected to bisulfite modification using the EpiTect Bisulfite Conversion Kit (Qiagen). In preliminary experiments, we verified that the number of cycles for semiquantitatively amplifying each promoter fragment with methylation-specific primers (MSPs) was in the linear range. The bisulfite-modified DNA was analyzed by PCR as follows: 1 cycle at 94°C for 1 min; 35 cycles at 94°C for 30 s, 59°C for 30 s, and 72°C for 1 min; and 1 cycle at 72°C for 5 min. Reactions were carried out at a final volume of 25 μL consisting of the following master mix: 50 ng of bisulfite-modified genomic DNA, 0.4 μL of JumpStart Taq DNA polymerase (Sigma-Aldrich), 2.5 μL of 10× PCR buffer, 3.5 μL of 25 mM MgCl2 (final concentration: 3.5 mmol/L), 0.5 μL of 10 mmol/L deoxyribonucleotide triphosphate mix (final concentration: 200 μmol/L), 1 μL each of forward and reverse primers, and water to bring the final volume to 25 μL. The PCR amplification products were separated on 2% agarose gels and visualized using ethidium bromide staining. PCR amplicons were of the expected size, and their authenticity was confirmed by direct sequencing. The primers (Sigma-Aldrich) used for DNA methylation studies are shown in Supplemental Table 1.
Statistical analysis.
Densitometries of CpG-methylated FXR after PCR amplification and FXR protein after Western blotting of samples from HCT-116 and HT-29 cells were performed using Kodak ID image EDAS 290 analysis software (Eastman Kodak). Statistical analyses of CpG methylation and expression data from mice and cell culture studies were performed by 1-factor ANOVA after assessing data normality using a Shapiro–Wilk test and variance homogeneity using Bartlett's test. After main effects were found to be significant at P ≤0.05, post hoc multiple comparisons among all means were conducted using Tukey's test.
Results
Apc deficiency correlates with lower Fxr expression in adjacent colonic mucosa and colon tumors.
Intestinal accumulation of BA, hyperlipidemia, and reduced FXR expression have been shown to accompany the development of intestinal tumors in Apc-deficient models (4, 29). To investigate the mechanisms that link Apc deficiency to reduced FXR expression, we first compared mRNA levels in nontumor (NT) proximal colonic mucosa of ApcMin/+ mice to those of control wild-type C57BL/6J mice. Fxr expression was 60% lower in NT mucosa from the ApcMin/+ mice than in colonic mucosa from control mice and was reduced an additional 30% in colon tumors of ApcMin/+ mice (Figure 1).
FIGURE 1.

Apc inactivation reduces Fxr expression in adjacent colonic mucosa and colon tumors of ApcMin/+ mice. Bars represent qRT-PCR quantitation (fold of C57BL/6J) of Fxr/Gapdh in NT colonic mucosa and colon tumors from ApcMin/+ mice. Values are means ± SEMs, n = 4 (mean of 6 replicates/mouse). Means without a common letter differ, P < 0.05. Apc, adenomatous polyposis coli; Fxr, farnesoid X receptor; NT, nontumor.
Constitutive CpG methylation of Fxrα3/4 promoter in Apc-deficient adjacent colonic mucosa and colon tumors.
To determine whether epigenetic mechanisms contributed to reducing Fxr expression in the colonic mucosa and tumors of ApcMin/+ mice, we studied changes in Fxrα3/4 CpG promoter methylation (Figure 2A, B). We selected the promoter region on exon-3 because it generates Fxrα3/4 transcripts, which are expressed in the intestine at higher levels compared with the Fxrα1/2 isoforms transcribed from exon-1 (1). In FAP patients, FXRα3/4 transcripts are markedly reduced compared with FXRα1/2 variants (30). In preliminary experiments, we confirmed that amplification with MSPs of Fxrα3/4 promoter fragments occurred in the linear range (Figure 2C). Accounting for the reduction in Fxr expression, CpG methylation of the Fxrα3/4 promoter was ∼0.4-fold higher in NT mucosa and ∼0.8-fold higher in ApcMin/+ mouse colonic tumors compared with levels found in wild-type C57BL/6J mice (Figure 3A). Using MSPs, we also performed direct-sequence analyses of bisulfonated genomic DNA obtained from 4 independent DNA clones derived from 4 separate ApcMin/+ tumors. We found that 13 CpG sites flanking the transcription start site on exon-3 (Figure 3B) were consistently methylated.
FIGURE 2.

Organization of the mouse nuclear receptor subfamily 1, group H, member 4 (Fxr gene name). (A) Top arrows indicate transcription start sites (+1) on exon-1 and -3. Bottom arrows indicate positions of oligonucleotides (−54/+416) around exon-3 used for Fxr CpG methylation studies. (B) Nucleotide sequence flanking exon-3. CpGs are highlighted in gray. The +1 indicates the transcription start site on exon-3. The underlined nucleotide sequence corresponds to exon-3. (C) PCR cycle number and MW control for amplification of Fxr-M. PCR bands were amplified from bisulfonated genomic DNA using mouse methylation-specific primers. Fxr, farnesoid X receptor; Fxr-M, methylated mouse Fxrα3/4 promoter; MW, molecular weight.
FIGURE 3.

Apc inactivation correlates with constitutive CpG methylation of Fxr in adjacent colonic mucosa and colon tumors of ApcMin/+ mice. (A) Bars represent qRT-PCR quantitation (fold of C57BL/6J) using methylation-specific primers of Fxr-M/β-actin in NT colonic mucosa and colon tumors from ApcMin/+ mice. Values are means ± SEMs, n = 4 (mean of 6 replicates/mouse). Means without a common letter differ, P < 0.05. (B) Position of CpGs in exon-3 of the mouse Fxr gene. Black circles indicate methylated and white circles indicate unmethylated CpGs of 4 clones with 10 replicates per clone from 4 independent colon tumors. Apc, adenomatous polyposis coli; Fxr, farnesoid X receptor; Fxr-M, Fxrα3/4 promoter methylation.
Apc deficiency affects the expression of FXR target genes in adjacent colonic mucosa and colon tumors.
The Shp and Ibabp genes are putative FXR transcriptional targets (1). Compared with wild-type C57BL/6J mice, Shp (Figure 4A) and Ibabp (Figure 4B) levels were reduced by 90–95% in ApcMin/+ NT mucosa and colon tumors. Conversely, cyclooxygenase-2 (Cox-2) expression (Figure 4C) in NT colon mucosa and tumors from ApcMin/+ compared with wild-type C57BL/6J mice was increased ∼0.9- and ∼2.5-fold, respectively. As a positive control, we found similar patterns of reduced Shp (Figure 4A) and Ibabp (Figure 4B) and increased Cox-2 (Figure 4C) expression in NT mucosa and colon tumors obtained from ApcMin/+Fxr−/− double knockout mice. Taken together, these animal data indicate that baseline Fxr expression was reduced in Apc-deficient colon cells and tumors in accordance with constitutive CpG methylation of the Fxrα3/4 promoter. Silencing of Fxr was associated with downregulation of FXR target genes (Shp and Ibabp) and upregulation of proinflammatory Cox-2.
FIGURE 4.
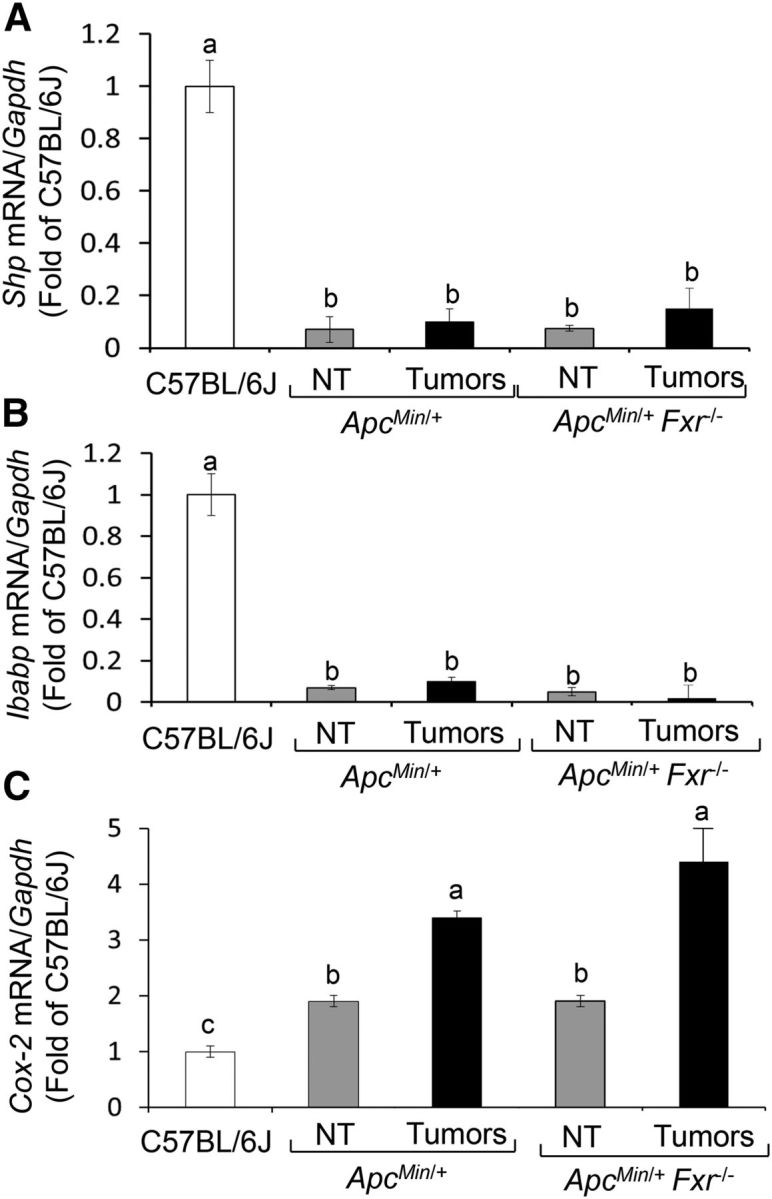
Apc inactivation leads to constitutive repression of Shp and Ibabp and activation of Cox-2 expression in adjacent colonic mucosa and colon tumors of ApcMin/+ mice. Bars represent qRT-PCR quantitation (fold of C57BL/6J) for (A) Shp, (B) Ibabp, and (C) Cox-2 corrected for Gapdh as an internal control in NT colonic mucosa and colon tumors from ApcMin/+ and ApcMin/+ Fxr−/− mice. Values are means ± SEMs, n = 4 (mean of 6 replicates/mouse). Means without a common letter differ, P < 0.05. Apc, adenomatous polyposis coli; Cox-2, cyclooxygenase-2; Fxr, farnesoid X receptor; Ibabp, ileal bile acid-binding protein; NT, nontumor; Shp, small heterodimer partner.
APC deficiency hampers the stimulation of FXR expression by DCA in human colon cancer cells in vitro.
To further examine the impact of APC inactivation on the regulation of FXR expression, we transfected HCT-116 cells with siAPC. Compared with cells transfected with nontarget silencing RNA, a large (∼80%) reduction in APC mRNA expression was accompanied by increased levels (∼0.4-fold) of c-MYC transcripts (Figure 5A). c-MYC is a downstream target for the activated Wnt/β-catenin pathway (17). As a positive control for tumor promotion by DCA in APC-deficient cells, we observed that the treatment of HT-29 colon cancer cells with DCA induced c-MYC protein levels (∼4.0-fold) (Figure 5B). In HCT-116 cells transfected with siAPC, we observed a ∼50% reduction in FXR mRNA levels, which were reduced by an additional 30% when exposed to DCA (Figure 5C).
FIGURE 5.

APC silencing represses expression of FXR in human colon cancer cells. (A) Bars represent qRT-PCR quantitation (fold of nontarget) from 2 separate experiments performed in triplicate for APC and c-MYC corrected for GAPDH mRNA as the internal control in human HCT-116 colon cancer cells transfected with nontarget and siAPC. Values are means ± SEMs. (B) Bands are representative immunocomplexes detected by Western blotting for c-MYC and β-ACTIN in HCT-29 cells cultured in control DMEM or DMEM supplemented with DCA (50 μM for 72 h). (C) Bars represent qRT-PCR quantitation (fold of nontarget) from 2 separate experiments performed in triplicate of FXR in human HCT-116 colon cancer cells transfected with nontarget and siAPC and in the presence of DCA (50 μM for 72 h). Values are means ± SEMs. *Different from nontarget, P < 0.05. APC, adenomatous polyposis coli; c-MYC, cellular myelocytomatosis; DCA, deoxycholic acid; FXR, farnesoid X receptor; siAPC, silencing RNA for APC.
To simulate the exposure of colon cells with wild-type APC to secondary BA in vitro, we treated human HCT-116 colon cancer cells with DCA. In response to the DCA treatment, FXR expression increased 1.5-fold in HCT-116 cells (Figure 6A). This was accompanied by reduced (∼50%) FXR CpG methylation (Figure 6B) and accumulation (1.0-fold) of the FXR protein (Figure 6C). Conversely, DCA treatment did not elicit significant changes in FXR CpG methylation (Figure 6B) and FXR protein (Figure 6C) concentrations in HT-29 cells. Overall, these cumulative in vitro findings suggested that in human colon cells harboring wild-type APC the DCA-induced expression of FXR was associated with a reduction in FXR CpG methylation. Conversely, in APC-deficient human colon cancer cells the FXR gene was refractory to DCA stimulation in association with increased expression of the c-MYC oncogene.
FIGURE 6.
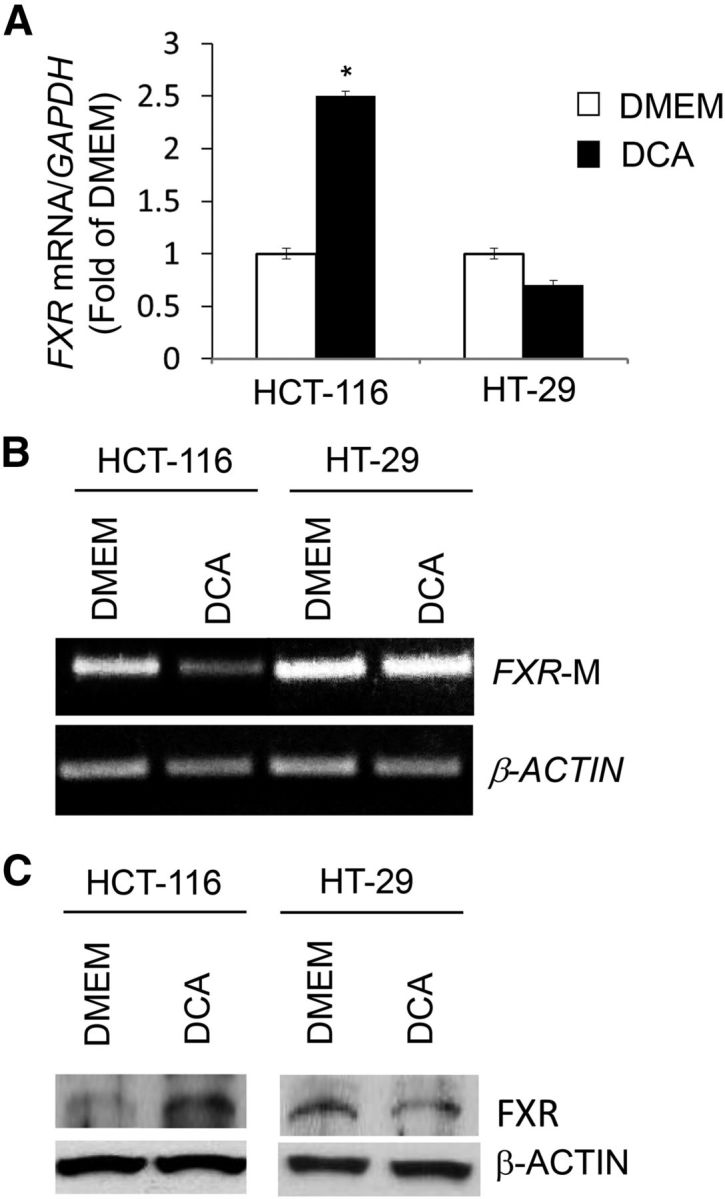
Differential regulation of FXR expression by secondary bile DCA in wild-type APC and APC-deficient human colon cancer cells. (A) Bars represent qRT-PCR quantitation (fold of DMEM) from 2 separate experiments performed in triplicate of FXR corrected for GAPDH as internal control in human HCT-116 (wild-type APC) and HT-29 (inactivated APC) colon cancer cells after 72 h of treatment with 50 μM DCA. Values are means ± SEMs. *Different from DMEM, P < 0.05. (B) Methylated PCR products were amplified with human FXR-M and β-ACTIN methylation-specific primers using as a template bisulfonated genomic DNA obtained from HCT-116 and HT-29 colon cancer cells. (C) Bands are representative immunocomplexes detected by Western blotting for FXR and β-ACTIN in HCT-116 and HT-29 cells cultured in control DMEM or DMEM supplemented with DCA. APC, adenomatous polyposis coli; DCA, deoxycholic acid; FXR, farnesoid X receptor; FXR-M, FXRα3/4 promoter methylation.
Discussion
Despite widespread screening and advances in treatment, colorectal cancer remains the second cause of cancer death in the United States (31). The FXR is a transcriptional regulator of several enterohepatic metabolic pathways (1, 32). Importantly, reduced FXR expression is associated with intestinal tumorigenesis in human subjects (3, 5) and animal models (4, 5).
In this study, the first objective was to examine whether changes in Fxr CpG methylation contributed to regulating Fxr expression in apparently normal colonic mucosa and colon tumors of ApcMin/+ mice. The ApcMin/+ mouse is a model of human FAP caused by mutations in the APC gene (4, 5). We found that the region of the Fxr gene flanking the transcription start site harbored in exon-3 (Fxrα3/4) was constitutively hypermethylated in nonneoplastic colonic mucosa and to a larger degree in colon tumors of ApcMin/+ mice. The increased CpG methylation of the Fxrα3/4 promoter correlated with the reduced expression of Shp and Ibabp and accumulation of Cox-2. The latter changes were also seen in colon tumors of ApcMin/+ Fxr−/− mice, which provided a positive control for the Fxr CpG methylation and Shp and Ibabp expression studies. The marked reduction in Shp and Ibabp expression in both nonneoplastic colonic mucosa, and colon tumors of ApcMin/+ mice were consistent with the fact both Shp and Ibabp genes are direct targets for transcriptional activation by FXR (1). Conversely, we attributed the activation of Cox-2 expression observed in ApcMin/+ and ApcMin/+ Fxr−/− mice to overriding of the negative feedback by FXR on the NF-κB/Cox-2 axis (33). Therefore, a possible implication of these findings is that changes in CpG methylation in the Fxr and Cox-2 genes could serve as sentinel biomarkers of intestinal inflammation and tumorigenesis associated with Apc inactivation. In support of this idea, early changes in CpG methylation profiles of genes involved in lipid metabolism and inflammation are being considered as epigenetic predictors of colon tumor development in humans (34).
The Fxr CpG methylation and expression data presented in this study complement those of a recent study (35) that documented reduced FXR expression in human precancerous lesions and colon tumors. The authors of the latter study, however, did not observe any changes in FXR promoter methylation at a distal CpG island comprising 11 CpGs and spanning the 5′ region upstream (∼−3.2 to −2.9 kb) from the transcription start site of exon-1. We focused our methylation studies on a proximal 470-bp region harboring 13 CpG dinucleotides and flanking the transcription start site of exon-3. Compared with FXRα1/2 variants, FXRα3/4 transcripts are markedly reduced in the tumors of FAP patients (30). Therefore, APC gene inactivation in intestinal cells may lead to preferential CpG methylation in the proximal FXRα3/4 promoter. This specificity may be related to the evidence that FXRα3/4 variants are transcribed at higher amounts in the intestine (1). Moreover, the alternative usage of transcription start sites on the FXR gene and expression of FXR variants (36, 37) may be related to differential regulation at various stages of colon tumorigenesis (35). Therefore, future studies should compare the effects of APC deficiency on CpG methylation and the expression of FXR splicing variants at various tumor stages (e.g., adenomas compared with adenocarcinomas) of colon tumor development.
The silencing of APC in human HCT-116 colon cancer cells abrogated basal FXR expression, which was reduced further upon cotreatment with DCA. These changes culminated with the upregulation of c-MYC, a downstream target for the Wnt/β-catenin pathway (17). In nonneoplastic mucosa of the ileum and proximal colon, APC and FXR expression has been shown to follow an increasing gradient from the bottom of the crypts to the top of the intestinal villi and luminal surface of the colonic crypts (3, 38). These expression patterns combined with published (4, 5) and our data suggest that APC may be required for properly regulating FXR expression and the differentiation of colonic cells (39). In support of this idea, we observed that the in vitro treatment of human HCT-116 colon cancer cells with physiological concentrations of DCA (40), a prototype secondary BA, induced FXR expression and reduced FXR CpG methylation. Although HCT-116 cells have been found to harbor 1 mutated allele for the catenin (cadherin-associated protein), β1, 88-kDa gene that encodes β-catenin, APC/β-catenin complexes are functional in these cells (20). In contrast, in human HT-29 colon cancer cells that harbor a mutated APC gene, modest changes in FXR expression or CpG methylation were observed in response to treatment with DCA, which, however, induced c-MYC expression, a downstream target of the Wnt/β-catenin pathway. These data are in accordance with those of earlier studies that reported constitutive activation of the Wnt/β-catenin pathway in the small intestine of ApcMin/+ mice (41) and in immune-deficient nude mice xenografted with HT-29 colon cancer cells (42).
Taken together, our mice data show that defective Apc expression induces CpG methylation in the Fxr gene and compromises the expression of downstream targets (e.g., Shp, Ibabp) necessary to maintain BA homeostasis. Moreover, reduced Fxr expression is accompanied by constitutive accumulation of proinflammatory Cox-2. We hypothesize that this environment may reinforce the tumor-promoting effects of secondary BAs such as DCA. The requirement for normal APC in activating FXR expression is corroborated by our in vitro observations with human colon cancer cells. Ongoing studies in our laboratory are exploring the signaling cascades through which APC inactivation modulates the placement of CpG methylation and other epigenetic marks (e.g., histone modifications) on the FXR gene and the potential effects of reduced FXR expression on the microbiome and production of secondary BAs (43). An important question raised by the current observations is whether individuals who carry mutated (familial) or inactivated (sporadic) APC and adhere to a HFD may be at higher risk of developing colon inflammation and cancer induced by secondary BAs via reduced FXR expression. Therefore, future studies should examine the differential effects of various HFDs (e.g., n–6 compared with n–3) on FXR CpG methylation in normal and APC-deficient colon models. A second question pertains to whether other epigenetic regulatory defects associate with tumor development in APC-deficient colon cells. Recent studies conducted in the ApcMin/+ model provided evidence that tumor-related hypermethylation appeared as a progressive event and reached higher levels in advanced tumor stages (44, 45). Consequently, progress in understanding the mechanisms responsible for DNA methylation dynamics in the Fxr and other genes may unravel how interactions between the inactivation of APC and exposure to dietary FAs influence the risk of inflammatory bowel diseases (46, 47) and colorectal tumors (42, 48) and thus offer new epigenetic targets for diagnosis and prevention.
Supplementary Material
Primers for qRT-PCR and CpG Methylation
Acknowledgments
OIS and AML conducted the research and analyzed the data; CF and JWS provided the ApcMin/+ and ApcMin/+ Fxr−/− control and tumor samples; OIS, PML, and DFR wrote the manuscript; and DFR had primary responsibility for final content. All authors contributed to project conception and have read and approved the final manuscript.
Abbreviations
- APC
adenomatous polyposis coli
- BA
bile acid
- c-MYC
cellular myelocytomatosis
- Cox-2
cyclooxygenase-2
- DCA
deoxycholic acid
- FAP
familial adenomatous polyposis
- FXR
farnesoid X receptor
- HFD
high-fat diet
- IBABP
ileal bile acid-binding protein
- MSP
methylation-specific primer
- NT
nontumor
- SHP
small heterodimer partner
- siAPC
silencing RNA for adenomatous polyposis coli
- Wnt
wingless-type mouse mammary tumor virus integration site family
Footnotes
Supported by a pilot project (PML and DFR) funded by the University of Arizona Cancer Center support grant P30CA23074.
References
- 1. Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal 2010;8:e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Gottardi A, Touri F, Maurer CA, Perez A, Maurhofer O, Ventre G, Bentzen CL, Niesor EJ, Dufour JF. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig Dis Sci 2004;49:982–9. [DOI] [PubMed] [Google Scholar]
- 3. Lax S, Schauer G, Prein K, Kapitan M, Silbert D, Berghold A, Berger A, Trauner M. Expression of the nuclear bile acid receptor/farnesoid X receptor is reduced in human colon carcinoma compared to nonneoplastic mucosa independent from site and may be associated with adverse prognosis. Int J Cancer 2012;130:2232–9. [DOI] [PubMed] [Google Scholar]
- 4. Maran RR, Thomas A, Roth M, Sheng Z, Esterly N, Pinson D, Gao X, Zhang Y, Ganapathy V, Gonzalez FJ, et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J Pharmacol Exp Ther 2009;328:469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Modica S, Murzilli S, Salvatore L, Schmidt DR, Moschetta A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res 2008;68:9589–94. [DOI] [PubMed] [Google Scholar]
- 6. Imray CH, Radley S, Davis A, Barker G, Hendrickse CW, Donovan IA, Lawson AM, Baker PR, Neoptolemos JP. Faecal unconjugated bile acids in patients with colorectal cancer or polyps. Gut 1992;33:1239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bayerdörffer E, Mannes GA, Richter WO, Ochsenkühn T, Wiebecke B, Köpcke W, Paumgartner G. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology 1993;104:145–51. [DOI] [PubMed] [Google Scholar]
- 8. Reddy BS, Watanabe K, Weisburger JH, Wynder EL. Promoting effect of bile acids in colon carcinogenesis in germ-free and conventional F344 rats. Cancer Res 1977;37:3238–42. [PubMed] [Google Scholar]
- 9. Rainey JB, Maeda M, Williams C, Williamson RC. The cocarcinogenic effect of intrarectal deoxycholate in rats is reduced by oral metronidazole. Br J Cancer 1984;49:631–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flynn C, Montrose DC, Swank DL, Nakanishi M, Ilsley JN, Rosenberg DW. Deoxycholic acid promotes the growth of colonic aberrant crypt foci. Mol Carcinog 2007;46:60–70. [DOI] [PubMed] [Google Scholar]
- 11. Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235–7. [DOI] [PubMed] [Google Scholar]
- 12. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al. Identification of FAP locus genes from chromosome 5q21. Science 1991;253:661–5. [DOI] [PubMed] [Google Scholar]
- 13. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997;275:1784–7. [DOI] [PubMed] [Google Scholar]
- 14. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275:1787–90. [DOI] [PubMed] [Google Scholar]
- 15. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999;398:422–6. [DOI] [PubMed] [Google Scholar]
- 16. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999;96:5522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–12. [DOI] [PubMed] [Google Scholar]
- 18. McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract 2008;204:479–90. [DOI] [PubMed] [Google Scholar]
- 19. Yamada Y, Mori H. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci 2007;98:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA 1997;94:10330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stenman LK, Holma R, Korpela R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J Gastroenterol 2012;18:923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bianchini F, Caderni G, Dolara P, Fantetti L, Kriebel D. Effect of dietary fat, starch and cellulose on fecal bile acids in mice. J Nutr 1989;119:1617–24. [DOI] [PubMed] [Google Scholar]
- 23. Suzuki R, Miyamoto S, Yasui Y, Sugie S, Tanaka T. Global gene expression analysis of the mouse colonic mucosa treated with azoxymethane and dextran sodium sulfate. BMC Cancer 2007;7:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000;102:731–44. [DOI] [PubMed] [Google Scholar]
- 25. Kemp MQ, Jeffy BD, Romagnolo DF. Conjugated linoleic acid inhibits cell proliferation through a p53-dependent mechanism: effects on the expression of G1-restriction points in breast and colon cancer cells. J Nutr 2003;133:3670–7. [DOI] [PubMed] [Google Scholar]
- 26. Kellermayer R, Dowd SE, Harris RA, Balasa A, Schaible TD, Wolcott RD, Tatevian N, Szigeti R, Li Z, Versalovic J, et al. Colonic mucosal DNA methylation, immune response, and microbiome patterns in Toll-like receptor 2-knockout mice. FASEB J 2011;25:1449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Papoutsis AJ, Lamore SD, Wondrak GT, Selmin OI, Romagnolo DF. Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J Nutr 2010;140:1607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF. Gestational exposure to theAhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoterhypermethylation and reduces BRCA-1 expression in mammary tissue of ratoffspring: Preventive effects of resveratrol. Mol Carcinog 2015;54:261–9. [DOI] [PubMed] [Google Scholar]
- 29. Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, Sugimura T, Wakabayashi K. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res 2003;63:6090–5. [PubMed] [Google Scholar]
- 30. Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M, Hla T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res 1995;55:3785–9. [PubMed] [Google Scholar]
- 31. National Cancer Institute Surveillance, epidemiology, and end results program [cited 2015 Apr 23]. Available from: http://seer.cancer.gov.
- 32. Modica S, Petruzzelli M, Bellafante E, Murzilli S, Salvatore L, Celli N, Di Tullio G, Palasciano G, Moustafa T, Halilbasic E, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012;142:355–65.e1–4. [DOI] [PubMed] [Google Scholar]
- 33. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silviera ML, Smith BP, Powell J, Sapienza C. Epigenetic differences in normal colon mucosa of cancer patients suggest altered dietary metabolic pathways. Cancer Prev Res (Phila) 2012;5:374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bailey AM, Zhan L, Maru D, Shureiqi I, Pickering CR, Kiriakova G, Izzo J, He N, Wei C, Baladandayuthapani V, et al. FXR silencing in human colon cancer by DNA methylation and KRAS signaling. Am J Physiol Gastrointest Liver Physiol 2014;306:G48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem 2003;278:104–10. [DOI] [PubMed] [Google Scholar]
- 37. Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, Gunyuzlu PL, Haws TF, Kassam A, Powell F, et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene 2002;290:35–43. [DOI] [PubMed] [Google Scholar]
- 38. Senda T, Iizuka-Kogo A, Onouchi T, Shimomura A. Adenomatous polyposis coli (APC) plays multiple roles in the intestinal and colorectal epithelia. Med Mol Morphol 2007;40:68–81. [DOI] [PubMed] [Google Scholar]
- 39. Degirolamo C, Modica S, Palasciano G, Moschetta A. Bile acids and colon cancer: solving the puzzle with nuclear receptors. Trends Mol Med 2011;17:564–72. [DOI] [PubMed] [Google Scholar]
- 40. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284:1365–8. [DOI] [PubMed] [Google Scholar]
- 41. Smith DL, Keshavan P, Avissar U, Ahmed K, Zucker SD. Sodium taurocholate inhibits intestinal adenoma formation in APCMin/+ mice, potentially through activation of the farnesoid X receptor. Carcinogenesis 2010;31:1100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang FY, Pai MH, Chiang EP. Consumption of high-fat diet induces tumor progression and epithelial-mesenchymal transition of colorectal cancer in a mouse xenograft model. J Nutr Biochem 2012;23:1302–13. [DOI] [PubMed] [Google Scholar]
- 43. Mai V, Colbert LH, Perkins SN, Schatzkin A, Hursting SD. Intestinal microbiota: a potential diet-responsive prevention target in ApcMin mice. Mol Carcinog 2007;46:42–8. [DOI] [PubMed] [Google Scholar]
- 44. Grimm C, Chavez L, Vilardell M, Farrall AL, Tierling S, Böhm JW, Grote P, Lienhard M, Dietrich J, Timmermann B, et al. DNA-methylome analysis of mouse intestinal adenoma identifies a tumour-specific signature that is partly conserved in human colon cancer. PLoS Genet 2013;9:e1003250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Forn M, Díez-Villanueva A, Merlos-Suárez A, Muñoz M, Lois S, Carriò E, Jordà M, Bigas A, Batlle E, Peinado MA. Overlapping DNA methylation dynamics in mouse intestinal cell differentiation and early stages of malignant progression. PLoS One 2015;10:e0123263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mills SC, Windsor AC, Knight SC. The potential interactions between polyunsaturated fatty acids and colonic inflammatory processes. Clin Exp Immunol 2005;142:216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tjonneland A, Overvad K, Bergmann MM, Nagel G, Linseisen J, Hallmans G, Palmqvist R, Sjodin H, Hagglund G, Berglund G, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut 2009;58:1606–11. [DOI] [PubMed] [Google Scholar]
- 48. Day SD, Enos RT, McClellan JL, Steiner JL, Velázquez KT, Murphy EA. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 2013;64:454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers for qRT-PCR and CpG Methylation