

Abstract
BACKGROUND
Allogeneic hematopoietic stem-cell transplantation for X-linked severe combined immunodeficiency (SCID-X1) often fails to reconstitute immunity associated with T cells, B cells, and natural killer (NK) cells when matched sibling donors are unavailable unless high-dose chemotherapy is given. In previous studies, autologous gene therapy with γ-retroviral vectors failed to reconstitute B-cell and NK-cell immunity and was complicated by vector-related leukemia.
METHODS
We performed a dual-center, phase 1–2 safety and efficacy study of a lentiviral vector to transfer IL2RG complementary DNA to bone marrow stem cells after low-exposure, targeted busulfan conditioning in eight infants with newly diagnosed SCID-X1.
RESULTS
Eight infants with SCID-X1 were followed for a median of 16.4 months. Bone marrow harvest, busulfan conditioning, and cell infusion had no unexpected side effects. In seven infants, the numbers of CD3+, CD4+, and naive CD4+ T cells and NK cells normalized by 3 to 4 months after infusion and were accompanied by vector marking in T cells, B cells, NK cells, myeloid cells, and bone marrow progenitors. The eighth infant had an insufficient T-cell count initially, but T cells developed in this infant after a boost of gene-corrected cells without busulfan conditioning. Previous infections cleared in all infants, and all continued to grow normally. IgM levels normalized in seven of the eight infants, of whom four discontinued intravenous immune globulin supplementation; three of these four in-fants had a response to vaccines. Vector insertion-site analysis was performed in seven infants and showed polyclonal patterns without clonal dominance in all seven.
CONCLUSIONS
Lentiviral vector gene therapy combined with low-exposure, targeted busulfan conditioning in infants with newly diagnosed SCID-X1 had low-grade acute toxic effects and resulted in multilineage engraftment of transduced cells, reconstitution of functional T cells and B cells, and normalization of NK-cell counts during a median follow-up of 16 months. (Funded by the American Lebanese Syrian Associated Charities and others; LVXSCID-ND ClinicalTrials.gov number, NCT01512888.)
X-LINKED SEVERE COMBINED IMMUNO-deficiency (SCID-X1) is a rare, life-threatening disorder caused by mutations in the gene that encodes the common γ-chain (IL2RG [GenBank accession number, D11086.1]), which is shared by multiple cytokine receptors necessary for the development and function of lymphocytes.1–3 Unless the condition is detected by newborn screening before the appearance of symptoms or identified on the basis of a positive family history, affected infants present with severe opportunistic infections during the first months of life because of defects in both cellular and humoral immunity. Laboratory studies typically show the lack of T cells, natural killer (NK) cells, and functional B cells. Procedures to restore immunity include either allogeneic hematopoietic stem-cell transplantation or autologous gene therapy.4–8 Hematopoietic stem-cell transplants from a matched sibling donor are effective but available for less than 20% of patients, and transplants from alternative donors are associated with an increased risk of graft-versus-host disease and incomplete immune reconstitution.9–14
Gene therapy is an experimental treatment that inserts a normal copy of the coding region of IL2RG into the genome of a patient’s own hematopoietic stem cells. Previous studies of gene therapy showed that first-generation γ-retroviral vectors restored T-cell immunity but resulted in vector-induced leukemia.15,16 Second-generation γ-retroviral vectors had an improved safety profile but did not restore humoral immunity or durable NK-cell production when used without conditioning chemotherapy.17 Our new lentiviral vector gene therapy combined with nonmyeloablative busulfan conditioning has been successful in restoring immunity in five patients 7 to 23 years of age in whom a previous allogeneic hematopoietic stem-cell transplantation for SCID-X1 had failed; two of the five patients gained normal B-cell function and independence from immune globulin infusions.18 We hypothesized that the combination of this lentiviral vector and low-exposure busulfan administered by means of pharmacokinetic dose targeting would be safe and effective as the primary treatment in infants with newly diagnosed SCID-X1. To test this hypothesis, we developed the Lentiviral Vector SCID-X1 Newly Diagnosed (LVXSCID-ND) dual-center, phase 1–2 safety and efficacy trial. We report the patient characteristics, vector marking (vector copy number per cell) in blood and bone marrow cells, immune reconstitution, and vector insertion-site patterns in the first eight consecutive patients.
METHODS
PATIENTS AND CLINICAL PROTOCOL
The protocol was approved by the Food and Drug Administration and by the institutional review boards at St. Jude Children’s Research Hospital and the University of California, San Francisco (UCSF) Benioff Children’s Hospital. Written informed consent was obtained from the legal guardians. Eight consecutive patients who lacked a matched sibling donor (four at St. Jude Children’s Research Hospital and four at UCSF Benioff Children’s Hospital) received their stem-cell product from September 2016 through March 2018. We report follow-up data through September 30, 2018 (median duration of follow-up, 16.4 months; range, 6 to 24).
Patients received one to two daily doses of busulfan intravenously to target a cumulative area under the curve (AUC) of 22 mg × hour per liter. The first dose was individualized on the basis of the weight and age of the patient with the use of a population-based pharmacokinetic model,19 and the second dose was adjusted on the basis of the first-dose pharmacokinetics. Clinical management was performed in accordance with local institutional guidelines regarding isolation, discharge to an outpatient setting, and transfer to local care. The authors vouch for the accuracy and completeness of the data and for the fidelity of the study to the protocol, available with the full text of this article at NEJM.org.
PRODUCT MANUFACTURING
The CL20-i4-EF1α-hγc-OPT vector was generated by a stable producer cell line at the Good Manufacturing Practice (GMP) production facility at St. Jude Children’s Research Hospital as previously described.20,21 Five independent vector batches were used in these studies. Bone marrow cells were collected in the operating room; cells that were collected at UCSF were shipped by courier to the St. Jude GMP production facility. Product manufacturing is described in detail in Figure S1 in the Supplementary Appendix, available at NEJM.org.
In brief, harvested marrow was depleted of erythrocytes and enriched for CD34+ cells with the use of a CliniMACS device (Miltenyi Biotec) according to the manufacturer’s instructions. The cells were prestimulated in the presence of RetroNectin (a 63-kD fragment of fibronectin that facilitates lentiviral binding and entry [Takara Bio USA]) and then transduced on days 2 and 3. Samples of transduced cells were tested while the bulk of the transduced-cell product was cryopreserved. When release testing criteria were met and the frozen cells returned to UCSF to treat the four patients there, busulfan was administered and the product was infused.
VECTOR COPY NUMBER AND INSERTION-SITE ANALYSIS
Peripheral-blood and bone marrow samples were sorted by means of flow cytometry into CD34+ progenitors and myeloid-cell (CD14+/15+), B-cell (CD19+), T-cell (CD3+), and NK-cell (CD3− CD56+) lineages, and genomic DNA was extracted. DNA samples were analyzed by quantitative polymerase chain reaction (PCR) with the use of a standard curve derived from a single-copy cell clone. The mean vector copy number per cell in the preinfusion transduced CD34+ cells and bone marrow CD34+ cells was measured in pooled myeloid colony-forming unit assays after 14 days of culture. Individual vector insertion sites were identified with the use of the quantitative shearing linear amplification (qsLAM) PCR method.22
STATISTICAL ANALYSIS
The primary objectives of the trial were to assess the safety, feasibility, and efficacy of lentiviral gene transfer in infants with newly diagnosed SCID-X1. Safety was defined as recovery of an absolute neutrophil count to a level of more than 500 cells per cubic millimeter by 42 days after busulfan infusion without any serious adverse events (grade ≥3, according to the adapted National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0). Feasibility was defined as having at least 1 million CD34+ cells per kilogram of body weight, with a vector copy number of at least 0.1 copies per cell. Efficacy was defined as clinically significant T-cell reconstitution within 52 weeks after gene transfer, as determined by meeting three of four criteria: proliferative response to phytohemagglutinin of at least 50% of the normal reference value for the laboratory, at least 1000 CD3+ T cells per cubic millimeter, at least 500 CD4+ T cells per cubic millimeter, and at least 200 naive CD4+CD45RA+ T cells per cubic millimeter. A Simon two-stage design was used to assess efficacy at an alpha level of 0.10, giving the trial 80% power to show that the true percentage of patients who had T-cell reconstitution after 1 year would be at least 70%.
RESULTS
PATIENT AND GRAFT CHARACTERISTICS
Eight consecutive infants (median age, 3.5 months) with newly diagnosed SCID-X1 who met the inclusion criteria were enrolled (Table 1). Before gene therapy, Patients 1, 7, and 8 had maternal T-cell engraftment (with 3916, 6, and <1 maternal CD3+ cells per cubic millimeter, respectively). Five patients had preexisting infections, including cytomegalovirus, disseminated bacille Calmette–Guérin due to neonatal bacille Calmette–Guérin vaccination, legionella, coronavirus, or asymptomatic rhinovirus (or combinations thereof).
Table 1.
Characteristics of the Patients.*
| Patient No. (Age at Gene Therapy) | IL2RG Mutation | Maternal T-Cell Engraftment | CD3+ Cell Count | CD19+ Cell Count | CD3− CD56+ Cell Count | Medical History | Busulfan Cumulative AUC | CD34+ Cell Dose | VCN of Graft | Follow-up Duration | Status at Last Follow-up† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| % of T cells | cells/mm3 | mg × hr/liter | no. × 10−6/kg | copies/cell | mo | ||||||
| 1 (6 mo) | c.720G→A p. Trp240X | 100 | 3916 | 1210 | 40 | Neutropenia, CMV, coronavirus | 20.0 | 4.46 | 0.16‡ | 24.9 | CMV and coronavirus resolved, autoimmune cytopenia had developed but resolved |
| 2 (3 mo) | c.270(−15)A→G IVS2 branch point A splice | 0 | 0 | 2340 | 13 | Rhinovirus | 22.5 | 8.67 | 1.13 | 23.1 | No longer receiving IV immune globulin, immunized§ |
| 3 (4 mo) | c.876(−1)G→A IVS5 splice | 0 | 10 | 1138 | 18 | Rhinovirus | 20.7 | 9.26 | 0.80 | 20.1 | No longer receiving IV immune globulin, immunized§ |
| 4 (14 mo) | c.562C→T p. Gln188X | 0 | 0 | 190 | 4 | Disseminated BCG, legionella, rhino‑ virus | 23.0 | 6.93 | 0.35 | 17.1 | Disseminated BCG and legionella resolved, no longer receiving IV immune globulin, immunized |
| 5 (3 mo) | c.677G→A p. Arg266His | 0 | 0 | 5832 | 1094 | None | 22.8 | 6.02 | 0.44 | 15.7 | Outpatient |
| 6 (11 mo) | c.903_910del p. Glu302Argf‑sX11 | 0 | 41 | 1376 | 19 | Disseminated BCG | 22.9 | 4.60 | 0.17 | 14.6 | Disseminated BCG resolved, no longer receiving IV immune globulin, immunized§ |
| 7 (2 (mo) | c.548delT p. Leu183Trpf‑sX90 | 42 | 15 | 1178 | 11 | Neutropenia | 20.4 | 15.10 | 1.10 | 11.1 | Outpatient |
| 8 (3 mo) | c.326_340del p.Glu109_ Ser113del | 1 | 4 | 2590 | 2 | Aphthous ulcers | 22.2 | 6.52 | 0.36 | 6.7 | Outpatient, ulcers resolved |
AUC denotes area under the curve, BCG bacille Calmette–Guérin, CMV cytomegalovirus, IV intravenous, and VCN vector copy number.
All patients are off protective precautions (i.e., not in protective isolation and not receiving prophylactic antimicrobial agents).
Patient 1 received a gene therapy boost without busulfan conditioning 12 months after the first infusion because of poor reconstitution; the CD34+ cell dose of the boost was 2.5×106 cells per kilogram of body weight, and the vector copy number was 0.22 copies per cell. Data for this patient after the gene therapy boost are provided in the Supplementary Appendix.
The patient had a response to vaccines.
Before infusion of transduced CD34+ cells, all patients received individualized doses of busulfan according to a pharmacokinetic model for busulfan clearance to target a cumulative AUC of 22 mg × hour per liter. The median busulfan cumulative AUC was 22.4 mg × hour per liter (range, 20 to 23), and overall exposure was within 10% of the intended target (Table S1 in the Supplementary Appendix). Hematopoietic recovery without severe adverse events other than hematologic toxic effects occurred by 3 to 4 weeks after busulfan infusion without blood product transfusions (Fig. S2 in the Supplementary Appendix). Mucositis occurred in two patients (Patients 5 and 6), although the cases were mild and resolved within 2 to 5 days with supportive care.
The median dose of processed CD34+ cells was 6.73 × 106 cells per kilogram of body weight (range, 4.46 × 106 to 15.10 × 106), and the median graft vector copy number was 0.40 copies per CD34+ cell (range, 0.16 to 1.13) (Table 1). All initial cell preparations met release specifications before busulfan administration.
All the patients were alive and well as outpatients at a median follow-up of 16.4 months (range, 6.7 to 24.9) and had normal growth with respect to weight and height (Fig. S3 in the Supplementary Appendix). Disseminated bacille Calmette–Guérin infections cleared in Patients 4 and 6 by 3 months after gene therapy, coincident with increasing T-cell counts. Cytomegalovirus infection cleared in Patient 1 after the gene therapy boost (Fig. S4C in the Supplementary Appendix); autoimmune thrombocytopenia and hemolytic anemia developed 1 month after the boost but fully resolved with the administration of glucocorticoids, rituximab, and intravenous immune globulin.
VECTOR MARKING IN BLOOD AND MARROW
Vector-marked peripheral-blood NK cells and myeloid cells first appeared approximately 4 weeks after infusion, whereas marked T cells and B cells were first detected at 2 to 3 months (Fig. 1A through 1D). By 4 months, peak vector copy numbers ranged from 1.30 to 2.40 copies per cell in CD3+ T cells and from 1.00 to 2.40 copies per cell in NK cells (Fig. 1A and 1C) in seven of the eight patients. The vector copy numbers were more variable in B cells (0.30 to 0.90 copies per cell) and myeloid cells (0.03 to 0.50 copies per cell) than in T cells (Fig. 1B and 1D). Marrow samples at 4 months showed vector copy numbers that ranged from 0.06 to 0.60 copies per cell in marrow CD34+ cells, from 0.02 to 0.60 copies per cell in myeloid colony-forming units, and from 0.30 to 1.00 copies per cell in marrow B cells (Fig. 1E). At 12 months, vector copy numbers in marrow CD34+ cells were 0.60 copies per cell in Patients 2 and 3 and 0.05, 0.10, and 0.07 copies per cell in Patients 4, 5, and 6, respectively (Fig. 1E). In all the patients, the vector copy numbers in marrow CD34+ cells correlated with peripheral-blood monocytes and with the vector copy number of the associated preinfusion graft (Fig. 1 and Table 1).
Figure 1. Vector Marking in Blood and Marrow Cells after Gene Therapy.
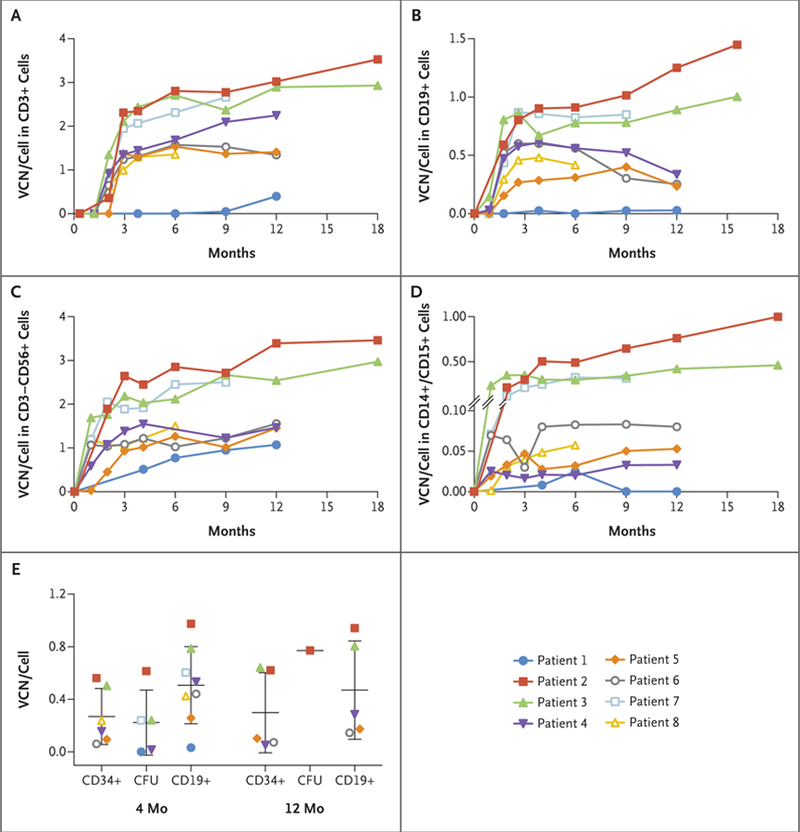
Panels A through D show the vector copy number (VCN) per cell in peripheral‑blood T‑cell (CD3+), B‑cell (CD19+), NK‑cell (CD3−CD56+), and myeloid‑cell (CD14+/CD15+) lineages, respectively, as sorted by flow cytometry, in Patients 1 through 8 at various time points after cell infusion. Panel E shows the results of cells obtained by bone marrow aspiration at 4 and 12 months after gene therapy. The mean vector copy number per cell of selected CD34+ and CD19+ cells is shown. In addition, the mean vector copy number per cell measured in pooled myeloid colony‑forming unit (CFU) assays after 14 days of culture is shown. The vector copy number is shown as the mean vector genome copy per cell in the sorted populations and was determined by quantitative polymerase chain reaction with the use of a standard curve derived from a transduced single‑copy clonal cell line.
Patient 1 showed persistent maternal T-cell engraftment with very low vector marking of T-cell, B-cell, and myeloid cell lineages during the first 12 months (Fig. 1). He received a boost of transduced CD34+ marrow cells without any conditioning at 12 months after the initial infusion, after which a clinically significant increase in vector-marked T cells was noted (Fig. S4A and S4B in the Supplementary Appendix).
IMMUNE RECONSTITUTION
T Cells and NK Cells
In Patients 2 through 8, normal numbers of circulating T cells were detected within 2 to 4 months after infusion (Fig. 2A through 2D). Absolute numbers of CD3+, CD4+, CD8+, and naive CD4+ T cells continued to rise until approximately 6 months and then plateaued to within normal ranges. T-cell–receptor excision circles increased to normal levels in these patients, indicating thymic differentiation of newly developed T cells (Fig. 2G). In vitro T-cell responses to the T-cell mitogen phytohemagglutinin were in the normal range 4 months after infusion (Fig. S5 in the Supplementary Appendix).
Figure 2. Immune Reconstitution after Gene Therapy.
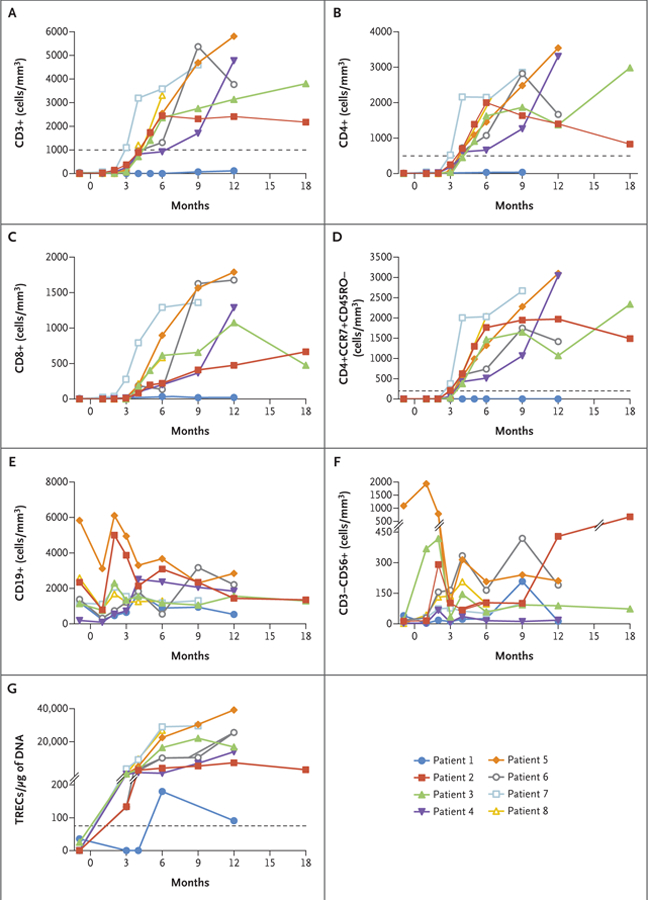
Panels A through F show absolute numbers of peripheral‑blood immune‑cell subsets, as determined by means of standard flow cytometry, over time after cell infusion. Dotted lines indicate values for T‑cell counts that are defined in the protocol as representing clinically significant reconstitution at 52 weeks after gene therapy. Panel G shows the quantity of DNA T‑cell–receptor excision circles (TRECs) in peripheral‑blood mononuclear cells, with a dotted line indicating the lower limit of the normal range. The values above the hash marks on the y axis range from 201 to 40,000.
Vβ spectratyping of T-cell receptors showed polyclonal diversity and normal distributions in all eight patients. All the patients had a normal T-cell–receptor Vβ complexity score except for Patient 1, who received a gene therapy boost. T-cell effector responses of naive CD8+ cells normalized after gene therapy in all three evaluated patients (Patients 3, 5, and 6) and were higher than in healthy adults, but this difference did not reach statistical significance. (Further details are provided in Figs. S6, S7, and S8A in the Supplementary Appendix.) The numbers of NK cells increased to within the normal range in five of the eight patients (Fig. 2F).
B Cells
B-cell counts were within the normal range by 2 months after busulfan infusion (Fig. 2E). IgM levels normalized in seven of the eight patients by 6 to 12 months after infusion (Fig. S8B in the Supplementary Appendix). Three months after discontinuing intravenous immune globulin supplementation, Patients 2, 3, 4, and 6 were vaccinated against tetanus, diphtheria, pertussis, polio, and pneumococcal polysaccharide at 12, 15, 13, and 9 months, respectively, after gene therapy. Protective antibody responses against polio were documented in three of four patients (Patients 2, 3, and 6) and against tetanus, diphtheria, pertussis, and pneumococcus in two of four patients (Patients 2 and 3), indicating the reconstitution of functional B cells (Fig. S8C, S8D, and S8E in the Supplementary Appendix).
VECTOR INTEGRATION SITE ANALYSIS
Vector integration site analysis of lineage-sorted blood cells obtained from Patients 1 through 7 at 6 to 21 months after infusion was performed with the use of a qsLAM PCR assay22; integration site analysis of the 6-month sample from Patient 8 is in progress, and the results were not available at the time of this report. Unique vector–genome junctions provide clonal markers for each originally transduced hematopoietic progenitor, and the frequency of each unique junction sequence is a measure of the clonal proliferation. This analysis showed a strong enrichment for vector integration sites in genes that are transcribed in blood cells and in gene exons and introns, as well as a relative paucity of vector integration sites in CpG islands (i.e., clusters of CpG dinucleotides) (Fig. S9 in the Supplementary Appendix). This pattern is consistent with the findings in a previous study involving patients with Wiskott–Aldrich syndrome who received lentiviral gene therapy.23
Vector integrations that perturb expression of genes in which products regulate growth can lead to clonal outgrowths and eventual leukemia and are indicated by a dominant integration-site or clone that becomes more predominant over time. In contrast, highly polyclonal patterns were noted in Patients 2 through 7, in whom no single vector integration site ever exceeded 5.5% of the total integrations (Fig. S10A in the Supplementary Appendix). A more restricted pattern was noted only in T cells and total nucleated cells from Patient 1 at 12 months; this sample was collected before the gene therapy boost, when the number of vector-marked autologous T cells was low (Fig. 2A). Greater numbers of transduced cells were present in NK cells and B cells in Patient 1, and these were associated with a more polyclonal pattern. Vector integration-site diversity improved in the T-cell compartment after the gene therapy boost at 21 months. (Further details are provided in Figs. S4F and S10A in the Supplementary Appendix.)
Common vector integration sites that are detected in cells from different lineages indicate transduction of pluripotent progenitors. Shared vector integration sites in diverse lineages were detected in all seven evaluated patients, with the number of shared sites ranging from 27 to 4707 per patient (Fig. S10B in the Supplementary Appendix). Patient 1 had 7 common vector integration sites that were shared in all four lineages (CD3+, CD19+, CD14+/15+, and CD56+), Patient 2 had 223, Patients 3 and 4 each had 10, Patient 5 had 25, Patient 6 had 7, and Patient 7 had 25; these findings provide evidence of the transduction of pluripotent progenitors.
Distinct but clustered vector integrations were seen in multiple patients, with three genes commonly targeted in Patients 1 through 7 and 2235 genes targeted in at least two of the seven evaluated patients (Fig. S10C in the Supplementary Appendix). These shared genes are enriched for cancer genes and include previously described recurrent vector integration sites for lentiviral vectors23 such as NF1, PTEN, STAT5B, and PACS1. The vector integration patterns for several of these “hot spots” have been mapped and are shown in Figure S11 in the Supplementary Appendix.
DISCUSSION
This study showed the successful use of a lentiviral vector combined with low-exposure, targeted busulfan conditioning as the primary treatment in infants with newly diagnosed SCID-X1. This approach led to broad immune reconstitution and efficient vector transduction in diverse lineages, including T cells, B cells, NK cells, myeloid cells, and bone marrow progenitors. This high-level marking was associated with evidence of B-cell reconstitution and restoration of humoral immunity, which has not been achieved in previously reported trials of gene therapy for infants with newly diagnosed SCID-X1. In addition, rapid reconstitution of T cells and NK cells was achieved in most cases, and the overall safety profile was favorable at the time of this interim analysis.
In previous trials that used a γ-retroviral vector without busulfan conditioning to treat newly diagnosed SCID-X1 in infants, durable immune reconstitution was limited to T cells, whereas myeloid-cell, B-cell, and NK-cell marking was relatively low, and patients did not recover humoral immunity.17,24,25 In contrast, we observed that marking was increased in all of these lineages (Fig. 1) and that recovery of B-cell function was sufficient to achieve independence from immune globulin supplementation in four patients at 15 to 23 months after gene therapy. Three of these four patients had an antibody response to vaccination, indicating good B-cell function.
The use of busulfan conditioning in allogeneic hematopoietic stem-cell transplantation in infants with all genetic forms of SCID has been controversial and deemed unnecessary in certain settings, because T-cell reconstitution is regularly achieved without conditioning and is sufficient for early patient survival.13,14,24 However, less than 10% of patients with SCID-X1 had reconstitution of functional B cells without conditioning after receiving a hematopoietic stem-cell transplant from a haploidentical or unrelated donor, a finding that is similar to the results obtained from previous SCID-X1 gene therapy trials.26,27 Moreover, in some patients with SCID-X1 who had undergone allogeneic donor transplantation, T-cell immunity waned over time, necessitating additional therapies, including a second bone marrow transplantation or gene therapy.18 Preclinical studies have highlighted that reconstitution of functional B cells can be achieved if a preconditioning regimen is combined with gene therapy.28,29 Our results show that busulfan facilitates durable engraftment of gene-corrected hematopoietic stem cells, allowing for long-term replenishment of all affected lymphocyte lineages in infants with newly diagnosed SCID-X1.18,30 The use of pharmacokinetic-guided administration of busulfan ensured consistent drug exposure, which is especially critical in infants and young children — a patient population in which metabolism and clearance of busulfan vary considerably.31 All the patients in our study had a cumulative AUC of busulfan within 10% of the targeted 22 mg × hour per liter, but the actual doses required to achieve this exposure varied from 2.59 to 5.67 mg per kilogram. This strategy resulted in no severe acute nonhematopoietic toxic effects associated with the expected level of reversible myelosuppression, and yet it succeeded in achieving engraftment in the marrow, myeloid-cell, T-cell, NK-cell, and B-cell compartments. Thus, a cumulative AUC of 22 mg × hour per liter, which is approximately 25% of the typical cumulative AUC used for allogeneic hematopoietic stem-cell transplantation, is well suited for SCID-X1 gene therapy in infants as young as 2 months of age.
Variable vector marking was noted in the nine infused grafts (Patient 1 received two grafts), with vector copy numbers ranging from 0.16 to 1.13 copies per cell and total infused cell numbers varying from 4.5×106 to 15.1×106 cells per kilogram. Higher marking levels in myeloid and B cells correlated with relatively high numbers of transduced CD34+ cells in the graft. Other potential sources of variability include the relative potency of different clinical grade vector lots on CD34+ cells and the quantitative degree of CD34+ expression in marrow cells, which was lower in Patients 1 and 6 and was associated with relatively low yields in these patients (Fig. S12 in the Supplementary Appendix). Despite this variability in the input grafts, the kinetics of T-cell reconstitution was similar in all patients who could be evaluated, except for Patient 1.
In conclusion, we found that the combination of a lentiviral vector construct and nonmyeloablative targeted busulfan conditioning in infants with newly diagnosed SCID-X1 resulted in multilineage engraftment of gene-marked cells, with prompt and durable normalization of T-cell immunity and high-level gene marking in B cells and NK cells, with normalization of IgM levels and the development of normal antibody responses. It is hoped that durable, complete adaptive immunity will be achieved in the majority of the patients over time. Successful manufacturing from a stable producer cell line and cryopreservation of the transduced-cell product would streamline the vector production process and facilitate efforts to commercialize product distribution through centralized manufacturing facilities. Long-term follow-up will be required to assess the durability of the immune reconstitution and the persistence of the transferred gene in hematopoietic and immune cells and to monitor the patients for potential late effects of busulfan conditioning and gene therapy.
Supplementary Material
Acknowledgments
Supported by the American Lebanese Syrian Associated Charities; by grants from the California Institute of Regenerative Medicine (CLIN2–09504), the National Heart, Lung, and Blood Institute (P01 HL053749), the National Cancer Institute (CA21765), the intramural program of the National Institute of Allergy and Infectious Diseases (NIAID) (Z01-AI-00988, to Drs. De Ravin and Malech), and the NIAID (U54-AI082973, to Drs. Cowan and Puck); and by the Assisi Foundation of Memphis.
We recognize the major contributions of our deceased colleague, Dr. Brian P. Sorrentino, who initiated this work and brought it to fruition with a successful clinical trial. We thank Dr. Arthur Nienhuis for his long-term support, inspiration, and discussions; the staff at the Department of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, and the staff at the Bone Marrow Transplant Program, University of California, San Francisco, Benioff Children’s Hospital, for their care of these children and support of this study; Dr. Don Kohn for his discussions about and help with establishing the bone marrow harvest procedure; Drs. Mary Ellen Conley and Andrew Scharenberg for their help and insights in planning and designing the study and Dr. Robert Throm from the Vector Production and Development laboratory at St. Jude Children’s Research Hospital for his discussions about and work in vector production and cell transduction; Drs. Taihe Lu and Soghra Fatima from the Department of Hematology at St. Jude Children’s Research Hospital and Dr. Jean-Yves Metais, Ms. Sara Schell, and Ms. MaCal Tuggle from the Department of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, for their technical assistance in analyzing patient samples; Dr. Thasia Leimig and Ms. Suzette Win-go from the Department of Therapeutics Production and Quality, St. Jude Children’s Research Hospital, for their work in cell production; Ms. Jane Stringfellow and Ms. Terri Davis for their administrative support of the St. Jude investigative team; Drs. Christine Narrin-Talbot and Wayne Wolfrey for their contributions to recruiting patients for the study; and, most of all, the families that participated in our study.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org. St. Jude Children’s Research Hospital has an existing exclusive license and ongoing partnership with Mustang Bio for the further clinical development and commercialization of this SCID-X1 gene therapy.
Contributor Information
E. Mamcarz, Departments of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN
S. Zhou, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
T. Lockey, Therapeutics Production and Quality, St. Jude Children’s Research Hospital, Memphis, TN
H. Abdelsamed, Departments of Immunology, St. Jude Children’s Research Hospital, Memphis, TN
S.J. Cross, Departments of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN;
G. Kang, Departments of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN
Z. Ma, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
J. Condori, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
J. Dowdy, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
B. Triplett, Departments of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN;
C. Li, Departments of Biostatistics, St. Jude Children’s Research Hospital, Memphis, TN
G. Maron, Infectious Diseases St. Jude Children’s Research Hospital, Memphis, TN
J.C. Aldave Becerra, Clinical Immunology Division, Hospital Nacional Edgardo Rebagliati Martins, Lima, Peru
J.A. Church, Department of Pediatrics, Allergy–Immunology Division, Children’s Hospital Los Angeles, Los Angele
E. Dokmeci, Department of Pediatrics, Pediatric Allergy and Immunology, University of New Mexico, Albuquerque
J.T. Love, University of Oklahoma Health Sciences Center, Tulsa
A.C. da Matta Ain, Departamento de Pediatria da Universidade de Taubaté, Conselho Nacional de Medicina, São Paulo.
H. van der Watt, Copper-field Childcare, Claremont, South Africa
X. Tang, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
W. Janssen, Departments of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN
B.Y. Ryu, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
S.S. De Ravin, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
M.J. Weiss, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN
B. Youngblood, Departments of Immunology, St. Jude Children’s Research Hospital, Memphis, TN
J.R. Long‑Boyle, Department of Pediatrics, Division of Pediatric Allergy–Immunology–Bone Marrow Transplantation, University of California, San Francisco (UCSF) Benioff Children’s Hospital, San Francisco both in California
S. Gottschalk, Departments of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN
M.M. Meagher, Therapeutics Production and Quality, St. Jude Children’s Research Hospital, Memphis, TN
H.L. Malech, Genetic Immunotherapy Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD
J.M. Puck, Department of Pediatrics, Division of Pediatric Allergy–Immunology–Bone Marrow Transplantation, University of California, San Francisco (UCSF) Benioff Children’s Hospital, San Francisco both in California
M.J. Cowan, Department of Pediatrics, Division of Pediatric Allergy–Immunology–Bone Marrow Transplantation, University of California, San Francisco (UCSF) Benioff Children’s Hospital, San Francisco both in California
B.P. Sorrentino, Departments of Hematology, St. Jude Children’s Research Hospital, Memphis, TN.
REFERENCES
- 1.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol 2004;22:625–55. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs S, Rensing-Ehl A, Erlacher M, et al. Patients with T+/low NK+ IL-2 receptor γ chain deficiency have differentially-impaired cytokine signaling resulting in severe combined immunodeficiency. Eur J Immunol 2014;44:3129–40. [DOI] [PubMed] [Google Scholar]
- 3.Puck JM, Deschênes SM, Porter JC, et al. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet 1993;2: 1099–104. [DOI] [PubMed] [Google Scholar]
- 4.Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 1999;340: 508–16. [DOI] [PubMed] [Google Scholar]
- 5.Gennery A Recent advances in treatment of severe primary immunodeficiencies. F1000Res 2015;4:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002;99:872–8. [DOI] [PubMed] [Google Scholar]
- 7.Thrasher AJ. Gene therapy for primary immunodeficiencies. Immunol Allergy Clin North Am 2008;28:457–71. [DOI] [PubMed] [Google Scholar]
- 8.Wahlstrom JT, Dvorak CC, Cowan MJ. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr Pediatr Rep 2015;3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowan MJ, Neven B, Cavazanna-Calvo M, Fischer A, Puck J. Hematopoietic stem cell transplantation for severe combined immunodeficiency diseases. Biol Blood Marrow Transplant 2008;14:Suppl 1:73–5. [DOI] [PubMed] [Google Scholar]
- 10.Gaspar HB, Qasim W, Davies EG, Rao K, Amrolia PJ, Veys P. How I treat severe combined immunodeficiency. Blood 2013; 122:3749–58. [DOI] [PubMed] [Google Scholar]
- 11.Haddad E, Logan BR, Griffith LM, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018;132:1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horn B, Cowan MJ. Unresolved issues in hematopoietic stem cell transplantation for severe combined immunodeficiency: need for safer conditioning and reduced late effects. J Allergy Clin Immunol 2013; 131:1306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pai S-Y, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014;371:434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Railey MD, Lokhnygina Y, Buckley RH. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy or post-transplant GVHD prophylaxis. J Pediatr 2009;155(6):834–840.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003; 302:415–9. [DOI] [PubMed] [Google Scholar]
- 16.Nam CH, Rabbitts TH. The role of LMO2 in development and in T cell leukemia after chromosomal translocation or retroviral insertion. Mol Ther 2006;13:15–25. [DOI] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 2014;371:1407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Ravin SS, Wu X, Moir S, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med 2016;8:335ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long-Boyle JR, Savic R, Yan S, et al. Population pharmacokinetics of busulfan in pediatric and young adult patients undergoing hematopoietic cell transplant: a model-based dosing algorithm for personalized therapy and implementation into routine clinical use. Ther Drug Monit 2015;37:236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Throm RE, Ouma AA, Zhou S, et al. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 2009;113:5104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou S, Mody D, DeRavin SS, et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood 2010;116:900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou S, Bonner MA, Wang YD, et al. Quantitative shearing linear amplification polymerase chain reaction: an improved method for quantifying lentiviral vector insertion sites in transplanted hematopoietic cell systems. Hum Gene Ther Methods 2015;26:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013;341:1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet 2004;364: 2181–7. [DOI] [PubMed] [Google Scholar]
- 25.Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med 2002;346: 1185–93. [DOI] [PubMed] [Google Scholar]
- 26.Dvorak CC, Cowan MJ. Hematopoietic stem cell transplantation for primary immunodeficiency disease. Bone Marrow Transplant 2008;41:119–26. [DOI] [PubMed] [Google Scholar]
- 27.Dvorak CC, Hassan A, Slatter MA, et al. Comparison of outcomes of hematopoietic stem cell transplantation without chemotherapy conditioning by using matched sibling and unrelated donors for treatment of severe combined immunodeficiency. J Allergy Clin Immunol 2014;134(4):935–943.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humbert O, Chan F, Rajawat YS, et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv 2018;2:987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LHuston MW, van Til NP, Visser TP, et al. Correction of murine SCID-X1 by lentiviral gene therapy using a codon-optimized IL2RG gene and minimal pretransplant conditioning. Mol Ther 2011;19: 1867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cowan MJ, Dvorak CC, Long-Boyle J. Opening marrow niches in patients under-going autologous hematopoietic stem cell gene therapy. Hematol Oncol Clin North Am 2017;31:809–22. [DOI] [PubMed] [Google Scholar]
- 31.Savic RM, Cowan MJ, Dvorak CC, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant 2013;19:1608–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.