

ABSTRACT
Profilin 1 (Pfn1), a regulator of actin polymerization, controls cell movement in a context‐dependent manner. Pfn1 supports the locomotion of most adherent cells by assisting actin‐filament elongation, as has been shown in skeletal progenitor cells in our previous study. However, because Pfn1 has also been known to inhibit migration of certain cells, including T cells, by suppressing branched‐end elongation of actin filaments, we hypothesized that its roles in osteoclasts may be different from that of osteoblasts. By investigating the osteoclasts in culture, we first verified that Pfn1‐knockdown (KD) enhances bone resorption in preosteoclastic RAW264.7 cells, despite having a comparable number and size of osteoclasts. Pfn1‐KD in bone marrow cells showed similar results. Mechanistically, Pfn1‐KD osteoclasts appeared more mobile than in controls. In vivo, the osteoclast‐specific conditional Pfn1‐deficient mice (Pfn1‐cKO) by CathepsinK‐Cre driver demonstrated postnatal skeletal phenotype, including dwarfism, craniofacial deformities, and long‐bone metaphyseal osteolytic expansion, by 8 weeks of age. Metaphyseal and diaphyseal femurs were drastically expanded with suppressed trabecular bone mass as indicated by μCT analysis. Histologically, TRAP‐positive osteoclasts were increased at endosteal metaphysis to diaphysis of Pfn1‐cKO mice. The enhanced movement of Pfn1‐cKO osteoclasts in culture was associated with a slight increase in cell size and podosome belt length, as well as an increase in bone‐resorbing activity. Our study, for the first time, demonstrated that Pfn1 has critical roles in inhibiting osteoclast motility and bone resorption, thereby contributing to essential roles in postnatal skeletal homeostasis. Our study also provides novel insight into understanding skeletal deformities in human disorders. © 2018 American Society for Bone and Mineral Research.
Keywords: OSTEOCLAST, GENETIC ANIMAL MODELS, DEVELOPMENTAL MODELING, BONE HISTOMORPHOMETRY, DISEASES AND DISORDERS OF/RELATED TO BONE
Introduction
Skeletal homeostasis is maintained by various factors, including hormones, cytokines, growth factors, and coupling factors.1, 2 Those factors influence cell–cell communication, signaling, gene expression, and behaviors including division, proliferation, vesicular transportation, and migration. By focusing on skeletal cell movement in growing mice,3, 4, 5, 6 we previously demonstrated that profilin1 (Pfn1), one of the major modulators of actin cytoskeleton, plays important roles during skeletogenesis and bone homeostasis.5, 6, 7 The conditional Pfn1‐deficient mice in limb mesenchyme had a failure in chest‐wall closure and endochondral ossification in long bones.5 Conditional Pfn1 deficiency in osteocytes resulted in impaired osteocytic dendrite formation and bone mass maintenance,6 consistent with the idea that Pfn1 enhances the movement of most mesenchymal cells by supporting the elongation of straight actin filaments assisted by formin.
However, pfn1 functions in modulating actin‐filament elongation are known to be controversial, depending on the cell‐type or cell‐context difference.8 In epithelial and cancer cells, Pfn1 is known to inhibit their movement,9 possibly through inhibiting branched‐end actin filaments10, 11, 12 that are necessary for structuring lamellipodia, podosomes, and invadopodias. Invadopodia is a podosome‐like process in cancer cells; thus, Pfn1‐deficient breast cancer cells are more invasive.9 Similarly, Pfn1‐deficient cytotoxic T cells are more active in movement and cancer‐killing functions than are Pfn1‐positive cells.13
Osteoclasts are critical players in bone remodeling and belong to the macrophage lineage cells. Their differentiation and activation requires cell–cell fusion, vesicle trafficking, transcytosis, and sealing zone formation, etc.—all of which require cytoskeletal reorganization.14, 15 Characteristically, the osteoclasts utilize a cellular process, named podosome, with specific arrangements for their movement or attachment, depending on the differentiation/activation status.16 Most characteristically, the activated osteoclasts at the terminal differentiation stage create the structure, the podosome belt, which is required for active bone resorption. Because the core structures of podosome and lamellipodia are postulated to be negatively regulated by Pfn1, it is of interest to test if the inhibition of Pfn1 in osteoclasts leads to increased cellular movement and to related behavior based on cytoskeletal changes.
Therefore, in this study, we investigated the Pfn1 function in osteoclasts by utilizing the preosteoclastic cell line and mice deficient for Pfn1, specifically in osteoclasts. By analyzing the phenotypic manifestations of mutant mice, we revealed the critical functions of Pfn1 in suppressing osteoclast movement and bone resorptive activity. In addition, we propose that observed skeletal phenotypes in these model mice might be related to the pathogenesis of certain osteolytic disorders that are possibly influenced by the modulation of the actin cytoskeleton.
Materials and Methods
Animals
Pfn1‐floxed (Pfn1 fl/fl) mice were generated previously.7 To obtain conditional mutant mice lacking Pfn1 in mature osteoclasts, Pfn1 fl/fl mice were crossed with Cathepsin K (CatK)‐Cre knocked‐in mice.17 Progenies were genotyped by genomic PCR. The neonatal, adolescent, and adult littermates were analyzed for the skeletal phenotypes at postnatal day 0 (P0), 4 weeks, and 8 weeks. Mice were housed under controlled conditions at 24°C on a 12‐hour light and 12‐hour dark cycle. All the experiments were approved by the animal welfare committee of Tokyo Medical and Dental University (#0170041A).
Radiological analysis
A skeletal soft X‐ray radiogram of each animal was captured under anesthesia at 40 kV, 12 mA for 5 s, using Softex apparatus (Softex Japan Co., Kobe, Japan). Three‐dimensional µCT images were obtained using the Scan‐Xmate‐E090 (Comscan Tecno, Sagamihara, Japan). The morphometric trabecular bone parameters such as bone volume/tissue volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), trabecular spacing (Tb.Spac), and trabecular separation (Tb.Sp) were analyzed on distal femurs, using Tri/3D‐Bon software (Ratoc System Engineering, Tokyo, Japan). Cortical bone volume, cortical thickness, and center‐line length were also analyzed as previously described,18 according to the guideline established by the ASBMR.19 To evaluate the topological differences of the cross‐sections, we calculated the ratios of the length on the short axis (SA) versus the long axis (LA) at the diaphysis.
Skeletal preparation
Neonatal skeletons stained with Alcian Blue and Alizarin Red, and stored in 80% glycerol in 0.2% KOH solution were photographed essentially according to previously described methods.20
Histological and histomorphometric analysis
Paraffin‐embedded decalcified sections were stained with hematoxylin and eosin or toluidine blue‐O (pH 4.5). To visulaize osteoclasts, sections were stained with substrate for tartrate‐resistant acid phosphatase (TRAP). Bone resorption parameters including osteoclast number/bone surface (Oc.N/BS) were analyzed based on the TRAP‐positive multinucleated cells on distal femoral sections.18 For some samples, nondecalcified femurs and skulls embedded in glycol methacrylate (GMA) were sectioned and stained. The analysis was based on the ASBMR guideline.21
Immunoblot analysis
Cultured cells were solubilized at each time point with SDS sample buffer containing 62.5mM Tris‐HCl (pH 6.8), 10% glycerol, 5% 2‐mercaptoethanol, and 2.25% SDS. The total cell lysates were separated by SDS‐polyacrylamide gel electrophoresis and transferred to an Amersham Hybond‐P PVDF membrane (GE Healthcare, Piscataway, NJ, USA), which were then treated with Blocking One P (Nacalai, San Diego, CA, USA) in Tris‐buffered saline containing Tween 20 for 60 min. The membranes were incubated with antibodies against Pfn1 (GeneTex, Irvine, CA, USA,) or GAPDH (14C10; Cell Signaling Technology, Beverly, MA, USA), washed, and incubated with horseradish peroxidase‐labeled antirabbit IgG (GE Healthcare). Band intensities were measured and analyzed using a LAS4000 imager with ImageQuant TL software (GE Healthcare).
Gene‐expression analysis
Total RNA was isolated from bone marrow cells and RAW264.7 cells using the Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. Reverse transcription (RT) was carried out using 1‐µg total RNA. Quantitative real‐time PCR (qPCR) was carried out using StepOne equipment (Applied Biosystems; Thermo Fisher Scientific). The aliquots of 10‐µL reagent mixture containing 2 µL of cDNA samples, 0.2µM forward and reverse primers, and 5 µL of FAST SYBR Green Master Mix (Applied Biosystems) were applied and analyzed. The primer sequences for the Pfn1 were as follows: Pfn1‐forward, 5'‐CCCCCACCGTTCCCTTTGGC‐3', Pfn1‐reverse, 5'‐GCCCCCAGCCCATGTGGTTT‐3'.
Cell culture
RAW264.7 cells were maintained in culture with Dulbecco's modified Eagle's medium or alpha‐MEM supplemented with 10% FBS by subconfluent stage. By replating the cells onto individual plates or wells, the osteoclastogenesis assay was conducted using RANKL (100 ng/mL; Oriental Yeast Co., Tokyo, Japan) as described previously.18, 22 The siRNA transfection was conducted using lipofectamine RNAiMax reagent (Life Technologies; Thermo Fisher Scientific), according to the manufacturer's standard protocol. The siRNA for the mouse Pfn1 (Ambion Silencer Select #4390771‐s71525, Thermo Fisher Scientific; Sense CAAUGGAUCUUCGUACCAATT, Antisense UUGGUACGAAGAUCCAUUGTA) and control siRNA (Ambion Silencer Negative Control No.1 siRNA) was purchased from the vendor (Life Technologies; Thermo Fisher Scientific).
Bone marrow cells obtained from WT and Pfn1‐cKO mouse femurs and tibias were plated on 100‐mm plastic dishes at around 4 × 105 cells/cm2 in alpha‐MEM supplemented with 10% FBS and penicillin/streptomycin. After 4 hours of incubation at 37 °C, 5% CO2, nonadherent bone marrow cells were replated on the assay plates at 4 × 105 cells/cm2. For osteoclastogenesis assay, bone marrow cells were cultured 48 hours in the presence of M‐CSF (10 ng/mL; R&D Systems, Minneapolis, MN, USA); then the medium was changed to that containing both RANKL (100 ng/mL) and M‐CSF. The osteoclast differentiation was monitored usually for 5 days, by periodically changing the medium. The osteoclast induction was also conducted by an optimized method.23, 24 Bone marrow cells were cultured on a 60‐mm plastic dish for 2 to 3 hours in alpha‐MEM containing 10% FBS. Nonadherent cells were reseeded in multiwell plates at 2.6 × 105 cells/cm2 and cultured for 24 hours in alpha‐MEM with 10% FBS and 1:50 CMG14‐12 culture supernatant containing M‐CSF. These cells were cultured for 72 hours with M‐CSF and RANKL (36 ng/mL). The cells were fixed and stained for TRAP activity. To quantify the TRAP‐positive multinucleated cells, microscopic images of entire wells were captured in tiled array by semiautomated microscope BZ‐X710 (Keyence, Osaka, Japan).
Immunofluorescent analysis of cultured osteoclasts
To analyze the morphological features of the wild‐type and Pfn1‐cKO osteoclasts, localization of the Pfn1 was detected by immunofluorescence, using rabbit anti‐Pfn1 antibodies (11680‐1‐AP; Proteintech‐Japan, Tokyo, and GTX102072; GeneTex, with identical results) and goat antirabbit IgG Allexa 546 (Life Technologies). Actin cytoskeleton and nuclei were visualized by Allexa Fluor 488 phalloidin (Life Technologies) and Hoechst 33342 (Dojindo, Kumamoto, Japan), respectively, as described in Lin et al.6
Pit formation assay
RAW264.7 cells plated at 5 × 103 cells/cm2 on a BD Bio Coat culture well (Osteologic System; BD Biosciences, Bedford, MA, USA) were cultured for 3 days in the presence of RANKL (100 ng/mL), then transfected with the siRNA against Pfn1 or negative control for 24 hours (until day 4). The RANKL‐containing (100 ng/mL) medium was changed every other day until evaluation. The observation was basically made at the end of day 5 (48 hours after transfection), but some experiments were done following a modified protocol with longer culture periods.
Bone marrow‐derived macrophages plated on BD Bio Coat at 5.3 × 104 cells/cm2 were immediately transfected with siRNA. By inducing the osteoclastogenesis with M‐CSF and RANKL, the observation was made usually at day 6 (150 hours after transfection). Bone marrow cells from Pfn1‐cKO and WT littermates were plated at 8.3 × 105 cells/cm2. By inducing the osteoclastogenesis with M‐CSF and RANKL, the observation was made at day 4 (96 hours after RANKL). For evaluation, the cells were removed by acid treatment, and the size of the resorption pits was analyzed using ImageJ software on the tiled phase‐contrast images captured by BZ‐X710 (Keyence).
Live‐cell imaging
RAW264.7 cells were plated on a 24‐well plate at 1 × 104 cells/well. After 24 hours, the cells were transfected with Pfn1‐ or control‐siRNA (Ambion; Thermo Fisher Scientific) using Lipofectamine RNAiMax reagent and cultured for 6 hours. Then, siRNA was washed out, and the cells were incubated with RANKL for 72 hours. Similarly, nonadherent bone‐marrow‐derived cells (5 × 105 cells/well) plated on a 24‐well plate were subjected to the osteoclastogenesis assay as described above.23 Live imaging was conducted from 48 to 72 hours after adding RANKL, using BioStation CT (Nikon, Melville, NY, USA). The images were captured every 13 min for RAW264.7 cells and every 11 min for bone marrow cells. The average distances and the velocity at each interval were quantified using the manual tracking and chemotaxis tool of the ImageJ software.
For live‐cell imaging of the modified wound‐healing assay, RAW264.7 cells were plated on glass‐bottom dishes (Matsunami, Bellingham, WA, USA) at 1 × 105 cells/cm2 in the presence of RANKL (100 ng/mL). To create a spotty wound on the bottom, a cloning cylinder was placed on the bottom. After 72 hours, the cells were transfected with Pfn1‐ or control‐siRNA. At 4 to 5 days, the cloning cylinders were removed to start live imaging using the laser‐scanning confocal microscope Fv‐10i (Olympus, Tokyo, Japan). Similarly, nonadherent bone‐marrow derived cells (1 × 106 cells/cm2) plated on the glass‐bottom dishes with cloning cylinders were subjected to osteoclastogenesis using M‐CSF (10 ng/mL) and RANKL (100 ng/mL). The cloning cylinders were removed at approximately 2 days to start capturing the images every 15 min for 24 hours.
Statistical analysis
Statistical analysis was performed on the data obtained by biological, histological, radiological, and physical measurements. Normal distribution was tested by the Kolmogorov‐Smirnov (K‐S) test, when applicable; the data were basically represented by mean ± SD. The Student's t test or the Mann‐Whitney test was applied to compare the two experimental groups.
Results
Pfn1‐KD affects osteoclast migration and matrix resorption
Pfn1 was expressed in osteoclasts, and its intracellular localization was detected by immunofluorescence (Supplemental Fig. 1A). To examine its cellular functions, we knocked down the Pfn1 mRNA in preosteoclastic RAW264.7 cells by siRNA transfection: The KD efficiency at 24 hours was 47.3 ± 2.52 % by qPCR analysis. In osteoclastogenesis by standard protocol, we found the number of TRAP‐positive cells was comparable between the Pfn1‐KD cells and control cells, whereas their size seemed to be slightly larger (Fig. 1 A, B). However, detailed analysis of their size distribution using fluorescent images did not indicate significant differences between them (Supplemental Fig. 2B). We did detect increased locomotion of Pfn1‐KD cells compared with that of controls by live‐cell imaging analysis (Fig. 1 F, G and Supplemental Movie 1). Similar results were obtained by modified scratch‐wound healing assay with live‐cell imaging (Supplemental Fig. 2C–E, Supplemental Movie 2). In addition, pit formation assay revealed that Pfn1‐KD cells tended to resorb more mineralized matrix by larger pits with comparable numbers per area (Fig. 1 C, D; 48 hours after transfection). The results suggest that the increased mobility of each osteoclast may amplify matrix‐resorbing activity. This is consistent with a recent report showing that chemokine pathways that activate RhoA and ROCK increase bone resorption in culture without increasing the osteoclast number.25 To test if the Pfn1‐KD osteoclasts are indeed more potent in bone resorption, we knocked down the Pfn1 on bone marrow cells. By evaluating the pit formation after 6 days of osteoclast induction, we found that the Pfn1‐KD osteoclasts resorb more mineralized matrix than those transfected with control siRNA (Supplemental Fig. 1B–E).
Figure 1.
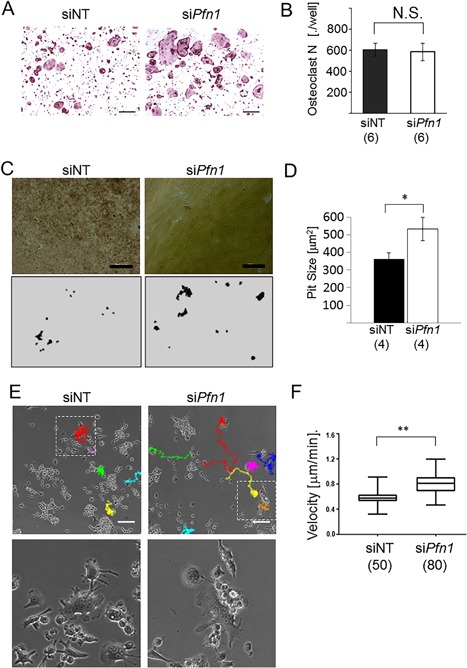
Migration and matrix‐resorption of the RAW264.7 cell‐derived Pfn1‐KD osteoclasts are increased. RAW264.7 cells transfected with siRNA as negative control (siNT) or against Pfn1 (siPfn1) were subjected to the osteoclastogenesis using RANKL. (A) Multinucleated (n ≥ 3) TRAP‐positive cells (Oc.N) were counted in all wells. Six wells per experimental group were evaluated. (B) Pit formation assay was conducted on artificial mineralized matrix by RAW264.7‐derived osteoclasts. After 5 days of culture (48 hours after transfection), cells were lysed and the microscopic images were obtained (upper panels). The traced images (bottom panels) were analyzed by ImageJ. N.S. = not significant. Scale bars: 100 µm. (C) Average pit size in area was analyzed based on the traced images (C, bottom). (D) Live imaging analysis was performed for 24 hours on the osteoclastogenesis assay using siPfn1 transfected RAW264.7 cells. Representative images at 24 hours after reseeding are shown with the trails of multinucleated cells. Bottom panels are the magnified images for the boxed areas in the upper panels. (E) The average migration velocity, measured every 13 min per cell, was compared between the control and Pfn1‐KD cells. Scale bars: 100 µm. Error bars: SDs. Student's t test was applied. *p < 0.05.
Growth failure arises postneonatally in Pfn1‐cKO mice
To investigate the roles of Pfn1 in osteoclasts in vivo, conditional Pfn1‐knockout (Pfn1‐cKO) mice were generated by crossing the Pfn1 fl/fl and CatK‐Cre knock‐in mice (CatK Cre/+). When the Pfn1 fl/fl females were crossed with the CatK Cre/+; Pfn1 fl/+ males or vice versa, the prevalence of the Pfn1‐cKO mice (Pfn1 fl/fl; CatK Cre/+) was approximately 25%. This natural segregation following the Mendelian law indicated that osteoclastic Pfn1 is dispensable for mouse embryogenesis. Neonatal skeletal preparation by Alizarin Red and Alcian Blue staining indicated no apparent difference between the Pfn1‐cKO and control mice (Pfn1 fl/fl; CatK +/+ mice: hereafter, described as WT; Supplemental Fig. 3).
However, at weaning, the mutant mice tended to be smaller in size, with a steeper nose than WT mice. The plain radiograms and 3D µCT images at 4 weeks revealed deformed limb long bones (Fig. 2 A–E) and craniofacial bones (Fig. 2 J). Morphometric analysis on trabecular and cortical femurs did not indicate significant differences, except for the shape of the diaphyseal cross‐sections at this age (Fig. 2 F–I).
Figure 2.
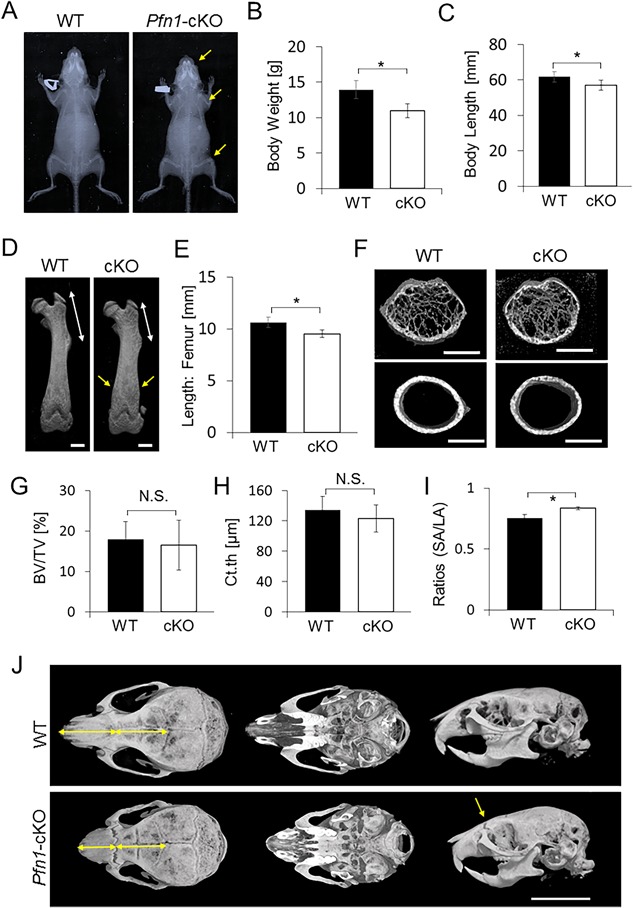
Postnatal skeletal growth is impaired in the Pfn1‐cKO mice at 4 weeks. (A) Representative plain radiograms of a male WT and the Pfn1‐cKO littermate at 4 weeks. Note that the mutant nose, the distal humerus, and the distal femurs (yellow arrows) are shorter than in WT mice. (B) Body weight was compared in WT and Pfn1‐cKO mice. (C) Body length (nose‐rump length) measured on plain radiograms was compared in WT and Pfn1‐cKO mice. (D) Representative reconstructed 3D‐µCT images of the femurs. Note that the mutant proximal femur (white arrow) is shorter than in WT mice. Note also the altered shape of the distal femur (yellow arrows). Scale bars: 1 mm. (E) Length of the femurs was measured based on the 3D reconstructed µCT images represented by panel D. (F) Representative cross‐sectional views of 3D µCT images of the diaphyseal femurs. Scale bars: 1 mm. (G–I) Trabecular bone parameters of distal femurs were analyzed based on the 3D µCT analysis. BV/TV = bone volume / tissue volume; Ct.th = cortical thickness; SA = shorter axis; LA = longer axis of cross‐sectional femoral diaphysis; N.S. = not significant. (J) Differential facial appearance in upper, bottom, and lateral views of WT and cKO skull were visualized by reconstructed 3D µCT images. Mutant nose is short and straight (yellow arrows). Scale bar: 8 mm. Error bars: SDs. The numbers of WT and Pfn1‐cKO mice were 6 and 5, respectively. Statistical analysis was conducted by Student's t test. *p < 0.05.
At 8 weeks, the skeletal phenotypes became more remarkable with shortened body and femur length (Fig. 3 A, E, H), as well as skull and long bone deformities in Pfn1‐cKO mice (Fig. 3 B–H). The shortened nose, maxilla, and distorted zygomatic arches (Fig. 3 B) could be the elements contributing to the altered facial appearance with a steep nose‐to‐head curvature on their profile (Fig. 3 B).
Figure 3.
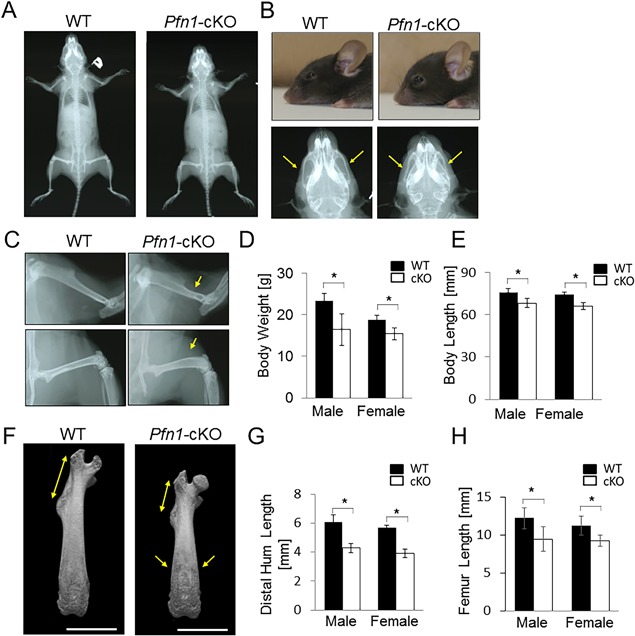
Skeletal craniofacial and limb growth is impaired in the Pfn1‐cKO mice. (A) Representative plain radiograms of a male WT and cKO littermate at 8 weeks. (B) Differential facial appearance was demonstrated by representative photo‐images of WT and cKO mice (upper panels). Magnified skull images from panel A indicate the shortened maxilla and nose as well as deformed zygomatic arches (arrows). (C) Skeletal radiograms of panel A were enlarged at femurs (bottom panels) and humerus (upper panels). Differential appearances were pointed by yellow arrows. (D) Comparison of body weight in WT and Pfn1‐cKO mice. Yellow arrows point to differential appearances. (E) Comparison of body length (Nose‐rump length) of WT and Pfn1‐cKO mice. (F) Representative 3D µCT images of the femurs. Scale bars: 4 mm. (G) The length of distal humerus from deltoid process to the end was quantified on the plain radiograms (as in panel C, upper panels). (H) Length of the femurs was measured based on the 3D reconstructed µCT images shown in panel F. Error bars: SDs. The number of samples for the WT and Pfn1‐cKO male and female were 15, 17, 12, and 14, respectively. Statistical analysis was conducted by Student's t test. *p < 0.05.
Anterior cranial and facial bones were hypoplastic in Pfn1‐cKO mice as found by 3D μCT
To analyze the skull deformities more quantitatively, we utilized the reconstructed 3D µCT images (Fig. 4 A). The quantification demonstrated a significant reduction of the midsagittal length of the nasal bone in Pfn1‐cKO mice compared with that of WT mice (Fig. 4 B; approximately 83% in male mice and 76% in female mice). The frontal bone was also slightly shorter in the male Pfn1‐cKO mice (Fig. 4 C; to approximately 91%), but temporal bones were similar. By looking at the cranial bottom‐side, the premaxilla and maxilla were remarkably hypoplastic (Fig. 4 A, E), whereas the sphenoidal and occipital bones were only moderately hypoplastic (Fig. 4 A, F, see also Supplemental Fig. 4D, E).
Figure 4.
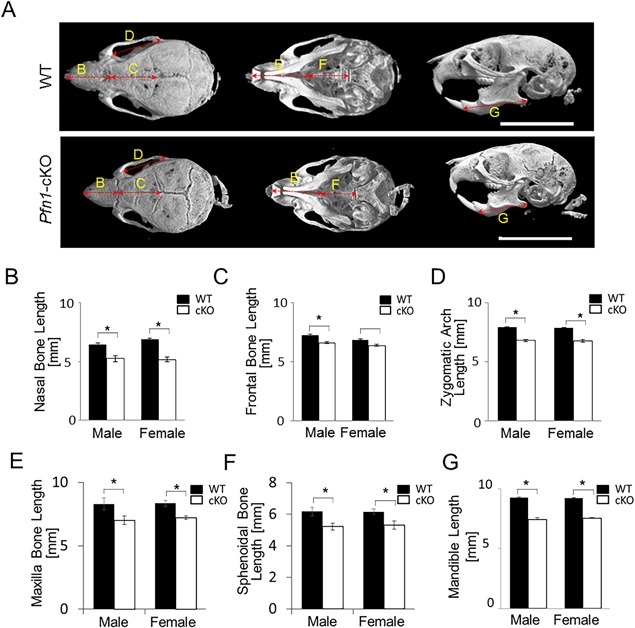
Facial bone‐ and cranial base‐hypoplasia revealed by 3D μCT analysis of the Pfn1‐cKO mouse skull. (A) Representative reconstructed 3D µCT images of the WT and cKO skull were visualized by upper (left), bottom (intermediate), and lateral projections (right). Note that the anterior skull including the maxilla and mandibula are hypoplastic in Pfn1‐cKO mice. The cranial base was hypoplastic, and the zygomatic arches were deformed in Pfn1‐cKO mice. Scale bars: 10 mm. (B–E) Length of major anatomical units was quantified three dimensionally, for the nasal bone length (B), frontal bone length (C), zygomatic arch length (D), maxilla length (E), sphenoidal bone length (F), and mandible length (G). The landmarks utilized for the measurements are indicated in Supplemental Fig. 2A, B, F, H. Error bars: SDs. Three randomly selected samples were utilized for the measurements in each group. Statistical analysis was conducted by Student's t test. *p < 0.05.
Interestingly, despite the longitudinal shortening of the frontal bones, the lateral width was significantly broader in the Pfn1‐cKO mice (Supplemental Fig. 4A, B; approximately 108% in male mice and 104% in female mice). Similarly, the zygomatic arches were shortened and broadened (Fig. 4 D and Supplemental Fig. 4C). We assumed the findings could indicate a mechanical weakness and imbalance among the cranial bones. This assumption was also consistent with the findings on lateral view of the reconstructed skull images: The calvarial bone curvature was flattened anteriorly and convex posteriorly, the facial bones, including the mandibula, were smaller in Pfn1‐cKO mice (Fig. 4 G and Supplemental Fig. 4F, G).
Long bones in extremities represent the Erlenmeyer‐flask deformity with osteolytic appearance
Because the site‐specific impairment of the cranial bone growth tended to be limited in the structures developmentally requiring chondrogenesis, we tested if there was a failure in cartilaginous bone growth in Pfn1‐cKO mice. However, despite the shortening of the entire bone (Figs. 2 and 3), no obvious change was detected in femoral and tibial growth‐plate chondrocytes and columnar structures in histological sections at 4 weeks (Supplemental Fig. 5) and 8 weeks (Fig. 5 A, B). Instead, we noticed that the curves of the growth plate tended to be flattened in the Pfn1‐cKO mice (this was confirmed by measurement; data not shown), possibly because of the expansion of bone marrow space. In parallel, they showed expanded distal femur metaphysis representing the Erlenmeyer‐flask deformity at complete penetration at each stage, with the findings of trabecular bone loss and expanded bone marrow cavity (Figs. 2 D–I and 5). These observations suggested that the Pfn1‐cKO skeletal deformity may be causally related to the increased resorption of the bone nearby growing cartilaginous structures. Indeed, the Erlenmeyer‐flask deformity is usually associated with osteolytic conditions in children, though osteopetrotic conditions would also contribute to the changes.(26) Thus, we analyzed if bone structures of Pfn1‐cKO mice at distal and midshaft femurs were consistent with this notion, by using 2D projection and 3D reconstruction of µCT images (Fig. 5 C, D). The altered cross‐sectional contour of the metaphysis (Fig. 5 C) and diaphyseal cortex (Fig. 5 D) were evident, where the Pfn1‐cKO femurs represented a circular shape, in contrast with the elliptical shape in WT femurs (Fig. 5 D). Significant differences were detected in the ratios of the SA versus LA length (Fig. 5 H; 0.79 ± 0.015 versus 0.69 ± 0.027 in males, 0.80 ± 0.026 versus 0.69 ± 0.025 in females). In addition, the trabecular bone parameters indicated the osteopenic conditions by lower BV/TV and Tb.th (Fig. 5 C, E, F). The paradoxical stability of the Tb.N, Tb.Sp, and Tb.Spac (Supplemental Table 1) could be because of the expanded trabecular bone area in the mutant bones as indicated by cross‐sectional images (Fig. 5 C). The cortical bone thickness at the diaphysis was also significantly smaller in Pfn1‐cKO femurs than in WT femurs (Fig. 5 G). Bone‐forming activity of the osteoblast seemed not to be so affected because the histomorphometric bone formation parameters in preliminary samples did not show any clear differences (data not shown). Therefore, the long‐bone deformity observed in the Pfn1‐cKO mice must be based on the osteolytic conditions in the metaphysis and diaphysis.
Figure 5.
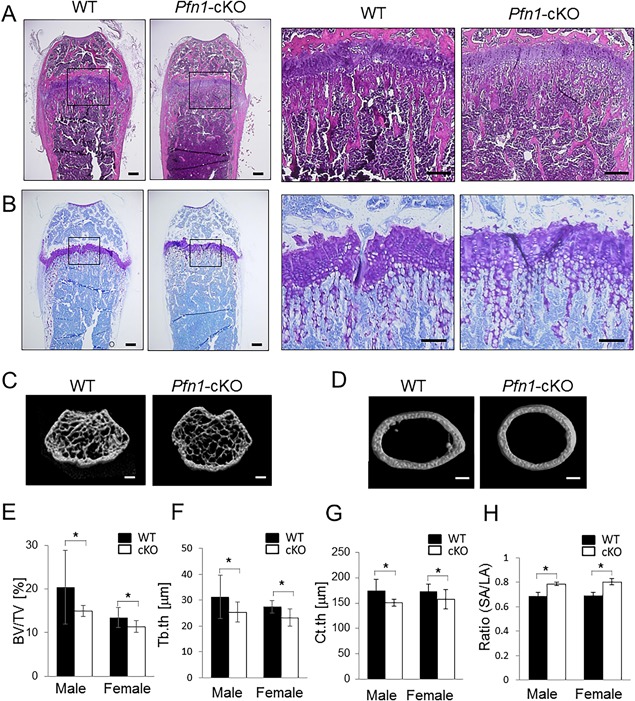
Long bones are deformed with osteolytic expansion. (A) Frontal section of the distal femurs at 8 weeks of age was compared using hematoxylin and eosin staining. Despite the differences in the bone shape, no significant difference was detected in the growth plate. Right panels represent magnified views of boxed areas in left panels. (B) Serial section was stained with toluidine blue. No obvious difference in columnar structure of the growth plate chondrocytes was detected (see also images of younger mice in Supplemental Fig. 4). (C–H) 3D µCT analysis was performed on WT and cKO mouse femurs. Representative images for distal femur trabeculae (C) and cortical bone structure at midshaft (D) are shown. Trabecular bone volume at distal femur (C) was quantified by bone volume/tissue volume (BV/TV; %) (E) and trabecular thickness (Tb.th; F). Cortical thickness (Ct.th) (G) was analyzed at midshaft. The ratio of shorter axis versus longer axis of the cross‐sectional cortex was compared (H). Scale bars: 200 μm (A,B), and 1 mm (C,D). Error bars: SDs. Statistical analysis was conducted by Student's t test. *p < 0.05.
We then examined if the osteoclastic bone resorption was indeed increased in the histological sections of Pfn1‐cKO mice (Fig. 6 A). TRAP staining indicated that the number of multinucleated osteoclasts was increased at the primary and adjacent secondary trabecular surface of the Pfn1‐cKO femurs (Fig. 6 B, D). More importantly, prominent numbers of multinucleated TRAP‐positive cells were detected in Pfn1‐cKO mice at the endosteal regions of the metaphysis to the diaphysis, whereas the osteoclast distribution is usually rare in WT mice (Fig. 6 C, E). In contrast, a few number of periosteal osteoclasts found in the WT metaphysis was absent in the Pfn1‐cKO femurs, possibly indicating the compensational protection against periosteal remodeling. We assumed such an imbalance led to the expanded metaphyseal deformity in the Pfn1‐cKO mice.
Figure 6.
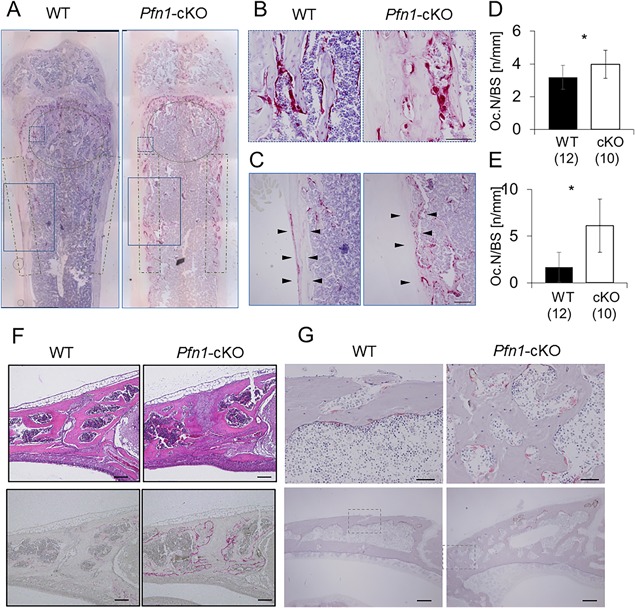
TRAP‐positive cells are increased at osteolytic expansion in limb long bones and cranial sutures. (A) Representative frontal sections of the distal femur were stained with TRAP activity. The areas circled or boxed with dot‐and‐dashed lines were subjected to the analysis of the osteoclast number per bone surface (Oc.N/BS; Fig. 6 D, E). Solid boxes and dashed boxes were magnified in 6B, C. (B) Magnified images of TRAP‐positive osteoclasts detected in dashed boxes in 6A (metaphyseal secondary trabeculae). (C) Magnified images of TRAP‐positive osteoclasts detected in solid boxes in 6A (endosteal bone surface). (D) The Oc.N/BS was counted at the primary and secondary spongiosa under the growth plate as indicated by solid boxes in panel A. (E) The Oc.N/BS was counted at the endosteal surface of the metaphysis as indicated by dot‐and‐dashed boxes in panel A. (F) Frontonasal suture of the WT and cKO mice was analyzed by sagittal histological sections stained with hematoxylin and eosin (upper panels) and TRAP staining (bottom panels). See also the µCT images (Figs. 2 J and 4A) for suture expansion. (G) TRAP‐positive osteoclasts were also visualized on GMA‐embedded sagittal sections. Upper panels are the magnified views of the boxed area in the bottom panels. Scale bars: 100 μm in B and C, 200 μm in F and G bottom, and 50 μm in G top. Error bars: SDs. Statistical analysis was conducted by Student's t test. *p < 0.05.
The osteolytic changes were also detected by skull µCT, especially the frontal and nasal bones with opened suture (Fig. 6 F). In histology, the suture in mutant mice was filled with thick fibrocartilaginous tissues, whereas the suture in wild‐type mice was almost closed. Correspondingly, the internal surface of the adjacent bones was strongly stained with residual TRAP activity when overstained (Fig. 6 F; bottom panel), and the multinucleated TRAP‐positive cells were increased (Fig. 6 G; plastic section). We assumed the excessive bone resorption may have competed against the growth of ossifying fibrous suture in this region.
Pfn1‐deficiency enhances osteoclast migration with slightly increased size
The increased osteoclast number in the metaphyseal endosteum of Pfn1‐cKO femurs may suggest an increased differentiation potential of preosteoclasts in bone marrow. However, the osteoclastogenesis assay using Pfn1‐cKO and WT bone marrow cells indicated that the number of TRAP‐positive osteoclasts was identical in Pfn1‐cKO and WT cultures (Fig. 7 A, B). Instead, we noticed the mutant osteoclasts were larger. A size‐distribution analysis of multinucleated osteoclasts using the Mann‐Whitney test indicated that the osteoclast size, in addition to the total podosome‐belt length, was significantly larger in Pfn1‐deficient cells (Fig. 7 G–I). The slightly increased osteoclast size may be related to the increased preosteoclast movement increasing the chance of fusion. To verify the increased movement of the Pfn1‐cKO osteoclasts, the locomotion of bone‐marrow‐derived Pfn1‐cKO osteoclasts was analyzed by live imaging (Fig. 7 C). Although the number of multinucleated cells was comparable between the WT and Pfn1‐cKO cells, the locomotive trails of the osteoclasts were significantly increased in the Pfn1‐cKO cells under this condition (Fig. 8 and Supplemental Movie 3). Pit formation assay indicated the increased matrix resorption by Pfn1‐cKO cells (Fig. 7 D–F). Therefore, these results show, for the first time, that Pfn1 has an essential role in the maintenance of osteoclast movement, and hence for proper skeletal development and homeostasis.
Figure 7.
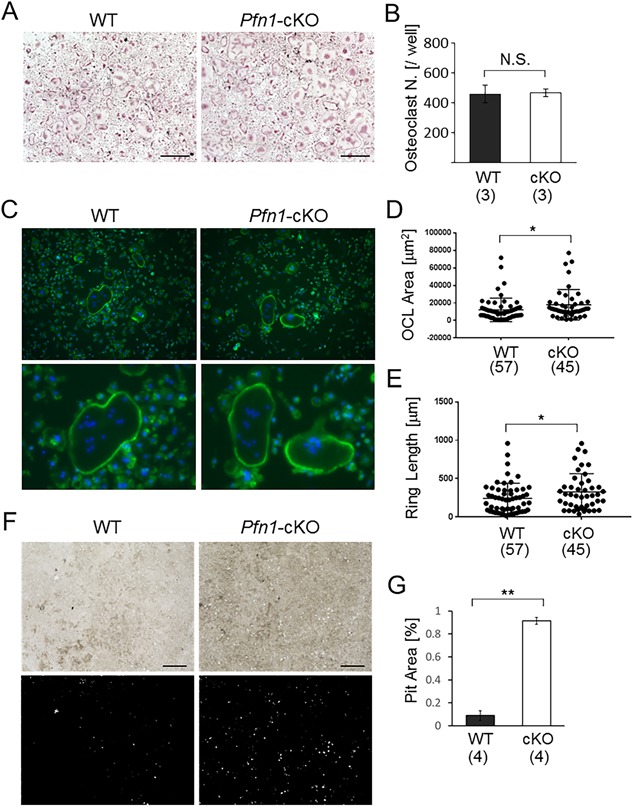
In vitro migration and maturation are enhanced in Pfn1‐deficient preosteoclasts. (A) The osteoclastogenesis of WT and Pfn1‐cKO bone marrow cells was evaluated by TRAP staining. Scale bar: 500 µm. (B) Multinucleated TRAP‐positive cells (Oc.N) were counted in all wells. Three wells per experimental group were evaluated in this experiment. Identical results were confirmed by experiments repeated more than 3 times. N.S. = not significant. (C) Representative Phalloidin staining of the osteoclasts. Magnified images of the osteoclasts are shown in the bottom panels. Scale bar: 100 µm. (D) Size of the cultured osteoclasts (OCL area; D) was quantified in the images represented by panel C. (E) Total podosome‐belt/ring length (ring length) was quantified similarly. (F) Pit formation assay was conducted using bone marrow cells derived from WT and Pfn1‐cKO mice. After 5 days of osteoclastogenesis on the plate, cells were lysed and the microscopic images were taken (upper panels). The images were analyzed on Image J software (bottom panels). (G) Entire pit area represented by panel F was quantified per well (24‐well plate) and compared in WT and Pfn1‐cKO cells. Error bars: SDs. Osteoclast number (B) and pit area (G) were statistically analyzed by Student's t test. Other parameters (D and E) were analyzed by Mann‐Whitney test. *p < 0.05, **p < 0.01.
Figure 8.
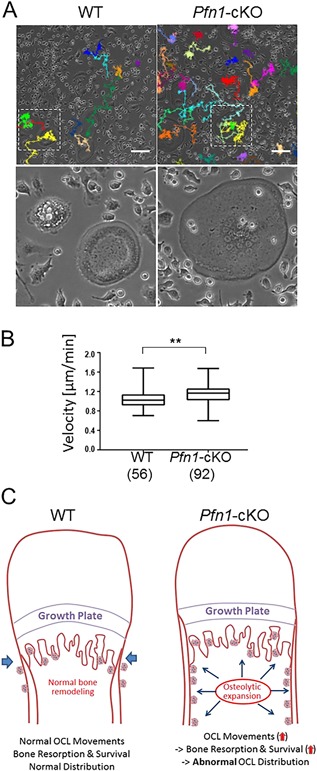
Pfn1‐cKO osteoclasts migrate faster than WT osteoclasts and may contribute to expanded bone resorption in vivo. (A) Live osteoclasts induced from WT and Pfn1‐cKO bone marrow cells were imaged and the trails of the multinucleated (n ≥ 3) cells were analyzed by ImageJ (upper panels). Bottom panels indicate the magnified views of the boxed area. (B) The average of migration velocities calculated at every 11 min per cell was compared in WT (n = 56) and Pfn1‐cKO cells (n = 92). **p < 0.01 by Student's t test. (C) Diagram representing the hypothetical mechanism of the Erlenmeyer‐flask deformity in Pfn1‐cKO mice.
Discussion
This study demonstrated that Pfn1 functions negatively in osteoclast movement and bone‐resorbing activity. A novel aspect of Pfn1 function was also demonstrated by postnatal growth failure in craniofacial and limb bones associated with an osteolytic expansion in osteoclast‐specific Pfn1‐deficient mice. Of note, the long bone deformity at metaphysis resembled the Erlenmeyer‐flask deformity that is known to be associated with several human disorders that result from dysregulated bone turnover during the growth phase.26 Although the exact molecular events altering the Pfn1‐deficient osteoclasts were not thoroughly defined in the current study, our study clearly indicates that the enhanced osteoclast movement based on altered cytoskeletal regulation resulted in an osteolytic Erlenmeyer‐flask deformity. Our findings provide a novel understanding of osteoclast function and related bone deformities, including the Erlenmeyer‐flask deformity.
The increased osteoclast movement observed in our Pfn1‐cKO mice is consistent with a recent theory that Pfn1 functions negatively in the assembly of branched actin networks required for the formation of lamellipodia and podosomes.11, 12, 13 Our findings are also consistent with previous reports suggesting a suppressive role for profilins on lamellipodia and invadopodia in cancer cells, contributing to their invasive features.14, 15, 27 The molecular mechanisms of how the profilins work in both enhancing and inhibiting cell movement still remain unclear.11, 12, 13 The classical understanding of the profilins is that profilins support the formin‐bound barbed‐end elongation of the actin filaments by efficiently adding the ATP‐bound actin monomers.28, 29 This molecular feature assists elongation of the nonbranched filopodial filaments contributing to mesenchymal cell movement. Mesenchymal cell movement is basically supported by Pfn1 in most situations. However, the different processes, namely the lamellipodia and podosomes, are structured mainly with branched filamentous networks.11, 12, 13, 30, 31 For these structures, the elongation relies on different molecules, including Ena/Vasp and Arp2/3, which have been shown to be inhibited by Pfn1.11, 12, 13 Thus, our observation provides another example of this inhibition by Pfn1. Osteoclast movements have been suggested to rely on this suppression by Pfn1. Interestingly, a recent study has found that cofilin, an actin‐binding protein that interacts with cortactin, is required for osteoclast podosome patterning.32 Pfn1 has been reported to be a binding factor for cortactin, which mediates smooth muscle contraction.33 Hence, it would be interesting to investigate if Pfn1 also exerts its interactive functions in the podosomes of the osteoclasts.
A study of how increased osteoclast movement could enhance osteolysis at the endosteal metaphysis and diaphysis in Pfn1‐cKO mice would also be of worth. In our cell culture experiments, the number of osteoclasts was not increased in contrast to the increased osteoclasts detected in vivo. In addition, only a slight increment was found in the size of the osteoclasts derived from RAW264.7 cells and bone marrow cells, which could be associated with increased migration theoretically by increasing the chance to fuse. Presumably, such an effect could be masked in culture conditions because their chance to meet the right fusion partners might be enhanced in culture conditions as compared with in vivo conditions. In another possibility, the locomotive function increased by the absence of Pfn1 in osteoclasts may have made it possible to travel from the initial site of osteoclastogenesis to distant areas that might have promoted a longer life span (Fig. 8 C). A recent live‐cell imaging study showed that moving osteoclasts are more potent in resorbing bone matrix,34 and may survive longer than resting cells. Thus, our observations could be explained by the increased endosteal bone resorption based on longer survival of more‐potent osteoclasts (Fig. 8 C). Accelerated endosteal osteolysis can be the cause of an expanded thinner bone collar. Periosteal expansion, which contributes to the characteristic Erlenmeyer‐flask deformity, could be explained by periosteal osteoblast compensation against mechanical impairment caused by endosteal bone resorption: The increased periosteal bone formation may have masked the periosteal osteoclasts in Pfn1‐cKO femurs that are usually found in WT femurs. Increased endosteal bone resorption would also explain the abnormal lateral broadening and impaired longitudinal growth of the facial and cranial bones in Pfn1‐cKO mice (Figs. 3 I, 4A, and 6D–F).
The Erlenmeyer‐flask deformity, a term describing a specific abnormality of the distal femur, is a clinical sign of the dysregulated long‐bone remodeling during longitudinal growth.26 Although various skeletal disorders, including osteopetrosis, could be associated with this abnormality, its predominant cause is increased bone resorption.26 The typical group of associated syndromes includes osteopenic disorders such as Engelmann disease and Melnick‐Needles syndrome; the unclassified group includes Gaucher disease and Nasu‐Hakola disease, whose characteristics include bone marrow expansion and infiltration. Thus, the phenotypic feature of the Pfn1‐cKO mice could be regarded as typical Erlenmeyer‐flask deformity based on the increased bone resorption at metaphysis.26 A recent study has found that macrophages utilize the MT1‐MMP for the turnover of podosomes as well as invadopodias.35 The Mt1‐mmp KO mice,36 like Mmp2‐deficient mice,37 showed osteolytic skeletal deformities resembling our Pfn1‐cKO mice. Because these mutant mice recapitulate the human disorder, multicentric osteolysis nodulosis arthropathy (MONA),38, 39 it might be of interest to investigate if the increased migration of MT1‐MMP‐deficient osteoclasts contributes to the osteolytic phenotype. Our Pfn1‐cKO mice could be used to investigate the pathogenesis of these related disorders. Our results also suggest the importance of the osteoclastic cytoskeleton in the disorders associated with Erlenmeyer‐flask deformity.
Lastly, the causative mechanisms of the short stature of the Pfn1‐cKO mice were not defined in the present study. Despite an apparent growth failure in proximal limb bones and the facial bones, the only detectable histological findings were the expanded distribution of the osteoclasts. In a previous study, Yang et al reported that the CatK‐Cre knock‐in mice that we utilized hereto delete the Pfn1‐flox allele in osteoclasts had aberrant periosteal Cre expression in the chondrogenic progenitor cells, specifically at the groove of Ranvier.40 However, despite displaying altered growth plate curvature, the chondrocytes in this region did not show any alteration in the columnar alignment that we observed histologically in chondrocyte‐specific Pfn1‐cKO mice in a previous study.7 The possible involvement of osteoblastic precursor cells and osteocytes in this region was not thoroughly investigated, but the absence of alterations in growth plate chondrocytes in dwarf mice is not surprising because this is often the case in the dysplastic mutant mice and human disorders associated with impaired bone turnover and short stature.41, 42 Short stature in osteopetrosis patients is a recurrent symptom, with no apparent pathological changes in growth plates. Similarly, most patients with osteodysplastic disorders—especially associated with Erlenmeyer‐flask deformity—are short in stature with no specific growth‐plate abnormalities. Melnick‐Needles syndrome is caused by the recurrent point mutations clustered at exon 22 of the filamin A gene (FLNA), one of the actin filament‐binding proteins.43 Therefore, whether the alteration of cytoskeletal protein functions in osteoclasts leads to growth failure in various mutant alleles in the conditional gene KO mice would be an interesting line of investigation.
In summary, we investigated the osteoclast‐specific functions of Pfn1 in mice and in cultured osteoclasts, and revealed its inhibitory function on osteoclast movement. The increased migration of osteoclasts because of Pfn1 deficiency resulted in an expansion of endosteal bone resorption at the metaphysis and diaphysis, leading to Erlenmeyer‐flask deformity. Our study explored a novel aspect of the molecular mechanisms that contribute to the pathogenesis of various skeletal disorders, which may lead to a better understanding and treatment of such disorders.
Disclosures
All authors declare that they have no conflict of interest.
Supporting information
Supporting Data S1.
Supporting Movie S1.
Supporting Movie S2.
Supporting Movie S3.
Acknowledgments
This study was supported by grants‐in‐aid from the Ministry of Education, Culture, Sports, Science and Technology (KAKENHI: 26253085). We thank Dr. Reinhard Fässler for his support of this research. We also thank Drs. Yuu Taguchi and Noriko Tokai at The Institute of Medical Science, The University of Tokyo, for their kind help with the osteoclastogenesis assays. Authors’ roles: MN, YE, and TH designed the study. RTB and MC provided the mutant mouse line. JS, SK, YI, and TH collected the data. TH, YE, and MN analyzed the data. JS, YE, and MN wrote the paper.
Jumpei Shirakawa and Shuhei Kajikawa contributed equally to the work.
Yoichi Ezura and Masaki Noda are co‐corresponding authors.
References
- 1. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Föger‐Samwald U, Ellinger I. Immunology of osteoporosis: a mini‐review. Gerontology. 2016;62(2):128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sims NA, Martin TJ. Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep. 2014;3:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aryal SAC, Miyai K, Hayata T, et al. Nck1 deficiency accelerates unloading‐induced bone loss. J Cell Physiol. 2013;228(7):1397–403. [DOI] [PubMed] [Google Scholar]
- 4. Aryal SAC, Miyai K, Izu Y, et al. Nck influences preosteoblastic/osteoblastic migration and bone mass. Proc Natl Acad Sci U S A. 2015;112(50):15432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyajima D, Hayata T, Suzuki T, et al. Profilin1 regulates sternum development and endochondral bone formation. J Biol Chem. 2012;287(40):33545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin W, Izu Y, Smriti A, et al. Profilin1 is expressed in osteocytes and regulates cell shape and migration. J Cell Physiol. 2018; 233 (1): 259–68. [DOI] [PubMed] [Google Scholar]
- 7. Böttcher RT, Wiesner S, Braun A, et al. Profilin 1 is required for abscission during late cytokinesis of chondrocytes. EMBO J. 2009;28:1157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding Z, Bae YH, Roy P. Molecular insights on context‐specific role of profilin‐1 in cell migration. Cell Adh Migr. 2012;6(5):442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Valenzuela‐Iglesias A, Sharma VP, Beaty BT, et al. Profilin1 regulates invadopodium maturation in human breast cancer cells. Eur J Cell Biol. 2015;94(2):78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henty‐Ridilla JL, Goode BL. Global resource distribution: allocation of actin building blocks by profilin. Dev Cell. 2015;32(1):5–6. [DOI] [PubMed] [Google Scholar]
- 11. Rotty JD, Wu C, Haynes EM, et al. Profilin‐1 serves as a gatekeeper for actin assembly by Arp2/3‐dependent and ‐independent pathways. Dev Cell. 2015;32(1):54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suarez C, Carroll RT, Burke TA, et al. Profilin regulates F‐actin network homeostasis by favoring formin over Arp2/3 complex. Dev Cell. 2015;32(1):43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schoppmeyer R, Zhao R, Cheng H, et al. Human profilin 1 is a negative regulator of CTL mediated cell‐killing and migration. Eur J Immunol. 2017;47(9):1562–72. [DOI] [PubMed] [Google Scholar]
- 14. Lakkakorpi PT, Wesolowski G, Zimolo Z, Rodan GA, Rodan SB. Phosphatidylinositol 3‐kinase association with the osteoclast cytoskeleton, and its involvement in osteoclast attachment and spreading. Exp Cell Res. 1997;237:296–306. [DOI] [PubMed] [Google Scholar]
- 15. Lakkakorpi PT, Väänänen HK. Kinetics of the osteoclast cytoskeleton during the resorption cycle in vitro. J Bone Miner Res. 1991;6(8):817–26. [DOI] [PubMed] [Google Scholar]
- 16. Georgess D, Machuca‐Gayer I, Blangy A, Jurdic P. Podosome organization drives osteoclast‐mediated bone resorption. Cell Adh Migr. 2014;8:192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–23. [DOI] [PubMed] [Google Scholar]
- 18. Mizoguchi F, Izu Y, Hayata T, et al. Osteoclast‐specific Dicer gene deficiency suppresses osteoclastic bone resorption. J Cell Biochem. 2010;109(5):866–75. [DOI] [PubMed] [Google Scholar]
- 19. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res. 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 20. Hayata T, Ezura Y, Asashima M, Nishinakamura R, Noda M. Dullard/Ctdnep1 regulates endochondral ossification via suppression of TGF‐β signaling. J Bone Miner Res. 2015;30(2):318–29. [DOI] [PubMed] [Google Scholar]
- 21. Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28(1):2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe C, Morita M, Hayata T, et al. Stability of mRNA influences osteoporotic bone mass via CNOT3. Proc Natl Acad Sci U S A. 2014;111(7):2692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kajikawa S, Taguchi Y, Hayata T, et al. Dok‐3 and Dok‐1/‐2 adaptors play distinctive roles in cell fusion and proliferation during osteoclastogenesis and cooperatively protect mice from osteopenia. Biochem Biophys Res Commun. 2018;498(4):967–74. [DOI] [PubMed] [Google Scholar]
- 24. Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–88. [DOI] [PubMed] [Google Scholar]
- 25. Lee J, Park C, Kim HJ, et al. Stimulation of osteoclast migration and bone resorption by C‐C chemokine ligands 19 and 21. Exp Mol Med. 2017;49:e358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faden MA, Krakow D, Ezgu F, Rimoin DL, Lachman RS. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Genet A. 2009;149A(6):1334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bae YH, Ding Z, Das T, Wells A, Gertler F, Roy P. Profilin1 regulates PI(3, 4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA‐MB‐231 cells. Proc Natl Acad Sci U S A. 2010;107(50):21547–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14(8):461–9. [DOI] [PubMed] [Google Scholar]
- 29. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9(6):446–54. [DOI] [PubMed] [Google Scholar]
- 30. Krause M, Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat Rev Mol Cell Biol. 2014;15(9):577–90. [DOI] [PubMed] [Google Scholar]
- 31. Devreotes P, Horwitz AR. Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol. 2015;7(8):a005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang R, Cleary RA, Wang T, Li J, Tang DD. The association of cortactin with profilin‐1 is critical for smooth muscle contraction. J Biol Chem. 2014;289(20):14157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zalli D, Neff L, Nagano K, et al. The actin‐binding protein cofilin and its interaction with cortactin are required for podosome patterning in osteoclasts and bone resorption in vivo and in vitro. J Bone Miner Res. 2016;31(9):1701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Merrild DM, Pirapaharan DC, Andreasen CM, et al. Pit‐ and trench‐forming osteoclasts: a distinction that matters. Bone Res. 2015;3:15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. El Azzouzi K, Wiesner C, Linder S. Metalloproteinase MT1‐MMP islets act as memory devices for podosome reemergence. J Cell Biol. 2016;213(1):109–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holmbeck K, Bianco P, Caterina J, et al. MT1‐MMP‐deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99(1):81–92. [DOI] [PubMed] [Google Scholar]
- 37. Inoue K, Mikuni‐Takagaki Y, Oikawa K, et al. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281(44):33814–24. [DOI] [PubMed] [Google Scholar]
- 38. Martignetti JA, Aqeel AA, Sewairi WA, et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28(3):261–5. [DOI] [PubMed] [Google Scholar]
- 39. Evans BR, Mosig RA, Lobl M, et al. Mutation of membrane type‐1 metalloproteinase, MT1‐MMP, causes the multicentric osteolysis and arthritis disease Winchester syndrome. Am J Hum Genet. 2012;91(3):572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang W, Wang J, Moore DC, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature. 2013;499:491–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geister AK, Camper SA. Advances in skeletal dysplasia genetics. Annu Rev Genomics Hum Genet 2015;16:199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Canalis E, Zanotti S. Hajdu‐Cheney syndrome: a review. Orphanet J Rare Dis. 2014;9:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robertson SP. Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick‐Needles syndrome. Eur J Hum Genet. 2007;15(1):3–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Data S1.
Supporting Movie S1.
Supporting Movie S2.
Supporting Movie S3.