

Abstract
Self-assembly of the microtubule-associated protein tau into neurotoxic oligomers, fibrils, and paired helical filaments, and cell-to-cell spreading of these pathological tau species are critical processes underlying the pathogenesis of Alzheimer’s disease and other tauopathies. Modulating the self-assembly process and inhibiting formation and spreading of such toxic species are promising strategies for therapy development. A challenge in investigating tau self-assembly in vitro is that, unlike most amyloidogenic proteins, tau does not aggregate in the absence of posttranslational modifications (PTM), aggregation inducers, or preformed seeds. The most common induction method is addition of polyanions, such as heparin; yet, this artificial system may not represent adequately tau self-assembly in vivo, which is driven by aberrant phosphorylation and other PTMs, potentially leading to in vitro data that do not reflect the behavior of tau and its interaction with modulators in vivo. To tackle these challenges, methods for in vitro phosphorylation of tau to produce aggregation-competent forms recently have been introduced (Despres et al. (2017) Proc. Natl. Acad. Sci. U.S.A., 114, 9080−9085). However, the oligomerization, seeding, and interaction with assembly modulators of the different forms of tau have not been studied to date. To address these knowledge gaps, we compared here side-by-side the self-assembly and seeding activity of heparin-induced tau with two forms of in vitro phosphorylated tau and tested how the molecular tweezer CLR01, a negatively charged compound, affected these processes. Tau was phosphorylated by incubation either with activated extracellular signal-regulated kinase 2 or with a whole rat brain extract. Seeding activity was measured using a fluorescence-resonance energy transfer-based biosensor-cell method. We also used solution-state NMR to investigate the binding sites of CLR01 on tau and how they were impacted by phosphorylation. Our systematic structure–activity relationship study demonstrates that heparin-induced tau behaves differently from in vitro phosphorylated tau. The aggregation rates of the different forms are distinct as is the intracellular localization of the induced aggregates, which resemble brain-derived tau strains suggesting that heparin-induced tau and in vitro phosphorylated tau have different conformations, properties, and activities. CLR01 inhibits aggregation and seeding of both heparin-induced and in vitro phosphorylated tau dose-dependently, although heparin induction interferes with the interaction between CLR01 and tau.
As one of the major molecular actors in Alzheimer’s disease (AD) and other tauopathies, including frontotemporal dementia, progressive supranuclear palsy, corticobasal syndrome, Pick’s disease, and chronic traumatic encephalopathy, the microtubule-associated protein tau has been investigated in the last few decades as an attractive, yet challenging, therapeutic target.1−3 In humans, tau exists as six alternatively spliced isoforms, which normally are bound to microtubules and are thought to stabilize the microtubule structure. In tauopathies, tau detaches from the microtubules and self-assembles into neurotoxic oligomers and fibrillar aggregates, which are found in the brain as neurofibrillary tangles (NFTs) and other types of deposits.4
NFTs, one of the two characteristic proteinaceous lesions of AD,4 faithfully correlate with clinical progression of the disease, in contrast to the other lesion, amyloid plaques, which are poorly associated with cognitive decline.5−7 Tau pathology propagates through the brain in a reproducible temporal and regional manner.8,9 In fact, immunostaining using monoclonal antibody (mAb) AT8, which binds specifically to hyperphosphorylated tau, is used for AD staging.10 Nonetheless, as in other proteinopathies, metastable tau oligomers have been suggested to be a major neurotoxic agent and possibly the main culprit responsible for impairing and eventually killing susceptible neurons.11
The main components of NFTs in AD are tau paired helical filaments (PHF) consisting of two helically twisted ribbons of 10–20 nm width with an 80 nm periodicity. In these insoluble structures, tau transforms from a soluble, natively disordered conformation into cross-β structures, typical of amyloid, which encompass primarily its microtubule-binding domain (MTBD, residues 244–372).12−15 A recent study using cryoelectron microscopy on PHFs and straight filaments (SF) isolated from patients with AD has defined the structure of tau fibrils and highlighted a distinct structural arrangement in PHF and SF.16 In AD brain fibrils, tau protofilaments adopt a cross-β/β-helix structure encompassing microtubule-binding repeats R3 and R4. The core of the fibrils is flanked by a “fuzzy coat” comprising the unfolded N- and C-terminal domains.17,18
In AD, all six isoforms of tau are found abnormally hyperphosphorylated in PHFs.19−22 The majority of the abnormal phosphorylation sites are distributed over ∼40 positions clustered in the proline-rich region (PRR) and C-terminal domains flanking the MTBD.22−24 Hyperphosphorylation of tau in these regions is thought to cause its pathologic aggregation,25−27 though the precise mechanism of this process remains elusive. Interestingly, certain phosphorylation sites, e.g., S262 in the MTBD and S214 in the PRR, have been found to inhibit aggregation.28−30
Many studies have characterized the self-assembly process of tau in vitro. However, because of the practical difficulty of obtaining purified hyperphosphorylated or otherwise post-translationally modified tau, in vitro aggregation of tau typically is studied using tau forms devoid of posttranslational modifications (PTMs), which do not aggregate spontaneously. Fragments of tau containing the MTBD or parts thereof are aggregation-prone, and their fibrils may serve as a template to promote the aggregation of full-length tau;15,31−35 yet, this strategy does not alleviate the concern about the lack of pathologically relevant PTMs. More commonly, in vitro aggregation assays use external inducers, including polyanions, such as heparin,36,37 RNA,38 and fatty acids, such as arachidonic acid,39,40 or small anionic molecules41 that stimulate the aggregation process through mechanisms that are not fully understood.42−45 Although external aggregation inducers may lead to formation of tau fibrils that can be morphologically similar to PHFs,18,36 the aggregation process and structural details of these fibrils may be distinct from those of disease-relevant tau aggregates.16,46 Along these lines, recently, tau fibrils formed in vitro by addition of heparin or RNA were found to contain substantial amounts of the inducers and to disaggregate upon treatment with heparinase or RNase, respectively.47 These findings are of particular concern in view of recent data demonstrating that conformationally distinct tau “strains” display substantially different biological and pathological behavior48,49 because they suggest that tau induced to aggregate by polyanions may not recapitulate adequately the structure of tau found in patient brains. In addition, the use of artificial aggregation inducers complicates the interpretation of experiments testing assembly modulators and inhibitors as potential therapeutic agents for tauopathies. An alternative approach is to produce in vitro phosphorylated tau for studies of aggregation, structure, pathological effects, and modulation of all of these processes. However, to date, only a few studies have used this strategy, and their focus was on the fibril formation and morphology.30,50,51 To our knowledge, tau oligomerization, the seeding activity of tau, and the inhibition of aggregation or seeding have not been studied using in vitro phosphorylated recombinant tau to date.
As a test modulator, we chose in this study the molecular tweezer CLR01. Molecular tweezers are broad-spectrum modulators of the aberrant self-assembly process of amyloid proteins.52,53 They act by moderate, labile binding to Lys, and to a lower extent to Arg residues through inclusion of the side chain in the cavity of the molecular tweezer, which leads to temporary masking of hydrophobic interactions in which these side chains participate and reversal of the positive charge of their end groups to a negative charge. This causes major perturbation of interactions involving Lys (or Arg), which play an important role in the abnormal self-assembly of amyloidogenic proteins. A lead molecular tweezer called CLR01, substituted by two negatively charged phosphate groups at the central benzene spacer-unit (Figure 4A), has been shown to modulate the oligomerization and aggregation of over a dozen disease-associated amyloidogenic proteins and to be an effective inhibitor of the toxicity of these proteins in cell culture and animal models of the associated diseases.52,53 However, relatively little is known about the interaction of molecular tweezers with tau.
Figure 4.
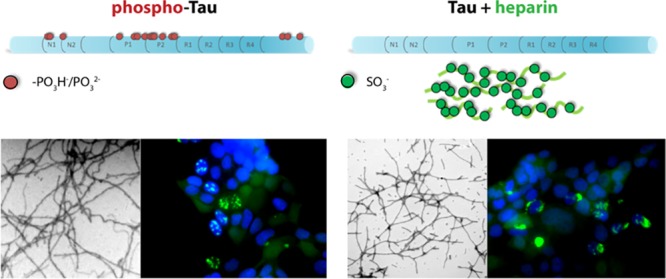
Inhibition of tau binding to heparin by CLR01. Binding of tau to immobilized heparin and its inhibition by CLR01 were measured using SPR. (A) A schematic structure of CLR01. The compound is partially protonated at physiologic pH. (B) SPR sensorgram of tau binding to heparin immobilized on a SPR chip. (C) Normalized tau binding in the presence of increasing concentrations of CLR01. (D) % Inhibition versus CLR01 concentration used for calculating the half-maximal inhibition (IC50).
Here, we conducted a systematic investigation of tau oligomerization, aggregation, and seeding, comparing heparin-induced tau with two forms of in vitro phosphorylated tau. We also tested how CLR01 interacted with each tau form and characterized the binding sites of CLR01 on tau. Our data suggest that the oligomerization and aggregation kinetics of heparin-induced tau are different from those of in vitro phosphorylated tau and that these different forms lead to different tau strains that can be detected in biosensor cells, as described previously for brain-derived tau aggregates supporting the notion that these forms of tau fibrils are structurally distinct.54 CLR01 interacts preferentially with Lys residues located in the MTBD and flanking regions and phosphorylation of tau has little effect on this interaction. CLR01 modulates the aggregation and seeding of the different forms of tau; yet, in most experiments, the dose–response relationship of CLR01 with heparin-induced tau and in vitro phosphorylated tau is different, supporting the notion that these forms of tau fibrils are distinct and suggesting that heparin induction interferes with the interaction of CLR01 with tau.
Results
Comparison of Heparin-Induced and in Vitro Phosphorylated Tau Self-Assembly
To establish baseline conditions for comparison of in vitro phosphorylated tau with heparin-induced tau, we incubated a mixture of 5 μM tau and 1.25 μM heparin at 37 °C and measured ThT fluorescence up to 100 h. As expected, we observed a typical sigmoidal increase in ThT fluorescence, beginning with a short lag phase (∼30 min), followed by a sharp increase in fluorescence over the next 6 h and then a slower increase up to a plateau at ∼12 h (Figure 1A). We derived the aggregation rate constant, k, by fitting the kinetic curves to a one-phase association equation (eq 1). Under these conditions, k was found to be 0.29 ± 0.02 h–1. Examination of the morphology at the end of the reaction by TEM showed abundant, relatively short fibrils (Figure 1B).
Figure 1.

Self-assembly of tau induced by heparin, p-tauERK and p-tau(S262A)RBE. (A) Time-dependent ThT fluorescence of 5 μM heparin-induced tau, 100 μM p-tauERK, or 30 μM p-tau(S262A)RBE. (B–D) Morphological examination by TEM at the end of the aggregation reaction (t = 100 h) of heparin-induced tau (B), p-tauERK (C) and p-tau(S262A)RBE (D). (E) Native PAGE analysis of 10 μM each of heparin (hep)-induced tau, p-tauERK, and p-tau(S262A)RBE at t = 0, 10, or 20 h of incubation under the same conditions. For heparin-induced tau, t = 0 is immediately after addition of heparin. Molecular weight markers are shown on the left. The mobilities of putative monomer (M), dimer (D), trimer (Tr), and tetramer (Te) are indicated on the right.
Despite site-specific, high-level phosphorylation at several sites, tau phosphorylated by ERK (p-tauERK) has been reported previously not to produce a detectable increase of the ThT fluorescence signal.29,30 In agreement with these previous observations, solutions of 100 μM p-tauERK did not aggregate up to 100 h (Figure 1A). TEM examination showed only nonfibrillar, amorphous aggregates (Figure 1C) as observed previously.29,30
The quantitative phosphorylation pattern of tau phosphorylated by a whole rat brain extract (RBE, p-tauRBE), as determined by NMR, has been reported to be similar to that of p-tauERK, but contains three additional phosphorylation sites—at S208 in the PRR, and at S262 and S356 in the MTBD (Table 1).29 Because phosphorylation of S262 is known to inhibit tau aggregation,28 S262 was substituted by Ala for generation of p-tau(S262A)RBE to prevent phosphorylation at this site. At 5–10 μM, the aggregation of p-tau(S262A)RBE was too slow to compare side-by-side with heparin-induced tau. However, at 30 μM, p-tau(S262A)RBE reactions showed a sigmoidal increase in ThT fluorescence, beginning with a short lag phase of ∼1 h, followed by an increase in fluorescence over 45 h to reach a plateau. Despite using a higher concentration, the aggregation kinetics of p-tau(S262A)RBE was substantially slower than that of heparin-induced unphosphorylated tau (Figure 1A). The rate constant, k, in this case was 0.066 ± 0.001 h–1. Examination of the fibril morphology at the end of the reaction by TEM showed abundant fibrils, which were thicker and longer than those of heparin-induced tau (Figure 1D, Supplementary Figure S1). As shown previously,30 the fibrils had both straight and squiggly morphology but did not show a helical twist (Supplementary Figure S1).
Table 1. Tau Variants Used in This Study.
| tau proteins | residues | phosphorylation sites |
|---|---|---|
| F4 | 208–324 | |
| tau | 1–441 | |
| p-tauERK | 1–441 | S46, T50, T69, T153, T175, T181, S191, S199, S202, T205, T231, S235, S396, S404, S422 |
| p-tau(S262A)RBE | 1–441, S262A | S46, T50, T69, T153, T175, T181, S191, S199, S202, T205, S208, T231, S235, S356, S396, S404, S422 |
The underlined residues correspond to phosphorylation sites uniquely found using the kinase activity in the rat brain extract.
We asked next whether intermediate oligomers forming during the aggregation process could be detected and how they compared among the three forms of tau. We addressed this question by analyzing aliquots from aggregation mixtures containing 10 μM of each tau form at 0, 10, and 20 h using native PAGE/Western blot probed with the antihuman tau mAb HT7. Interestingly, we found a distinct behavior for each form (Figure 1E). At t = 0 h, i.e., just after addition of heparin to unphosphorylated tau, the protein appeared as a smear over nearly the entire length of the gel. A band at ∼70 kDa, presumably a monomer, could be discerned, as well as a lower band, at ∼60 kDa, likely representing a truncation/degradation product. At t = 10 h, most of the protein seemed to “disappear,” likely due to aggregation preventing it from entering the gel, and only relatively faint smears with apparent mobility corresponding to 140–1000 kDa were observed. At t = 20 h relatively little protein was apparent, suggesting further aggregation. At this time point, possibly due to the relatively low amount of apparent protein, faint smeary bands corresponding to putative monomer through tetramer were observed.
In agreement with the lack of observed fibril formation in the case of p-tauERK, this protein showed several bands, including putative monomer through tetramer, a higher molecular weight smear at 800–1000 kDa, and a band at ∼1100 kDa, likely a high-molecular-weight oligomer. At 10 and 20 h, the putative trimer, tetramer, and high-molecular-weight smear at 800–1000 kDa seemed to convert to the high-molecular-weight oligomer at ∼1100 kDa, whereas little or no change was observed in the putative monomer and dimer abundance.
The behavior of p-tau(S262A)RBE, which unlike p-tauERK did form β-sheet-rich fibrils, was more similar to that of heparin-induced unphosphorylated tau than to p-tauERK. At t = 0 h, the appearance was smeary with discernible putative monomer and dimer bands that migrated slightly slower than their unphosphorylated counterparts, whereas at 10 and 20 h, most of the signal gradually disappeared and lower molecular weight species appeared more abundant at 20 h than at 10 h. In all cases, the solutions/suspensions were mixed thoroughly before loading on the gel to ensure that potential fibrils/aggregates precipitated at the bottom of the tube would be included. However, these large species apparently were too large to enter the gel and therefore were not observed. Alternatively, they may have been present but did not react with HT7.
Comparison of Heparin-Induced and in Vitro Phosphorylated Tau Seeding in Biosensor Cells
Toxic tau species are thought to pass from affected cells to synaptically connected unaffected cells, where the seeds serve as templates that corrupt normal tau proteins and cause them to aggregate.62,63 To facilitate characterization of the seeds, Diamond’s group has developed biosensor cells that allow detecting and quantifying tau seeding using a sensitive fluorescence-resonance energy transfer (FRET) assay.64 The assay has been shown to have a wide dynamic range, allowing comparison of samples with vastly different levels of tau seeding and distinguishing among samples showing subtle differences.63
Here, we used this assay to compare the seeding capability of the three tau variants. We began with testing the seeding activity of fibrils formed by heparin-induced tau or p-tau(S262A)RBE, which were sheered by sonication for 10 min and mixed with the transfection agent, lipofectamine, for effective transduction into the biosensor cells, as described previously.64 We examined the cells qualitatively by fluorescence microscopy for the presence of observable puncta and quantitatively for the CFP–YFP FRET signal using flow cytometry.
In untreated cells, only diffuse fluorescence was observed (Figure 2A), whereas bright puncta were detected following addition of either type of fibrils (Figure 2B,C) indicating seeding of tau aggregation. Quantitative analysis by flow cytometry showed that both forms induced a robust seeding response. The magnitude of the response was higher overall for heparin-induced tau, though the difference reached statistical significance only at the highest concentration (1 μM, Figure 2D). Interestingly, high-magnification examination revealed distinct morphology and localization of the aggregates. Aggregates induced by heparin-induced tau were larger and mainly cytosolic or perinuclear (Figure 2E), whereas those induced by p-tau(S262A)RBE were smaller and colocalized with nuclei. These observations suggest that although both forms of tau form ThT-positive fibrils the arrangements of the tau molecules in these fibrils are different and represent distinct strains based on their seeding activity and aggregate morphology in the biosensor cells.
Figure 2.
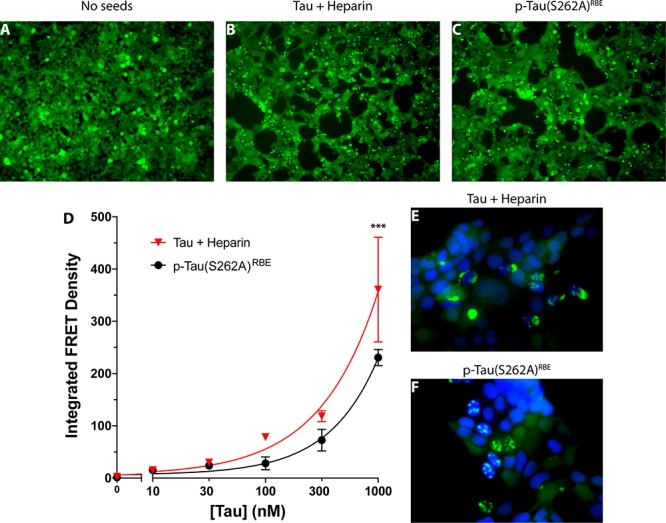
Induction of tau seeding by aggregated tau in biosensor cells. (A–C) Fluorescence microscopy images of tau biosensor cells. (A) Negative control cells incubated with media alone. (B) Cells incubated with heparin-induced tau fibrils. (C) Cells incubated with p-tau(S262A)RBE fibrils. (D) Dose–response analysis of the seeding activity (integrated FRET density) of each tau form. (E, F) High-magnification fluorescence microscopy images of cells incubated with 1 μM of either heparin-induced tau fibrils (E) or p-tau(S262A)RBE fibrils (F). Bright YFP fluorescence indicates tau aggregates. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, blue).
Because previously both tau fibrils and oligomers as small as a trimer were shown to act as seeds,64 we tested also whether the three tau variants could seed tau aggregation immediately after dissolution, when presumably only monomers and putative oligomers are present. Cells treated with freshly dissolved unphosphorylated tau in the absence (Figure 3A) or presence of heparin (Figure 3B) did not show formation of puncta. The latter condition produced a minimal FRET signal (Figure 3E). In contrast, freshly dissolved 1-μM p-tauERK showed abundant bright puncta (Figure 3C) and induced a strong, dose-dependent seeding response (Figure 3E). Similarly, freshly dissolved 1-μM p-tau(S262A)RBE promoted formation of bright puncta (Figure 3D) and caused dose-dependent tau aggregation in the biosensor cells, albeit slightly weaker than p-tauERK (Figure 3E). Overall, the seeding activity of the freshly dissolved phosphorylated tau proteins was ∼4-fold weaker than that of the fibrils (cf. Figures 2D and 3E). These data suggested that unlike unphosphorylated tau, p-tauERK and p-tau(S262A)RBE contained seed-competent tau monomers or preformed seeds, or formed seeds, presumably oligomers, during the experiment.65 High-resolution images showed that the aggregate morphology and localization of freshly dissolved p-tauERK (Figure 3F) and p-tau(S262A)RBE (Figure 3G) resembled those of fibrillar p-tau(S262A)RBE (Figure 2F), i.e., were small relative to the cell body and colocalized partially with nuclei, but not of heparin-induced tau fibrils (Figure 2E).
Figure 3.
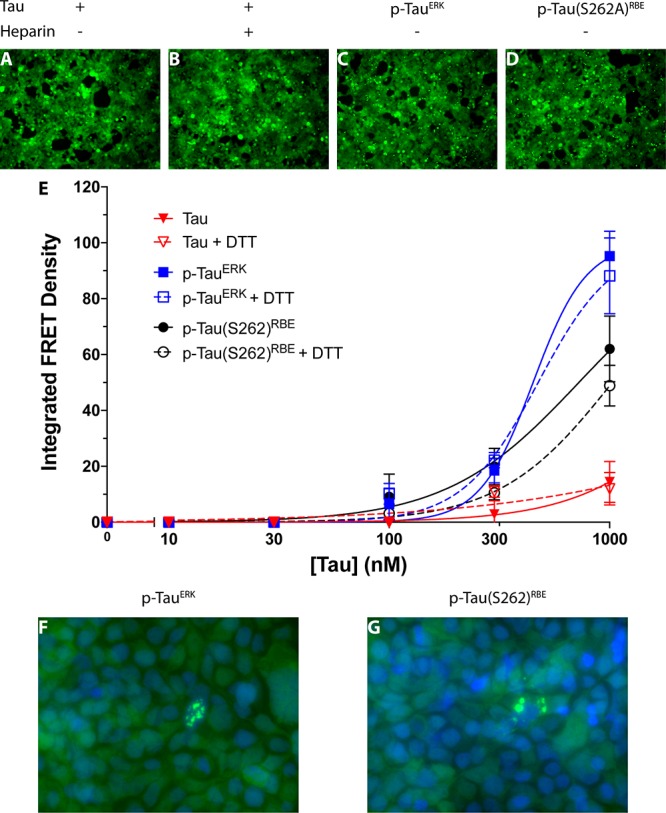
Induction of tau seeding by freshly dissolved tau in biosensor cells. (A–D) Fluorescence microscopy images of tau biosensor cells incubated with 1 μM freshly dissolved tau (A), tau + heparin (B), p-tauERK (C), or p-tau(S262A)RBE (D). (E) Dose–response analysis of the seeding activity (integrated FRET density) of each tau form in the absence or presence of DTT. (F, G) High-magnification fluorescence microscopy images of cells incubated with 300 nM freshly dissolved p-tauERK (F) or p-tau(S262A)RBE (G). Bright YFP fluorescence indicates tau aggregates. Nuclei were stained with DAPI (blue).
Formation of a small population of aberrant, cross-linked tau dimers via an intermolecular disulfide bond has been reported previously to promote seeding by the FTD-linked P301L tau isoform.66 To test whether the seeding in our experiments was due to formation of disulfide-cross-linked dimers (or other oligomers), we reduced the protein by addition of 6-fold excess DTT and repeated the seeding measurements. The final concentration of DTT in the cell culture media, 6 μM, did not cause any apparent change to the cells’ morphology or viability. The measurements did not show significant differences between the seeding of either tau variant in the presence of DTT (Figure 3E), suggesting that aberrant intermolecular disulfide-mediated oligomerization did not contribute to the observed seeding.
Taken together, the results suggest that unphosphorylated tau needs to be induced to form fibrils in order to become seeding-competent. In contrast, in vitro phosphorylated tau is seeding-competent already when freshly dissolved, presumably due to the presence of oligomers (Figure 1E). However, upon fibril formation, the seeding activity of p-tau(S262A)RBE increases by ∼4-fold. p-tauERK and p-tau(S262A)RBE appear to form a similar strain, which is different from the one formed by heparin-induced tau. The strain formed by p-tau(S262A)RBE seems to be maintained in both the freshly dissolved and fibrillar states.
CLR01 Inhibits the Aggregation of Heparin-Induced and in Vitro Phosphorylated Tau with Distinct Dose-Dependencies
Similar to anionic aggregation inducers, such as heparin or arachidonic acid, CLR01 (Figure 4A) binds to positively charged residues in tau. This competition for the same binding sites makes data interpretation difficult because CLR01 binding could inhibit tau aggregation by two independent mechanisms: (1) competition with the inducer for the Lys/Arg residues, thereby attenuating the induction of tau aggregation; and (2) direct interference with intermolecular interactions among tau molecules. Therefore, we sought to distinguish between these two mechanisms and to compare between this nonideal system and a simpler system in which CLR01 interacts with phosphorylated tau.
To determine the extent to which CLR01 competes with heparin for tau binding, we used SPR. Heparin was immobilized on a SPR chip, and tau binding to the immobilized heparin was found to occur with KD = 10.9 nM (Figure 4B), in agreement with recent results obtained using ELISA.67 Similar experiments using solutions in which tau was mixed with CLR01 at different ratios then were performed, with the expectation that when the heparin binding sites in tau are occupied by CLR01, tau binding to the surface-immobilized heparin should decrease, resulting in a reduction in the magnitude of the SPR signal. This well-established method has been used previously for characterizing heparin–protein interaction.68,69 The analysis showed that CLR01 competed with heparin for binding to tau (Figure 4C) but, in agreement with its highly labile binding, was a relatively weak competitor. The IC50 for the inhibition of 0.5 μM tau binding to immobilized heparin by CLR01 was found to be 20 ± 1 μM (Figure 4D); i.e., 40-fold excess CLR01 relative to tau was needed for half-maximal inhibition.
Next, to test the effect of CLR01 on heparin-induced aggregation of tau, the unphosphorylated protein was incubated at 5 μM in the presence of 1.25 μM heparin, and CLR01 was added at concentrations ranging from 2.5 to 40 μM. CLR01 caused a dose-dependent decrease in the aggregation rate constant, k, and inhibited the aggregation completely at 6-fold excess (Figure 5A). A plot of percentage of inhibition (calculated using eq 2) versus [CLR01] showed that the molecular tweezer inhibited the aggregation of heparin-induced tau with a half-maximal inhibitory concentration (IC50) of 11.3 ± 0.6 μM (Figure 5B). Considering that both primary and secondary nucleation events may affect tau aggregation, we also attempted to use the method of Cohen et al.,70,71 which takes into account both nucleation mechanisms. We found little or no contribution of secondary nucleation, minimal changes in the data fitting, and a nearly identical IC50 value, 11.8 ± 0.7 μM, using this method. Because inhibition of aggregation does not always produce the same effect on the slope of the ThT curve and the final plateau value, we calculated an IC50 value also based on the final plateau and found it to be in the same range, 19 ± 0.6 μM (Supplementary Figure S2A). In this experiment, only 2−3-fold excess CLR01 was sufficient for half-maximal inhibition, as opposed to the 40-fold excess needed for half-maximal inhibition of tau binding to heparin. Therefore, we conclude that the main inhibitory effect of CLR01 on tau aggregation is mediated by direct interference with tau self-assembly rather than by inhibition of heparin binding.
Figure 5.

Inhibition of tau aggregation by CLR01. (A) Time-dependent ThT fluorescence in aggregation reactions containing 5 μM tau, 1.25 μM heparin, and increasing concentrations of CLR01 showing replicates and fitted association curves. (B) Dose–response analysis of the fluorescence data in panel A was calculated by extracting the kinetic constant k from fitted curves and normalizing to k0, the kinetic constant in the absence of CLR01. (C) Time-dependent ThT fluorescence in aggregation reactions containing 30 μM p-tau(S262A)RBE and increasing concentrations of CLR01 showing replicates and fitted association curves. “Tau-S262A” is a negative control containing this variant of tau in an unphosphorylated form. (D) Dose–response analysis of the fluorescence data in panel C was calculated by extracting the kinetic constant k from fitted curves and normalizing to k0, the kinetic constant in the absence of CLR01.
Because of the different aggregation kinetics of heparin-induced tau and p-tau(S262A)RBE (Figure 1A), side-by-side comparison at the same concentration was not practical. Thus, to measure the effect of CLR01 on the aggregation of p-tau(S262A)RBE, the protein was incubated at 30 μM in the presence of increasing CLR01 concentrations. Interestingly, at substoichiometric concentrations, CLR01 appeared to have a mixed effect on the aggregation kinetics of p-tau(S262A)RBE—initially, the kinetics was accelerated, whereas after ∼24 h, the aggregation rate was reduced compared to reactions in the absence of CLR01 (Figure 5C). In contrast, at 2- or 4-fold excess, CLR01 inhibited the aggregation completely (Figure 5C,D and Supplementary Figure S2B). These results further demonstrate that heparin-induced tau aggregation is dissimilar to the aggregation of hyperphosphorylated tau and in particular may yield misleading results when analyzing aggregation modulators, such as CLR01.
ThT fluorescence measures the formation of the cross-β structure when amyloidogenic proteins aggregate, but the increase in ThT fluorescence does not always correlate with fibril formation.72−74 To assess whether in this case the initial acceleration in β-sheet formation reflected formation of fibrils, we examined the morphology of each preparation at the end of the aggregation reaction by TEM. The morphological examination showed that addition of CLR01 resulted in a dose-dependent inhibition of fibril formation by both heparin-induced tau and p-tau(S262A)RBE (Figure 6, Supplementary Figure S3). In the case of heparin-induced tau, thin, straight or squiggly fibrils could still be observed up to a tau/CLR01 concentration ratio of 1:4, respectively, whereas at higher concentrations the morphology was amorphous (Supplementary Figure 3A). Fibrils of p-tau(S262A)RBE were substantially reduced at a p-tau(S262A)RBE/CLR01 concentration ratio 1:0.25 and eliminated at a concentration ratio of 1:0.5 or higher (Supplementary Figure 3B), demonstrating that the acceleration of the ThT fluorescence increase at substoichiometric CLR01 concentrations did not reflect fibril formation. These data are in agreement with an increase in ThT fluorescence without fibril formation by other proteins in the presence of CLR01;75,76 yet the nature of the early ThT-positive structures forming in the presence of CLR01 is not well-understood.
Figure 6.

Representative TEM images taken at the end of the aggregation reactions shown in Figure 5. (A) Images of 5 μM heparin-induced tau in the absence or presence of the indicated tau/CLR01 concentration ratios. (B) Images of 30 μM p-tau(S262A)RBE in the absence or presence of the indicated p-tau(S262A)RBE/CLR01 concentration ratios.
CLR01 Inhibits Tau Seeding in Biosensor Cells
Next, we asked whether CLR01 could inhibit the seeding activity of tau fibrils. To answer the question, we incubated the cells with CLR01 concentrations ranging from 10 nM to 50 μM for 16 h to allow uptake of the molecular tweezer into the cells. Then, we added 150 nM heparin-induced tau or p-tau(S262A)RBE fibrils, which had been sonicated for 10 min prior to addition to the cells and measured the seeding activity after additional 24 h of incubation.
The seeding activity of heparin-induced tau fibrils was again higher than that of p-tau(S262A)RBE (Figure 7A). Confocal microscopy confirmed the formation of abundant seeds in cells treated only with heparin-induced tau fibrils and not with CLR01 (Figure 7B), whereas no seeds could be seen in cells incubated with CLR01 prior to addition of tau seeds (Figure 7C). The presence of CLR01 in cells treated with heparin-induced tau up to a 1:1 tau/CLR01 concentration ratio, respectively, increased the seeding activity compared to cells treated with the same seeds in the absence of CLR01, whereas at higher concentrations, CLR01 inhibited tau seeding. In contrast to the mixed effect of CLR01 on the seeding of heparin-induced tau fibrils, the molecular tweezer inhibited the seeding of p-tau(S262A)RBE seeds dose-dependently with IC50 = 660 ± 140 nM (Figure 7A), further demonstrating the distinct behavior of the two types of tau aggregates and the problematic nature of using heparin-induced tau aggregates for testing aggregation and/or seeding modulators, such as CLR01.
Figure 7.
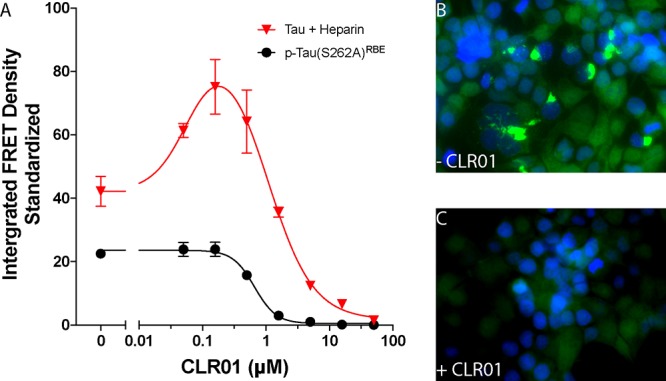
CLR01 inhibits tau seeding in biosensor cells. Cells were incubated for 16 h in the absence or presence of increasing CLR01 concentrations, followed by addition of seeds. After additional 24 h of incubation, seeding was visualized by fluorescence microscopy and quantified using flow cytometry. (A) Dose–response analysis of the seeding activity (integrated FRET density) of each tau form in the absence or presence of CLR01. (B, C) High-magnification fluorescence microscopy images of cells incubated with heparin-induced tau fibrils after pretreatment with media alone (B) or 50 μM CLR01 (C). Bright YFP fluorescence indicates tau aggregates. Nuclei were stained with DAPI (blue).
A potential concern with interpretation of this experiment is that CLR01 could act directly on the seeds themselves and induce their dissociation once they get internalized by the biosensor cells. However, the labile binding of CLR01 would not be expected to affect such fibrils, unless they are incubated with a large excess of the molecular tweezer for long periods, as has been shown previously for other amyloidogenic proteins.75,77,78 Indeed, morphological examination showed that when tau fibrils were incubated with CLR01 for 24 h in vitro, CLR01 did not cause appreciable changes in the fibril structure (Supplementary Figure S4).
NMR-Based Mapping of CLR01 Binding to the Different Tau Variants
To gain further insight into the way CLR01 interacts with tau, we used solution-state NMR. Full-length tau contains 44 Lys residues, each of which is a potential binding site for CLR01. However, the affinity of CLR01 for each Lys residue depends both on the flanking amino acid sequences and on the local and global conformation. Lys residues also are binding sites for heparin.42,79,80 Previous NMR mapping of tau–heparin interaction revealed that large heparins bind to most of the Lys residues in the MTBD and PRR, and to a low extent also to Lys residues in the extreme N-terminus.81 Using small heparins, specific hotspots have been identified allowing discrimination between sites with different binding affinities.42 Because binding of either CLR01 or heparin would be expected to cause perturbation of Lys residues, we did not attempt to use NMR for the characterization of the competition between the two molecules for binding to tau. Rather, we used NMR to determine whether phosphorylation alters the binding of CLR01 to tau.
Unlike heparin, CLR01 binds one Lys residue at a time by including the Lys side chain inside the cavity of the molecular tweezer, thus forming hydrophobic interactions with the tweezer’s “arms” and electrostatic interactions between the ε-NH3+ group of Lys and the phosphate groups of the molecular tweezer. As CLR01 is a symmetric compound containing two phosphate groups, if another positively charged residue, such as Lys or Arg is nearby, it could form additional electrostatic interactions with the second phosphate group of CLR01, but those would be substantially weaker because only one Lys side chain can be included inside the cavity. Given this binding mode, we asked first if CLR01 had a preference for selective binding to certain Lys residues versus others. CLR01 also binds to Arg residues, but with 5–10-times lower affinity than to Lys residues.55,82,83 Therefore, we focused most of our analysis on Lys only using selective 15N-Lys labeling of tau, which helps reducing signal crowding in two-dimensional 1H–15N HSQC spectra. The assignment of Lys resonances in the 1H–15N HSQC spectrum of 15N-Lys-labeled tau has been described previously.42,81
We analyzed unphosphorylated tau first to establish a baseline. We titrated 200 μM 15Nα,ε-Lys-labeled tau with increasing concentrations of CLR01 ranging from 20 to 400 μM and recorded a 1H–15N HSQC spectrum for each titration point. As the concentration of CLR01 increased, we observed minimal chemical shift variations, but the HSQC signal intensity decreased gradually (Figure 8A; Supplementary Figure S5). Because of the dynamic nature of its on–off exchange, CLR01 binding induces a severe broadening of Lys resonances associated with signal intensity decrease, as has been reported previously in Aβ.75 We observed a similar behavior in a short peptide derived from tau (data not shown). In 1H spectra, we observed a substantial decrease in 1H resonance intensities of aliphatic protons in Lys residues, whereas other 1H resonances decreased only by <5% at the highest concentration of CLR01 (Figure 8B), confirming specific interactions of CLR01 with Lys residues. For example, ∼50% of the Lys Hε signal at 2.99 ppm disappeared at the highest tau/CLR01 ratio 1:2, respectively (Figure 8B). At this ratio, the CLR01/Lys ratio is 1:22. Thus, the strong decrease in the overall signal of Lys demonstrates the fast on–off rate of CLR01 binding to Lys. Interactions of CLR01 with more than one Lys residues at a time, leading to further signal broadening, also could account for the large signal decrease at low CLR01/Lys ratios.
Figure 8.

NMR mapping of CLR01 binding to tau and F4. (A) NMR mapping of 15N-Lys-labeled tau in the absence (black spectrum) or presence (blue spectrum) of a 2-fold excess CLR01. (B) 1H spectra of 15N-Lys-labeled tau with increasing concentration of CLR01. The Hε resonance of Lys side chains at 2.99 ppm is depicted in the inset showing a selective decrease of 1H signals of Lys residues. A similar intensity decrease was found for Lys Hδ resonances at 1.65 ppm. (C, D) Mapping of resonance intensity of Lys residues along the 15N-Lys-tau (C) or 15N–F4 (D) sequences for different protein/CLR01 ratios. The region in gray in (C) corresponds to the F4 sequence.
Mapping of the resonance intensity variations using the two-dimensional 1H–15N HSQC spectra indicated that CLR01 bound preferentially to Lys residues in the MTBD and to a lower extent in the PRR. Most of the binding occurred in the region between K224 and K395 with the exception of K130 for which strong binding was found. Interestingly, little binding was detected to the Lys residues at the N- and C-termini (Figure 8C). A possible explanation for this preference is that the extreme N- and C-terminal regions are more acidic than the remaining of the protein (Supplementary Figure S6), which may lead to electrostatic repulsion of the negatively charged CLR01.
To gain additional insight into the binding of CLR01 within its main region of interaction in tau, we mapped its binding on the fragment F4, which encompasses residues 208–324 (Figure 8D and Supplementary Figure S5). F4 maintains many of the physiologic and pathologic functions of full-length tau, e.g., microtubule binding and the capacity to form PHF-like fibrils.84,85 F4 was produced with uniform 15N-labeling allowing mapping of intensity changes for all the residues of the fragment. The titration was performed using the same increasing concentrations of CLR01 and under the same conditions used for full-length tau. F4 contains 17 of the 44 Lys residues of full-length tau, out of a total of 117 residues (i.e., Lys residues comprise 14.5% of the F4 sequence, compared to 10% of full-length tau) and 5 Arg residues (4.3% of the F4 sequence) all located in the PRR, at positions 209, 211, 221, 230, and 242. R209 and R211 were not detected in the HSQC spectrum, and only low-intensity variations, between 12% and 25% intensity loss, were observed for the other three Arg residues (Figure 8D) indicating weak binding of CLR01 to these Arg residues, in line with data described previously.55,82,83
A gradual intensity loss was observed for some, but not all of the Lys residues (Figure 8D). Residues in the extreme N-terminus, e.g., S210, which was conjugated to a (His)6-tag, and His residues, e.g., H268 in the F4 sequence, showed substantial intensity changes, which likely reflected the high sensitivity of the His signal to small pH variations rather than CLR01 binding. Previously, the affinity of CLR01 for His was determined to be only ∼10% of the affinity for Lys.55
Except for these residues, K224, K225, K240, K259, and K280 showed substantial intensity decrease, up to 50%, at equimolar concentration of CLR01 (Figure 8D). The most pronounced intensity decrease, 60 and 80%, respectively, was found for K290 and K321, both within KCGS motifs. However, it is difficult to determine whether this strong decrease represents the high sensitivity of Cys residues to changes in the environment or an actual higher affinity of CLR01 for the Lys residues in these positions. The data are in good agreement with a recent report, which found using top-down mass spectrometry that a primary binding site for CLR01 in full-length tau was in a segment spanning residues K294–K331.86 Importantly, Lys residues in F4 showed a comparable intensity decrease to their counterparts in full-length tau at the same protein/CLR01 concentration ratios (Supplementary Figure S5C), suggesting that the changes reflected the genuine affinity of CLR01 for these particular Lys residues.
After establishing the main binding region and specific primary binding sites of CLR01 on unphosphorylated tau, we asked how phosphorylation might affect CLR01 binding. We investigated the binding of CLR01 to p-tauERK and p-tau(S262A)RBE by NMR in a manner similar to the unphosphorylated protein. First, we performed experiments using uniformly 15N,13C-labeled tau and selectively 15Nα,ε/13C-Lys,13C-labeled p-tauERK, to determine the phosphorylation pattern (Supplementary Figures S7 and S8) and facilitate the mapping of resonance intensity upon CLR01 titration, to determine the phosphorylation pattern (Supplementary Figure S7). The findings are summarized in Table 1.
Spectral heterogeneity occurs upon incomplete phosphorylation of several Ser/Thr residues leading to a splitting of protein resonances for the phosphorylation sites and surrounding residues. Assignment of 1H–15N resonances of 15Nα,ε/13C-Lys,13C-labeled p-tauERK was performed using a three-dimensional HN(CO)CACB experiment, which allows assigning Lys resonances that shift upon phosphorylation due to their proximity to phosphorylation sites (Supplementary Figure S7). Examination of the 1H–15N HSQC NMR spectra of Lys-labeled p-tauERK revealed distinct behavior of certain Lys resonances. Some Lys resonances were split into two or more peaks due to phosphorylation heterogeneity, including K67, K190, K224, K225, and K234, and low-intensity resonances of the nonphosphorylated form were still detected. In contrast, for K44, K150, K174, K180, K240, and K395, only peaks corresponding to the phosphorylated state were detected, as shown for K240, Figure 9A,B, indicating a high phosphorylation level for sites in the vicinity of these Lys residues (Supplementary Figure S7, Table 1).
Figure 9.

Binding site analysis of CLR01 on p-tauERK. (A, B) Superimposition of 1H–15N HSQC spectra of selectively 15N-Lys-labeled p-tauERK (A) and unphosphorylated tau (B) at 200 μM in the absence (black) or presence of 200 μM CLR01 (blue). Resonances of K240 corresponding to phosphorylated forms in panel A are annotated by K240–P1 and K240–P2. Note that the unphosphorylated form of K240 shown in panel B is absent in p-tauERK. The location of this missing resonance is indicated in panel A by “x”. (C) Variation of K240 resonance intensity upon increasing CLR01 concentrations for the unphosphorylated form in tau (open squares) and both phosphorylated forms in p-tauERK shown in panel A (filled diamonds and triangles). (D) Changes in resonance intensity of Lys signals in p-tauERK upon CLR01 binding for protein/CLR01 ratios ranging from 1:0.1 to 1:2. For Lys residues close to phosphorylated residues, only intensity corresponding to the resonance in the phosphorylated form is indicated. Nt – N-terminus, Ct – C-terminus.
The resonance intensity changes upon CLR01 binding to p-tauERK (Figure 9C) suggested that phosphorylation had only a minor effect on the binding. The binding pattern of CLR01 to p-tauERK resembled the binding to unphosphorylated tau, targeting primarily the MTBD and to a lesser extent the flanking regions (Figure 9C and Supplementary Figure S9). Only binding to the PRR was slightly reduced (Supplementary Figures S9 and S10). This observation was consistent with the location of most of CLR01’s binding sites in the MTBD, as opposed to most of the phosphorylation sites in the PRR and the extreme N- and C-termini. The only exceptions were K150, K174, and K240, which are adjacent to the phosphorylation sites T153, T175, and T231/S235, respectively. Binding of CLR01 to these three Lys residues was significantly reduced upon phosphorylation (Figure 9C,D and Supplementary Figures S9 and S10).
The binding sites found for CLR01 on p-tau(S262A)RBE were similar to those in unphosphorylated tau and p-tauERK (Supplementary Figures S9 and S10). Together, these data indicate that tau phosphorylation does not affect CLR01 binding substantially, allowing the molecular tweezer to remain an effective inhibitor of tau aggregation, oligomerization, and seeding.
Discussion
In Alzheimer’s disease and other tauopathies, aberrant post-translational modification of tau leads to its self-assembly into neurotoxic oligomers and fibrils. Of the PTMs studied to date, hyperphosphorylation has been found most frequently and likely is the main PTM instigating aberrant tau self-assembly. The requirement of post-translational modifications for induction of aggregation makes studying this process in vitro or in cell culture difficult. To create experimental systems allowing such studies, researchers long have been using heparin and other polyanions to induce tau aggregation. These artificial models are particularly convenient for screening tau aggregation inhibitors and disaggregation inducers;87 yet, concerns about the artificial nature of such systems, and in particular about potential interference by the polyanions with testing potential modulators and inhibitors of aberrant tau self-assembly, have been increasing. Recently, cryo-EM structures showed that heparin-induced 4R and 3R tau fibrils are structurally distinct from those in Alzheimer’s and Pick’s disease questioning the relevance of such in vitro assays.88 Furthermore, Fichou et al. have reported that heparin and RNA used to induce fibril formation coassemble with tau in vitro.47 Heparin-induced tau fibrils used in in vitro seeding assays seeded freshly dissolved tau only in the presence of RNA, whereas mouse-brain-derived tau fibrils could be used as seeds in the absence of cofactors in the first-generation seeding reaction and required RNA as a cofactor to increase the seeding activity of the second-generation reaction. On the basis of these findings, they suggested that in vivo, an unknown cofactor might be necessary for tau aggregation. Our data suggest that such cofactors are not necessary for tau fibrillation and that hyperphosphorylation of tau is sufficient for induction of fibril formation. In view of recent advances in generating in vitro phosphorylated tau,29,30 here we set out to compare side-by-side this newer form of aggregation-prone tau with the common method of tau induced to aggregate by addition of heparin.
Induction of tau aggregation by addition of heparin allowed using a lower concentration of the protein and resulted in faster aggregation kinetics than those measured using p-tau(S262A)RBE (Figure 1A). The morphology of the resulting fibrils was different—p-tau(S262A)RBE fibrils were both thicker and longer than those of heparin-induced tau (cf. Figure 1, panels B and D), demonstrating that the two forms were structurally distinct.
Unlike p-tau(S262A)RBE, p-tauERK did not form fibrils. NMR analysis showed that the two forms of in vitro phosphorylated wild-type tau shared 15 common phosphorylated sites and differed in only three sites—S208, S262, and S356—which were present in RBE- but not in ERK-phosphorylated tau.29 Comparison of these two forms suggested that the phosphorylation of S208 was the key site necessary for aggregation, as described by Despres et al.,30 because phosphorylation of S262 and S356 had been reported previously to inhibit aggregation.28 Therefore, S262, a site that gets phosphorylated in vitro at a high level by RBE (in contrast to S356), was substituted by Ala in subsequent experiments to obtain an aggregation-prone isoform. Interestingly, comparison of the oligomerization behavior of these two forms showed that the aggregation-resistant p-tauERK formed a variety of small (dimer through tetramer), medium (apparent mobility corresponding to 800–1000 kDa), and large (apparent mobility corresponding to 1100 kDa) oligomers, whereas the aggregation-prone p-tau(S262A)RBE comprised a mixture in which individual oligomers were less discernible and high-molecular-weight species were prominent. Following incubation, most of the p-tau(S262A)RBE preparation was no longer visible in native PAGE/Western blot analysis (Figure 1E), presumably due to formation of large aggregates that did not enter the gel.
Using the tau biosensor cell line developed by Diamond and co-workers, we found, as expected, that tau fibrils seeded intracellular tau aggregation, regardless of whether they were made of heparin-induced tau or p-tau(S262A)RBE (Figure 2A–D). We were surprised, however, to discover that the morphology and cellular distribution of the two forms suggested that they belonged to distinct strains. Heparin-induced tau fibrils induced formation primarily of relatively large cytoplasmic/perinuclear aggregates (Figure 2E), whereas p-tau(S262A)RBE seeded small, primarily nuclear aggregates (Figure 2F). These morphologies resembled the “ordered” and “speckles” strains described recently by Kaufman et al.54 That study found that tau from different sources, including human AD brain, cell lines from patients with different tauopathies, brain extract from the P301S mouse model, and recombinant tau gave rise to various strains distinguished by the appearance of the aggregates in the biosensor cells. Our data suggest that heparin-induced tau fibrils prepared under the conditions described here and p-tau(S262A)RBE may allow obtaining discrete strains reproducibly using recombinant tau for further structural and biological studies. In agreement with previous reports suggesting that tau seeding did not require fibril formation and could be mediated by oligomers, we found that both forms of freshly dissolved in vitro phosphorylated tau, p-tauERK and p-tau(S262A)RBE, though importantly not unphosphorylated tau, seeded tau aggregation in the biosensor cells.
Before the current study, only two pieces of information were available regarding the effect of CLR01 on tau. Using the ThT fluorescence assay, equimolar concentration of CLR01 was shown to inhibit completely the aggregation of the shortest form of tau, 0N3R, which was induced to aggregate by arachidonic acid.75 In the presence of arachidonic acid, tau aggregates into amorphous, rather than fibrillar aggregates, and therefore the effect of CLR01 on fibril formation was not assessed in that study. The impact of CLR01 on tau aggregates in vivo was examined in the triple-transgenic AD mouse model expressing mutant forms of tau, amyloid β-protein precursor, and presenilin-1. These mice accumulate both amyloid plaques and NFTs in their brain with aging. Treatment of 14–15-month-old mice, who already had both types of lesions, with a low dose of CLR01 led to a substantial reduction in both amyloid plaques and NFTs.90 Because tau pathology is thought to be secondary to the neurotoxic effects of amyloid β-protein (Aβ), the study in the triple-transgenic mice could not reveal whether the decrease in neurofibrillary tangles was due to a direct effect of CLR01 on tau or an indirect effect due to inhibition of Aβ toxicity. Though the current study does not address this question directly, it reveals that CLR01 effectively inhibits tau aggregation and intracellular seeding. The data also demonstrate that the choice of the experimental system for studying the effect of the modulator is crucial.
When tau was induced to aggregate by heparin, CLR01 could interfere both with the induction itself and with the intermolecular interactions among tau monomers that mediate their further aggregation. Comparison of direct inhibition of tau-heparin binding using SPR (Figure 4) and inhibition of tau aggregation using ThT fluorescence (Figure 5) suggested that the main inhibitory activity of CLR01 was effected on the self-assembly of tau polypeptides rather than on tau–heparin binding. Thus, the concentration of CLR01 needed for half-maximal inhibition of tau aggregation was only twice the concentration of tau, whereas 20-fold excess CLR01 was needed for half-maximal inhibition of tau binding to heparin. Consistent with the interpretation of efficient inhibition of tau self-assembly, CLR01 also efficiently inhibited the aggregation of p-tau(S262A)RBE. Interestingly, however, the molecular tweezer had a more complex effect on the aggregation of the protein in this case. At low concentrations, an initial acceleration of ThT fluorescence increase was observed, whereas later in the aggregation reaction, the rate of increase and the final fluorescence values were lower than that of p-tau(S262A)RBE alone. Examination of the morphology at the end of the reaction showed dose-dependent inhibition of fibril formation (Figure 6), suggesting that the initial increase in ThT fluorescence did not reflect formation of tau fibrils, but rather of a nonfibrillar, yet presumably β-sheet-rich structure. Similar nonfibrillar structures displaying increased ThT fluorescence have been observed before in the presence of CLR01 for other amyloidogenic proteins, including insulin, β2-microglobulin, transthyretin, and p53 mutants.75,76 The details of these structures are not known, but they seem to form when CLR01 interacts with structured, rather than unstructured, amyloidogenic proteins. If this trend holds true here too, it would suggest that phosphorylation of tau may induce formation of a more stable structure than that of unphosphorylated tau. Interestingly, formation of a partially structured conformation of tau recently has been suggested to transform tau from a benign to a seeding-competent form,65 in agreement with this idea. NMR studies also have highlighted phosphorylation-induced conformational changes in the PRR around the binding phospho-epitopes of antibodies AT8 (pS202/pT205)91 and AT180 (pT231/pS235)92,93 in which phosphorylation stabilizes transient secondary structures. The double phosphorylation at the AT8 epitope was shown to induce a turn-like structure which protects tau from aggregation, while a triple phosphorylation which includes pS208 destabilizes this conformation and stimulates aggregation of phosphorylated tau.30
CLR01 binds to surface-exposed residues94 and therefore in natively unstructured proteins would be expected to bind most or all of the Lys residues. However, previous studies with Aβ75 and α-synuclein95 have shown that certain residues are preferred over others as binding sites. Here, we found that CLR01 binds preferentially to Lys residues in the MTBD (Figure 8, Supplementary Figure S5), possibly due to a high concentration of acidic residues at the N- and C-terminal regions that may repel the negatively charged molecular tweezer (Supplementary Figure S6). An alternative explanation is involvement of Lys residues in those regions in salt bridges or other structural elements that prevent them from interacting with CLR01. However, there is little evidence supporting the existence of such structural elements, and the observation that the interaction of CLR01 with the Lys residues in tau was not affected substantially by phosphorylation (Figure 9, Supplementary Figures S9 and S10), which may increase structural order, suggests against this explanation.
Our study demonstrates that heparin-induced tau aggregation yields distinct aggregation kinetics, oligomerization patterns, and seeding behavior from those of in vitro phosphorylated tau, which we hypothesize is a closer mimic of the hyperphosphorylated tau in toxic oligomers and NFTs in AD and other tauopathies. If our hypothesis is correct, interpretation of studies using tau induced to self-assemble by addition of heparin or other polyanions should be done with caution. Testing modulators and inhibitors of tau self-assembly and seeding in the presence of aggregation inducers is particularly problematic due to the competition between the inducer and the modulator for binding to tau. In vitro phosphorylation of tau alleviates this problem and provides a better mimic of disease-associated hyperphosphorylated tau. Our data also suggest that CLR01 is an effective inhibitor of tau self-assembly and seeding. As CLR01 also has been shown to be a potent inhibitor of Aβ self-assembly and toxicity in vitro and in vivo,75,90,96,97 it may be a particularly attractive lead for developing disease-modifying therapy thanks to its ability to inhibit both of the offending proteins in AD simultaneously.
Materials and Methods
Heparin sodium salt from porcine intestinal mucosa was purchased from Sigma or Celsus Laboratories (Cincinnati, OH). Thioflavin T, okadaic acid, Pefabloc, and all other chemicals, unless indicated otherwise, were purchased from Sigma. For expression of isotopically labeled tau, bacterial culture media were reconstituted using 13C6-glucose 15NH4Cl, 15N/13C-ISOGRO, and 15N2,13C6-l-lysine hydrochloride from Sigma-Aldrich (Isotec). Complete EDTA-free protease-inhibitor cocktail was purchased from Roche. BCA colorimetric assay was from Pierce. CLR01 was prepared as described previously55 and purified as a sodium salt.
Production and Purification of Recombinant Tau in Isotopically Labeled and Unlabeled Forms
The 2N4R tau isoform (residues 1–441 of human MAPT) in its wild-type or S262A form, and the fragment F4 (tau residues 208–324) carrying a C322S substitution were produced in the E. coli strain BL21(DE3) carrying a pET15b recombinant plasmid (Novagen). Residue numbering of F4 is based on the 2N4R sequence. The wild-type mapt gene was inserted into NcoI/XhoI cloning sites. The S262A substitution was introduced by site-directed mutagenesis of the wild-type construct. For uniform 15N-labeling, cells were grown at 37 °C in M9 minimal medium containing 4 g/L glucose, 1 g/L 15N-NH4Cl, 0.5 g/L 15N-Isogro (Sigma), 1 mM MgSO4, 100 μM CaCl2, MEM vitamin cocktail, and 100 mg/L ampicillin. Expression was induced by addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside for 3 h at 37 °C. For selective 15Nα,ε,13C-Lys, and uniform 13C-labeling, the same growth medium was used except that it contained 1 g/L of unlabeled NH4Cl and 2 g/L 13C6-glucose and 150 mg 15N2,13C-Lys were added 20 min before induction. Cells were harvested by centrifugation at 5000g for 30 min, and the pellet was resuspended in 50 mM NaH2PO4/Na2HPO4, pH 6.2, 2.5 mM EDTA, 2 mM DTT, and 0.5% Triton X-100 supplemented with a Complete protease-inhibitor cocktail. The lysate was obtained by homogenizing this suspension using a high-pressure homogenizer followed by centrifugation at 30000g for 30 min. The extract was incubated at 75 °C for 15 min to precipitate most of the bacterial proteins from the soluble extract as a first purification step. The soluble proteins were separated from the precipitate by centrifugation at 4000g for 20 min, and the desired protein was purified by cation-exchange chromatography (HiTrap SP HP 5 mL, GE Healthcare). The column was equilibrated in 50 mM NaH2PO4/Na2HPO4, pH 6.4, 2 mM EDTA (buffer A). The bacterial extract was loaded onto the column, and then the column was washed with buffer A. The protein was eluted with a gradient from 0% to 50% buffer B (buffer A supplemented with 2 M NaCl) over 20 mL (i.e., 4 column volumes). Elution fractions were analyzed by SDS-PAGE, and fractions containing the pure protein were pooled together for buffer-exchange in 50 mM ammonium bicarbonate (HiPrep 26/10 desalting, GE Healthcare) prior to lyophilization. Lyophilized proteins were stored at −20 °C or −80 °C until further use.56 The recombinant tau proteins used in this study are listed in Table 1 with an indication of phosphorylation sites provided by the kinase activity of either activated ERK2 (ERK2*) or a rat brain extract (RBE). These phosphorylation patterns are consistent with those described previously.29,30
In Vitro Phosphorylation of Tau by Activated ERK2
Recombinant ERK2 and a constitutively active MEK3 variant, a mitogen-activated protein-kinase kinase that phosphorylates and activates ERK2, were produced as described previously.29,56,5715N- or 15N2, 13C-Lys/13C-tau were dissolved at 100 μM with 1 μM of ERK2 and ∼0.1 μM of MEK3 in 400 μL of buffer containing 50 mM HEPES-KOH, pH 8.0, 12.5 mM MgCl2, 50 mM NaCl, 2 mM EDTA, 1 mM DTT, 1 mM EGTA, and 12.5 mM ATP. The mixtures were incubated at 37 °C overnight. Enzymatic reactions were stopped by incubating the reaction mixtures at 75 °C for 15 min followed by centrifugation at 16000g for 20 min. Then, the supernates were buffer-exchanged in 50 mM ammonium bicarbonate and lyophilized. Prior to further analyses, phosphorylation was checked qualitatively by a shift in the apparent mobility of the protein band on SDS-PAGE. Then, the phosphorylated proteins were analyzed by high-resolution NMR spectroscopy to determine the phosphorylation patterns and potential protein degradation in a quantitative manner.29,57
In Vitro Phosphorylation of Tau by Rat Brain Extract (RBE)
RBE was prepared from adult Sprague–Dawley rats by homogenizing whole brains (∼2 g) in 5 mL of homogenizing buffer, containing 10 mM Tris-HCl, pH 7.4, 5 mM EGTA, 2 mM DTT, 1 μM okadaic acid (OA), 20 μg/mL leupeptin, and 40 mM Pefabloc. Insoluble material was precipitated by ultracentrifugation at 100000g and 4 °C for 1 h. The supernate was directly used for its kinase activity. Total protein concentration was estimated at 11 mg mL–1 by a BCA colorimetric assay. 15N,13C-tau-S262A was dissolved at 10 μM in 2.5 mL of phosphorylation buffer, containing 40 mM HEPES, pH 7.3, 2 mM MgCl2, 5 mM EGTA, 2 mM DTT, 2 mM ATP, and 1 μM OA, supplemented with a Complete protease-inhibitor cocktail. The protein was incubated with 500 μL of brain extract at 37 °C for 24 h. The enzymatic reaction was stopped by heating the mixture at 75 °C for 15 min followed by centrifugation at 16000g for 20 min. Then, the supernate was buffer-exchanged in 50 mM ammonium bicarbonate, and the protein was lyophilized. Qualitative evaluation of protein phosphorylation was performed by SDS-PAGE, and then the phosphorylation sites were determined by NMR analyses, as described above.
In Vitro Aggregation Assay and Transmission Electron Microscopy (TEM)
For aggregation assays, unphosphorylated tau was mixed with heparin at a ratio of 4:1, respectively, in a buffer containing 25 mM NaH2PO4/Na2HPO4, pH 6.6, 25 mM NaCl, 2.5 mM EDTA, and 0.33 mM DTT at a final protein concentration of 5 μM. Reactions carried out in the absence of heparin were used as a negative control. ThT was prepared freshly at 5 mM and added at a final concentration of 50 μM before measurements. ThT emission was detected at 490 nm after excitation at 440 nm in 96-well plates on a plate reader (PHERAStar, BMG LABTECH GmbH, Ortenberg, Germany). Kinetic curves were fitted to a plateau followed by a single exponential association equation (eq 1).
| 1 |
where t0 is the lag-time when nucleation occurs, k is the association rate constant, Y0 is the value of the plateau at the initial step when t ≤ t0, Ymax is the maximal value of the plateau at the end of the elongation step. Inhibition of aggregation by CLR01 was tested under the same conditions by diluting a 10 mM stock solution of the molecular tweezer in triply distilled water into the reaction mixture at different concentrations. Concentrations of CLR01 ranging from 1.25 to 20 μM were evaluated by 20 min step kinetics for a total of 92 h.
IC50 values were derived by fitting the percentage of inhibition (eq 2) as a function of CLR01 concentration.
| 2 |
where k is the association rate constants at a given concentration of CLR01 and k0 is the rate constant in the absence of CLR01.
After completion of the aggregation, the morphology of the protein alone or of Tau:CLR01 mixtures was examined by TEM. For these experiments, 10-μL aliquots of the aggregation reactions were applied to 400-meshed hexagonal Formvar-coated copper grids for 90 s. The liquid was wicked off using a filter paper, and the grids were washed thrice with ultrapure water, stained with 10 μL of 2% uranyl acetate for 60 s, and wicked off using a filter paper. TEM was performed using a HITACHI H7500 microscope at 80 kV.
Aggregation assays using tau phosphorylated by RBE (p-tau(S262A)RBE) were performed at 30 μM of the protein in a buffer containing 100 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.9, 2 mM EGTA, 1 mM MgCl2, 20 mM NaF, 0.33 mM DTT, and 50 μM ThT. p-tau(S262A)RBE was incubated without agitation for 5 days at 37 °C in the absence or presence of 7.5, 15, 22.5, 30, 60, or 120 μM CLR01, and aggregation kinetics was followed by ThT fluorescence. At the end of incubation, samples were analyzed by TEM as described above.
Preparation of Tau Seeds
To evaluate the seeding capability of tau, the different variants either were freshly dissolved and tested immediately or incubated at 37 °C for 5–10 days to form fibrils before addition to biosensor cells. Unphosphorylated tau was incubated in the presence of heparin or in the absence of heparin as a negative control, whereas tau phosphorylated by ERK2 (p-TauERK) or p-tau(S262A)RBE was incubated in the absence of heparin. Before preparing transduction complexes, the proteins were sonicated for 10 min in a bath sonicator. Transduction complexes were made by combining a solution of 8.75 μL Opti-MEM (Gibco) and 1.25 μL of lipofectamine 2000 (Invitrogen) with a solution/suspension of tau seeds in Opti-MEM at the desired concentration, determined using the BCA colorimetric assay for a total volume of 20 μL per well. The concentration was calculated based on tau monomer. Transduction complexes were incubated at RT for 20 min before adding to cells.
Transduction of Tau Seeds in Biosensor Cells
A monoclonal, FRET-based biosensor cell line58 was maintained as described in instructions from ATCC (CRL-3275). Briefly, the cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, ATCC-30-2002) supplemented with 2 mM l-alanyl-l-glutamine and 10% fetal bovine serum (FBS) under a 5% CO2 atmosphere at 37 °C. These cells stably express the tau MTBD containing the disease-associated substitution P301S and fused to either cyan fluorescent protein (CFP) or yellow fluorescent protein (YFP).58 The cells were plated at 35 000 cells per well in 96-well plates (Sigma-Aldrich). Eighteen hours later, at ∼60% confluency, the cells were transduced with tau seeds and incubated with the seeds for 24–48 h prior to flow cytometry analysis.
FRET Flow Cytometry
Cells were incubated in 50 μL of TrypLE (Cat. 12605010, ThermoFisher) per well for 5 min at 37 °C followed by addition of 150 μL of DMEM supplemented with 2 mM l-alanyl-l-glutamine and 10% fetal bovine serum to stop the reaction. The cells were transferred to a round-bottom 96-well plate (Cat. 07-200-89, ThermoFisher), centrifuged at 400g for 5 min, and then resuspended in 200 μL of flow cytometry buffer comprising 1% FBS and 1-mM EDTA in Hank’s balanced salt solution (GIBCO). A SORP BD Biosciences HTLSRII analytic flow cytometer was used for FRET flow cytometry measurements. To measure CFP and FRET, cells were excited using a 405 nm laser, and fluorescence was captured using 405/50 nm and 525/50 nm filters, respectively. To measure YFP, cells were excited using a 488 nm laser, and fluorescence was captured with a 525/50 nm filter. To quantify FRET, we used a gating strategy similar to that described previously.58 Briefly, CFP bleed-through into the YFP and FRET channels was compensated for using FACSDiva 8.0.1 software from BD Biosciences. Because some YFP-only cells exhibit emission in the FRET channel, we introduced an additional gate to exclude from analysis cells that generate a false-positive signal in the FRET channel (i.e., false FRET gate). Subsequently, we created a final bivariate plot of FRET vs CFP and introduced a triangular gate to assess the number of FRET-positive cells. This FRET gate was adjusted to biosensor cells treated with lipofectamine but no tau seeds, which are thus FRET-negative. This allows for direct visualization of sensitized acceptor emission arising from excitation of the CFP donor at 405 nm. The integrated FRET density, defined as the percentage of FRET-positive cells multiplied by the median fluorescence intensity of FRET-positive cells, was used for all analyses. For each experiment, 20 000 cells per replicate were analyzed, and each condition was analyzed in quadruplicate. Data analysis was performed using Flow Jo v10 (Treestar).
Native PAGE/Western Blots
Recombinant protein aliquots from seeding experiments were mixed with 4X NativePAGE sample buffer, containing 50 mM Bis-Tris, 6 N HCl, 50 mM NaCl, 10% v/v glycerol, and 0.001% w/v Ponceau S, pH 7.2. Ten microliters sample per well were loaded onto Novex NativePAGE 3–12%-gradient Bis-Tris gels (Thermo Fisher). The proteins were fractionated for 90–115 min at 150 V constant voltage using a XCell SureLock Mini-Cell system (ThermoFisher). The proteins then were transferred to a PVDF membrane for 1 h at 25 V constant voltage using a XCell II Blot Module (ThermoFisher). Membranes then were incubated in 20 mL of 8% acetic acid for 15 min to fix the proteins. The blots were probed with antihuman tau mAb HT7 diluted 1:1000 in 5% (v/v) nonfat dry milk in TBST. ECL Plus (GE Healthcare) was used to visualize the bands in a Syngene PXi multiapplication gel imaging system, and band intensity was quantified densitometrically using ImageJ.59,60
Surface Plasmon Resonance
Biotinylated heparin was prepared by reaction with amine-PEG3-Biotin (Pierce, Rockford, IL) in the presence of NaCNBH3 as described previously.61 The biotinylated heparin was immobilized on a streptavidin (SA) chip (GE Healthcare) based on the manufacturer’s protocol. Briefly, 20 μL of 0.1 mg mL–1 of the biotinylated heparin in HBS-EP running buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, and 0.005% surfactant P20, pH 7.4) were injected over flow cells 2, 3, and 4 of the SA chip at a flow rate of 10 μL/min. The successful immobilization of heparin was confirmed by the observation of a ∼200-resonance-unit (RU) increase in the sensor chip. The control flow cell was prepared by a 1 min injection of saturated biotin. Tau was dissolved in HBS-EP buffer, and different dilutions of the protein solution were injected at a flow rate of 30 μL/min. At the end of the sample injection, the same buffer was flowed over the sensor surface to facilitate dissociation. After a 3 min dissociation period, the sensor surface was regenerated by injection of 30 μL of 2 M NaCl. The response sensograms were monitored as a function of time at 25 °C. For testing the inhibition of the tau–heparin interaction by CLR01, 500 nM of tau was mixed with 0, 5, 12.5, 25, or 50 μM CLR01. Then, each mixture was injected over the heparin chip at a flow rate of 30 μL/min. At the end of the sample injection, the same buffer was flowed over the sensor surface to facilitate dissociation and the sensor surface was regenerated as described above. The highest response was obtained for calculation of normalized binding inhibition.
NMR Spectroscopy
Spectra were acquired at 293 K on a Bruker 900-MHz spectrometer equipped with a triple-resonance cryogenic probe head. 15N-Lys-labeled tau was dissolved at 200 μM in NMR sample buffer, containing 50 mM NaH2PO4/Na2HPO4, pH 6.4, 25 mM NaCl, 2.5 mM EDTA, 1 mM DTT, and 10% D2O in 3 mm NMR tubes. The proton frequency was calibrated with 1 mM sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 as a reference. 1H–15N HSQC spectra were recorded for 15N- and 15N,13C-labeled samples with 64 scans per increment, and 3072 and 512 points in the 1H and 15N dimensions, respectively.
The phosphorylation patterns of p-tauERK, p-tauRBE, and p-tau(S262A)RBE were analyzed as described previously.56,57 The patterns (Table 1) were similar to those characterized previously by Despres et al.30 and Qi et al.29 For 15N,13C-Lys,13C-labeled p-tau assignment, a three-dimensional HN(CO)CACB experiment was performed with 16 scans per increment. Spectral widths were 16.0, 23.0, and 61.0 ppm in the 1H, 15N, and 13C dimensions with 2048, 64, and 120 sample points, respectively.
For the NMR mapping of protein resonance intensities upon CLR01 binding, the intensity of each resonance in the 1H–15N HSQC spectrum in the presence of CLR01 (I) was determined and compared to the resonance intensity in the absence of CLR01 (I0). The normalized intensities I/I0 were plotted as a function of the protein sequence.
For aggregation assays, protein concentrations were normalized based on their 1D proton spectra (zggpw5, Bruker) acquired at 600 MHz with 64 scans, 16 dummy scans and 16 384 points for a spectral width of 13.97 ppm centered on 4.70 ppm.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- DAPI
4′,6-diamidino-2-phenylindole
- ERK2
extracellular signal-regulated kinase 2
- HSQC
heteronuclear single-quantum coherence
- MEK3
mitogen-activated protein-kinase kinase 3
- MTBD
microtubule-binding domain
- NFTs
neurofibrillary tangles
- PHF
paired helical filaments
- PTM
posttranslational modification
- RBE
rat brain extract
- SPR
surface plasmon resonance
- TEM
transmission electron microscopy
- ThT
thioflavin T
- TOCSY
total correlation spectroscopy
- FRET
fluorescence-resonance energy transfer
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00325.
Supplementary Figure 1: Morphological examination of p-tau(S262A)RBE fibrils. Supplementary Figure 2: Comparison of fitting the plateau vs the rate constant k. Supplementary Figure 3: Representative TEM images taken at the end of the aggregation reactions. Supplementary Figure 4: Transmission electron microscopy of p-tau(S262A)RBE fibrils in the absence or presence of CLR01. Supplementary Figure 5: Titration of full-length tau and the F4 fragment with CLR01. Supplementary Figure 6: Mean charge along the tau sequence. Supplementary Figure 7: Phosphorylation of 15N-lysine, 13C-tau by ERK2. Supplementary Figure 8: 1H–15N HSQC spectra of p-tau(S262A)RBE phosphorylated by a rat brain extract. Supplementary Figure 9: Comparison of signal intensity variations in p-tauERK, p-tau(S262A)RBE, and nonphosphorylated tau. Supplementary Figure 10: Ratio of signal intensity of phosphorylated tau (PDF)
Author Present Address
■ (G.L.) Laboratoire d’Ingénierie des Systèmes Biologiques et des Procédés, University of Toulouse, CNRS, INRA, INSA, Toulouse, France.
Author Contributions
◆ C.D. and J.D. contributed equally.
The NMR facilities were funded by the Région Nord, CNRS, Pasteur Institute of Lille, European Community (FEDER), French Research Ministry, and the University of Sciences and Technologies of Lille. We acknowledge support from the TGE RMN THC (FR-3050, C.S.-N., C.D., F.X.C., I.L.), FRABio (FR 3688, C.S.-N., C.D., F.X.C., I.L.), Lille NMR, and RPE Health and Biology core facility. Our research was supported by grants from the Laboratory of Excellence DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to Alzheimer’s disease, C.S.N., C.D., F.X.C., I.L.), NIH/NIA Grants R01 AG050721 and RF1 AG054000 (G.B.), a voucher from NIH/NCRR UL1TR000124 – UCLA Clinical and Translational Science Institute (G.B.), Cure Alzheimer’s Fund Grant 20152631 (G.B.), and Cure PSP grant 600-6-15 (G.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- Ballatore C.; Lee V. M.-Y.; Trojanowski J. Q. (2007) Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 8, 663–672. 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Götz J.; Ittner A.; Ittner L. M. (2012) Tau-Targeted Treatment Strategies in Alzheimer’s Disease: Tau-Targeted Treatment of Alzheimer’s Disease. Br. J. Pharmacol. 165, 1246–1259. 10.1111/j.1476-5381.2011.01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M.; Avila J. (2014) New Perspectives on the Role of Tau in Alzheimer’s Disease. Implications for Therapy. Biochem. Pharmacol. 88, 540–547. 10.1016/j.bcp.2014.01.013. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Mandelkow E. (2016) Tau in Physiology and Pathology. Nat. Rev. Neurosci. 17, 5–21. 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Buée L.; Bussière T.; Buée-Scherrer V.; Delacourte A.; Hof P. R. (2000) Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Rev. 33, 95–130. 10.1016/S0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Braak H.; Braak E. (1991) Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 82, 239–259. 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H.; Alafuzoff I.; Arzberger T.; Kretzschmar H.; Del Tredici K. (2006) Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 112, 389–404. 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustinack J. C.; Schneider A.; Mandelkow E. M.; Hyman B. T. (2002) Specific Tau Phosphorylation Sites Correlate with Severity of Neuronal Cytopathology in Alzheimer’s Disease. Acta Neuropathol. 103, 26–35. 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Kimura T.; Ono T.; Takamatsu J.; Yamamoto H.; Ikegami K.; Kondo A.; Hasegawa M.; Ihara Y.; Miyamoto E.; Miyakawa T. (2004) Sequential Changes of Tau-Site-Specific Phosphorylation during Development of Paired Helical Filaments. Dementia Geriatr. Cognit. Disord. 7, 177–181. 10.1159/000106875. [DOI] [PubMed] [Google Scholar]
- Braak H.; Braak E. (1995) Staging of Alzheimer’s Disease-Related Neurofibrillary Changes. Neurobiol. Aging 16, 271–278. 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Gerson J. E.; Castillo-Carranza D. L.; Kayed R. (2014) Advances in Therapeutics for Neurodegenerative Tauopathies: Moving toward the Specific Targeting of the Most Toxic Tau Species. ACS Chem. Neurosci. 5, 752–769. 10.1021/cn500143n. [DOI] [PubMed] [Google Scholar]
- Landau M.; Sawaya M. R.; Faull K. F.; Laganowsky A.; Jiang L.; Sievers S. A.; Liu J.; Barrio J. R.; Eisenberg D. (2011) Towards a Pharmacophore for Amyloid. PLoS Biol. 9, e1001080. 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriman J.; Serpell L. C.; Oberg K. A.; Fink A. L.; Goedert M.; Crowther R. A. (2003) Tau Filaments from Human Brain and from in Vitro Assembly of Recombinant Protein Show Cross-Beta Structure. Proc. Natl. Acad. Sci. U. S. A. 100, 9034–9038. 10.1073/pnas.1530287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S.; Davies P.; Mandelkow E. (2004) Tau Paired Helical Filaments from Alzheimer’s Disease Brain and Assembled in Vitro Are Based on Beta-Structure in the Core Domain. Biochemistry 43, 1694–1703. 10.1021/bi0357006. [DOI] [PubMed] [Google Scholar]
- Wischik C. M.; Novak M.; Edwards P. C.; Klug A.; Tichelaar W.; Crowther R. A. (1988) Structural Characterization of the Core of the Paired Helical Filament of Alzheimer Disease. Proc. Natl. Acad. Sci. U. S. A. 85, 4884–4888. 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick A. W. P.; Falcon B.; He S.; Murzin A. G.; Murshudov G.; Garringer H. J.; Crowther R. A.; Ghetti B.; Goedert M.; Scheres S. H. W. (2017) Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 547, 185–190. 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S.; Medalsy I. D.; Mandelkow E.; Müller D. J. (2013) The Fuzzy Coat of Pathological Human Tau Fibrils Is a Two-Layered Polyelectrolyte Brush. Proc. Natl. Acad. Sci. U. S. A. 110, E313–E321. 10.1073/pnas.1212100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G.; Cairns N. J.; Crowther R. A. (1992) Tau Proteins of Alzheimer Paired Helical Filaments: Abnormal Phosphorylation of All Six Brain Isoforms. Neuron 8, 159–168. 10.1016/0896-6273(92)90117-V. [DOI] [PubMed] [Google Scholar]
- Köpke E.; Tung Y. C.; Shaikh S.; Alonso A. C.; Iqbal K.; Grundke-Iqbal I. (1993) Microtubule-Associated Protein Tau. Abnormal Phosphorylation of a Non-Paired Helical Filament Pool in Alzheimer Disease. J. Biol. Chem. 268, 24374–24384. [PubMed] [Google Scholar]
- Ksiezak-Reding H.; Liu W. K.; Yen S. H. (1992) Phosphate Analysis and Dephosphorylation of Modified Tau Associated with Paired Helical Filaments. Brain Res. 597, 209–219. 10.1016/0006-8993(92)91476-U. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M.; Hasegawa M.; Takio K.; Suzuki M.; Yoshida H.; Watanabe A.; Titani K.; Ihara Y. (1995) Hyperphosphorylation of Tau in PHF. Neurobiol. Aging 16, 365–371. 10.1016/0197-4580(95)00027-C. [DOI] [PubMed] [Google Scholar]; discussion 371–380.
- Morishima-Kawashima M.; Hasegawa M.; Takio K.; Suzuki M.; Yoshida H.; Titani K.; Ihara Y. (1995) Proline-Directed and Non-Proline-Directed Phosphorylation of PHF-Tau. J. Biol. Chem. 270, 823–829. 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- Hanger D. P.; Betts J. C.; Loviny T. L.; Blackstock W. P.; Anderton B. H. (1998) New Phosphorylation Sites Identified in Hyperphosphorylated Tau (Paired Helical Filament-Tau) from Alzheimer’s Disease Brain Using Nanoelectrospray Mass Spectrometry. J. Neurochem. 71, 2465–2476. 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- Hanger D. P.; Byers H. L.; Wray S.; Leung K.-Y.; Saxton M. J.; Seereeram A.; Reynolds C. H.; Ward M. A.; Anderton B. H. (2007) Novel Phosphorylation Sites in Tau from Alzheimer Brain Support a Role for Casein Kinase 1 in Disease Pathogenesis. J. Biol. Chem. 282, 23645–23654. 10.1074/jbc.M703269200. [DOI] [PubMed] [Google Scholar]
- Hanger D. P.; Hughes K.; Woodgett J. R.; Brion J. P.; Anderton B. H. (1992) Glycogen Synthase Kinase-3 Induces Alzheimer’s Disease-like Phosphorylation of Tau: Generation of Paired Helical Filament Epitopes and Neuronal Localisation of the Kinase. Neurosci. Lett. 147, 58–62. 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Biernat J.; Mandelkow E. M.; Schröter C.; Lichtenberg-Kraag B.; Steiner B.; Berling B.; Meyer H.; Mercken M.; Vandermeeren A.; Goedert M. (1992) The Switch of Tau Protein to an Alzheimer-like State Includes the Phosphorylation of Two Serine-Proline Motifs Upstream of the Microtubule Binding Region. EMBO J. 11, 1593–1597. 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Jakes R.; Crowther R. A.; Cohen P.; Vanmechelen E.; Vandermeeren M.; Cras P. (1994) Epitope Mapping of Monoclonal Antibodies to the Paired Helical Filaments of Alzheimer’s Disease: Identification of Phosphorylation Sites in Tau Protein. Biochem. J. 301, 871–877. 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A.; Biernat J.; von Bergen M.; Mandelkow E.; Mandelkow E. M. (1999) Phosphorylation That Detaches Tau Protein from Microtubules (Ser262, Ser214) Also Protects It against Aggregation into Alzheimer Paired Helical Filaments. Biochemistry 38, 3549–3558. 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- Qi H.; Prabakaran S.; Cantrelle F.-X.; Chambraud B.; Gunawardena J.; Lippens G.; Landrieu I. (2016) Characterization of Neuronal Tau Protein as a Target of Extracellular Signal-Regulated Kinase. J. Biol. Chem. 291, 7742–7753. 10.1074/jbc.M115.700914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despres C.; Byrne C.; Qi H.; Cantrelle F.-X.; Huvent I.; Chambraud B.; Baulieu E.-E.; Jacquot Y.; Landrieu I.; Lippens G.; Smet-Nocca C. (2017) Identification of the Tau Phosphorylation Pattern That Drives Its Aggregation. Proc. Natl. Acad. Sci. U. S. A. 114, 9080–9085. 10.1073/pnas.1708448114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik C. M.; Novak M.; Thogersen H. C.; Edwards P. C.; Runswick M. J.; Jakes R.; Walker J. E.; Milstein C.; Roth M.; Klug A. (1988) Isolation of a Fragment of Tau Derived from the Core of the Paired Helical Filament of Alzheimer Disease. Proc. Natl. Acad. Sci. U. S. A. 85, 4506–4510. 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard J. E.; Horsley D.; Wischik C. M.; Harrington C. R. (2017) Assays for the Screening and Characterization of Tau Aggregation Inhibitors. Methods Mol. Biol. 1523, 129–140. 10.1007/978-1-4939-6598-4_8. [DOI] [PubMed] [Google Scholar]
- Novak M.; Kabat J.; Wischik C. M. (1993) Molecular Characterization of the Minimal Protease Resistant Tau Unit of the Alzheimer’s Disease Paired Helical Filament. EMBO J. 12, 365–370. 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik C. M.; Edwards P. C.; Lai R. Y.; Roth M.; Harrington C. R. (1996) Selective Inhibition of Alzheimer Disease-like Tau Aggregation by Phenothiazines. Proc. Natl. Acad. Sci. U. S. A. 93, 11213–11218. 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille H.; Drewes G.; Biernat J.; Mandelkow E. M.; Mandelkow E. (1992) Alzheimer-like Paired Helical Filaments and Antiparallel Dimers Formed from Microtubule-Associated Protein Tau in Vitro. J. Cell Biol. 118, 573–584. 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M.; Jakes R.; Spillantini M. G.; Hasegawa M.; Smith M. J.; Crowther R. A. (1996) Assembly of Microtubule-Associated Protein Tau into Alzheimer-like Filaments Induced by Sulphated Glycosaminoglycans. Nature 383, 550–553. 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- Pérez M.; Valpuesta J. M.; Medina M.; Montejo de Garcini E.; Avila J. (1996) Polymerization of Tau into Filaments in the Presence of Heparin: The Minimal Sequence Required for Tau-Tau Interaction. J. Neurochem. 67, 1183–1190. 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- Kampers T.; Friedhoff P.; Biernat J.; Mandelkow E. M.; Mandelkow E. (1996) RNA Stimulates Aggregation of Microtubule-Associated Protein Tau into Alzheimer-like Paired Helical Filaments. FEBS Lett. 399, 344–349. 10.1016/S0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Wilson D. M.; Binder L. I. (1997) Free Fatty Acids Stimulate the Polymerization of Tau and Amyloid Beta Peptides. In Vitro Evidence for a Common Effector of Pathogenesis in Alzheimer’s Disease. Am. J. Pathol. 150, 2181–2195. [PMC free article] [PubMed] [Google Scholar]
- Chirita C. N.; Necula M.; Kuret J. (2003) Anionic Micelles and Vesicles Induce Tau Fibrillization in Vitro. J. Biol. Chem. 278, 25644–25650. 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- Nanavaty N.; Lin L.; Hinckley S. H.; Kuret J. (2017) Detection and Quantification Methods for Fibrillar Products of In Vitro Tau Aggregation Assays. Methods Mol. Biol. 1523, 101–111. 10.1007/978-1-4939-6598-4_6. [DOI] [PubMed] [Google Scholar]
- Sibille N.; Sillen A.; Leroy A.; Wieruszeski J. M.; Mulloy B.; Landrieu I.; Lippens G. (2006) Structural Impact of Heparin Binding to Full-Length Tau as Studied by NMR Spectroscopy. Biochemistry 45, 12560–12572. 10.1021/bi060964o. [DOI] [PubMed] [Google Scholar]
- Eschmann N. A.; Georgieva E. R.; Ganguly P.; Borbat P. P.; Rappaport M. D.; Akdogan Y.; Freed J. H.; Shea J.-E.; Han S. (2017) Signature of an Aggregation-Prone Conformation of Tau. Sci. Rep. 7, 44739. 10.1038/srep44739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S.; Rhoades E. (2012) Identification of an Aggregation-Prone Structure of Tau. J. Am. Chem. Soc. 134, 16607–16613. 10.1021/ja305206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuret J.; Congdon E. E.; Li G.; Yin H.; Yu X.; Zhong Q. (2005) Evaluating Triggers and Enhancers of Tau Fibrillization. Microsc. Res. Tech. 67, 141–155. 10.1002/jemt.20187. [DOI] [PubMed] [Google Scholar]
- Fichou Y.; Vigers M.; Goring A. K.; Eschmann N. A.; Han S. (2018) Heparin-Induced Tau Filaments Are Structurally Heterogeneous and Differ from Alzheimer’s Disease Filaments. Chem. Commun. 54, 4573–4576. 10.1039/C8CC01355A. [DOI] [PubMed] [Google Scholar]
- Fichou Y.; Lin Y.; Rauch J. N.; Vigers M.; Zeng Z.; Srivastava M.; Keller T. J.; Freed J. H.; Kosik K. S.; Han S. (2018) Cofactors Are Essential Constituents of Stable and Seeding-Active Tau Fibrils. Proc. Natl. Acad. Sci. U. S. A. 115, 13234–13239. 10.1073/pnas.1810058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D. W.; Kaufman S. K.; DeVos S. L.; Sharma A. M.; Mirbaha H.; Li A.; Barker S. J.; Foley A. C.; Thorpe J. R.; Serpell L. C.; Miller T. M.; Grinberg L. T.; Seeley W. W.; Diamond M. I. (2014) Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 82, 1271–1288. 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe I. (2016) Neurodegenerative Disease: New in Vivo Evidence That Different Tau Strains Cause Different Diseases. Nat. Rev. Neurol. 12, 677. 10.1038/nrneurol.2016.178. [DOI] [PubMed] [Google Scholar]
- Crowther R. A.; Olesen O. F.; Smith M. J.; Jakes R.; Goedert M. (1994) Assembly of Alzheimer-like Filaments from Full-Length Tau Protein. FEBS Lett. 337, 135–138. 10.1016/0014-5793(94)80260-2. [DOI] [PubMed] [Google Scholar]
- Alonso A.; Zaidi T.; Novak M.; Grundke-Iqbal I.; Iqbal K. (2001) Hyperphosphorylation Induces Self-Assembly of Tau into Tangles of Paired Helical Filaments/Straight Filaments. Proc. Natl. Acad. Sci. U. S. A. 98, 6923–6928. 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar A.; Bitan G. (2014) Disrupting Self-Assembly and Toxicity of Amyloidogenic Protein Oligomers by “Molecular Tweezers” - from the Test Tube to Animal Models. Curr. Pharm. Des. 20, 2469–2483. 10.2174/13816128113199990496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader T.; Bitan G.; Klärner F.-G. (2016) Molecular Tweezers for Lysine and Arginine - Powerful Inhibitors of Pathologic Protein Aggregation. Chem. Commun. 52, 11318–11334. 10.1039/C6CC04640A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman S. K.; Sanders D. W.; Thomas T. L.; Ruchinskas A. J.; Vaquer-Alicea J.; Sharma A. M.; Miller T. M.; Diamond M. I. (2016) Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 92, 796–812. 10.1016/j.neuron.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbiersky P.; Bastkowski F.; Klarner F. G.; Schrader T. (2008) Molecular Clip and Tweezer Introduce New Mechanisms of Enzyme Inhibition. J. Am. Chem. Soc. 130, 9824–9828. 10.1021/ja801441j. [DOI] [PubMed] [Google Scholar]
- Qi H., Despres C., Prabakaran S., Cantrelle F.-X., Chambraud B., Gunawardena J., Lippens G., Smet-Nocca C., and Landrieu I. (2017) The Study of Posttranslational Modifications of Tau Protein by Nuclear Magnetic Resonance Spectroscopy: Phosphorylation of Tau Protein by ERK2 Recombinant Kinase and Rat Brain Extract, and Acetylation by Recombinant Creb-Binding Protein, in Tau Protein: Methods and Protocols (Smet-Nocca C., Ed.) pp 179–213, Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- Danis C.; Despres C.; Bessa L. M.; Malki I.; Merzougui H.; Huvent I.; Qi H.; Lippens G.; Cantrelle F.-X.; Schneider R.; Hanoulle X.; Smet-Nocca C.; Landrieu I. (2016) Nuclear Magnetic Resonance Spectroscopy for the Identification of Multiple Phosphorylations of Intrinsically Disordered Proteins. J. Visualized Exp. 118, e55001. 10.3791/55001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B.; Furman J. L.; Mahan T. E.; Yamasaki T. R.; Mirbaha H.; Eades W. C.; Belaygorod L.; Cairns N. J.; Holtzman D. M.; Diamond M. I. (2014) Proteopathic Tau Seeding Predicts Tauopathy in Vivo. Proc. Natl. Acad. Sci. U. S. A. 111, E4376–4385. 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramoff M. D.; Magalhães P. J.; Ram S. J. (2004) Image Processing with ImageJ. Biophotonics Int. 11, 36–42. [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. Y.; Zhao J.; Liu X.; Fraser K.; Lin L.; Zhang X.; Zhang F.; Dordick J. S.; Linhardt R. J. (2017) Interaction of Zika Virus Envelope Protein with Glycosaminoglycans. Biochemistry 56, 1151–1162. 10.1021/acs.biochem.6b01056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B.; Diamond M. I. (2012) Cellular Mechanisms of Protein Aggregate Propagation. Curr. Opin. Neurol. 25, 721–726. 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B.; Diamond M. I. (2014) Prion-like Properties of Tau Protein: The Importance of Extracellular Tau as a Therapeutic Target. J. Biol. Chem. 289, 19855–19861. 10.1074/jbc.R114.549295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfoury N.; Holmes B. B.; Jiang H.; Holtzman D. M.; Diamond M. I. (2012) Trans-Cellular Propagation of Tau Aggregation by Fibrillar Species. J. Biol. Chem. 287, 19440–19451. 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H.; Chen D.; Morazova O. A.; Ruff K. M.; Sharma A. M.; Liu X.; Goodarzi M.; Pappu R. V.; Colby D. W.; Mirzaei H.; Joachimiak L. A.; Diamond M. I. (2018) Inert and Seed-Competent Tau Monomers Suggest Structural Origins of Aggregation. eLife 7, e36584. 10.7554/eLife.36584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lim S.; Haque M. M.; Ryoo N.; Hong H. S.; Rhim H.; Lee D.-E.; Chang Y.-T.; Lee J.-S.; Cheong E.; Kim D. J.; Kim Y. K. (2015) Identification of Disulfide Cross-Linked Tau Dimer Responsible for Tau Propagation. Sci. Rep. 5, 15231. 10.1038/srep15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch J. N.; Chen J. J.; Sorum A. W.; Miller G. M.; Sharf T.; See S. K.; Hsieh-Wilson L. C.; Kampmann M.; Kosik K. S. (2018) Tau Internalization Is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci. Rep. 8, 6382. 10.1038/s41598-018-24904-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Zhang Z.; Lin X.; Beenken A.; Eliseenkova A. V.; Mohammadi M.; Linhardt R. J. (2009) Compositional Analysis of Heparin/Heparan Sulfate Interacting with Fibroblast Growth Factor.Fibroblast Growth Factor Receptor Complexes. Biochemistry 48, 8379–8386. 10.1021/bi9006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Liu X.; Kao C.; Zhang E.; Li Q.; Zhang F.; Linhardt R. J. (2016) Kinetic and Structural Studies of Interactions between Glycosaminoglycans and Langerin. Biochemistry 55, 4552–4559. 10.1021/acs.biochem.6b00555. [DOI] [PubMed] [Google Scholar]
- Cohen S. I. A.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2011) Nucleated Polymerization with Secondary Pathways. II. Determination of Self-Consistent Solutions to Growth Processes Described by Non-Linear Master Equations. J. Chem. Phys. 135, 065106. 10.1063/1.3608917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. (2013) Proliferation of Amyloid- 42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. U. S. A. 110, 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson L. P.; Smith N. W.; Dzyuba S. V. (2012) Dye-Binding Assays for Evaluation of the Effects of Small Molecule Inhibitors on Amyloid (Aβ) Self-Assembly. ACS Chem. Neurosci. 3, 807–819. 10.1021/cn300076x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. G.; Wu C.; Hannaberry E.; Watson M. D.; Shea J.-E.; Raleigh D. P. (2016) Analysis of the Amyloidogenic Potential of Pufferfish (Takifugu Rubripes) Islet Amyloid Polypeptide Highlights the Limitations of Thioflavin-T Assays and the Difficulties in Defining Amyloidogenicity. Biochemistry 55, 510–518. 10.1021/acs.biochem.5b01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonov I.; Korkotian E.; Born B.; Feldman Y.; Bitler A.; Rahimi F.; Li H.; Bitan G.; Sagi I. (2012) Zn2+-Aβ40 Complexes Form Metastable Quasi-Spherical Oligomers That Are Cytotoxic to Cultured Hippocampal Neurons. J. Biol. Chem. 287, 20555–20564. 10.1074/jbc.M112.344036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Lopes D. H.; Du Z.; Pang E. S.; Shanmugam A.; Lomakin A.; Talbiersky P.; Tennstaedt A.; McDaniel K.; Bakshi R.; Kuo P. Y.; Ehrmann M.; Benedek G. B.; Loo J. A.; Klarner F. G.; Schrader T.; Wang C.; Bitan G. (2011) Lysine-Specific Molecular Tweezers Are Broad-Spectrum Inhibitors of Assembly and Toxicity of Amyloid Proteins. J. Am. Chem. Soc. 133, 16958–16969. 10.1021/ja206279b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog G.; Shmueli M. D.; Levy L.; Engel L.; Gazit E.; Klärner F.-G.; Schrader T.; Bitan G.; Segal D. (2015) The Lys-Specific Molecular Tweezer, CLR01, Modulates Aggregation of the Mutant P53 DNA Binding Domain and Inhibits Its Toxicity. Biochemistry 54, 3729–3738. 10.1021/bi501092p. [DOI] [PubMed] [Google Scholar]
- Prabhudesai S.; Sinha S.; Attar A.; Kotagiri A.; Fitzmaurice A. G.; Lakshmanan R.; Ivanova M. I.; Loo J. A.; Klärner F.-G.; Schrader T.; Stahl M.; Bitan G.; Bronstein J. M. (2012) A Novel “Molecular Tweezer” Inhibitor of α-Synuclein Neurotoxicity in Vitro and in Vivo. Neurotherapeutics 9, 464–476. 10.1007/s13311-012-0105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes D. H. J.; Attar A.; Nair G.; Hayden E. Y.; Du Z.; McDaniel K.; Dutt S.; Bravo-Rodriguez K.; Mittal S.; Klärner F.-G.; Wang C.; Sanchez-Garcia E.; Schrader T.; Bitan G.; Bandmann H. (2015) Molecular Tweezers Inhibit Islet Amyloid Polypeptide Assembly and Toxicity by a New Mechanism. ACS Chem. Biol. 10, 1555–1569. 10.1021/acschembio.5b00146. [DOI] [PubMed] [Google Scholar]
- Jeganathan S.; von Bergen M.; Mandelkow E.-M.; Mandelkow E. (2008) The Natively Unfolded Character of Tau and Its Aggregation to Alzheimer-like Paired Helical Filaments. Biochemistry 47, 10526–10539. 10.1021/bi800783d. [DOI] [PubMed] [Google Scholar]
- Trzeciakiewicz H.; Esteves-Villanueva J. O.; Carlin N.; Martić S. (2015) Electrochemistry of Heparin Binding to Tau Protein on Au Surfaces. Electrochim. Acta 162, 24–30. 10.1016/j.electacta.2014.08.101. [DOI] [Google Scholar]
- Kamah A.; Huvent I.; Cantrelle F.-X.; Qi H.; Lippens G.; Landrieu I.; Smet-Nocca C. (2014) Nuclear Magnetic Resonance Analysis of the Acetylation Pattern of the Neuronal Tau Protein. Biochemistry 53, 3020–3032. 10.1021/bi500006v. [DOI] [PubMed] [Google Scholar]
- Dutt S.; Wilch C.; Gersthagen T.; Talbiersky P.; Bravo-Rodriguez K.; Hanni M.; Sánchez-García E.; Ochsenfeld C.; Klärner F.-G.; Schrader T. (2013) Molecular Tweezers with Varying Anions: A Comparative Study. J. Org. Chem. 78, 6721–6734. 10.1021/jo4009673. [DOI] [PubMed] [Google Scholar]
- Dutt S.; Wilch C.; Gersthagen T.; Wölper C.; Sowislok A. A.; Klärner F.-G.; Schrader T. (2013) Linker Effects on Amino Acid and Peptide Recognition by Molecular Tweezers: Recognition by Molecular Tweezers. Eur. J. Org. Chem. 2013, 7705–7714. 10.1002/ejoc.201301211. [DOI] [Google Scholar]
- Fauquant C.; Redeker V.; Landrieu I.; Wieruszeski J. M.; Verdegem D.; Laprevote O.; Lippens G.; Gigant B.; Knossow M. (2011) Systematic Identification of Tubulin-Interacting Fragments of the Microtubule-Associated Protein Tau Leads to a Highly Efficient Promoter of Microtubule Assembly. J. Biol. Chem. 286, 33358–33368. 10.1074/jbc.M111.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvent I.; Kamah A.; Cantrelle F.-X.; Barois N.; Slomianny C.; Smet-Nocca C.; Landrieu I.; Lippens G. (2014) A Functional Fragment of Tau Forms Fibers without the Need for an Intermolecular Cysteine Bridge. Biochem. Biophys. Res. Commun. 445, 299–303. 10.1016/j.bbrc.2014.01.161. [DOI] [PubMed] [Google Scholar]
- Nshanian M.; Lantz C.; Wongkongkathep P.; Schrader T.; Klärner F.-G.; Blümke A.; Despres C.; Ehrmann M.; Smet-Nocca C.; Bitan G.; Loo J. A. (2019) Native Top-Down Mass Spectrometry and Ion Mobility Spectrometry of the Interaction of Tau Protein with a Molecular Tweezer Assembly Modulator. J. Am. Soc. Mass Spectrom. 30, 16–23. 10.1007/s13361-018-2027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulic B.; Pickhardt M.; Schmidt B.; Mandelkow E.-M.; Waldmann H.; Mandelkow E. (2009) Development of Tau Aggregation Inhibitors for Alzheimer’s Disease. Angew. Chem., Int. Ed. 48, 1740–1752. 10.1002/anie.200802621. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Falcon B.; Murzin A. G.; Fan J.; Crowther R. A.; Goedert M.; Scheres S. H. (2019) Heparin-Induced Tau Filaments Are Polymorphic and Differ from Those in Alzheimer’s and Pick’s Diseases. eLife 8, e43584. 10.7554/eLife.43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Shepherd J. D.; Murphy M. P.; Golde T. E.; Kayed R.; Metherate R.; Mattson M. P.; Akbari Y.; LaFerla F. M. (2003) Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 39, 409–421. 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Attar A.; Ripoli C.; Riccardi E.; Maiti P.; Li Puma D. D.; Liu T.; Hayes J.; Jones M. R.; Lichti-Kaiser K.; Yang F.; Gale G. D.; Tseng C. H.; Tan M.; Xie C. W.; Straudinger J. L.; Klarner F. G.; Schrader T.; Frautschy S. A.; Grassi C.; Bitan G. (2012) Protection of Primary Neurons and Mouse Brain from Alzheimer’s Pathology by Molecular Tweezers. Brain 135, 3735–3748. 10.1093/brain/aws289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N. S.; Landrieu I.; Byrne C.; Kukic P.; Amniai L.; Cantrelle F.-X.; Wieruszeski J.-M.; Mancera R. L.; Jacquot Y.; Lippens G. (2015) A Phosphorylation-Induced Turn Defines the Alzheimer’s Disease AT8 Antibody Epitope on the Tau Protein. Angew. Chem., Int. Ed. 54, 6819–6823. 10.1002/anie.201501898. [DOI] [PubMed] [Google Scholar]
- Sibille N.; Huvent I.; Fauquant C.; Verdegem D.; Amniai L.; Leroy A.; Wieruszeski J. M.; Lippens G.; Landrieu I. (2012) Structural Characterization by Nuclear Magnetic Resonance of the Impact of Phosphorylation in the Proline-Rich Region of the Disordered Tau Protein. Proteins: Struct., Funct., Genet. 80, 454–462. 10.1002/prot.23210. [DOI] [PubMed] [Google Scholar]
- Schwalbe M.; Kadavath H.; Biernat J.; Ozenne V.; Blackledge M.; Mandelkow E.; Zweckstetter M. (2015) Structural Impact of Tau Phosphorylation at Threonine 231. Structure 23, 1448–1458. 10.1016/j.str.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Bier D.; Rose R.; Bravo-Rodriguez K.; Bartel M.; Ramirez-Anguita J. M.; Dutt S.; Wilch C.; Klärner F.-G.; Sanchez-Garcia E.; Schrader T.; Ottmann C. (2013) Molecular Tweezers Modulate 14–3-3 Protein-Protein Interactions. Nat. Chem. 5, 234–239. 10.1038/nchem.1570. [DOI] [PubMed] [Google Scholar]
- Acharya S.; Safaie B. M.; Wongkongkathep P.; Ivanova M. I.; Attar A.; Klärner F.-G.; Schrader T.; Loo J. A.; Bitan G.; Lapidus L. J. (2014) Molecular Basis for Preventing α-Synuclein Aggregation by a Molecular Tweezer. J. Biol. Chem. 289, 10727–10737. 10.1074/jbc.M113.524520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Du Z.; Maiti P.; Klärner F.-G.; Schrader T.; Wang C.; Bitan G. (2012) Comparison of Three Amyloid Assembly Inhibitors: The Sugar Scyllo-Inositol, the Polyphenol Epigallocatechin Gallate, and the Molecular Tweezer CLR01. ACS Chem. Neurosci. 3, 451–458. 10.1021/cn200133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Liu D.; Klärner F.-G.; Schrader T.; Bitan G.; Bowers M. T. (2015) Amyloid β-Protein Assembly: The Effect of Molecular Tweezers CLR01 and CLR03. J. Phys. Chem. B 119, 4831–4841. 10.1021/acs.jpcb.5b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.