

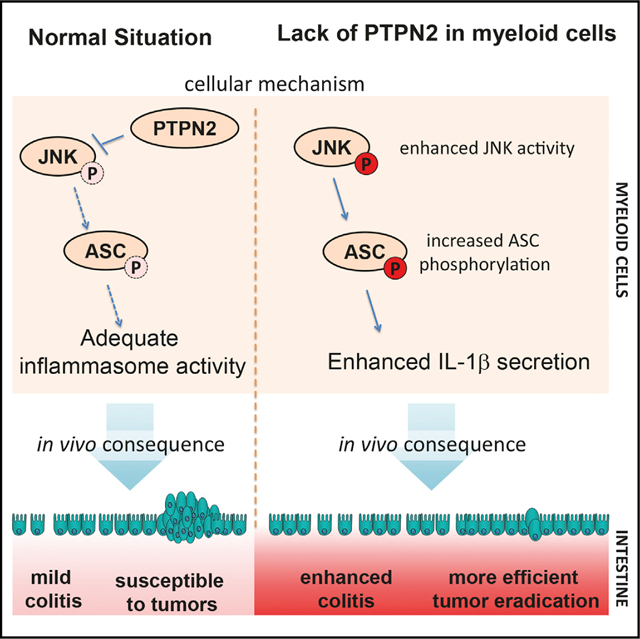
SUMMARY
Variants in the gene locus encoding protein tyrosine phosphatase non-receptor type 2 (PTPN2) are associated with inflammatory disorders, including inflammatory bowel diseases, rheumatoid arthritis, and type 1 diabetes. The anti-inflammatory role of PTPN2 is highlighted by the fact that PTPN2-deficient mice die a few weeks after birth because of systemic inflammation and severe colitis. However, the tissues, cells, and molecular mechanisms that contribute to this phenotype remain unclear. Here, we demonstrate that myeloid cell-specific deletion of PTPN2 in mice (PTPN2-LysMCre) promotes intestinal inflammation but protects from colitis-associated tumor formation in an IL-1β-dependent manner. Elevated levels of mature IL-1β production in PTPN2-LysMCre mice are a consequence of increased inflammasome assembly due to elevated phosphorylation of the inflammasome adaptor molecule ASC. Thus, we have identified a dual role for myeloid PTPN2 in directly regulating inflammasome activation and IL-1β production to suppress pro-inflammatory responses during colitis but promote intestinal tumor development.
In Brief
Spalinger et al. find that macrophage-specific loss of the IBD-risk gene PTPN2 promotes the onset of intestinal inflammation but protects from colitis-associated tumor formation in an inflammasome-and IL-1 ß-dependent manner.PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer
Graphical Abstract
INTRODUCTION
In recent decades, inflammatory bowel disease (IBD), with its sub-forms ulcerative colitis (UC) and Crohn’s disease (CD), has emerged as an increasing health problem in developed countries. There is evidence that genetic as well as environmental factors contribute to disease pathophysiology, and large genome-wide association studies have associated variants in more than 200 genetic loci with IBD (Wellcome Trust Case Control Consortium, 2007; Liu et al., 2015). Two of these variants are located within the gene encoding protein tyrosine phosphatase non-receptor type 2 (PTPN2; also known as T cell protein tyrosine phosphatase [TC-PTP]) (Wellcome Trust Case Control Consortium, 2007; Glas et al., 2012; Scharl et al., 2012), which negatively regulates pro-inflammatory signaling cascades (Aradi et al., 2015; Scharl et al., 2010, 2011, 2012). Of note, variants within the PTPN2 gene are associated not only with IBD but also with other inflammatory disorders, such as rheumatoid arthritis and type 1 diabetes (Wellcome Trust Case Control Consortium, 2007; Todd et al., 2007). Ptpn2-deficient (Ptpn2–/–) mice suffer from severe systemic inflammation and die 4–5 weeks after birth because of multiple organ dysfunction as well as severe intestinal and systemic inflammation (You-Ten et al., 1997; Heinonen et al., 2004). However, the tissue- and cell-specific contribution to this phenotype remains poorly understood. T cell-specific loss of PTPN2 results in increased numbers of effector and memory CD8+T cells, promotes autoimmune syndrome, and increases susceptibility to intestinal inflammation (Spalinger et al., 2015; Wiede et al., 2011). Loss of PTPN2 in intestinal epithelial cells (lECs) has profound effects on cytokine secretion and barrier function in vitro (Scharl et al., 2011) but does not significantly affect intestinal inflammation in vivo (Kasper et al., 2016). Cell culture studies in myeloid cells demonstrated that loss of PTPN2 enhances interferon (IFN)-γ, interleukin (IL)-6, and IL-1β secretion (Aradi et al., 2015; Scharl et al., 2010, 2011, 2012). Initial studies characterizing Ptpn2–/– mice identified that loss of PTPN2 impaired the bone marrow microenvironment (You-Ten et al., 1997) and made splenic macrophages more responsive to lipopolysaccharide (LPS) ex vivo (Heinonen et al., 2004). This suggests that myeloid cell function is disturbed in Ptpn2–/– mice, but the in vivo importance of PTPN2 function in myeloid cells has not been addressed thus far.
In the microbe-rich environment of the intestine, resident myeloid cells exert important immune-regulatory functions (i.e., removal of dead cells, elimination of invading pathogens, and the production of the anti-inflammatory cytokines TGF-β1 and IL-10; reviewed in Zigmond and Jung, 2013). During inflammation, blood monocyte-derived macrophages, which predominantly secrete pro-inflammatory cytokines (e.g., IL-1β, TNF, and IL-6), accumulate in the tissue (Rivollier et al., 2012; Zigmond and Jung, 2013).
The inflammasome products IL-1β and IL-18 are particularly important for initiating early immune reactions against invading pathogens (Martinon et al., 2002). Although pro-inflammatory in nature, the role of inflammasomes in intestinal pathologies remains controversial: IL-1β and IL-18 levels are highly increased in active lesions in IBD patients (Street et al., 2004; Li et al., 2004), and some studies have demonstrated that mice deficient in the inflammasome receptor NLRP3, the adaptor ASC, or IL-1β are protected from experimental colitis and induction and progression of colorectal carcinoma (CRC) (Bauer et al., 2010; Allen et al., 2010). However, other studies have shown the opposite, with NLRP3-, ASC-, and caspase-1-deficient mice being highly susceptible to colitis induction (Hirota et al., 2011; Zaki et al., 2010).
Here, we investigated the functional importance of PTPN2 in myeloid cells in vivo with respect to modulating intestinal inflammation and tumorigenesis. We demonstrate that PTPN2 is an important regulator of inflammasome activation, and its loss in myeloid cells promotes intestinal inflammation but, counterintuitively, protects from colitis-associated tumors in an inflammasome- and IL-1β-dependent manner.
RESULTS
Deletion of PTPN2 in Myeloid Cells
To address the role of PTPN2 in myeloid cells, mice with a floxed Ptpn2 gene (Ptpn2fl/fl mice [Spalinger et al., 2015], referred to as wild-type [WT] throughout) were crossed with mice expressing Cre-recombinase under the lysozyme promoter (Clausen et al., 1999) to generate mice lacking PTPN2 in myeloid cells (PTPN2-LysMCre mice). In contrast to CD3+ T cells, B220+ B cells, or F4/80+ macrophages from WT littermates, F4/80+ cells from PTPN2-LysMCre mice did not express PTPN2 (Figures S1A and S1B), confirming efficient deletion of Ptpn2 in macrophages. Similar results were obtained using immunofluo-rescent staining of intestinal samples for F4/80 and PTPN2 (Figure S1C). Next, we analyzed mononuclear cell populations in the lamina propria, mesenteric lymph nodes, and the spleen for the extent of Ptpn2 deletion. As expected, deletion was efficient in macrophages and monocytes, while there was no reduction of Ptpn2 expression among CD11c+ dendritic cells (Figures S2A and S2B). We did observe reduced Ptpn2 expression in sorted neutrophils (Figures S2A and S2B), which is consistent with published reports of the LysMCre mouse (Clausen et al., 1999). Despite the decrease of Ptpn2 in neutrophils, we continued our studies using the LysMCre strain because other macrophage-targeting Cre strains (i.e., the F4/80-Cre line) target only tissue-resident but not recently recruited monocyte-derived macrophages (Schaller et al., 2002; Abram et al., 2014). We also found reduced proportions of monocytes in the spleen of PTPN2-LysMCre mice but not in mesenteric lymph nodes or the lamina propria (Figures S2A and S2B). Furthermore, numbers of CD103+ dendritic cells (DCs) were enhanced in mesenteric lymph nodes but reduced in the lamina propria of PTPN2-LysMCre mice (Figures S2A and S2B). Of note, we did not observe differences in the development of bone marrow-derived macrophages (BMDMs) or major histocompatibility complex (MHC)-II induction upon treatment with LPS (Figure S2C).
Loss of PTPN2 in Myeloid Cells Aggravates Colitis
To identify the consequences of myeloid cell-specific deletion of PTPN2 on intestinal inflammation, we induced acute colitis in PTPN2-LysMCre mice and WT littermates by administration of 2.5% dextran sulfate sodium (DSS) in drinking water for 7 days. Although DSS-induced body weight loss and colon shortening did not differ significantly from WT mice (Figures 1A and 1B), PTPN2-LysMCre mice suffered from pronounced colitis, as observed in colonoscopy, by increased levels of myeloperoxidase (MPO), increased spleen weight, and increased histology scores in the terminal colon (Figures 1C–1F). PTPN2-LysMCre mice were also more susceptible to inflammation in the more disease-relevant, chronic DSS colitis model. These mice showed delayed recovery after each DSS cycle, increased spleen weight, increased MPO, and pronounced shortening of the colon. However, unlike in the acute DSS colitis model, histology scores were not different in PTPN2-LysMCre mice compared with WT mice in chronic DSS colitis (Figure S3). Although DSS colitis does involve macrophage activation, it is a T cell-independent chemical injury model. To assess the role of PTPN2 in myeloid cells in an immune cell-driven colitis model, we crossed PTPN2-LysMCre mice onto the IL-10–/– background. We observed that several inflammatory parameters, including incidences of rectal prolapse, endoscopy scores, MPO levels, spleen weight, and histology scores in adult (50 days old) and aged (150 days old) mice dramatically increased in IL-10–/– mice lacking PTPN2 in myeloid cells (IL-10xPTPN2-LysMCre mice) compared with IL-10–/– mice (Figure 2). Overall, these data clearly indicate that PTPN2-LysMCre mice demonstrate enhanced susceptibility to colitis in a variety of experimental models.
Figure 1. Loss of PTPN2 in Myeloid Cells Promotes Acute Intestinal Inflammation.
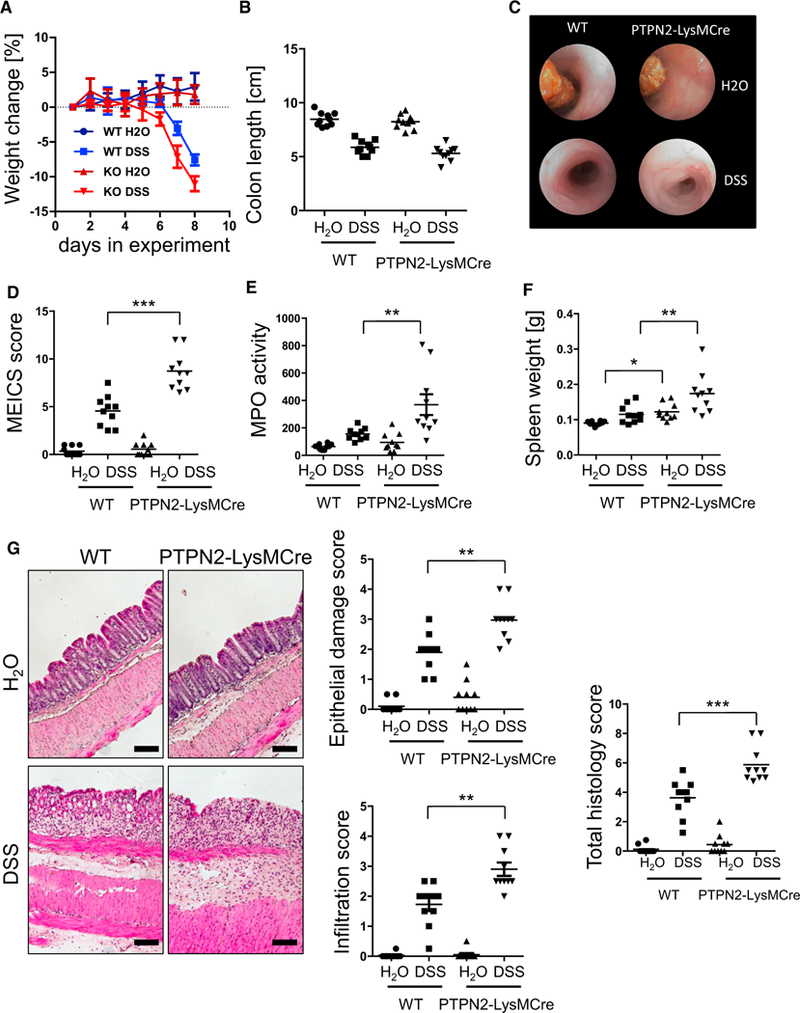
Acute colitis was induced in WT and PTPN2-LysMCre mice.
(A–G) Shown are weight change (A), colon length (B), representative pictures from colonoscopy (C) and respective statistical analysis (D), myeloperoxidase (MPO) activity (E), spleen weight (F), and representative pictures and scoring of epithelial damage and inflammatory infiltration in the terminal colon (G).
Depicted are pooled results from two independent experiments with four to six mice per group. Asterisks denote significant differences (*p < 0.05, **p < 0.01, and ***p < 0.001; Mann-Whitney U test with Bonferroni correction). Scale bars represent 100 μm. Error bars represent means ± SD. See Figures S1–S3 for characterization of PTPN2-LysMCre mice.
Figure 2. IL-10–/– Mice Lacking PTPN2 in Myeloid Cells Develop More Severe Colitis.
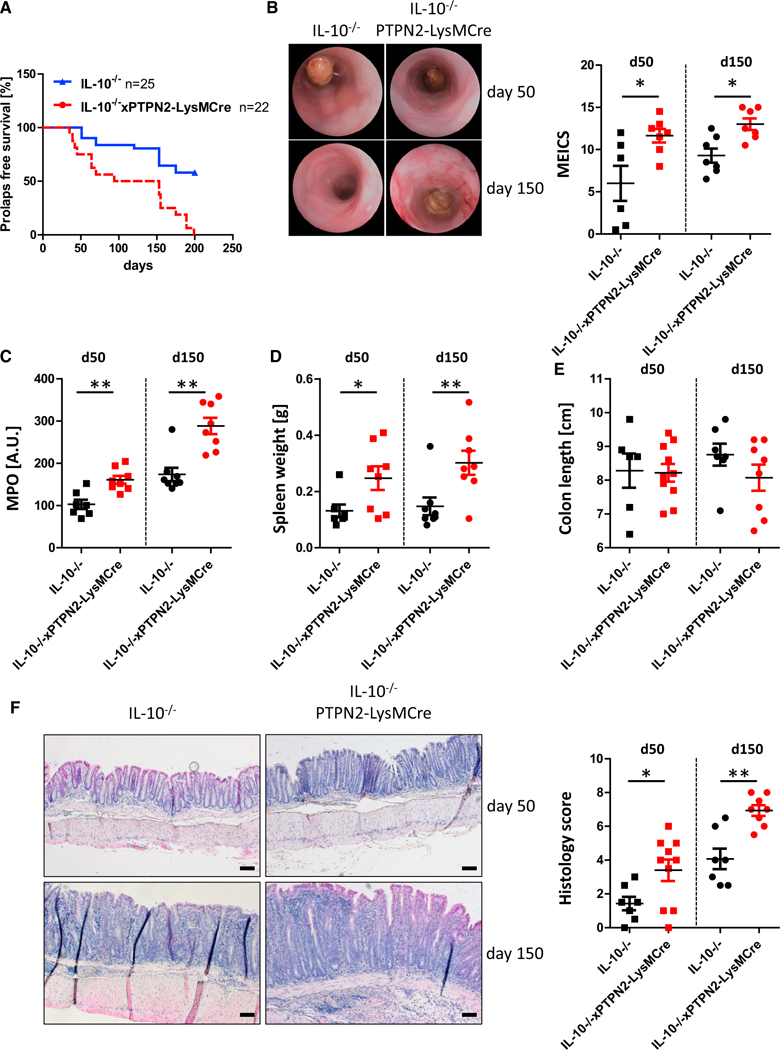
(A) IL-10–/– mice lacking PTPN2 in myeloid cells (IL-10xPTPN2-LysMCre) and their IL-10–/– littermates were monitored for 200 days for prolapse incidence. At 50 and 150 days, mice that did not develop prolapses were analyzed for colitis severity parameters.
(B–F) Shown are representative pictures from colonoscopy and respective statistical analysis (B), myeloperoxidase (MPO) activity (C), spleen weight (D), colon length (E), and representative pictures and scoring of epithelial damage and inflammatory infiltration in the terminal colon (F).
Depicted are results from seven or eight mice per group. Asterisks denote significant differences (*p < 0.05 and **p < 0.01; Mann-Whitney U test with Bonferroni correction). Scale bars represent 100 mm. Error bars represent means ± SD.
Reduced Tumor Load in PTPN2-LysMCre Mice
Intestinal inflammation can trigger intestinal hyperplasia in response to colitis, and indeed we observed increased levels of proliferating (Ki67+) cells in DSS-treated PTPN2-LysMCre mice (Figure 3A). Because enhanced proliferation might promote tumor formation and progression, we next investigated if PTPN2-LysMCre mice are more prone to inflammation-driven tumorigenesis by challenging them with the AOM/DSS model of colorectal carcinogenesis. WT mice developed several medium-sized to large tumors, but tumors were less frequent and much smaller in PTPN2-LysMCre littermates (Figures 3B–3D). This indicates that myeloid cell-specific loss of PTPN2 promotes proliferation but protects against induction of CRC. Of interest, mice lacking PTPN2 in IEC (PTPN2-VilCre mice; Kasper et al., 2016) also showed reduced tumor burden compared with WT littermates (Figures 3E–3G), although this phenotype was less pronounced than in PTPN2-LysMCre mice.
Figure 3. PTPN2-LysMCre Mice Are Protected from Colitis-Associated Tumor Induction.
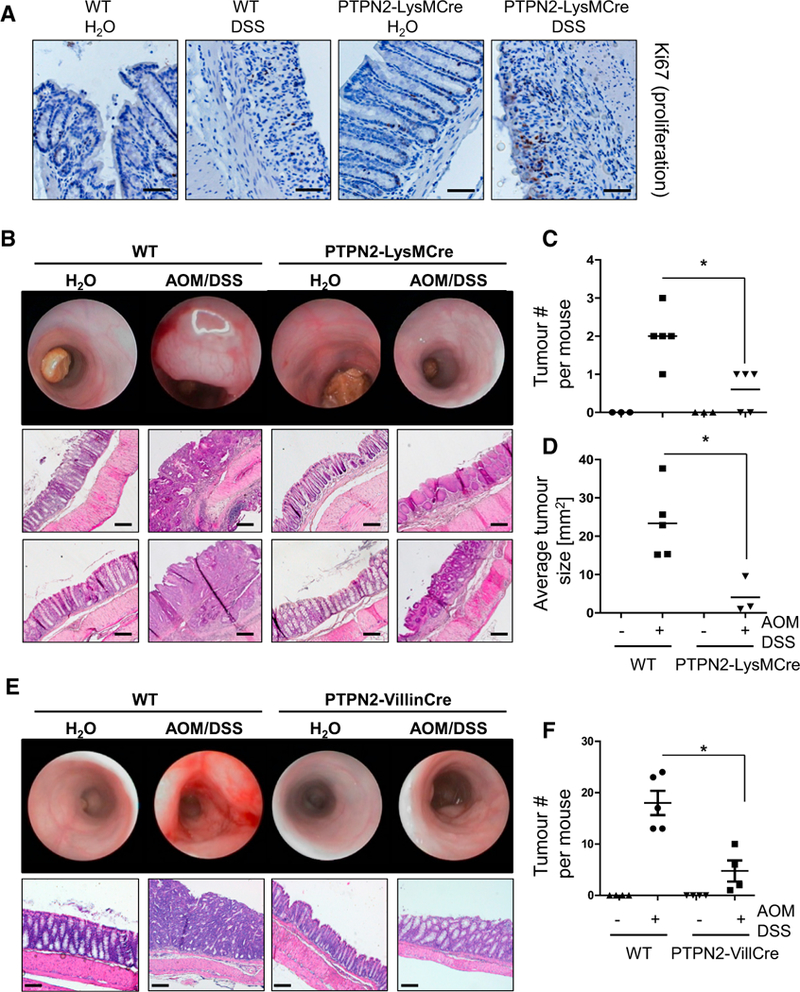
(A) Chronic colitis was induced in WT and PTPN2-LysMCre mice, and sections from the terminal colon were stained for the proliferation marker Ki67.
(B–F) Colitis-associated tumors were induced in WT and PTPN2-LysMCre mice (B–D) or mice lacking PTPN2 in intestinal epithelial cells (PTPN2-VillinCre mice; E and F). Depicted are representative pictures of colonoscopy and histology of the terminal colon (B and E), observed tumor number in dissected colon (C and F), and (D) average tumor size in each mouse.
Data are representative of one of two independent experiments with three to five mice per group. Asterisks denote statistical significance (*p < 0.05; Mann-Whitney U test with Bonferroni correction). Scale bars represent 100 μm. Error bars represent means ± SD. See Figures S4 and S5 for cytokine levels and T cells in PTPN2-LysMCre mice in the DSS and DSS/AOM models.
Altered Cytokine Profile in the Inflamed Intestine upon Myeloid Cell-Specific Deletion of PTPN2
To understand the mechanism by which loss of PTPN2 in myeloid cells exacerbates colitis but restricts inflammation-driven CRC formation, we measured the expression of pro- and anti-inflammatory cytokines in colon tissue. PTPN2-LysMCre mice expressed enhanced mRNA levels of the pro-inflammatory cytokines IL-1β, IL-6, IFN-γ, IL-18, MIP-2, and IL-17 upon induction of colitis and after AOM/DSS tumor induction (Figure S4). On the other hand, DSS-mediated induction of IL-10 was reduced, while levels of TGF-β1, TNF, IL-13, IL-22, and IL-4 were not altered in a consistent pattern (Figure S4).
Increased levels of the T helper (Th) 1 and Th17 hallmark cytokines IFN-γ and IL-17 prompted us to further address whether loss of PTPN2 in macrophages affects Th1 and Th17 cell abundance, but there was no difference in CD4+ cells or in proportions of IFN-γ+, IL-17+, or FoxP3+ CD4+ T cells (Figure S5). Hence we conclude that loss of PTPN2 in macrophages does not lead to an overt change in Th cell proportions in the intestine.
Increased Inflammasome Activation in Macrophages Lacking PTPN2
Levels of mature IL-1β, IL-18, and cas-pase-1, key mediators of inflammasome activation, were increased in PTPN2-LysMCre mice during colitis and AOM/ DSS treatment (Figures 4A and S6). Of note, in isolated IECs from PTPN2-VilCre mice, we also found increased levels of mature IL-1β, IL-18, and caspase-1, while in whole colon pieces we only observed a trend toward enhanced inflammasome activity. Therefore, we next investigated if the loss of PTPN2 in myeloid cells directly affected inflammasome activity or whether the altered levels of inflammasome markers were secondary to elevated inflammation in these mice. PTPN2-deficient BMDMs, isolated from PTPN2-LysMCre mice, secreted increased levels of IL-1β in response to NLRP3 inflammasome activators (i.e., muramyl dipeptide [MDP], MSU, ATP, nigericin) and to the AIM2 activator double-stranded DNA (dsDNA), but IL-1β production in response to NLRC4 activation with flagellin was not affected (Figure 4B). Consistent with our findings in mice and cell culture experiments, levels of mature IL-1β and IL-18 were enhanced in the serum of patients carrying an IBD-associated loss-of-function variant in PTPN2, while levels of TNF, IL-8, and IL-10 were not altered (Figure 4C). Of note, mRNA expression of IL1B and IL18 was also slightly increased in biopsies from inflamed regions of the intestine in those patients (Figure 4D).
Figure 4. Loss of PTPN2 Promotes ASC-Dependent Inflammasome Activation.
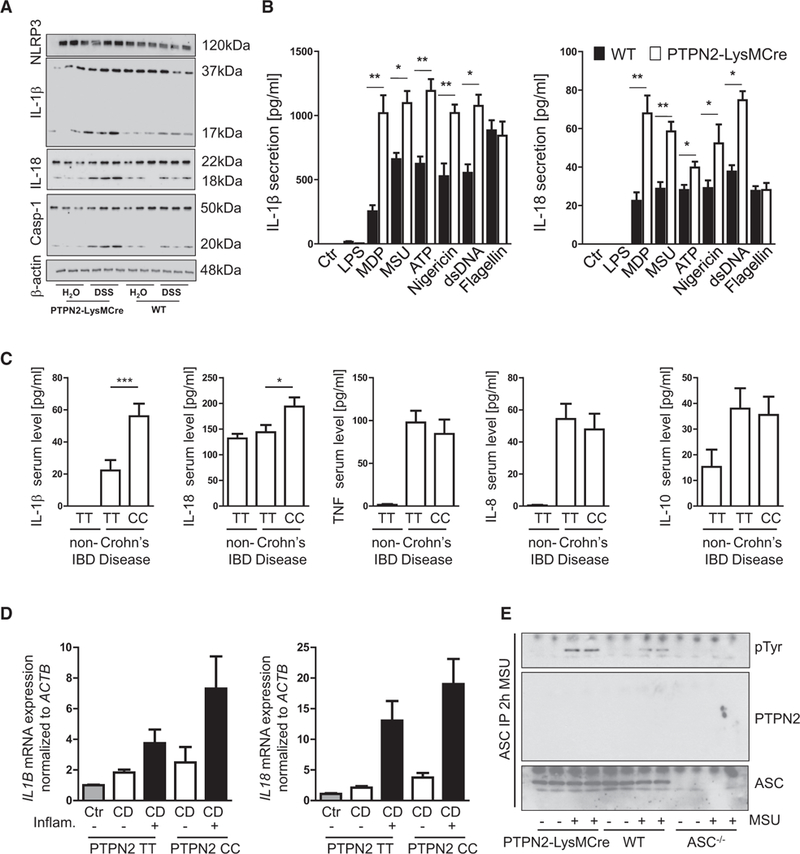
(A) Colon pieces from WT and PTPN2-LysMCre mice with acute DSS colitis were analyzed by western blot for NLRP3, IL-1β, caspase-1, and IL-18 expression and maturation.
(B) BMDMs from WT and PTPN2-LysMCre mice were treated with indicated inflammasome activators and cell culture supernatants analyzed for IL-1β and IL-18 secretion.
(C) Serum samples from control (non-IBD) patients (n = 15) or CD patients homozygous either for the PTPN2 WT allele (PTPN2 TT, n = 8) or the CD-associated PTPN2 allele (PTPN2 CC, n = 8) were analyzed for presence of IL-1β and IL-18.
(D) Intestinal biopsies from non-inflamed and inflamed regions were taken from the same patients as in (C) and analyzed for IL1b and IL18 mRNA expression.
(E) BMDMs from WT, PTPN2-LysMCre, and ASC–/– mice were left untreated or activated with MSU for 2 hr. ASC was precipitated and analyzed for tyrosine phosphorylation and co-precipitation of PTPN2.
Results are representative of at least two different experiments, each with three to five mice per group (A) or three or four biological replicates(B and E). Asterisks denote significant different results (*p < 0.05 and **p < 0.01; Student’s t test with Bonferroni correction). Error bars represent means ± SD. See also Figure S6.
In contrast to NLRC4, NLRP3 and AIM2 require the adaptor molecule ASC for inflammasome assembly, although more recent findings indicate that ASC might also be involved in efficient NLRC4-mediated caspase-1 activation. ASC is tyrosine phosphorylated upon inflammasome activation, which iscrucial for the formation of ASC specks, cytosolic structures that mediate IL-1β cleavage upon inflammasome induction (Hara et al., 2013). Because PTPN2 is a tyrosine phosphatase, we hypothesized that it might dephosphorylate ASC and in this way control inflammasome activity. When precipitating ASC from BMDMs and THP-1 cells, we indeed found enhanced ASC phosphorylation in PTPN2-deficient cells (Figure 4E), but there was no direct interaction between PTPN2 and ASC or other select inflammasome components, NLRP3 or caspase-1 (Figures 4E and S6G).
Enhanced Inflammasome Activation Requires JNK Activity
ASC phosphorylation is mediated via the kinases JNK and Syk (Hara et al., 2013; Lin et al., 2015), and JNK can serve as a substrate for PTPN2 (Tiganis et al., 1999). Indeed, we found increased levels of tyrosine phosphorylated JNK and Syk in MSU-treated PTPN2-deficient BMDMs (Figure 5A). To assess whether PTPN2-mediated changes in inflammasome activity are dependent on changes in Syk, JNK, or p38 activity, we pre-treated BMDMs with the Syk inhibitor R406, the JNK inhibitor SP600125, or the p38 inhibitor SB203580 prior to inflammasome activation. In WT BMDMs, inhibition of Syk or JNK markedly reduced IL-1β secretion. PTPN2-deficient BMDMs, however, still secreted IL-1β upon inhibition of Syk, while inhibition of JNK abrogated inflammasome activation and ASC phosphorylation (Figures 5B–5D). Comparable results were obtained when BMDMs were treated with Syk- or JNK-specific small interfering RNA (siRNA) (Figures S7A–S7C). The p38 inhibitor had no effect on MSU-stimulated IL-1b secretion in WT or knockout (KO) BMDMs (Figures 5B and 5C). Taken together, this indicates that PTPN2 regulates inflammasome activation by controlling ASC phosphorylation in a JNK-dependent manner. Of note, similar effects on inflammasome activation were obtained when we used intestinal epithelial cells (Figures S7D and S7E); hence we conclude that PTPN2-mediated control of the inflammasome is a general mechanism and not confined to macrophages or myeloid cells.
Figure 5. Inhibition of JNK Prevents Inflammasome Activation in PTPN2-LysMCre Mice.
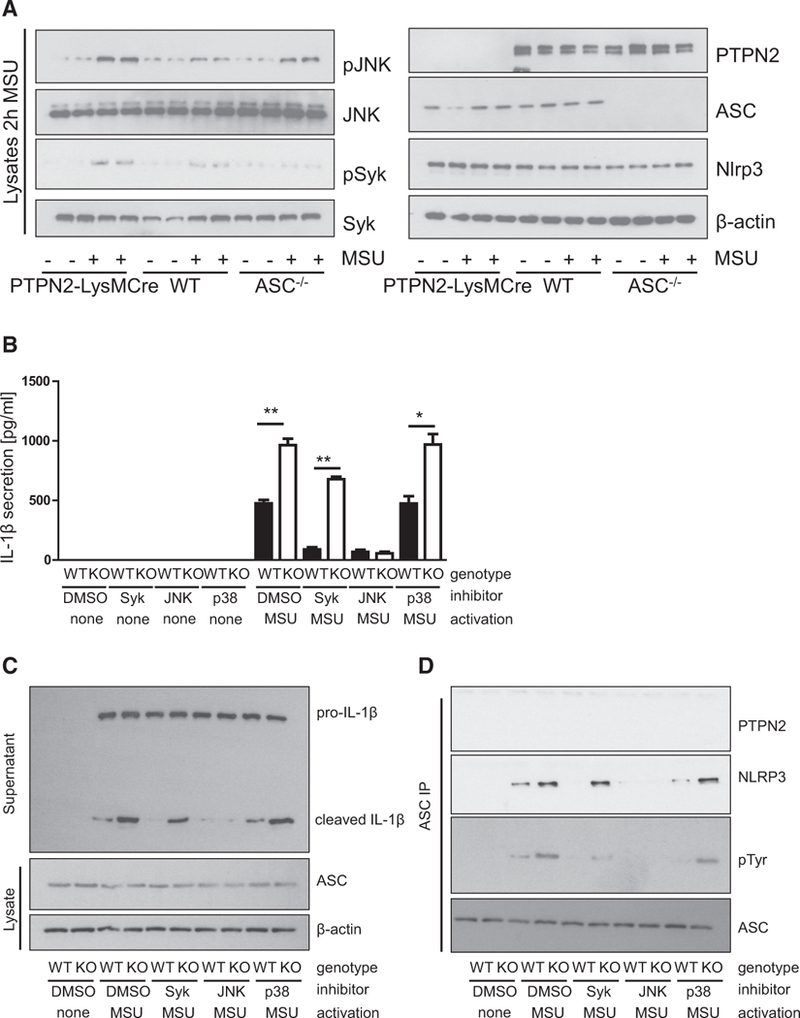
(A) BMDMs from WT, PTPN2-LysMCre, and ASC–/– mice were left untreated or activated with MSU for 2 hr and lysates analyzed for JNK and Syk phosphorylation and expression of PTPN2, ASC, and NLRP3. Each lane represents an individual mouse.
(B and C) BMDMs from WT and PTPN2-LysMCre (KO) mice were treated with Syk, JNK, or p38 inhibitors 1 hr prior to activation with MSU. Analysis of the cell culture supernatant for IL-1β secretion (B) and maturation (C) by ELISA and western blot, respectively.
(D) Precipitation of ASC from the cell lysate and analysis for tyrosine phosphorylation and co-precipitated NLRP3 and PTPN2.
Asterisks denote significant differences (*p < 0.05 and **p < 0.01; Student’s t test with Bonferroni correction). Error bars represent means ± SD. See also Figure S7.
Inhibition of IL-1β Protects against Aggravated Colitis in PTPN2-LysMCre Mice but Increases Tumor Burden
Having shown that PTPN2 controls inflammasome assembly, we next assessed whether changes in inflammasome activation are involved in promoting acute inflammation and/or protection from tumor formation. To interrupt inflammasome activation in vivo, WT control and PTPN2-LysMCre mice were administered a vaccine against IL-1β consisting of recombinant murine IL-1β coupled to virus-like particles (VLPs) of the bacteriophage Qβ, as previously described (Spohn et al., 2008). In response to acute and chronic DSS colitis, PTPN2-LysMCre mice pre-treated with control VLPs exhibited more severe colitis compared with respective WT controls. However, IL-1β-VLP pre-treated PTPN2-LysMCre mice showed only mild colitis, comparable with WT animals, indicating that inflammasome inhibition protected against the exaggerated inflammatory response to DSS in PTPN2-LysMCre mice (Figures 6A–6C [acute colitis], Figures 6D–6F [chronic colitis], and Figure S8).
Figure 6. Inhibition of IL-1 b Rescues PTPN2-LysMCre Mice from Increased Colitis.
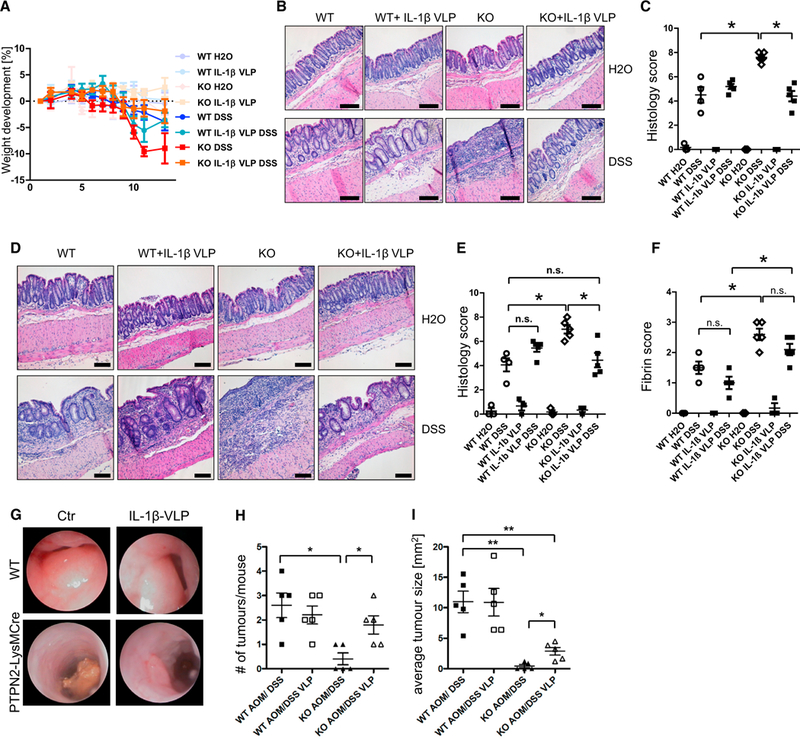
WT and PTPN72-LysMCre(KO) mice were immunized with IL-1β-coupled Qβ virus-like particles (IL-1β VLP) or with control Qβ virus-like particles 5,3, and 1 week prior to start of colitis/tumor induction.
(A–C) Weight development (A), representative pictures of H&E-stained pieces of the terminal colon (B), and results of histological scoring of intestinal pathology
(C) from mice with acute DSS colitis.
(D–F) Representative pictures from colonoscopy (D), statistical analysis of histopathology (E), and fibrin score from mice with chronic DSS colitis (F).
(G–I) Representative pictures of colonoscopy (G), observed tumor number in dissected colon (H), and average tumor size (I) in each mouse from mice subjected to AOM/DSS treatment.
Results are representative of one of two independent experiments with three to five mice per group. Asterisks denote statistical significance (*p < 0.05, **p < 0.01, and *** = p< 0.001; Mann-Whitney U test with Bonferroni correction). Scale bars represent 100 mm. Error bars represent means ± SD. See also Figures S8 and S9.
In the AOM/DSS tumor model, inhibition of IL-1β by IL-1 p-VLP pre-treatment in PTPN2-LysMCre mice enhanced tumor burden compared with control VLP-treated PTPN2-LysMCre mice, reaching a similar number of tumors as observed in WT animals (Figures 6G and 6H). However, tumors in IL-1β-VLP-treated PTPN2-LysMCre mice were smaller than those observed in WT mice and WT mice treated with IL-1β-VLPs (Figure 6I). Similar effects were observed when we crossed PTPN2-LysMCre mice with ASC-deficient mice as an alternative approach to interrupting inflammasome activation (not shown). Thus, inflammasome inhibition increased tumor burden in AOM/DSS challenged PTPN2-LysMCre mice. Of note, inhibition of IL-1β normalized serum cytokine levels of IL-1β, IL-1α, and IL-18 and decreased mRNA levels of Ilia, Il1b, and Il18 in PTPN2-LysMCre mice (Figures S9A and S9B), indicating that increased levels of IL-1β mediate the exaggerated expression of those cytokines.
Enhanced Serum Levels but Reduced Membrane-Associated IL-1α in PTPN2-LysMCre Mice
In addition to enhanced IL-1β, we also observed increased serum levels of the structurally related cytokine IL-1α in PTPN2-LysMCre mice during acute DSS-induced colitis (Figure 7A). Because IL-1α also exists in a membrane-associated form, we next addressed whether in PTPN2-LysMCre mice membrane-associated IL-1α is also affected. Of interest, membrane associated IL-1α was reduced in macrophages and IECs isolated from PTPN2-LysMCre mice (Figure 7B), while mRNA levels of calpain, which mediates shedding of membrane-associated IL-1α (Carruth et al., 1991), were unchanged (Figure 7C). Inhibition of IL-1α, using vaccination with IL-1α-VLPs, did not affect colitis severity (Figures 7D and 7E). However, IL-1α-VLPs did reduce tumor burden and size in WT but had no effect on tumorigenesis in PTPN2-LysMCre mice (Figures 7G–7I). Taken together, our data show that inhibition of IL-1β, but not IL-1α, can protect PTPN2-LysMCre mice from enhanced colitis but at the same time re-establishes susceptibility to colitis-associated tumor induction.
Figure 7. Inhibition of IL-1α Protects WT Mice from Tumor Induction.
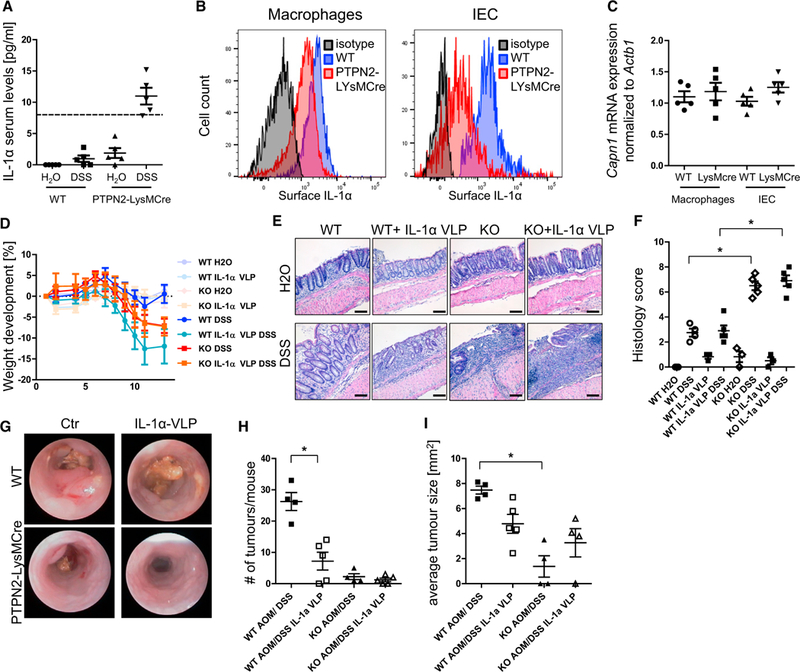
(A) Serum from WT and PTPN92-LysMCre mice with chronic DSS colitis was analyzed for IL-1α levels. Dashed line, detection limit of the ELISA.
(B and C) Peritoneal macrophages and intestinal epithelial cells (lECs) from WT and PTPN2-LysMCre mice were analyzed for IL1a surface expression (B) and calpain mRNA levels (C).
(D–F) WT and PTPN2-LysMCre mice were immunized with IL-1α-coupled Qβ virus-like particles (IL-1α VLP) or with control Qβ virus-like particles 5,3, and 1 week prior to start of colitis and/or tumor induction. (D) Weight development of mice in acute colitis, (E) representative pictures of H&E-stained slides, and
(F) quantification of histology score of terminal colon pieces from WT and PTPN2-LysMCre mice subjected to chronic colitis.
(G-I) Representative pictures of colonoscopy (G), observed tumor number in dissected colon (H), and average tumor size (I) in each mouse in the AOM/DSS tumor model.
Results are representative of one of two independent experiments with three to five mice per group. Asterisks denote statistical significance (*p < 0.05; MannWhitney U test with Bonferroni correction). Scale bars represent 100 mm. Error bars represent means ± SD.
DISCUSSION
In this study, we demonstrate that PTPN2 regulates inflammasome activity by restricting phosphorylation of the key inflammasome assembly protein ASC. The physiologic importance of this regulatory mechanism for intestinal health is highlighted by the fact that lack of PTPN2 in macrophages results in more severe colitis, which can be prevented by inhibition of IL1-β. However, inhibition of IL-1β also increased susceptibility to tumor induction in PTPN2-LysMCre mice. This indicates the context-dependent nature of inflammasome activity as well as a clear pathophysiological relevance of PTPN2 loss in myeloid cells.
Myeloid cell numbers in the intestine were not altered in PTPN2-LysMCre mice, and there was no effect on the development of BMDMs. This is of interest, because PTPN2 has been implicated to play a role in hematopoiesis (You-Ten et al., 1997), and it has been described that bone marrow cells from Ptpn2–/– mice are more sensitive to stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) and give rise to more macrophages (Simoncic et al., 2006). However, another report demonstrated that changes in hematopoiesis might be due to alterations in bone marrow stromal cell function and does not necessarily reflect an immune cell-intrinsic effect on hematopoiesis (Bourdeau et al., 2007). Even though loss of PTPN2 did not affect monocyte or macrophage numbers in the intestine, it did have important consequences for intestinal health by promoting colitis and affecting inflammasome activation. The role of inflammasomes in intestinal health is controversial, indicating pro-versus anti-inflammatory effects (Bauer et al., 2010, 2012; Bersudsky et al., 2014; Hirota et al., 2011; Ruiz et al., 2017; Zaki et al., 2010). To a certain extent, these contradictions can be explained by differences in the intestinal microbiota or the use of littermate versus non-littermate controls (Bauer et al., 2012). To limit the confounding factor of microbiota, we used littermates for our experiments and caged WT and PTPN2-LysMCre together until the start of the experiment to minimize microbiota-associated differences in all experimental groups.
Besides its influence on anti-microbial properties, lack of inflammasome activity can impair effective host defense against invading pathogens during colitis, ultimately resulting in enhanced disease severity. On the other hand, over-activation of inflammasomes, and subsequently excess secretion of mature IL-1β, can mediate tissue damage and promote intestinal inflammation. Our results fit well with this dual-role paradigm for the inflammasome: inhibition of IL-1β in WT animals, reflecting models with impaired inflammasome activation, results in slightly enhanced severity of DSS-induced colitis. On the other hand, PTPN2-LysMCre mice exhibit increased, and therefore pathogenic, levels of IL-1β- In this highly inflammatory context, inhibition of (excessive) IL-1β had clear beneficial effects with respect to reducing inflammation. It is striking that PTPN2-LysMCre mice, despite enhanced inflammation, exhibit significantly reduced tumor burden in the AOM/DSS model of inflammation-driven tumorigenesis. Of interest, we also observed reduced tumor burden in mice lacking PTPN2 in IECs, indicating that this effect is not restricted to loss of PTPN2 in myeloid cells. Taken together, our findings offer additional insights into a recent publication indicating that PTPN2 promotes evasion from immune cell-mediated cancer therapy (Manguso et al., 2017).
Recently, we described that PTPN22, a phosphatase closely related to PTPN2, promotes NLRP3 inflammasome activity via de-phosphorylation of NLRP3 (Spalinger et al., 2016). It might seem surprising that two very similar molecules regulate the inflammasome in an opposite manner. However, PTPN22 interacts directly with NLRP3, while PTPN2 acts on pathways upstream of inflammasome activation (i.e., via regulation of the kinases responsible for ASC phosphorylation). Our findings regarding JNK/Syk activity upstream of ASC are in agreement with the previous work of Hara et al. (2013) and Lin et al. (2015). Although these publications described the mechanism of how ASC is phosphorylated, our results demonstrate how activation of one of the two kinases responsible for ASC phosphorylation, namely JNK, is restricted by PTPN2 to prevent excessive inflammasome induction.
There are reports showing important anti-tumor roles of IL-1β, as IL-1β activation drives the development of anti-tumor T cells (Ghiringhelli et al., 2009), and inflammasomes negatively regulate tumorigenesis in colitis-associated cancer models (Allen et al., 2010; Zaki et al., 2011). Conversely, evidence of IL-1β as a driver in tumor development is abundant: IL-1β promotes epithelial-to-mesenchymal transition (Li et al., 2012), a process involved in tumor development and metastasis (Nieto et al., 2016); IL-1β, alone or in concert with IL-6, drives tumor angiogenesis (Voronov et al., 2003); and several studies have shown a tumor-promoting role for IL-1β/inflammasomes in the context of colitis-associated tumors (Allen et al., 2010; Bauer et al., 2012). Our results demonstrate that IL-1β is involved in the protection against CRC development observed in PTPN2-LysMCre mice, as inhibition of IL-1β increased susceptibility to tumor development. However, it is also clear that there must be other factors involved: upon IL-1β inhibition, tumor numbers in PTPN2-LysMCre mice were comparable with those in WT animals, but tumor size was still drastically reduced. Reduced tumor size might result from decreased proliferation, but because we observed enhanced proliferation in the epithelium of PTPN2-LysMCre mice, it is questionable as to whether reduced proliferation is the mechanism resulting in reduced tumor size. Moreover, apoptosis levels were similar in WT and PTPN2-LysMCre mice, suggesting that elevated apoptosis is not responsible for decreased tumor size in PTPN2-LysMCre mice. In summary, it is still unclear how loss of PTPN2 in macrophages protects against colitis-associated epithelial tumors, and it will be important to evaluate this in more detail in future studies.
The IL-1 family of cytokines consists of several members, including IL-1α. Although IL-1α and IL-1β share little sequence homology, they are structurally related and both activate the same receptor, that is, the IL-1 receptor type I (IL-1R1) (March et al., 1985). Similar to IL-1β, IL-1α is expressed as a pro-form upon Toll-like receptor (TLR) activation. IL-1β, however, needs to be cleaved by the inflammasome/caspase-1 to be secreted in its active form. In contrast, IL-1α is already active in its pro-form, which can be released by cell death. Also, TLR stimulation alone is sufficient to induce cell surface expression of IL-1α, which is biologically active and can be shed by calpains to yield soluble IL-1α (Kobayashi et al., 1990). Additionally, activation of the inflammasome leads to IL-1α release (Fettelschoss et al.,(2011). There are reports demonstrating that IL-1α promotes intestinal inflammation in cell culture studies (Scarpa et al., 2015) and in models of acute colitis (Bersudsky et al., 2014). However, in our hands, inhibition of IL-1α had no overt effect on colitis severity. Interestingly, however, inhibition of IL-1α resulted in a drastically reduced tumor burden in WT animals. These mouse data are in line with the observation that cancer progression and metastasis correlate with IL-1α expression (reviewed by Dinarello, 2014), and treatment with monoclonal anti-IL-1α antibody improves the prognosis of cancer patients (Hong et al., 2014). Although IL-1α was very effective in reducing tumorigenesis in WT mice, the already small tumor numbers in PTPN2-LysMCre mice could not be decreased further. It is possible that the decreased levels of membrane-bound IL-1α might contribute to the reduced tumor load observed in PTPN2-LysMCre mice, further studies are required to make this determination.
In addition to altered inflammasome-associated cytokine levels (IL-1β, IL-18, and IL-1α), the Th1- and Th17-associated cytokines IL-17, IL-6, IL-23, and IFN-γ were enhanced in PTPN2-LysMCre mice, whereas expression of Th2-associated cytokines IL-4 and IL-13 was decreased. Nevertheless, there were no changes in the proportion of Th cell subsets isolated from the colon or mesenteric lymph nodes. This does not necessarily mean that in certain regions or in the tissue directly surrounding tumors the function and proportions of T cell subsets remain unaltered, but in their overall abundance, T cells seem not to be affected upon loss of PTPN2 in macrophages.
In summary, we describe an essential role for myeloid cell-specific PTPN2 in maintaining intestinal homeostasis in the face of inflammatory challenge. Our results clearly demonstrate the pathophysiologic relevance of PTPN2 dysfunction and subsequently altered inflammasome activation in vivo. We have previously shown that loss or dysfunction of PTPN2 in T cells is also involved in the pathogenesis of colitis, while loss of PTPN2 in IECs had little effect on intestinal inflammation. Dysfunction of PTPN2 in T cells drives the secretion of IFNγ and IL-17, which promote IL-18 and IL-1β secretion, respectively. Given that in the clinical setting, loss-of-function variants affect all cell types; dysfunctional PTPN2 in T cells might potentiate the effect of dysfunctional PTPN2 in macrophages and in other cell types (Figure S9C). This is underscored by the fact that none of the reported mouse strains with a tissue-specific PTPN2 deletion shows the severe inflammatory phenotype observed in constitutive PTPN2 KO mice. Because of its important anti-inflammatory role, PTPN2 might provide an interesting target for future treatment options in IBD or other inflammatory disorders. However, our data also point to the need to recognize the importance of context-dependent targeting of PTPN2 signaling given the differential roles observed for myeloid expressed PTPN2 in inflammation versus inflammation-driven tumorigenesis.
EXPERIMENTAL PROCEDURES
Mice, Colitis, and Colitis-Associated Tumor Models
All animal experiments were conducted according to Swiss animal welfare legislation, and the local veterinary office (Veterinaramt des Kantons Zurich) approved all procedures. All mice used for the studies were homozygous for the floxed Ptpn2 gene and either heterozygous for the LysMCre construct (PTPN2-LysMCre) or not expressing LysMCre (WT). Adult (10–12 weeks old) female littermates housed in a specific pathogen-free (SPF) facility were used for all studies. Colitis was induced by DSS administration (Becker et al., 2006; Obermeier et al., 1999), as detailed in Supplemental Experimental Procedures, and by backcrossing the mice onto the IL-10–/– background. For induction of CRC, azoxymethane (AOM; 10 mg/kg body weight; Sigma-Aldrich) was injected on day 1 and day 8 of each DSS cycle during chronic DSS colitis induction. Colonoscopy was performed as described and scored using the murine endoscopic index of colitis severity (MEICS) scoring system (Becker et al., 2006). Histological scoring for inflammatory infiltration and epithelial cell damage was performed on H&E-stained section of the most distal 1 cm of the colon (Becker et al., 2006; Obermeier et al., 1999).
Immunization Using IL-1β and IL-1α Qβ VLPs
Fifty micrograms of IL-1α-Qβ, IL-1β-Qβ, or Qβ-VLP was prepared and diluted in PBS as previously described (Spohn et al., 2008) and injected subcutaneously 5, 3, and 1 week prior to the start of colitis induction. For chronic colitis and colitis-associated tumor models, a boost immunization was performed during the recovery phase of the second DSS cycle. Levels of anti-IL-1α and anti-IL-1β antibodies in the serum were analyzed as previously described (Spohn et al., 2008) to confirm immunization effectiveness.
H&E, Immunohistochemistry, and Immunofluorescent Staining
Standard protocols were used for H&E, immunohistochemistry, and immunofluorescent staining. Sections of the most distal part of the colon were used unless otherwise stated. Details on the methods used are given in Supplemental Experimental Procedures.
Western Blot Analysis, Immunoprecipitation, ELISA, Flow Cytometry, and RNA Isolation/RT-PCR
Western blot analysis, immunoprecipitation, flow cytometry, and RNA isolation/RT-PCR were performed according to standard protocols. ELISAs were obtained from commercial suppliers and performed according to the manufacturer’s instructions. Details can be found in Supplemental Experimental Procedures.
Cell Isolation and Cell Culture Experiments
For sorting of immune cells from the spleen, single-cell suspensions were stained with an appropriate antibody mix and B220+, F4/80+, and CD3+ cells sorted using a BD Aria III cell sorter. BMDCs were derived from the bone marrow and peritoneal macrophages were isolated as previously described (Spalinger et al., 2016). Further details are given in Supplemental Experimental Procedures. THP-1 and HT-29 cells were obtained from DSMZ and cultured as previously described (Spalinger et al., 2016).
For inflammasome activation, THP-1 cells were differentiated into macrophages by treatment with 50 nM PMA (phorbol 12-myristate 13-acetate; Sigma-Aldrich) for 3 hr. All cells were primed for 12 hr with ultra-pure lipopoly-saccharide (upLPS; Invivogen) prior to inflammasome activation, unless otherwise stated. Mono-sodium urate (MSU), ATP, flagellin, and MDP were obtained from Invivogen. MDP was transfected into the cells using FuGene transfection reagent (Promega). Flagellin and dsDNA were transfected using DOTAP transfection reagent (Roche).
Inhibitors and siRNA Treatment
For experiments with kinase inhibition, cells were incubated with Syk inhibitor R406 (1 μM; Sigma-Aldrich), JNK inhibitor SP600125 (40 μM; Sigma-Aldrich), or p38 inhibitor SB203580 (10 μM; Sigma-Aldrich) for 1 hr prior to treatment with MSU (100 ng/mL; Invivogen) (Hara et al., 2013). Transfection was carried out as detailed in Supplemental Experimental Procedures. After the transfection, cells were cultured for 24 hr in RPMI supplemented with 10% fetal calf serum (FCS) prior to activation, as described above.
Patient Serum and Biopsies
Serum and intestinal biopsies were obtained from previously genotyped (Scharl et al., 2012) CD patients (WT [TT]: n = 15, age range 27–66 years, mean age 47.4 ± 13.5 years; variant [CC]: n = 8, age range 28–69 years, mean age 43.8 ± 18.3 years) from the Swiss IBD cohort and from healthy controls (n = 15, age range 37–61 years, mean age 49.1 ± 9.5 years). Written informed consent was obtained before sample collection and the local Ethics Committee of the Canton Zürich approved the studies (Approval No. EK-1755 and EK-1316). Patients were matched for age, gender, and disease activity.
Statistical Analysis
Data are presented as mean ± SD for one of at least two independent experiments, each with three to six mice or biological replicas as indicated in the figure legends. Statistical analysis was performed using ANOVA followed by the Mann-Whitney U or Student’s t test as appropriate. Bonferroni correction was used to correct for multiple testing as appropriate. p values < 0.05 were considered to indicate significance.
Supplementary Material
Highlights
Increased colitis but fewer tumors upon PTPN2 loss in myeloid cells
Excessive inflammasome activation and IL-1β release in PTPN2-deficient macrophages
PTPN2 regulates phosphorylation of the inflammasome adaptor molecule ASC
Rescue of the colitis/tumor phenotype upon inhibition of IL-1β
ACKNOWLEDGMENTS
We thank all patients and the members of the Swiss IBD cohort for their commitment. This research was supported by research grants from the Swiss National Science Foundation to M.S. (grants314730-146204,314730_166381/1, 314730_166381/2, and CRSII3_154488/1), to G.R. for the Swiss IBD cohort (grant 3347C0-108792), and to T.M.K. (grant CRSII3_154490); the NIH (grant 2R01DK091281) and the Crohn’s and Colitis Foundation (Senior Research Award) to D.F.M.; the Promedica Foundation and the Hartmann-Muller Foundation to M.S.; and the European Crohn’s and Colitis Organisation and the Holcim Foundation to M.R.S. The founding institutions had no role in study design, analysis, or interpretation of the results.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and nine figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.052.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, and Ting JP (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aradi B, Kato M, Filkova M, Karouzakis E, Klein K, Scharl M, Kolling C, Michel BA, Gay RE, Buzas EI, et al. (2015). Protein tyrosine phosphatase nonreceptor type 2 (PTPN2), an important regulator of IL-6 production in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 67, 2624–2633. [DOI] [PubMed] [Google Scholar]
- Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, and Schnurr M (2010). Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome.Gut 59, 1192–1199. [DOI] [PubMed] [Google Scholar]
- Bauer C, Duewell P, Lehr HA, Endres S, and Schnurr M (2012). Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig. Dis. 30 (Suppl 1), 82–90. [DOI] [PubMed] [Google Scholar]
- Becker C, Fantini MC, and Neurath MF (2006). High resolution colonoscopy in live mice. Nat. Protoc. 1, 2900–2904. [DOI] [PubMed] [Google Scholar]
- Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I, Aychek T, Dinarello CA, et al. (2014). Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 63, 598–609. [DOI] [PubMed] [Google Scholar]
- Bourdeau A, Dubé N, Heinonen KM, Théberge JF, Doody KM, and Tremblay ML (2007). TC-PTP-deficient bone marrow stromal cells fail to support normal B lymphopoiesis due to abnormal secretion of interferon-gamma. Blood 109, 4220–228. [DOI] [PubMed] [Google Scholar]
- Carruth LM, Demczuk S, and Mizel SB (1991). Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J. Biol.Chem. 266, 12162–12167. [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Forster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2014). Interleukin-1α neutralisation in patients with cancer. Lancet Oncol. 15, 552–553. [DOI] [PubMed] [Google Scholar]
- Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, and Kundig TM (2011). Inflammasome activation and IL-1β target IL-1α for secretion as opposed to surface expression. Proc. Natl. Acad. Sci. USA 108, 18055–18060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Glas J, Wagner J, Seiderer J, Olszak T, Wetzke M, Beigel F, Tillack C, Stallhofer J, Friedrich M, Steib C, et al. (2012). PTPN2 gene variants are associated with susceptibility to both Crohn’s disease and ulcerative colitis supporting a common genetic disease background. PLoS ONE 7, e33682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, and Mitsuyama M (2013). Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 74, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, and Lapp WS (2004). T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 703, 3457–3464. [DOI] [PubMed] [Google Scholar]
- Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, et al. (2011). NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 77, 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS, Piha-Paul S, Wheler JJ, Fu S, Tsimberidou AM, et al. (2014). MABp1,a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 75, 656–666. [DOI] [PubMed] [Google Scholar]
- Kasper SH, Spalinger MR, Leonardi I, Gerstgrasser A, Raselli T, Got-tier C, Atrott K, Frey-Wagner I, Fischbeck-Terhalle A, Rogler G, and Scharl M (2016). Deficiency of protein tyrosine phosphatase non-receptor type 2 in intestinal epithelial cells has no appreciable impact on dextran sulphate sodium colitis severity but promotes wound healing. Digestion 93, 249–259. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, and Matsushima K (1990). Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc. Natl. Acad. Sci. USA 87, 5548–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Moran T, Swanson E, Julian C, Harris J, Bonen DK, Hedl M, Nicolae DL, Abraham C, and Cho JH (2004). Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Hum. Mol. Genet. 73, 1715–1725. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang L, Pappan L, Galliher-Beckley A, and Shi J (2012). IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 77, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Huang DY, Wang JS, Lin YL, Hsieh SL, Huang KC, and Lin WW (2015). Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J. Leukoc. Biol. 97, 825–835. [DOI] [PubMed] [Google Scholar]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. ; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547,413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March CJ, Mosley B, Larsen A, Cerretti DP, Braedt G, Price V, Gillis S, Henney CS, Kronheim SR, Grabstein K, et al. (1985). Cloning, sequence and expression oftwo distinct human interleukin-1 complementary DNAs. Nature 315, 641–647. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–26. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Obermeier F, Kojouharoff G, Hans W, Scholmerich J, Gross V, and Falk W (1999). Interferon-gamma (IFN-gamma)-and tumour necrosisfactor(TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 116, 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivollier A, He J, Kole A, Valatas V, and Kelsall BL (2012). Inflammation switches the differentiation program of Ly6Chi monocytesfrom antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 209, 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz PA, Moron B, Becker HM, Lang S, Atrott K, Spalinger MR, Scharl M, Wojtal KA, Fischbeck-Terhalle A, Frey-Wagner I, et al. (2017). Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut 66, 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa M, Kessler S, Sadler T, West G, Homer C, McDonald C, de la Motte C, Fiocchi C, and Stylianou E (2015). The epithelial danger signal IL-1β is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am. J. Pathol. 185, 1624–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller E, Macfarlane AJ, Rupec RA, Gordon S, McKnight AJ, and Pfeffer K (2002). Inactivation of the F4/80 glycoprotein in the mouse germ line. Mol. Cell. Biol. 22, 8035–8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharl M, Hruz P, and McCole DF (2010). Protein tyrosine phosphatase non-receptor type 2 regulates IFN-γ-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 16, 2055–2064. [DOI] [PubMed] [Google Scholar]
- Scharl M, McCole DF, Weber A, Vavricka SR, Frei P, Kellermeier S, Pesch T, Fried M, and Rogler G (2011). Protein tyrosine phosphatase N2 regulates TNFα-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 60, 189–197. [DOI] [PubMed] [Google Scholar]
- Scharl M, Mwinyi J, Fischbeck A, Leucht K, Eloranta JJ, Arikkat J, Pesch T, Kellermeier S, Mair A, Kullak-Ublick GA, et al. (2012). Crohn’s disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm. Bowel Dis. 18, 900–912. [DOI] [PubMed] [Google Scholar]
- Simoncic PD, Bourdeau A, Lee-Loy A, Rohrschneider LR, Tremblay ML, Stanley ER, and McGlade CJ (2006). T-cell protein tyrosine phosphatase (Tcptp) is a negative regulator of colony-stimulating factor 1 signaling and macrophage differentiation. Mol. Cell. Biol. 26, 4149–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalinger MR, Kasper S, Chassard C, Raselli T, Frey-Wagner I, Gottier C, Lang S, Atrott K, Vavricka SR, Mair F, et al. (2015). PTPN2 controls differentiation of CD4 T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 8, 918–929. [DOI] [PubMed] [Google Scholar]
- Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Gutte PM, Grütter MG, Beer H-D, et al. (2016). NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Invest. 126, 1783–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spohn G, Keller I, Beck M, Grest P, Jennings GT, and Bachmann MF (2008). Active immunization with IL-1 displayed on virus-like particles protects from autoimmune arthritis. Eur. J. Immunol. 38, 877–887. [DOI] [PubMed] [Google Scholar]
- Street ME, de’Angelis G, Camacho-Hübner C, Giovannelli G, Ziveri MA, Bacchini PL, Bernasconi S, Sansebastiano G, and Savage MO (2004). Relationships between serum IGF-1, IGFBP-2, interleukin-1beta and interleukin-6 in inflammatory bowel disease. Horm. Res. 61, 159–164. [DOI] [PubMed] [Google Scholar]
- Tiganis T, Kemp BE, and Tonks NK (1999). The protein-tyrosine phosphatase TCPTP regulates epidermal growth factor receptor-mediated and phosphatidylinositol 3-kinase-dependent signaling. J. Biol. Chem. 274, 27768–27775. [DOI] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. ; Genetics of Type 1 Diabetes in Finland; Wellcome Trust Case Control Consortium (2007). Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 39, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, Dinarello CA, and Apte RN (2003). IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 100, 2645–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, Tremblay ML, Russell SM, Godfrey DI, and Tiganis T (2011). T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Invest. 121, 4758–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You-Ten KE, Muise ES, Itié A, Michaliszyn E, Wagner J, Jothy S, Lapp WS, and Tremblay ML (1997). Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 186, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, and Kanneganti TD (2010).The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Lamkanfi M, and Kanneganti TD (2011). Inflammasomes and Intestinal Tumorigenesis. Drug Discov. Today Dis. Mech. 8, e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond E, and Jung S (2013). Intestinal macrophages: well educated exceptions from the rule. Trends Immunol. 34, 162–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.