

Abstract
Iron diazenido species (Fe(NNH)) have been proposed as the earliest intermediates of catalytic N2-to-NH3 conversion (N2RR) mediated by synthetic iron complexes, and relatedly as intermediates of N2RR by nitrogenase enzymes. However, direct identification of such iron species, either during or independent of catalysis, has proven challenging owing to their high degree of instability. The isolation of more stable silylated diazenido analogs, Fe(NNSiR3), and also of further downstream intermediates (e.g., Fe(NNH2)), nonetheless points to Fe(NNH) as the key first intermediate of protonation in synthetic systems. Herein we show that low temperature protonation of a terminally bound Fe-N2− species, supported by a bulky trisphosphinoborane ligand (ArP3B), generates an S = 1/2 terminal Fe(NNH) species that can be detected and characterized by continuous-wave (CW) and pulse EPR techniques. The 1H-hyperfine for ArP3BFe(NNH) derived from the presented ENDOR studies is diagnostic for the distally-bound H-atom (aiso = 16.5 MHz). The Fe(NNH) species evolves further to cationic [Fe(NNH2)]+ in the presence of additional acid, the latter being related to a previously characterized [Fe(NNH2)]+ intermediate of N2RR mediated by a far less encumbered iron tris(phosphine)borane catalyst. While catalysis is suppressed in the present sterically very crowded system, N2-to-NH3 conversion can nevertheless be demonstrated. These observations in sum add support to the idea that Fe(NNH) plays a central role as the earliest intermediate of Fe-mediated N2RR in a synthetic system.
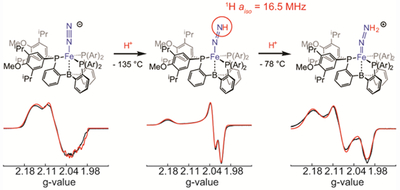
Graphical Abstract
INTRODUCTION
The mechanism of nitrogen fixation in nitrogenase enzymes has fascinated researchers since the discovery and characterization of the iron-molybdenum-cofactor (FeMoco), which is the active site for substrate binding and reduction in FeMo-nitrogenase.1,2,3 While a host of scenarios continue to be considered, hypotheses surrounding the mechanism of the key bond breaking and making steps are often framed in the context of two limiting pathways.4 One is the distal pathway, where successive delivery of H-atom equivalents occurs at Nβ, leading to an early N-N bond cleavage step with liberation of the first ammonia equivalent. The second is an alternating pathway, wherein the delivery of H-atom equivalants alternates between Nβ and Nα and late-stage N-N cleavage leads to the first NH3 equivalent (Figure 1).
Figure 1.

Outline of limiting pathways for distal and alternating scenarios for N2-to-NH3 conversion.
Model chemistry from Chatt, Hidai, and others on group VI coordination complexes focused early attention on the distal mechanism, and seminal studies of a well-defined molybdenum tris(amido)amine catalyst system by Schrock cemented the viability of such a pathway for a single Mo active site.5,6,7,8 The development of improved Mo catalysts continues at a robust pace.9
Structural and spectroscopic studies of nitrogenase enzymes have, however, implicated one or two iron centers as the initial site/s of N2 binding and subsequent reduction steps,4,10 consistent with the fact that molybedum is not an essential metal for nitrogenase activity.2 In this context, several iron model systems have shown catalytic activity for N2-to-NH3 conversion (N2RR).11
Recent mechanistic studies from our lab using a tris(phosphine)borane iron (P3BFe) catalyst system have established the viability of a distal iron pathway for NH3 generation, releasing a terminal nitride intermediate ([P3BFeIV≡N]+).12 However, related mechanistic studies we have undertaken have revealed the possibility that multiple pathways may be operative in the iron catalyst systems, including hybrid crossover pathways.13 This idea is consistent with the competitive generation of N2H4 in both stoichiometric and catalytic yields.11e,13,14
These complexities notwithstanding, a critical first intermediate that serves as the linchpin to later stage intermediates along various trajectories is the terminal diazenido, Fe-NNH (Figure 1). This type of species may be regarded as a nitrogenous analogue of an iron hydroperoxo, Fe-OOH, the latter playing a central role in enzymes such as cytochrome P450’s and bleomycin via reductive protonation of O2.15 Fe-NNH species are expected to be even less stable, and current precedent for their reliable characterization using any transition metal is limited to three examples featuring either Mo or W.
Schrock and Yandulov unambiguously identified, based on 1H NMR, 15N NMR, IR, and X-ray crystallographic data, examples of comparatively stable Mo- and W-NNH species.16 Earlier evidence for such species was first reported by Chatt and coworkers,17 which was later validated by more careful vibrational studies from Lehnert and Tuczek for the complex (dppe)W(F)(NNH) (dppe = bis(diphenyl)phospinoethane).18 Lehnert and Tuczek also showed that some of the early assignments of “M-NNH” (M = Mo/W) species were misleading, owing to alternative M(H)(N2) isomers being highly favored, or in equilibrium with the M-NNH form, and also due to misinterpretation of complex vibrational data. Indeed, there have been long standing challenges associated with the use of vibrational data to reliably assign terminal M-NNH species, and their N-H vibrations in particular.18
There is also the mention of a cationic trans-(H)(DMeOPrPE)2Fe(HN=NH)+ species formed as part of a reaction mixture by Tyler and coworkers from the addition of hydrazine to (H)(DMeOPrPE)2Fe(Cl)+; within the same mixture, it is speculated that a resonance observed at 13.8 ppm in the 1H NMR spectrum may be attributable to (H)Fe(NNH), possibly generated via deprotonation of (H)Fe(HN=NH)+ by available hydrazine.19 Further data are not available.20
We have previously shown that the addition of stoichiometric acid, even at low temperature (−135 °C to −78 °C), to P3EFe-N2− species (E = B, Si, C) known to be active for NH3 generation, leads instead to net oxidation and liberation of 0.5 equiv of H2 (Eqn 1). Given that Fe-NNH species are predicted to have extremely weak N-H bonds,11g,21,22 such reactivity is perhaps unsurprising. The presence of additional acid can lead to the formation of P3EFe-NNH2+ (established for E = B, Si),11a,13,23 presumably via trapping of Fe-NNH prior to bimolecular H2 release (Eqn 2). This observation, along with related studies preparing kinetically stabilized, terminal Fe-NNR species (R = alkyl, silyl) by alkylation/silylation of Fe-N2− precursors,24,25 strongly implicates Fe-NNH intermediates.
| (Eqn 1) |
| (Eqn 2) |
To overcome the inherent challenges posed by direct detection of an Fe-NNH species in a system active for NH3 generation from N2, one with a comparatively very weak and therefore highly reactive N-H bond,26 we have undertaken the design of a sterically encumbered trisphosphine(borane)-iron system and have studied the protonation of its Fe-N2− derivative at extremely low temperature by continuous wave (CW) and pulse EPR spectroscopy, including electron-nuclear double resonance (ENDOR) and hyperfine sublevel correlation (HYSCORE) spectroscopies.27 Herein we describe the direct characterization of ArP3BFe-NNH (where Ar = 3,5-diisopropyl-4-methoxyphenyl), and its evolution to the next intermediate state, ArP3BFe-NNH2+. In addition to bearing on the mechanism of Fe-mediated nitrogen fixation by synthetic systems, these data provide the first detailed EPR signatures for an Fe-NNH species, using methods similar to those being employed to probe for intermediates of the FeMoco. These data should therefore prove valuable in constraining future assignments of plausible intermediates within the enzymatic system/s.
RESULTS AND DISCUSSION
To attenuate the rate of bimolecular degradation of a terminal Fe-NNH species, we targeted an electron rich, tris(diarylphosphino)borane system (ArP3B) (where Ar = 3,5-diisopropyl-4-methoxyphenyl) as a ligand framework that might provide sufficient vertical steric bulk to create a protective binding pocket for an NNH ligand. While aryl (instead of alkyl) substituents on the tris(phosphine)borane-iron system have not been compatible with N2RR catalysis, we reasoned that inclusion of isopropyl and methoxy substituents on the aryl rings would render the phosphines sufficiently electron-donating to be able to observe successful N2 protonation, and possibly even NH3 formation.
The new ArP3B ligand was assembled from previously reported orthobromophenyldichlorophosphine, 4-bromo-2,6-diisopropyl-1-methoxybenzene, and B(OMe)3 (Scheme 1).28,29 Metalation of ArP3B with FeBr2 in THF followed by sodium-mercury amalgam reduction in benzene afforded, following work-up, dark brown iron(I) ArP3BFeBr as an S = 3/2 dark brown microcrystalline solid (58.9 %, Scheme 1). The solid-state X-ray crystal structure of ArP3BFeBr is shown in Figure 2 (top), revealing a pseudo three-fold symmetric environment with the ligand binding in the expected tetrapodal fashion. Further reduction of ArP3BFeBr with sodium-mercury amalgam in benzene produced diamagnetic iron(0) ArP3BFe-N2, isolated as a dark green solid in 79.5 % yield. The diamagnetic nature of ArP3BFe-N2, compared with the S = 1 spin state of P3BFe-N2, derives from an additional interaction of the iron center with pi-density from one of the phenylene linkers (Figure 2, bottom), leading to a closed-shell 18-electron configuration.30 Access to the desired S = 1/2 anion ArP3BFe-N2− proceeded smoothly by additional reduction of ArP3BFe-N2 over sodium-mercury amalgam in THF.
Scheme 1.

Synthesis of ArP3B, metalation, and generation of ArP3BFe-N20/−
Figure 2.

ORTEP representation of (top) ArP3BFeBr (H atoms omitted for clarity), and (bottom) ArP3BFe(N2). Thermal ellipsoids are drawn at the 50% probability level.
Treatment of the solution with two equivalents of 12-crown-4 (12-C-4) and work-up provided [ArP3BFe(N2)][Na(12-C-4)2] in 90% yield as a red solid. While we were unable to obtain single crystals of [ArP3BFe(N2)][Na(12-C-4)2] suitable for solid-state XRD analysis, a DFT optimized geometry for ArP3BFe-N2− is shown in Figure 3 that illustrates the high degree of steric shrouding of the coordinated N2 ligand. Isostructural and isoelectronic iPrP3BFe-N2− has been characterized in the solid state.31 The N2 stretching frequency observed for ArP3BFe(N2) of 2014 cm−1 is very similar to that of the parent iPrP3BFe(N2) system (2011 cm−1) with isopropyl substituents on the phosphines. However, comparison of the N2 stretching frequencies of [ArP3BFe(N2)][Na(12-C-4)2] and [ArP3BFe(N2)][Na(12-C-4)2] (1937 cm−1 and 1905 cm−1, respectively) qualitatively suggests that the bulky arylphosphine ligand does not facilitate activation of the N2 ligand to quite the same extent that the parent ligand (iPrP3B) does at the redox state poised for protonation ([Fe-N2]−).31
Figure 3.

Space-filling model of the DFT optimized structure of ArP3BFe-N2−, using the full ArP3B ligand (TPSS, def2-TZVP on Fe, def2-SVP on C, H, P, B, O, N).
To assess the value of the ArP3BFe-system as a model for probing intermediates of catalytic N2-to-NH3 conversion, we examined whether NH3 could be generated by the addition of acid to [ArP3BFe(N2)][Na(12-C-4)2] in the absence of reductant. The NH3 yields obtained established the viability of N2-to-NH3 conversion (~0.25 equiv in the presence of 2 – 5 equiv acid) and show that early stage protonation is likely feasible. Nevertheless, the bulky ArP3BFe-system is inactive as a catalyst when both acid and reductant are present (using either HBArF24 in combination with KC8, or CoCp*2 with [Ph2NH2][OTf]). This is not unexpected. The extreme degree of steric bulk, and the fact that a phenyl-derivative of the parent system, [PhP3BFe(N2)][Na(12-C-4)2], is also inactive as a catalyst, suggest the ArP3BFe-system would be unlikely to be catalytically competent.32
We next investigated the reactivity of [ArP3BFe-N2][Na(12-C-4)2] with HBArF24•2Et2O (BArF24 = tetra-(3,5-bistrifluoromethylphenyl)borate) by continuous-wave (CW) X-band (~9.4 GHz) EPR spectroscopy (Figure 4).
Figure 4.

Reaction progress of [ArP3BFe(N2)][Na(12-C-4)2] with excess HBArF24•2Et2O in 2-Me-THF, as monitored by CW X-band EPR. Simulations for discrete species are shown in red. Spectra show the progression from: (A) Pure [ArP3BFe(N2)][Na(12-C-4)2]. (B) Mixture of [ArP3BFe(N2)][Na(12-C-4)2] and ArP3BFe(NNH), on addition of 1 equiv HBArF24•2Et2O for 15 min at 138 K. (C) Same as in spectrum B, but instead with 2.3 equiv HBArF24•2Et2O present, showing full conversion to ArP3BFe(NNH). An identical spectrum was obtained on mixing for 30 minutes in the presence of 1 equiv HBArF24•2Et2O. (D) Reaction mixture from trace C, after warming to 195 K for 30 seconds and then rapidly freeze quenching in liquid N2. Trace shows a mixture of ArP3BFe(NNH) and [ArP3BFe(NNH2)][BArF24]. (E) Reaction mixture from trace D after warming to 195 K for 90 seconds, showing complete conversion to [ArP3BFe(NNH2)][BArF24]. Reactions with up to 5 equiv HBArF24•2Et2O provided identical spectra. See Tables 1, 2, and 4 for relevant simulation parameters. Acquisition parameters for all spectra: temperature = 77 K; MW frequency = 9.44 GHz; MW power = 6.44 mW; modulation amplitude = 0.1 mT; conversion time = 5.12 ms. Simulations parameters for the signals in A C and E (with broadening parameters HStrain = [50 50 170] MHz and gStrain = [0.001, 0.02, 0.03]) can be found in Tables 1, 2, and 4, respectively.
Reactions between [ArP3BFe(N2)][Na(12-C-4)2], whose X-band EPR spectrum is shown in Figure 4A, and HBArF24•2Et2O were performed in EPR tubes in thawing 2-Me-THF solutions and mechanically mixed with a stainless steel needle. Mixing of [ArP3BFe(N2)][Na(12-C-4)2] with one equivalent of HBArF24•2Et2O in thawing 2-Me-THF for 15 minutes generated a new S = ½ species with a nearly axial signal with g = [2.021, 2.023, 2.177] (Figure 4B), assigned as the desired ArP3BFe(NNH). Mixing instead for 30 minutes, or mixing for 15 minutes but with an excess of acid (2.3 equiv), produces a clean S = ½ signal for ArP3BFe(NNH) (Figure 4C). This signal decays rapidly upon warming to −78 °C to generate a mixture of species, of which the major product is ArP3BFe(N2).
Warming in situ generated ArP3BFe(NNH) to −78 °C for 30 seconds in the presence of excess acid, followed by re-cooling the sample resulted in the growth of a new and more rhombic S = ½ signal with g = [2.004, 2.086, 2.176] (Figure 4D), corresponding to the doubly protonated species, [ArP3BFe(NNH2)][BArF24]. Warming for 90 instead of 30 seconds produces this signal cleanly, with full decay of the signal for ArP3BFe(NNH) (Figure 4E). Hence, the X-band EPR spectra collected in Figure 4 show the progression of the earliest stages of protonation of [ArP3BFe-N2]− via ArP3BFe(NNH) and [ArP3BFe(NNH2)]+.
To further corroborate these assignments, and to obtain more detailed information regarding the electronic structures of ArP3BFe(NNH) and [ArP3BFe(NNH2)][BArF24], pulse ENDOR and HYSCORE were applied to samples of [ArP3BFe(N2)]−, ArP3BFe(NNH), and [ArP3BFe(NNH2)][BArF24], as well as their isotopically enriched analogs generated using DBArF24•2Et2O, or beginning from the 15N-labelled anion [ArP3BFe(15N2)]− . We also prepared and similarly characterized ArP3BFe(NNSiMe3) as a stable isoelectronic and isostructural analogue of ArP3BFe(NNH).
For comparison to the protonated species of interest, the field-dependent Q-band ENDOR spectra of [ArP3BFe(N2)][Na(12-C-4)2] were collected. They are well simulated with hyperfine coupling to 11B and three unique 31P environments (Figure 5A and 5B, Table 1). The electron-spin-echo (ESE)-detected EPR spectrum is shown in Figure 5C.
Figure 5.

(A) Field-dependent Q-Band 11B-Davies ENDOR spectra of [ArP3BFe(N2)][Na(12-C-4)2] with simulation of 11B features (red). (B) Field-dependent Q-Band 31P-Davies ENDOR spectra of [ArP3BFe(N2)][Na(12-C-4)2] with simulation of individual 31P hyperfine couplings. Intense features near c.a. 50 MHz arise from weakly coupled ligand/solvent protons. (C) Electron-spin-echo(ESE)-detected EPR spectrum with ENDOR field positions marked in red. Acquisition parameters: temperature = 15 K; MW frequency = 34.124 GHz; τ = 300 ns; MW pulse length (π/2, π) = 40 ns, 80 ns; RF pulse length = 40 μs; shot repetition time (srt) = 5 ms.
Table 1.
g-Values and Nuclear hyperfine couplings (in MHz) derived from CW-EPR, ENDOR and HYSCORE for [ArP3BFe(N2)][Na(12-C-4)2]. *
| g | 2.014 | 2.074 | 2.144 | |
|---|---|---|---|---|
| Nucleus | A1 | A2 | A3 | aiso |
| 11B | 15.2 | 15.3 | 15.3 | 15.3 |
| 31P(1) | 41 | 20 | 18 | 26.3 |
| 31P(2) | 63 | 48 | 42 | 51 |
| 31P(3) | 123 | 111 | 110 | 115 |
| 14Nα | −1.4 | −2.3 | 0.7 | −1.0 |
In order to determine hyperfine couplings to 14/15N, we undertook HYSCORE studies, which generally are quite sensitive to highly anisotropic hyperfine couplings and quadrupolar nuclei, and offer equivalent information on hyperfine couplings and nuclear quadrupole coupling parameters for 14/15N in comparison to ENDOR spectroscopy. Isotopic labelling with 15N2 aided in independent determination of the hyperfine coupling constants for 15/14N and quadrupole parameters for 14N via HYSCORE spectroscopy at X-band (Figure 6; also see SI for other field positions). The combined HYSCORE data sets for natural abundance and 15N-enriched [ArP3BFe(N2)][Na(12-C-4)2] were well simulated by a single class of 14/15N, with A(14N) = [−1.4, −2.3, 0.7] MHz, nuclear quadrupole coupling constant e2Qq/h = 3.4 MHz, and very low estimation of the electric field gradient (e.f.g.) asymmetry parameter η = 0.1, consistent with the presence of axially symmetric charge density about N expected for an Fe-N≡N moiety. It is important to note that we do not have direct experimental evidence from the data for the absolute sign of the 14/15N hyperfine, but the experimental data and simulations are sensitive to the relative sign of the principle components of the hyperfine tensor.
Figure 6.

X-Band HYSCORE spectra (top) of natural abundance (left) and 15N enriched (right) [ArP3BFe(N2)][Na(12-C-4)2] with comparison (bottom) of experimental data (grey) to simulations of hyperfine couplings to 14/15Nα (red) using parameters in Table 1. Acquisition parameters: temperature = 20 K; magnetic field = 325.3 mT; microwave frequency = 9.411 GHz (14N), 9.424 GHz (15N); MW pulse length (π/2, π) = 8 ns, 16 ns; τ = 142 ns; t1 = t2 = 100 ns; Δt1 = Δt2 = 16 ns; shot repetition time (srt) = 1 ms).
Pulse EPR characterization of the proposed ArP3BFe(NNH) intermediate was pursued next. The viscous nature of the thawing 2-Me-THF and significant thermal sensitivity of the reaction precluded generation of this species in the relatively fragile, small diameter tubes used for Q-band ENDOR spectroscopy (O.D. = 1.6 mm), and X-band ENDOR spectroscopy was therefore employed instead. The field-dependent X-band ENDOR spectra of the Fe(NNH) species are well-simulated with hyperfine coupling to 11B, two unique 31P environments (one strongly coupled, and two equivalent, more weakly coupled nuclei), and a single acid-derived 1H nucleus (see Figure 7 and Table 2). Isotopic labelling using 2H-enriched HBArF24•2Et2O as the acid gave ArP3BFe(NND), which appears identical to ArP3BFe(NNH) by CW X-band EPR; however, subtraction of the ENDOR spectra of ArP3BFe(NND) from those of ArP3BFe(NNH) reveals coupling to a single acid-derived 1H nucleus with A(1H) = [19.3, 18.3, 12] MHz, (Figure 7B). X-band HYSCORE spectra of analagously prepared ArP3BFe(NNH) and ArP3BFe(NND) samples confirm the presence of this single class of acid-derived 1H (see SI). Specifically, the features from this acid-derived 1H that are well simulated using the same parameters determined from ENDOR are absent in the HYSCORE of ArP3BFe(NND). Additionally, 2H-1H difference spectra exhibit features arising from a single class of 2H coupling that is well simulated by scaling the previously determined 1H HFI by the proportion of 2H/1H gyromagnetic ratios (2Hγ/1Hγ = 0.154) such that A(2H) = [2.97, 2.82, 1.85] MHz, and relatively small 2H nuclear quadrupole parameters, e2Qq/h = 0.25 MHz and η = 0.1, as expected for deuterium in N-H bonds.33
Figure 7.

(A) Field-Dependent X-Band Davies ENDOR spectra of ArP3BFe(NNH) with simulation of individual hyperfine couplings. Acquisition parameters: temperature = 10 K; MW frequency = 9.72 GHz; τ = 240 ns; MW pulse length (π/2, π) = 20 ns, 40 ns; RF pulse length = 15 μs; srt = 5 ms. (B) Difference spectra of ArP3BFe(NNH) and ArP3BFe(NND) (black), with simulations of the acid-derived 1H hyperfine coupling (red). Difference spectra presented were smoothed using a 3-point Savitzky-Golay filter. The narrow peak at c.a. 10 MHz in these difference spectra are due to residual intensity from the 11B ENDOR signal. (C) X-band ESE-detected EPR spectrum with ENDOR field positions marked in red.
Table 2.
g-Values and Nuclear Hyperfine Couplings (in MHz) Derived from CW- EPR, ENDOR and HYSCORE for (ArP3B)Fe(NNH).*
| g | 2.021 | 2.023 | 2.177 | |
|---|---|---|---|---|
| Nucleus | A1 | A2 | A3 | aiso |
| 1H | 19.3 | 18.3 | 12.0 | 16.5 |
| 11B | 12 | 11.3 | 10.5 | 11.3 |
| 31P(1,2) | 22 | 17 | 17 | 18.7 |
| 31P(3) | 85 | 65 | 72 | 74 |
| 14Nα | −2.5 | −7.1 | −1.8 | −3.8 |
| 14Nβ | −2.9 | −1.8 | 0.7 | −1.3 |
The 1H hyperfine coupling derived from ENDOR and HYSCORE spectroscopy can be decomposed into its isotropic (aiso) and anisotropic components (Aaniso), giving aiso = 16.5 MHz and Aaniso = [2.8, 1.8, −4.5]. Utilizing the point-dipole approximation, the minimum distance of the 1H nucleus from the unpaired spin can be estimated using equation 3 and solving for r by taking the largest value of Aaniso as twice the dipolar component t of the 1H hyperfine, such that Aaniso = [−T(1−δ), −T(1+δ), 2T], where δ represents the rhombicity of Aaniso. This places the lower limit for the distance of the 1H from the metal center at 3.31 Å (i.e. r ≥ 3.31 Å). To evaluate whether this distance is consistent with the assignment of ArP3BFe(NNH), we performed a DFT geometry optimization on a model structure using the TPSS functional, the def2-TZVP basis set for Fe, and def2-SVP for all other atoms (see SI). This combination of functional and basis set have been previously used by our group in the study of the related P3BFe systems.11g,22 The distance between Fe and the proton on the β-nitrogen, predicted by the optimized structure, is ~3.52 Å, consistent with the estimated r ≥ 3.31 Å using Eqn 3.
| (Eqn 3) |
The relatively large value of aiso = 16.5 for this Fe-NNH proton, in comparison to the aforementioned structurally analogous iron hydroperoxo (Fe-OOH) intermediate in Cytochrome P450 (A1 ≈ 8.2 MHz via Q-band ENDOR)15c can likely be rationalized by the presence of spin density in Fe-Nα pi-bonds, facilitating polarization of the 1H s-orbital through hyperconjugation.
X-Band HYSCORE spectra of natural abundance ArP3BFe(NNH) and 15N-enriched ArP3BFe(NNH), along with corresponding simulations, are provided in Figure 8 (vide infra). In this case, simulations of the combined HYSCORE data of ArP3BFe(NNH) required consideration of two distinct classes of 14/15N nuclei, with A(14Nα) = [−2.5, −7.1, −1.8] and A(14Nβ) = [−2.9, −1.8, 0.7] MHz. Assignments of these couplings as Nα and Nβ is made on the basis of the magnitude of their isotropic hyperfine (aiso), which is generally sensitive to bonding covalency with the center of spin density. Simulations indicate these two nitrogens exhibit similar nuclear quadrupole parameters, with e2Qq/h (14Nα) = 2.0 MHz, e2Qq/h (14Nβ) = 1.8 MHz, and η(14Nα) = η(14Nβ) = 0.6.
Figure 8.

X-Band HYSCORE spectra (top) of natural abundance (left) and 15N enriched (right) ArP3BFe(NNH) with comparison (bottom) of experimental data (grey) to simulations of hyperfine couplings to 14/15Nα (red), 14/15Nβ (blue) and 11B (green) using parameters in Table 2. Acquisition parameters: temperature = 20 K; magnetic field = 330.1 mT; microwave frequency = 9.392 GHz; MW pulse length (π/2, π) = 8 ns, 16 ns; τ = 142 ns; t1 = t2 = 100 ns; Δt1 = Δt2 = 16 ns; shot repetition time (srt) = 1 ms).
As noted above, we also undertook the generation of the stable silylated complex ArP3BFe(NNSiMe3) by analogy to derivatives prepared previously.24 Vigorously stirring ArP3BFeBr in the presence of 1 equiv Me3SiCl and 3 equiv Na/Hg amalgam in THF followed by work-up afforded ArP3BFe(NNSiMe3) as a greenish-brown solid (νNN = 1717 cm−1; thin film). In a manner similar to ArP3BFe(NNH), the CW X-band EPR spectrum of ArP3BFe(NNSiMe3) features a near axial g-tensor with well-resolved hyperfine coupling to a single 31P atom evident at the high-field edge of the spectrum (see SI). Likewise, comparative ENDOR (Figure 9) and HYSCORE (Figure 10) data confirm similar trends in the 31P, 11B and 14N hyperfine coupling tensors to ArP3BFe(NNH); two weakly coupled 31P nuclei, with one more strongly coupled, and rather isotropic 11B and two distinct 14N nuclei (Table 3). Q-band HYSCORE was also performed on this species, confirming the presence of these two distinct 14N couplings (see SI).
Figure 9.

Field-Dependent Q-Band Davies ENDOR spectra of ArP3BFe(NNSiMe3). (A) Simulation of the 11B hyperfine coupling simulated in red. Asterisks indicate features arising from the 3rd harmonic of the higher frequency 11B ENDOR peak. (B) Simulation of individual 31P hyperfine couplings. Intense features near c.a. 49-52 MHz stem from weak 1H couplings from ligand or solvent protons. (C) Q-band ESE-detected field sweep with field positions at which the ENDOR spectra were acquired marked in red. Acquisition parameters: temperature = 15 K; MW frequency = 34.040 GHz; MW pulse length (π/2, π) = 20 ns, 40 ns;τ = 132 ns; RF pulse length = 15 μs; srt = 5 ms).
Figure 10.

(top) X-Band HYSCORE spectrum of ArP3BFe(NNSiMe3), with comparison (bottom) of experimental data to simulations (grey) of 14Nα (red), 14Nβ (blue) and 11B (green), using the simulation parameters in Table 3. Acquisition parameters: temperature = 20 K; magnetic field = 334.0 mT; microwave frequency = 9.392 GHz; MW pulse length (π/2, π) = 8 ns, 16 ns; τ = 140 ns; t1 = t2 = 100 ns; Δt1 = Δt2 = 16 ns; shot repetition time (srt) = 1 ms).
Table 3.
g-Values and Nuclear Hyperfine Couplings (in MHz) Derived from CW-EPR, ENDOR and HYSCORE for ArP3BFe(NNSiMe3). *
| g | 2.009 | 2.030 | 2.234 | |
|---|---|---|---|---|
| Nucleus | A1 | A2 | A3 | aiso |
| 11B | 12.5 | 12.7 | 13.5 | 12.9 |
| 31P(1) | 25 | 16 | 26 | 22.3 |
| 31P(2) | 36 | 26 | 26 | 29.3 |
| 31P(3) | 111 | 90 | 92 | 97.7 |
| 14Nα | −5.5 | −7.6 | −1.6 | −4.9 |
| 14Nβ | −4.0 | −4.5 | −1.8 | −3.4 |
Importantly, simulations of the 14N HYSCORE data for these two complexes indicate that the nuclear quadrupole coupling constant and electric field gradient asymmetry parameters of the two classes of detected 14N nuclei of ArP3BFe(NNSiMe3) and ArP3BFe(NNH) are the same, with e2Qq/h (14Nα) = 2.0 MHz, e2Qq/h (14Nβ) = 1.8 MHz, and η(14Nα) = η(14Nβ) = 0.6, corroborating similarity in bonding environments and symmetry of charge about the N nuclei in each complex. This confirms that the silyl-diazenido analog is an appropriate electronic structure model for the more reactive ArP3BFe(NNH).
To confirm the assignment of [ArP3BFe(NNH2)][BArF24] as the product of the reaction of [ArP3BFe(N2)][Na(12-C-4)2] with excess acid, we collected Q-band ENDOR spectra to probe the 11B, 31P and 1H hyperfine interactions. For the assignment of this species, we benefit from comparative EPR spectral data already available for the parent [P3BFe(NNH2)][BArF24].23
The field-dependent Q-Band ENDOR spectra of [ArP3BFe(NNH2)][BArF24] are well modeled with hyperfine couplings to 11B, three nearly equivalent 31P nuclei, and two 1H nuclei, with one being strongly coupled and one more weakly coupled (Figures 11 and Table 4). X-band HYSCORE spectra of the similarly prepared [ArP3BFe(NNH2)][BArF24] and [ArP3BFe(NND2)][BArF24] samples confirm the presence of these two classes of acid-derived 1H (see SI).
Figure 11.

Field-Dependent Q-Band Davies ENDOR spectra of [ArP3BFe(NNH2)][BArF24]. (A) Simulations of the 11B hyperfine coupling in red using simulation parameters in Table 4. (B) Simulations of the individual 31P and 1H hyperfine couplings, using simulation parameters in Table 4. (C) Difference spectra of 1H and 2H [ArP3BFe(NNH2)][BArF24] (black) with individual simulations of the two acid-derived 1H hyperfine couplings. Difference spectra presented were smoothed using a 3 point Savitzky-Golay filter. (D) Q-band ESE-detected EPR spectrum with ENDOR field positions marked in red. The asterisk indicates a small background signal arising from a component of the EPR cavity. Acquisition parameters: temperature = 10 K; MW Frequency = 34.121 GHz; τ = 240 ns; MW pulse length (π/2, π) = 20 ns, 40 ns; RF pulse length = 15 μs; srt = 5ms).
Table 4:
g-Values and Nuclear Hyperfine Couplings (in MHz) derived from CW-EPR, ENDOR and HYSCORE for [ArP3BFe(NNH2)][BArF24].*
| g | 2.004 | 2.087 | 2.176 | |
|---|---|---|---|---|
| Nucleus | A1 | A2 | A3 | aiso |
| 1Ha | 16 | 21 | 27 | 21.3 |
| 1Hb | 10 | 13.5 | 15 | 12.8 |
| 11B | 9.5 | 7.5 | 6.1 | 7.7 |
| 31P(1) | 40 | 16 | 23 | 26.3 |
| 31P(2) | 46 | 47 | 47 | 46.7 |
| 31P(3) | 64 | 78 | 65 | 69 |
| 14Nα | −0.9 | −7.1 | 0.1 | −2.6 |
Note: 1Ha includes an Euler rotation of the hyperfine tensor relative to the molecular frame of (α, β, γ) = [0, 35, 0]°.
See figures 11, 12. Approximate error estimates for A values determined from simulations are: 1H (A ± 5 %); 11B (A ± 2 %); 31P (A ± 5 %); 14/15Nα (A ± 10 %).
These parameters are similar to those for the analogous [P3BFe(NNH2)][BArF24] previously characterized by similar methods; however, the H-atoms on Nβ in [ArP3BFe(NNH2)][BArF24] exhibit somewhat larger isotropic hyperfine couplings23, potentially due to a greater extent of polarization of the 1H s-orbitals via hyperconjugation with spin density localized in Fe-Nα pi-symmetery orbitals in-plane with the Nβ-H bonds. Decomposition of the observed 1H hyperfine couplings into their isotropic and anisotropic components provides aiso = 21.3 MHz, Aaniso = [−5.3, −0.3, 5.7] MHz for the more strongly coupled 1H, and aiso = 12.8 MHz, Aaniso = [−2.8, 0.7, 2.2] for the more weakly coupled 1H.
Again from equation 3, the estimated minimum distances r for the two protons are: r(1) ≥ 3.07 Å for the more strongly coupled 1H, and r(2) ≥ 3.89 Å for the more weakly coupled 1H. These distances are consistent with both protons being localized on Nβ, and are in reasonably good agreement with the predicted Fe-H distances from a DFT optimized structure of [ArP3BFe(NNH2)][BArF24]: 3.20 Å and 3.73 Å, respectively. In a manner similar to the previously characterized [P3BFe(NNH2)][BArF24], an Euler rotation (β = 35°) of the hyperfine tensor of the more strongly coupled proton was necessary to simulate the ENDOR spectra, consistent with a non-linear Fe-N-NH2 moiety with an Fe-N-N bond angle of ~ 140°, also consistent with the DFT optimized structure.
X-Band HYSCORE spectra of natural abundance [ArP3BFe(NNH2)][BArF24] and its 15N-enriched derivative, along with corresponding simulations are provided in Figure 12. For this species, only a single coupling to 14/15N with A(14Nα) = [−0.9, 7.1, 0.1] MHz, e2Qq/h (14Nα) = 1.7 MHz, and η(14Nα) = 0.7 is neccessary to simulate the experimental data. This was also the case for the previously characterized [P3BFe(NNH2)][BArF24], with only a single class of nitrogen detected via Q-band ENDOR and electron spin echo envelope modulation (ESEEM).23 Q-band HYSCORE was also performed on natural abundance [P3BFe(NNH2)][BArF24] in order to evaluate the possibility of coupling to a second class of 14N, but this data was also well simulated using the same 14N simulation parameters derived from X-band HYSCORE (see SI).
Figure 12.

X-Band HYSCORE spectra (top) of natural abundance (left) and 15N enriched (right) [ArP3BFe(NNH2)][BArF24] with comparison (bottom) of experimental data (grey) to simulations of 14/15Nα (red), and 11B (green) using parameters in Table 4. Acquisition parameters: temperature = 20 K; magnetic field = 324.1 mT; microwave frequency = 9.424 GHz (14N), 9.423 GHz (15N); MW pulse length (π/2, π) = 8 ns, 16 ns; τ = 142 ns; t1 = t2 = 100 ns; Δt1 = Δt2 = 16 ns; shot repetition time (srt) = 1 ms.
Information about the electronic structure of the NNHx (x = 0 1, 2) moiety can be gleaned from examination of the nuclear hyperfine couplings, nuclear quadrupole couplings (e2Qq/h) and electric field gradient asymmetry parameters (η) for Nα across the series of [ArP3BFe(N2)]−, ArP3BFe(NNH) and [ArP3BFe(NNH2)]+. These parameters are of particular interest because of the scarcity of data on these types of iron compounds, and their potential value in the identification of catalytically relevant intermediates in synthetic and biological nitrogen fixation systems.
To determine the hyperfine couplings to nitrogen, as well as the nuclear quadrupole parameters e2Qq/h and η for the 14N atoms in the N2, NNH, and NNH2 ligands, we relied on comparison of the X-Band HYSCORE spectra of samples of [ArP3BFe(N2)][Na(12-C-4)2], ArP3BFe(NNH), and [ArP3BFe(NNH2)][BArF24] with that of their 15N-labeled samples (Figures 6, 8, 10, and 12, and Tables 5 and 6). Simulation of the 15N HYSCORE allowed for the 15N HFI to be determined in the absence of any complications form the influence of nuclear quadrupole interactions. Accounting for the ratio of 14/15N gyromagnetic ratios (14Nγ/15Nγ = − 0.7219), the 14N HFI could be fixed, allowing the nuclear quadrupole interactions to be estimated independently via simulation of the 14N HYSCORE spectra. The combined HYSCORE data sets reveal that the hyperfine tensors assigned to Nα in each case exhibit significant anisotropy, comparable in magnitude to the isotropic component in each complex. Specifically, aiso = −1.0 MHz for [ArP3BFe(N2)][Na(12-C-4)2]; aiso = −3.8 MHz for ArP3BFe(NNH); aiso = −2.6 MHz for [ArP3BFe(NNH2)][BArF24].
Table 5:
14N Hyperfine Coupling Parameters (in MHz) for Nα in the (ArP3B)Fe(NαNβHx) series.
| Compound | A(14N) | aiso | Aaniso |
|---|---|---|---|
| [ArP3BFe(N2)]− | [−1.4, −2.3, 0.7] | −1.0 | [−0.4, −1.3, 1.7] |
| ArP3BFe(NNH) | [−2.5, −7.1, −1.8] | −3.8 | [1.3, −3.3, 2] |
| ArP3BFe(NNSiMe3) | [−5.5, −7.6, −1.6] | −4.9 | [−0.6, −2.7, 3.3] |
| [ArP3BFe(NNH2)]+ | [−0.9, −7.1, 0.1] | −2.6 | [1.7, −4.5, 2.7] |
| [P3BFe(NNH2)]+† | [−6.2, −7.2, −4.3] | −5.9 | [−0.3, −1.3, 1.6] |
See ref 23
Table 6:
14N Nuclear Quadrupole Parameters derived from ENDOR and HYSCORE for Nα.
| Compound | e2Qq/h (MHz) | η |
|---|---|---|
| [ArP3BFe(N2)]− | 3.4 | 0.1 |
| (ArP3B)Fe(NNH) | 2.0 | 0.6 |
| ArP3BFe(NNSiMe3) | 2.0 | 0.6 |
| [ArP3BFe(NNH2)]+ | 1.7 | 0.7 |
| [P3BFe(NNH2)]+ † | 1.74 | 0.64 |
See ref 23. Approximate error estimates for e2Qq/h and η are ± 0.2 and ± 0.1 MHz, respectively.
The magnitude of the anisotropic contributions to the 14N hyperfine increases as a function of degree of protonation. Additionally, for each of the complexes measured in this series, Aaniso exhibits significant rhombic symmetry, and in the case of the ArP3BFe(NNH), ArP3BFe(NNSiMe3), and [ArP3BFe(NNH2)]+ species, is too large to be purely accounted for by the through-space dipolar interaction between Nα and the spin density localized on Fe. Utilizing the point dipole approximation and Fe-N distances from the geometry-optimized DFT structures, the estimated 14Nα dipolar hyperfine magnitudes for the FeNNx series of complexes examined here are all within the range of 1.01 < t < 1.188 MHz. The higher magnitude of Aaniso values for the ArP3BFe(NNH) and [ArP3BFe(NNH2)][BArF24] indicates at least partial, if modest, delocalization of spin onto p-orbitals at Nα upon protonation. The significant rhombicity of the hyperfine tensors for all species examined here suggests modest localization of unpaired spin density in multiple orthogonal p-orbitals at Nα, which increases as a function of protonation.
The 14N nuclear quadrupole interaction measured through HYSCORE arises from the interaction between the 14N quadrupole moment with the inhomogeneous electric field produced by electrons in p-orbitals localized at the nucleus. As such, this interaction provides a point-specific measure of the axial electric field gradient magnitude (parametrized by e2Qq/h) and its symmetry, parametrized by η, which ranges from 0 for pure axial symmetry to 1 for full rhombic symmetry. Starting from [ArP3BFe(N2)]−, the trend in the observed 14Nα quadrupole parameters for the series can be discussed in terms of the Townes-Dailey model,34 in which these quadrupole parameters are primarily determined by differences in the relative electron ocupancy in the nitrogen 2pσ orbital (denoted N3) and the average of the densities in the two orthogonal 2pπ orbitals (N1, N2), such that |e2qQ/e2qQ0| = |N3 − (N1 + N2)/2|, where e2qQ0 = −(8-10) MHz for a single electron in a 2p orbital for the sp hybridized Nα. Differences in the relative charge densities of the two orthogonal 2pπ orbitals at 14Nα contributes to the electric field gradient asymmetry η. With this simple picture in mind, the downward trend (Table 6) in the magnitude of e2Qq/h as a function of protonation of the terminal N atom can be rationalized by a decrease in in N-N sigma bonding relative to Fe-N sigma-bonding and/or Fe-N pi bonding. The subsequent increase in η suggests a significant imbalance between the charge density in the two orthogonal 14Nα 2pπ orbitals, as would be expected to accompany loss of a single Fe-N pi-bond upon protonation, though a loss of linearity of the Fe-N-N moiety would also be expected to contribute significantly.
Interestingly, the most significant change in the 14Nα quadrupole interaction occurs after the first protonation, with a reduction of e2Qq/h by ~ 40% and a significant increase in η from 0.1 to 0.6, with a much more modest reduction of e2Qq/h and increase in η upon the second protonation. This observation suggests that there is fairly little change in the bonding environment at Nα in going from NNH to NNH2 in comparison to the initial protonation. Also, the measured 14N quadrupole coupling parameters for (ArP3BFe(NNH) and the silyl-diazenido analogue ArP3BFe(NNSiMe3) are identical, reflecting very similar bonding environment at Nα and Nβ for these two species, respectively. Finally, the similarity of e2Qq/h = 1.7 MHz and η = 0.7 for [ArP3BFe(NNH2)][BArF24] to the previously reported values of e2Qq/h = 1.74 MHz and η = 0.64 for Nα in [P3BFe(NNH2)][BArF24], suggests that the bonding character/electronic structure at Nα is comparable in both compounds, further cementing the assignment of [ArP3BFe(NNH2)][BArF24] (Table 6).23,35
CONCLUSIONS
We have described the first characterization of a terminally bonded Fe(NNH) species, generated by protonation of Fe-N2−. Fe-NNH intermediates provide a linchpin between the limiting mechanisms of Fe-mediated N2-to-NH3 conversion, and feature unusually weak and hence reactive N-H bonds, with estimated BDFEN-H values less than 35 kcal/mol for the types species discussed here. As a result, Fe(NNH) species had proven very difficult to detect up to now. CW and pulse EPR methods have been shown to be highly effective in the characterization of reactive S = ½ states of the FeMoco, and such tools, coupled with a sterically encumbering tris(phosphine)borane ligand and very low temperatures, enabled us to detect and assign ArP3BFe(NNH) and observe its evolution to doubly protonated ArP3BFe(NNH2)+. The latter species is a presumed on-path intermediate for the structurally related and catalytically active P3BFe system. The synthesis and characterization of a stable ArP3BFe(NNSiMe3) complex, which serves as an isoelectronic and kinetically stabilized relative of ArP3BFe(NNH), adds additional support our assignment.
The 1H-hyperfine for ArP3BFe(NNH) derived from the presented ENDOR studies is diagnostic for the distally-bound H-atom (aiso = 16.5 MHz). The 14Nα quadrupole interaction, extracted from HYSCORE data for each species discussed, shows significant change in bonding at Nα upon the first protonation step (Fe-N2− → Fe(NNH)) with a ~ 40% decrease in e2Qq/h consistent with loss of N-N sigma-bonding and increase in Fe-N sigma bonding, and the significant increase in the electric field gradient asymmetery η with differential charge densities in 2px,y orbitals at C and loss of Fe-N-N linearity. This bonding symmetry at Nα appears to be largely maintained at the second protonation step that produces Fe(NNH2)+.
By analogy to the better studied reductive protonation of oxygen at iron introduced earlier, the S = ½ Fe(NNH) species can be considered as an N2-derived analogue of iron(III) hydroperoxide species, FeIII(OOH), well known in heme and non-heme iron systems in the context of oxygen binding and reductive protonation. EPR methods were also crucial to the characterization these FeIII(OOH) species, and in particular in identifying the H-atom bound to the the distal O-atom.15 While the S = 1/2 iron center in ArP3BFe(NNH) can be most simply formulated as monovalent (ignoring the sigma backbond to boron), it can also be regarded as trivalent (including the sigma backbond to boron).30 The latter description underscores the analogy with FeIII(OOH). The present Fe(NNH) system is highly covalent and hence valence assignments, while helpful in considering this conceptual analogy, need to be interpreted with caution. Additional studies are of interest to map the electronic structures of Fe(NNH) species as a function of coordination number, geometry, and ligand field strength.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (R01-070757). The EPR facility at the California Institute of Technology has been supported by the NSF via its MRI program (NSF-1531940), and the DOW Next Generation Educator Fund. We thank Dirk Schild for performing a BDFEN-H calculation on trans-(H)(DMeOPrPE)2Fe(N=NH), and Dr. Jonathan Rittle for contributions to the ligand synthesis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Procedures and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Burgess BK; Lowe DJ Mechanism of Molybdenum Nitrogenase. Chem. Rev 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- 2.Eady RR Structure–Function Relationships of Alternative Nitrogenases. Chem. Rev 1996, 96, 3013–3030. [DOI] [PubMed] [Google Scholar]
- 3.Howard JB; Rees DC How many metals does it take to fix N2? A mechanistic overview of biological nitrogen fixation . Proc. Natl. Acad. Sci 2006, 103, 17088–17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffman BM; Lukoyanov D; Yang Z; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatt J; Dilworth JR; Richards RL Recent advances in the chemistry of nitrogen fixation. Chem. Rev 1978, 78, 589–625. [Google Scholar]
- 6.Hidai M; Mizobe Y Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev 1995, 95, 1115–1133. [Google Scholar]
- 7.Yandulov DV; Schrock RR Studies Relevant to Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum Triamidoamine Complexes. Inorg. Chem 2005, 44, 1103–1117. [DOI] [PubMed] [Google Scholar]
- 8.Yandulov DV; Schrock RR Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [DOI] [PubMed] [Google Scholar]
- 9.(a) Arashiba K; Miyake Y; Nishibayashi Y A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Commun 2010, 3, 120–125. [DOI] [PubMed] [Google Scholar]; (b) Nishibayashi Y Development of catalytic nitrogen fixation using transition metal–dinitrogen complexes under mild reaction conditions. Dalton Trans. 2018, 47, 11290–11297. [DOI] [PubMed] [Google Scholar]
- 10.(a) Spatzal T; Perez K; Einsle O; Howard JB; Rees DC Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morrison CN; Hoy JA; Zhang L; Einsle O; Rees DC Substrate Pathways in the Nitrogenase MoFe Protein by Experimental Identification of Small Molecule Binding Sites. Biochemistry 2015, 54, 2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–1489. [DOI] [PubMed] [Google Scholar]
- 11.For examples of synthetic Fe catalysts for N2RR see:; (a) Anderson JS; Rittle J; Peters JC Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Creutz SE; Peters JC Catalytic Reduction of N2 to NH3 by an Fe–N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc 2014, 136, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ung G; Peters JC Low-Temperature N2 Binding to Two-Coordinate L2Fe0 Enables Reductive Trapping of L2FeN2− and NH3 Generation. Angew. Chem. Int. Ed 2015, 54, 532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Del Castillo TJ; Thompson NB; Peters JC A Synthetic Single-Site Fe Nitrogenase: High Turnover, Freeze-Quench 57Fe Mössbauer Data, and a Hydride Resting State. J. Am. Chem. Soc 2016, 138, 5341–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kuriyama S;Arashiba K;Nakajima K;Matsuo Y;Tanaka H;Ishii K;Yoshizawa K;Nishibayashi Y Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand. Nat. Commun 2016, 7, 12181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Buscagan TM; Oyala PH; Peters JC N2-to-NH3 Conversion by a triphos–Iron Catalyst and Enhanced Turnover under Photolysis. Angew. Chem. Int. Ed 2017, 56, 6921–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chalkley MJ; Del Castillo TJ; Matson BD; Roddy JP; Peters JC Catalytic N2-to-NH3 Conversion by Fe at Lower Driving Force: A Proposed Role for Metallocene-Mediated PCET. ACS Cent. Sci. 2017, 3, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson NB; Green MT; Peters JC Nitrogen Fixation via a Terminal Fe(IV) Nitride. J. Am. Chem. Soc 2017, 139, 15312–15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rittle J; Peters JC An Fe-N2 Complex That Generates Hydrazine and Ammonia via Fe═NNH2: Demonstrating a Hybrid Distal-to-Alternating Pathway for N2 Reduction. J. Am. Chem. Soc 2016, 138, 4243–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Mankad NP; Whited MT; Peters JC Terminal FeI–N2 and FeII⋯H–C Interactions Supported by Tris(phosphino)silyl Ligands. Angew Chem. Int. Ed 2007, 46, 5768–5771. [DOI] [PubMed] [Google Scholar]; (b) Hill PJ; Doyle LR; Crawford AD; Myers WK; Ashley AE Selective Catalytic Reduction of N2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc 2016, 138, 13521–13524. [DOI] [PubMed] [Google Scholar]
- 15.See for example:; (a) Neese F; Zaleski JM; Loeb Zaleski K; Solomon EI Electronic Structure of Activated Bleomycin: Oxygen Intermediates in Heme versus Non-Heme Iron. J. Am. Chem. Soc 2000, 122, 11703–11724. [Google Scholar]; (b) Solomon EI; Wong SD; Liu LV; Decker A; Chow MS Peroxo and oxo intermediates in mononuclear nonheme iron enzymes and related active sites. Curr. Op. Chem. Bio. 2009, 13, 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Davydov R; Macdonald IDG; Makris TM; Sligar SG; Hoffman BM EPR and ENDOR of Catalytic Intermediates in Cryoreduced Native and Mutant Oxy-Cytochromes P450cam: Mutation-Induced Changes in the Proton Delivery System. J. Am. Chem. Soc 1999, 121, 10654–10655. [Google Scholar]; (d) Tajima K A possible model of a hemoprotein-hydrogen peroxide complex. Inorg. Chim. Acta 1989, 163, 115–122. [Google Scholar]; (e) Veselov A; Burger RM; Scholes CP Q-band Electron Nuclear Double Resonance of Ferric Bleomycin and Activated Bleomycin Complexes with DNA: Fe(III) Hyperfine Interaction with 31P and DNA-Induced Perturbation to Bleomycin Structure. J. Am. Chem. Soc 1998, 120, 1030–1033. [Google Scholar]; (f) Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Costas M; Mehn MP; Jensen MP; Que L Dioxygen Activation at Mononucleaer Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]; (h) Liang Y; Wei J; Qiu X; Jiao N Homogeneous Oxygenase Catalysis. Chem. Rev 2018, 118, 4912–4945. [DOI] [PubMed] [Google Scholar]; (i) Denisov IG; Makris TM; Sligar SG; Schlichtin I Structure and Chemistry of Cyctochrome P450. Chem. Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]; (j) Oloo WN; Que L Bioinspired Nonheme Iron Catalysts for C-H and C=C bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res 2015, 48, 2612–2621. [DOI] [PubMed] [Google Scholar]
- 16.(a) Yandulov DV; Schrock RR; Rheingold AL; Ceccarelli C; Davis WM Synthesis and Reactions of Molybdenum Triamidoamine Complexes Containing Hexaisopropylterphenyl Substituents. Inorg. Chem 2003, 42, 796–813. [DOI] [PubMed] [Google Scholar]; (b) Yandulov DV; Schrock RR Synthesis of tungsten complexes that contain hexaisopropylterphenyl-substituted triamidoamine ligands, and reactions relevant to the reduction of dinitrogen to ammonia. Can. J. Chem 2005, 83, 341–357. [Google Scholar]
- 17.Chatt J; Pearman AJ; Richards RL Diazenido-complexes of molybdenum and tungsten. J. Chem. Soc., Dalton Trans. 1976, 1520–1524. [Google Scholar]
- 18.Lehnert N; Tuczek F The Reduction Pathway of End-on Coordinated Dinitrogen. I. Vibrational Spectra of Mo/W–N2, –NNH, and –NNH2 Complexes and Quantum Chemistry Assisted Normal Coordinate Analysis. Inorg. Chem 1999, 38, 1659–1670. [DOI] [PubMed] [Google Scholar]
- 19.Crossland JL; Balesdent CG; Tyler DR Coordination of a Complete Series of N2 Reduction Intermediates (N2H2, N2H4, and NH3) to an Iron Phosphine Scaffold. Inorg. Chem 2012, 51, 439–445. [DOI] [PubMed] [Google Scholar]
- 20.To evaluate the plausibility of this assignment, we calculated the N-H BDFE of the hypothetical trans-(H)(DMeOPrPE)2Fe(N=NH) structure using methods described elsewhere (see reference 22). Our calculation predicts a weak BDFEN-H of only 27.3 kcal/mol, suggesting such a species would be highly unstable in solution at room temperature and is therefore unlikely to correspond to the 1H NMR resonance assigned to it.
- 21.Rittle J; Peters JC N–H Bond Dissociation Enthalpies and Facile H Atom Transfers for Early Intermediates of Fe–N2 and Fe–CN Reductions. J. Am. Chem. Soc 2017, 139, 3161–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matson BD; Peters JC Fe-Mediated HER vs N2RR: Exploring Factors That Contribute to Selectivity in P3EFe(N2) (E = B, Si, C) Catalyst Model Systems. ACS Catal. 2018, 8, 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson JS; Cutsail GE; Rittle J; Connor BA; Gunderson WA; Zhang L; Hoffman BM; Peters JC Characterization of an Fe≡N–NH2 Intermediate Relevant to Catalytic N2 Reduction to NH3. J. Am. Chem. Soc 2015, 137, 7803–7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For a pertinent review, see:; Dilworth JR Diazene, diazenido, isodiazene and hydrazido complexes. Coord. Chem. Rev 2017, 330, 53–94. [Google Scholar]
- 25.For specific examples, see:; (a) Betley TA; Peters JC . Dinitrogen Chemistry from Trigonally Coordinated Iron and Cobalt Platforms. J. Am. Chem. Soc 2003, 125, 10782–10783. [DOI] [PubMed] [Google Scholar]; (b) Lee Y; Mankad NP; Peters JC Triggering N2 uptake via redox-induced expulsion of coordinated NH3 and N2 silylation at trigonal bipyramidal iron. Nat. Chem 2010, 2, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moret ME; Peters JC N2 Functionalization at Iron Metallaboratranes. J. Am. Chem. Soc, 2011, 133, 18118–18121. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Piascik AD; Hill PJ; Crawford AD; Doyle LR; Green JC; Ashley AE Cationic silyldiazenido complexes of the Fe(diphosphine)2(N2) platform: structural and electronic models for an elusive first intermediate in N2 fixation. Chem. Commun 2017, 7657–7660. Also see: 11c. [DOI] [PubMed] [Google Scholar]
- 26.For comparison, we have estimated the N-H BDFE for P3BFe-NNH as 31.2 kcal/mol, compared with 49.7 kcal/mol for Schrock’s original (HIPT)Mo-NNH species. See:; Chalkley MJ; Del Castillo TJ; Matson BD; Peters JC. Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring pKa Effects and Demonstrating Electrocatalysis. J. Am. Chem. Soc 2018, 140, 6122–6129. Also see ref 11g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Hoffman BM Electron-nuclear double resonance spectroscopy (and electron spin-echo envelope modulation spectroscopy) in bioinorganic chemistry. Proc. Natl. Acad. Sci. 2003, 100, 3575–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Calle C; Sreekanth A; Fedin M; Forrer J; Garcia-Rubio I; Gromov I; Hinderberger D; Kasumaj B; Léger P; Mancosu B; Mitrikas G; Santangelo M; Stoll S; Schweiger A; Tschaggelar R; Harmer J Pulse EPR Methods for Studying Chemical and Biological Samples Containing Transition Metals. Helv. Chim. Acta 2006, 89, 2495–2521. [Google Scholar]; (c) van Gastel M Pulsed EPR spectroscopy. Photosynth. Res 2009, 102, 367–373. [DOI] [PubMed] [Google Scholar]; (d) Cox N; Nalepa A; Pandelia ME; Lubitz W; Savitsky A Pulse Double-Resonance EPR Techniques for the Study of Metallobiomolecules. Methods Enzymol. 2015, 563, 211–249. [DOI] [PubMed] [Google Scholar]
- 28.Chiswell B; Venanzi LM Some manganese(I) complexes with multidentate substituted phosphine ligands. J. Chem. Soc. A: Inorg., Phys., Theor 1966, 417–419. [Google Scholar]
- 29.Provencher L; Wynn H; Jones JB; Krawczyk AR Enzymes in organic synthesis 51. Probing the dimensions of the large hydrophobic pocket of the active site of pig liver esterase. Tetrahedron: Asymmetry 1993, 4, 2025–2040. [Google Scholar]
- 30.Using the neutral ligand formalism for counting valence electrons in ArP3BFe(N2): 18-electrons (3 x 2 e− from P donors; 2 e− from N2; 8 e− from Fe; 2 e− from an arene-π interaction). Also note that while ArP3BFe(N2) is appropriately assigned as iron(0), regarding the tris(phosphine)borane as a neutral ligand, if one considers a sigma backbond from iron-to-boron, the iron center is then assigned as divalent. Likwise, ArP3BFe(N2)− can be assigned as monovalent at iron if a sigma backbond is assumed.
- 31.Moret M-E; Peters JC Terminal Iron Dinitrogen and Iron Imide Complexes Supported by a Tris(phosphino)borane Ligand. Angew. Chem. Int. Ed 2011, 50, 2063–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Note that the N2 ligand in [ArP3BFe(N2)][Na(12-C-4)2] is significantly less activated than that of the parent system (ν(NN) = 1937 cm−1 for ArP3B versus 1905 cm−1 for P3B), which generally correlates with attenuated NH3 yields. Also, ArP3BFe(N2) is structurally (and electronically) distinct from P3BFe(N2), owing to the additional arene interaction (Figure 2). This is manifest in an irreversible ArP3BFe-N2/ArP3BFe-N2− reduction event (see SI), which may also short-circuit catalytic N2-to-NH3 conversion in preference to HER.
- 33.From a linear correlation of 2H e2Qq/h for deuterium nuclei in N+-D…O bonds with the hydrogen bond strength (parametrized by 1/R(D…O)3), Hunt et al. estimated that the 2H e2Qq/h for an isolated (non-hydrogen bonded) N+-D group should be ~ 0.25 MHz, in good agreement with the values estimated from the 2H HYSCORE simulations for the Nβ-D nuclei in this study. See:; Hunt MJ; Mackay AL Deuterium and Nitrogen Pure Quadrupole Resonance in Deuterated Amino Acids. Journ. Mag. Res 1973, 15, 402–414. [Google Scholar]
- 34.Townes CH; Dailey BP Determination of Electronic Structure of Molecules from Nuclear Quadrupole Effects. J. Chem. Phys 1949, 17, 782–796. [Google Scholar]
- 35.Lucken EAC Nuclear Quadrupole Coupling Constants; Academic Press: London, 1969. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.