

Time-resolved x-ray scattering reveals light-induced signal transduction in insect cryptochromes.
Abstract
Cryptochromes are blue-light photoreceptor proteins, which provide input to circadian clocks. The cryptochrome from Drosophila melanogaster (DmCry) modulates the degradation of Timeless and itself. It is unclear how light absorption by the chromophore and the subsequent redox reactions trigger these events. Here, we use nano- to millisecond time-resolved x-ray solution scattering to reveal the light-activated conformational changes in DmCry and the related (6-4) photolyase. DmCry undergoes a series of structural changes, culminating in the release of the carboxyl-terminal tail (CTT). The photolyase has a simpler structural response. We find that the CTT release in DmCry depends on pH. Mutation of a conserved histidine, important for the biochemical activity of DmCry, does not affect transduction of the structural signal to the CTT. Instead, molecular dynamics simulations suggest that it stabilizes the CTT in the resting-state conformation. Our structural photocycle unravels the first molecular events of signal transduction in an animal cryptochrome.
INTRODUCTION
The cryptochrome protein family is widespread in plants and animals (1, 2). Cryptochromes are important for the entrainment of the circadian rhythm (3–5) and have also been suggested to be involved in magnetoreception (6–9). The blue-light–sensing flavoproteins stem from the DNA repair enzymes known as photolyases but have since evolved to blue-light photoreceptors with little or no DNA repair ability (10). Cryptochromes and photolyases have a noncovalently bound light- and redox-active flavin adenine dinucleotide (FAD) as an essential chromophore. The FAD can be reduced twice, and whereas fully reduced FAD is required for DNA repair in DNA photolyases, the singly reduced semiquinone FAD triggers photoreceptor activity in cryptochromes (10, 11). Structurally, photolyases and cryptochromes are highly similar, but cryptochromes contain additional amino acids at their C-terminal end (1). The length of this C-terminal tail (CTT) varies between different cryptochromes, from tens to several hundreds of amino acids (1, 2).
The fruit fly Drosophila melanogaster contains one type I insect cryptochrome (DmCry). This protein is the primary light receptor of the D. melanogaster circadian clock (3). In the organism, it coordinates interactions between Timeless (TIM) and the E3-ubiquitin ligase Jetlag (JET) in a light-dependent manner. The interaction between DmCry, TIM, and JET depends on conformational changes that occur at the CTT of DmCry (12). It appears that the CTT blocks the interaction surface in the dark, and deletion of the CTT abolishes the light dependence of these interactions (13, 14). In DmCry, the CTT is about 20 amino acids long, and in the dark, the CTT forms a 10-residue helix that is bound to the photolyase homology domain, placing phenylalanine 534 of the conserved Phe-Phe-Trp (FFW) motif at the position where photolyases bind DNA (15, 16).
Blue-light absorption triggers FAD excitation and nanosecond electron transport along four tryptophan residues to generate a radical pair FAD•−⋯W394•+ (Fig. 1) (17, 18). After a few microseconds, W394•+ is deprotonated (19), and on a millisecond time scale, the FAD•−⋯W394• radical pair recombines. If the intermediate W394• radical is reduced by an external electron donor before recombination, then the remaining flavin radical is reoxidized over the course of several minutes, depending on the concentration of electron acceptors such as oxygen. Although the anion radical (FAD•−) is the predominant photoproduct, some neutral radical (FADH•) is also formed (20).
Fig. 1. Cryptochrome structure and photoreduction.
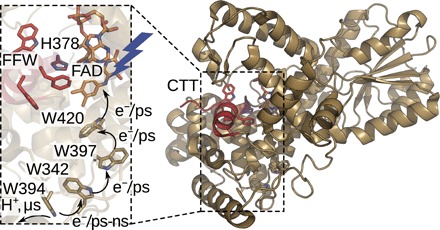
The overall structure of DmCry [Protein Data Bank (PDB) ID: 4GU5]. The inset depicts an enlarged view of the area around the chromophore, H378, the FFW motif, and the tryptophan tetrad.
Despite this knowledge about the redox reactions of the FAD chromophore, reliable information about protein structural changes is sparse. After continuous light excitation, limited proteolysis assays and small-angle x-ray scattering (SAXS) measurements have suggested that the CTT of DmCry undocks or gets unstructured to open up a binding site for TIM (21). It appears that the FFW motif is directly involved in this process. The molecular rearrangements that lead from light absorption to release of the CTT remain less clear. A focal point of interest is a conserved histidine (378 in DmCry), which is located between the isoalloxazine moiety of the chromophore and the CTT (Fig. 1) (16, 22). In (6-4) photolyases, the equivalent histidine has been shown to be crucial for DNA repair activity (23, 24) and light-induced conformational changes (25). A recent study of DmCry suggested that flavin reduction causes proton uptake at H378 and that the induced changes in the hydrogen bonding network result in the detachment of the CTT (20). All of this information was obtained at photoequilibrium. Hence, it is currently not even clear at which point during the photoreaction the signaling state confirmation is reached. To make progress in understanding the mechanism of signal transduction in cryptochromes, it is necessary to investigate the protein using techniques that can resolve structural changes as a function of time.
Here, we use nano- to millisecond time-resolved x-ray solution scattering (TRXSS) (26–32) and molecular dynamics (MD) simulations to investigate the light-induced conformational changes in DmCry after light excitation. Both the wild-type protein and a histidine 378 to alanine (H378A) mutant were investigated. As control experiment, we also investigated the (6-4) photolyase from Xenopus laevis (XlPho). XlPho is structurally similar to DmCry and undergoes similar light-induced electron transfer reactions, but it lacks the CTT.
RESULTS
TRXSS reveals a series of light-induced conformational changes from nano- to milliseconds
We used TRXSS to probe the conformational changes during photoactivation of DmCry. In the experiment, the protein solution is excited by a nanosecond laser pulse, and the x-ray solution scattering is recorded at a series of defined delay times. We recorded TRXSS data covering delay times from 10 ns to 10 ms for DmCry, from 3 μs to 10 ms for DmCry(H378A), and from 100 ns to 10 ms for XlPho (Fig. 2). Difference scattering (light-dark) signals were observed for all three samples. The shapes of the signals for the two cryptochromes evolve over time with two changes of sign for the slope at q < 0.1 Å−1 (q is the modulus of the scattering vector defined as q = 4πsin(θ)/λ, where 2θ is the scattering angle and λ is the x-ray wavelength) observed at 3 μs and between 3 and 10 ms and the grow-in of a positive peak at q = 1 Å−1 at approximately 1 ms. In contrast, the data for XlPho show only one signal at q < 0.1 Å−1, which rises at 300 ns and decays between 1 and 3 ms.
Fig. 2. TRXSS data.
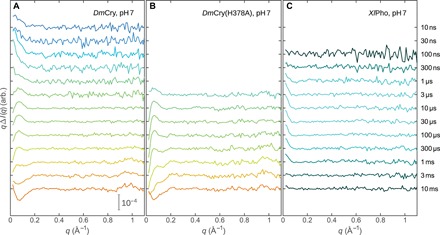
TRXSS data covering delay times from 10 ns to 10 ms for wild-type DmCry (A), for the DmCry(H378A) mutant (B), and for XlPho (C). Coloring follows the transient population of the different intermediates shown in Fig. 3. Different delay time ranges were probed for the samples because of restrictions in beamtime and available sample volumes.
We decomposed the difference scattering signal into time-independent species-associated difference scattering curves, assuming a sequence of transitions following first-order kinetics (Fig. 3 and Table 1). Five species were required to model the data for DmCry. Fewer intermediates resulted in systematic deviations between the model and the data, whereas additional components resulted in unreasonable values for the refined rate constants or did not notably improve the fit (fig. S1 and table S1). The five species will be referred to as DmCryα, DmCryβ, DmCryγ, DmCryδ, and DmCryε. The data for DmCry(H378A) could be modeled using four species. The scattering profiles of the four intermediates closely resembled DmCryβ to DmCryε of the wild-type cryptochrome (Fig. 3, E and F). The difference scattering of the β intermediate of DmCry(H378A) may be convoluted with scattering for an α state, which is not resolved because of the limited time range probed. The time-resolved x-ray scattering of XlPho was well described by the rise and the decay of a single intermediate state.
Fig. 3. Kinetic analysis of TRXSS data.
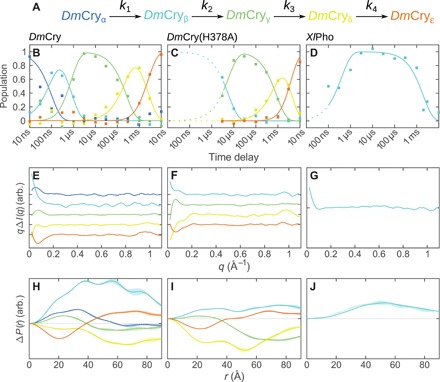
(A) The kinetic model used for the analysis of the TRXSS data. Time-dependent populations of the different structural intermediates are shown in (B) to (D) for DmCry wild type (B), DmCry(H378A) (C), and XlPho (D). The fitted model is shown as lines. The squares represent the optimized populations obtained by fitting the experimental difference scattering curves with the species-associated difference scattering curves in (E) to (G). The model outside the probed time region is shown as dotted lines. Species-associated difference scattering curves for DmCry (E), DmCry(H378A) (F), and XlPho (G). Change in the pair distance distribution [P(r)] for DmCry (H), DmCry(H378A) (I), and XlPho (J). The shaded area represents ± 1 SD. The first intermediate state of DmCry(H378A) is not well resolved because of the limited time range probed and appears to be a mixture of DmCryα and DmCryβ.
Table 1. Rate constants for structural transitions by TRXSS.
| TRXSS (pH 7) | k1 (μs−1) | k2 (μs−1) | k3 (ms−1) | k4 (ms−1) |
| DmCry | 10.05 ± 0.64 | 1.30 ± 0.06 | 3.86 ± 0.30 | 0.40 ± 0.03 |
| DmCry(H378A) | no data | 0.17 ± 0.01 | 1.15 ± 0.07 | 0.30 ± 0.04 |
Note that the transient difference scattering signal observed for XlPho closely resembles one of the early DmCry states, namely, DmCryβ (Fig. 3, E and G). Since the two proteins are structurally comparable and undergo similar electron transfer reactions after illumination (1), it is likely that the XlPho intermediate and DmCryβ represent analogous structural changes.
TRXSS measures the change in distances between the scattering electrons in the sample. Generally, large-scale conformational rearrangements, such as domain movement, partial unfolding, mass transit, or changes in the hydration layer, are observed at low q (q < 0.1 Å−1) (26, 29, 31, 33, 34). Clear difference signals can be observed at low q for all delay times. We estimated the change of the radius of gyration (ΔRg; Eq. 3) to be less than ±0.2 Å for all states (table S2). This indicates concerted but small changes to the global protein structure. Signals in the intermediate q range (0.2 < q < 1 Å−1) are associated with tertiary structural changes (26, 27, 30, 31). All three samples exhibit fairly flat difference scattering in the intermediate q range, thereby suggesting that there is no substantial change of the tertiary structure of the protein. We observe a peak at q = 1 Å−1 for DmCry and DmCry(H378A), which appears with the DmCryδ state. The q range and the magnitude of the signal suggest that it originates from a coordinated change of interatomic distances around 6 Å (r = 2π/q). Considering the pitch of α helices of 5.4 Å, the signal would be consistent with structural changes of the partly α-helical CTT. This assignment is supported by the fact that the peak is absent in XlPho, which lacks the CTT.
To gain more structural insight, we computed the difference pair distance distribution [ΔP(r), Fig. 3, H to J] from the difference scattering curves [ΔI(q)] using a sine Fourier transform (Eq. 2). The ΔP(r) curve corresponding to DmCryγ shows a decrease of long (>40 Å) and an increase of short distances, which suggest a compacted state. DmCryε has more long and less short distances, characteristics of an elongated structure. This is also confirmed by ΔRg, which we calculated to be +0.09 and +0.07 Å for DmCryε and DmCry(H378A)ε, respectively (table S2). We conclude that DmCryε has an extended conformation with a detached CTT (21).
The ΔP(r) of DmCryα, DmCryβ, and DmCryδ is positive or negative at all distances. This cannot be explained by a structural change solely within the protein, where an increase in long distances would always be accompanied by a corresponding decrease in short distances and vice versa. The protein, however, is surrounded by a solvent layer, which has a different density to that of the bulk solvent. The density of this layer could increase or decrease, leading to all-positive or all-negative ΔP(r) functions.
We confirmed that this is a reasonable explanation by calculating the x-ray scattering of the DmCry crystal structure (22) with CRYSOL (35). We varied the contrast of the hydration layer corresponding to addition or removal of 20 water molecules, and we referenced the data to scattering computed for the default value of 0.03 e−/Å3 (Fig. 4B). The resulting P(r) curves reveal an increase or a decrease in all distances (Fig. 4C). We therefore conclude that there is a significant contribution of such solvent layer remodeling for DmCryα, DmCryβ, and DmCryδ. Considering that substantial charge redistribution occurs over the course of the photoreaction, changes in the ion cloud (36) or in the dynamics of the surface water seem reasonable. Overall, the structural analysis indicates that the tertiary structure of the DmCry is stable over the course of the photoreaction but that a number of local and small structural changes lead to formation of the final conformational state DmCryε.
Fig. 4. Steady-state difference SAXS, modeling of hydration layer, FFW flexibility in MD simulations, and active-site hydrogen bonding network.

(A) Difference scattering from a steady-state SAXS measurement compared to DmCryε. Note that it is the regularized difference intensity that is shown for the SAXS measurement. Predicted difference scattering for DmCry crystal structure (PDB ID: 4GU5) by only varying the hydration layer contrast by the equivalent of a few water molecules (B) and the corresponding change in P(r) (C). Simulations with the oxidized chromophore parameters display overall lower FFW RMSD compared to simulations with the reduced chromophore parameters (D and E). Arrows indicate shift and widening of the RMSD distribution when going from an oxidized to reduced chromophore. When the chromophore is oxidized, H378 preferentially forms a hydrogen bond with W536 (F), but when the FAD becomes reduced, H378 preferentially binds to the FAD (G). arb., arbitrary unit.
The formation of the signaling state is pH dependent
To find out which of the states corresponds to the signaling conformation, we conducted a regular SAXS experiment in which DmCry was kept either in the dark or under continuous blue-light exposure (fig. S2 and table S3). The results are in agreement with previously published SAXS data from dark- and light-adapted DmCry (21). The difference between our “light” and “dark” SAXS curves [ΔI(q)] is highly similar to the scattering pattern detected for the last state, DmCryε (Fig. 4A). We therefore consider this state, which is reached within 10 ms of photoexcitation, to be the signaling state.
In the previous SAXS study, the light state was associated with a detached CTT (21). Our data are in agreement with this assignment. An extended conformation in DmCryε is indicated by the increase in Rg (table S2) and by the ΔP(r) function (Fig. 3, H and I). Moreover, the peak at q = 1 Å−1 in DmCryδ and DmCryε and the absence of a scattering signal in XlPho equivalent to that of DmCryε are consistent with conformational changes at the CTT (Fig. 3, E and F).
To further investigate the signal transduction pathway, we recorded TRXSS data of DmCry and DmCry(H378A) in a more basic solution with pH 9 (fig. S4, A and B). We decomposed the data using a sequential model as described for the measurements at pH 7 (table S4). By comparison of TRXSS patterns and ΔP(r) functions of the intermediates at pH 9 to those obtained at pH 7, we identified states at pH 9, which were equivalent to DmCryβ, DmCryγ, and DmCryδ at pH 7. We could not identify an equivalent to DmCryα possibly because of altered fast kinetics at pH 9, which could shift the appearance of this intermediate outside the experimental time window.
An equivalent of the final conformational state DmCryε was not observed at pH 9. The final x-ray scattering pattern at pH 9 contains the positive peak at q = 1 Å−1, but the signals at q < 0.2 Å−1 differed from those at pH 7. The ΔP(r) of the final states (Fig. 3, H and I, and fig. S4, G and H) reveals that the positive feature at long distances, indicating an extension of the protein, is absent at pH 9. Because of technical limitations, we could only probe delay times up to 10 ms, and it is possible that an equivalent to DmCryε is developed outside the probed time window at pH 9. In either case, the data show that the formation of the signaling state DmCryε depends on pH and requires uptake of a proton.
The role of H378 in DmCry signal transduction
Placed directly between the isoalloxazine moiety of the chromophore and the CTT, the conserved H378 has been implicated in pH-dependent signal transduction between the chromophore and the CTT (20, 23, 24). We therefore recorded the TRXSS data on DmCry(H378A), but to our surprise, we found that the TRXSS patterns and kinetics in DmCry and DmCry(H378A) are highly similar (Fig. 3, E and F, and Table 1). The ΔP(r) curves for DmCry(H378A)γ–ε show this most clearly, as they exhibit the same patterns of positive and negative signals as for DmCry (Fig. 3, H and I). We note that the states α and β are not resolved for DmCry(H378A) because of the limited time range of the data. In both samples, the signaling state is reached within 10 ms. This confirms that the H378A mutation hardly alters the structural changes after blue-light photoexcitation. Moreover, our TRXSS data show that even at pH 9, DmCry and DmCry(H378A) display highly similar structural rearrangements (fig. S4). Thus, we conclude that protonation reactions of H378 are not required for the pH-dependent formation of the signaling state.
To investigate the role of H378 in DmCry in greater detail, we performed classical MD simulations of the H378A mutant and the wild-type protein, with the FAD modeled with parameters for the oxidized (FAD) and semireduced (FAD•−) states. H378 was protonated at the Nε (N3) position as suggested by the crystal structures (16, 22) and Ganguly et al. (20). Four trajectories, each 250 ns long, were generated for each situation and will be referred to as WTFADox1-4, WTFADred1-4, H378AFADox1-4, and H378AFADred1-4. The mobility of the CTT was evaluated by the root mean square deviation (RMSD) of the FFW motif with respect to the crystal structure. As in (20), we removed rotational and translational degrees of freedom before calculating the RMSD by aligning the protein backbone on the quartile of residues that exhibited the lowest root mean square fluctuations (RMSFs).
For the wild-type protein, we find that the mobility of the FFW is elevated when FAD is reduced compared to when it is oxidized (compare the blue to the red line in Fig. 4D and fig. S3, A and B). This trend has been observed previously (20). We also find that the hydrogen bonding network between the FAD, H378, and W536 changes in response to the additional charge on the FAD. When FAD is oxidized, H378 predominantly hydrogen-bonds to W536 of the CTT (Fig. 4F and fig. S3E), and when FAD is reduced, the Nδ of H378 bonds to the ribose hydroxyl (OS2) of the chromophore (Fig. 4G and fig. S3E). We note that the described trends are observed in most trajectories but that WTFADred3 is an exception as H378 forms a hydrogen bond to W536, as in the oxidized trajectories. Also, the CTT completely detaches in WTFADred4 after 60 ns, and no hydrogen bond is observed (fig. S3, B and E).
The observations from the DmCry trajectories could be seen as an indication that H378 is actively involved in signal transduction. However, when we repeated the simulation after mutation of H378 into an alanine, we observed an upshift of the FFW-RMSD distribution, similar to the trend observed for the wild-type protein (compare the blue line and the red line in Fig. 4E and fig. S3, C and D). This underpins our conclusions from the TRXSS data and suggests that H378 is not the sole critical factor for relaying the change of redox state of the FAD chromophore to the CTT.
When comparing the mobility of DmCry and DmCry(H378A) with FAD in the oxidized (resting) state, we find that the peak of the FFW RMSD distribution remains unchanged at 1.2 Å between the samples but that the distribution is wider in the case of DmCry(H378A) (compare blue lines in Fig. 4, D and E). For one of the mutant simulations (H378AFADox1), we observe pronounced CTT detachment, manifesting as a secondary peak around 6 Å RMSD. Together, these results suggest that the main role of H378 is to reduce the dynamics of the CTT when FAD is in the resting (oxidized) configuration. It achieves this by anchoring the CTT to the photolyase homology domain. Tight attachment of the CTT may be critical in vivo as it would prevent the cryptochrome from leaking signals in the dark.
DISCUSSION
Here, we show (i) that blue-light absorption triggers a series of conformational rearrangements in DmCry, resulting in the formation of the signaling state and detachment of the CTT on a millisecond time scale, (ii) that the last transition into the signaling state depends on pH, and (iii) that the conserved H378 is not required for signal transduction and is not the sole cause of the pH dependence. Instead, we propose that it stabilizes the CTT in the resting-state conformation.
Structural changes are already observed within 10 ns of excitation, and the protein undergoes a sequence of conformational rearrangements before reaching the signaling conformation after a few milliseconds. The protein retains its overall globular shape during the photoreaction, and changes in the water or ion density around the protein occur. The resulting photoreaction scheme of DmCry contains more structural than spectroscopic intermediates (Fig. 5A). This is not surprising, considering the fact that ultraviolet-visible (UV-vis) spectroscopy reports selectively on the redox state and the immediate surroundings of the FAD chromophore but that TRXSS is sensitive to structural changes in the entire protein as well as the surrounding hydration layer. Structural intermediates have also been identified in a plant cryptochrome by step-scan infrared spectroscopy (37), which also probes structural changes in the entire protein.
Fig. 5. Structural photocycle for DmCry.

(A) Proposed structural photocycle for DmCry based on TRXSS data, shown as the inner cycle in red. The outer cycle in blue is the photochemical photocycle as determined by transient absorption (TA) spectroscopy. (B) The proposed structural rearrangements associated with the DmCryγ to DmCryε states.
Comparing kinetics from TRXSS and transient optical spectroscopy [see Fig. 5A and (17, 19)] reveals that formation of the FAD•−⋯W394•+ radical pair triggers the start of conformational changes of the protein. However, the rate constant for deprotonation of the tryptophanyl cation radical does not seem to correlate directly with the TRXSS kinetics (table S5). The rate constant for recombination is slightly lower than the formation rate of the signaling state as measured by TRXSS (Table 1 and table S4; see Materials and Methods for a detailed discussion). This supports our view that the protein structural changes, which are probed by TRXSS, are caused by changes in the oxidation state of FAD but that they otherwise are kinetically uncoupled from the reactions of the radical pair.
Our data support the notion that the signaling state DmCryε has a detached CTT (12, 16, 21). The transition into the final state is slowed down or prevented at elevated pH (fig. S4 and table S4). We therefore conclude that a proton is taken up by the protein to facilitate this transition (Fig. 5). Currently, we are not able to identify the residue at which the protonation reaction occurs. H378 is located directly between the chromophore and CTT and has been proposed to undergo a protonation reaction during photoactivation of DmCry (20). However, this proposal is not consistent with our TRXSS data as the H378A mutation does not alter the formation of the signaling state at two different pH values. If H378 would be the primary residue responsible for the pH dependence, then we should have observed a change in rate constant for the formation of the signaling state upon removal of the residue by mutation. Alternative candidates are the residues S526 and E530, which coordinate a network of hydrogen bonds, important for structural activation of DmCry (16). It is possible that this network may be disrupted by the protonation of one of the involved residues. To investigate this issue further, it would be good to conduct experiments, which can probe the protonation state of H378 and other residues directly.
As an alternative role for H378, we conclude from our MD simulations that H378 stabilizes the CTT in the resting state. This result is in agreement with activity assays in cell cultures of different H378 mutants of DmCry, from which similar conclusions were drawn (20). Thus, in the dark, the CTT is docked to the photolyase homology domain through hydrogen bonds between H378 and the CTT (Fig. 4, D to G). Stabilization of the CTT in the resting state may ensure that DmCry does not leak any signal in the dark, a feature that may be as important as efficient photoactivation. In the circadian clock in D. melanogaster, low levels of the TIM:PER (Period) complex have to be able to effectively block the expression of themselves in the cell nucleus at late night (3). If DmCry would have a residual dark activity, then it would mark TIM for degradation in the dark, hindering the clock cycle.
So how does the signal relay from the chromophore to the CTT? We consider the possibility that CTT detachment is caused by long-range Coulomb interactions between the semireduced FAD•− and residues in the CTT. This is supported by the similarity of the structural changes that we detected when comparing wild-type DmCry and the H378A mutant. If the interaction between the FAD and the CTT is direct and through space, then the type of amino acid residing in between can be expected to play a minor role for signal transduction. Another alternative is that the signal is transduced from the chromophore to the CTT by a collective action of several amino acids that reside between the two sites. The alternatives are not mutually exclusive, and both are consistent with the cellular activity assays of various H378 mutations of DmCry, where it was found that none of the mutants completely disabled the light-induced change in activity of the protein (20).
Adding to the current understanding of cryptochromes, our findings provide hallmarks of the conformational photoactivation of DmCry (Fig. 5B). H378 stabilizes the resting-state conformation by tightly binding the CTT to the photolyase homology domain (gray structure). When photoexcited by blue light, the FAD•−⋯W394• radical pair is formed within a few microseconds. Structurally, the protein is in the state DmCryγ at this point of the reaction (green structure), which is slightly contracted compared to the resting state (Fig. 3E). After about 0.2 ms, the next state (DmCryδ) is reached, and we propose that the CTT loosens up or becomes unstructured as a direct consequence of the charge on the FAD (yellow structure). The loosening up of the CTT is a prerequisite for protonation of a yet unknown residue. This protonation reaction causes the CTT to completely detach from the N-terminal part of the protein (orange structure). The signaling conformation (DmCryε) is formed, primed for binding of TIM.
MATERIALS AND METHODS
Protein production and purification
The N-terminally His-tagged DmCry was produced in Escherichia coli BL21 cells following published procedures (19). E. coli cells were grown overnight at 303 K in the dark, and the protein synthesis was induced at an A600 (absorbance at 600 nm) of 0.4 to 0.6 by using 1 mM isopropyl-β-d-thiogalactopyranoside. After cell disruption, the lysate in buffer A [50 mM Hepes (pH 7.0), 100 mM NaCl, and 10% (v/v) glycerol] was initially purified on a nickel-Sepharose column using binding buffer [50 mM Hepes (pH 7.0), 200 mM NaCl, 10% (v/v) glycerol, and 20 mM imidazole] and elution buffer [50 mM Hepes (pH 7.0), 200 mM NaCl, 20% (v/v) glycerol, and 500 mM imidazole]. Additional purification steps were carried out using gel filtration chromatography with buffer [50 mM Hepes (pH 7.0), 100 mM NaCl, and 20% (v/v) glycerol] and anion exchange chromatography using binding buffer [50 mM Hepes (pH 7.0), 50 mM NaCl, and 10% (v/v) glycerol] and elution buffer [50 mM Hepes (pH 7.0), 500 mM NaCl, and 10% (v/v) glycerol]. Yellow-colored DmCry-containing fractions were pooled and subjected to desalting column and concentration with microconcentrators. The histidine-to-alanine mutation was performed using the FastCloning method (38) with two overlapping primers in combination with a high-fidelity polymerase (KAPA HiFi/PEQLAB Biotechnologie) and verified by sequencing. The pET28a vector containing the mutated gene was transformed into E. coli SoluBL21 cells. DmCry(H378A) was produced in the same way as the wild-type protein. XlPho was produced using published procedures (39). A buffer consisting of 50 mM Hepes (pH 7.0 or pH 9.0), 100 mM NaCl, 5 mM ferricyanide, and 10% (v/v) glycerol was used in TRXSS experiments. The same buffer was used in the transient absorption experiments, and the ferricyanide concentration was reduced to 1 mM to account for the fivefold reduced protein concentration. In the conventional SAXS measurements, a buffer at pH 7 without ferricyanide and with a glycerol concentration of 20% (v/v) was used. Before any experiment, samples were filtered through a 0.2-μm filter.
Nonreducing SDS–polyacrylamide gel electrophoresis
An SDS–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing conditions (no β-mercaptoethanol was added to the loading buffer) was performed to exclude the presence of any multmerization reactions due to disulfide linkages in any of the experiments. Both DmCry and DmCry(H378A) were investigated, and two parameters were varied: Samples were either kept in 50 mM Hepes (pH 7.0), 100 mM NaCl, and 10% (v/v) glycerol or, in the same buffer, supplemented with a five-times molar excess of ferricyanide. In addition, both proteins were kept either in the dark or were loaded on the gel directly after 5 min of blue light illumination using a lamp power of 9.7 mWcm−2. As can be seen in fig. S5A, all band patterns were highly similar, and neither ferricyanide nor blue light illumination resulted in the formation of DmCry dimers or other degradation products.
Transient absorption measurements
Transient absorption measurements were performed as described in (19). In detail, time-resolved optical absorption spectroscopy was performed at 277 K with a commercially available laser flash photolysis spectrometer (LP920K, Edinburgh Instruments Ltd., Kirkton Campus, UK), and data were recorded with a digital oscilloscope (TDS-3012C, Tektronix, Beaverton, OR). The protein sample was placed in a synthetic quartz (Suprasil) semimicro cell (108F-QS, Hellma, Muellheim, Germany). The temperature was regulated to 277 ± 1 K by a temperature controller (Alpha RA 8, LAUDA, Lauda-Koenigshofen, Germany). Optical excitation was carried out using an OPO system (OPO PLUS, Continuum, San Jose, CA) pumped with a Nd:YAG laser (Surelite I, Continuum) at a wavelength of 460 nm, a pulse width of 6 ns, and a pulse energy of 4.0 ± 0.2 mJ. The repetition rate of the spectrometer was set to 6.67 mHz. To account for background signals, transients were measured alternately with and without optical excitation and used for calculation of difference absorbance spectra with Beer-Lambert’s law. We recorded two datasets per sample, one on a microsecond time scale to probe the deprotonation of the terminal tryptophan and the other on a millisecond time scale to probe the recombination of the radical pair.
Conventional SAXS data collection
SAXS data were collected at BM29 at the European Synchrotron Radiation facility at protein concentrations of 0.5, 1, 2, and 4 mg/ml. The measurements were performed at 10°C. The x-ray energy was 12.5 keV, and the detector used was Pilatus 1M. For each concentration, 10 images with 1-s x-ray exposure were collected with 10 images of the buffer before and after. Two sets of measurements were performed, one with the chromophore in the oxidized state and the other with the chromophore in the reduced anion radical state; this was achieved by illumination with 455-nm light and verified by UV-vis spectroscopy. The dark measurements were performed under red light, and the light-state measurements were performed under blue light. The data were averaged and subtracted using software at the beamline. Low-concentration (0.5 mg/ml) and high-concentration (4 mg/ml) data were merged at 0.05 < q < 0.10 Å−1. Additional data processing was performed using the ATSAS package (40), and the molecular weight was estimated as described by Rambo and Tainer (41).
TRXSS data collection
Time-resolved scattering experiments were performed at the BioCARS beamline at the Advanced Photon Source (APS). All measurements were performed at ambient temperatures (ca. 22°C). The protein concentration was 24 to 30 mg/ml (0.4 to 0.6 mM), and the buffer was supplemented with 5 mM potassium ferricyanide (ca. 10 times molar excess) to ensure that the chromophore is reoxidized during the experiment. With transient optical absorption spectroscopy, we established that the millisecond recombination of the radical pair did not change significantly by adding ferricyanide (table S5). To evaluate whether the high concentrations used for the TRXSS measurements distorted the difference scattering curve, the packing structure factor S(q) (see fig. S5B) was estimated on the basis of a dilution series (24, 12, and 6 mg/ml and extrapolated to infinite dilution). No significant variation of this was observed (⩽±5% at each point). We used the pink beam profile with a nominal x-ray energy of 12 keV, and the x-rays were focused to a ca. 50 μm by 50 μm spot. The sample was excited using a 475-nm 7-ns laser pulse with an energy density of 2 mJ/mm2; the laser spot size was adjusted so that it engulfed the x-ray spot. The x-rays and laser were overlapped on a 1-mm-diameter quartz capillary, holding the sample. The protein was delivered through inert polyether ether ketone tubing using a peristaltic pump at a constant pump speed of 2 μl/s. To collect data at different time points after excitation, the temporal offset between laser and x-rays was adjusted. Every fourth image recorded corresponded to a negative time delay (the laser pulse arrives after the x-ray pulse). The fingerprint of solvent heating was recorded by adding azo dye (0.5 mg/ml; Fast Yellow, Sigma-Aldrich) to the buffer solution. This dye absorbs the same wavelength as the protein sample, but most of the energy is transferred to the solvent as heat. This experiment was performed in the same way as just described. Detector images were radially integrated using our own software and normalized to the average scattering in the region 1.97 < q < 2.17 Å−1, where water scattering displays an isosbestic point with respect to heating. Absolute scattering data deviating from the median by more than 3 SDs and difference data deviating more than 2 SDs from the median in the region 2 < q < 2.5 Å−1 and 1.5 < q < 2.1 Å−1, respectively, were regarded as outliers and were removed. In total, more than 90% of the images were kept. To generate difference scattering curves, the data of the closest (before and after) negative time delay were averaged and subtracted from each scattering curve. Difference scattering curves corresponding to the same x-ray laser delay time were averaged together. The contribution of the solvent heating was subtracted by scaling the pure heating curve to each time delay in the range 1.7 < q < 3 Å−1.
Kinetic decomposition and basic structural modeling of the scattering data
The x-ray solution difference scattering data were considered to be described as a linear combination of several time-independent species-associated difference scattering curves (Fig. 3 and fig. S1). A sequence of transitions described by first-order reactions was used to model the time-dependent concentration of the different intermediate states. The general case for five components (A to E) is described by Eqs. 1A to 1E. The species-associated difference scattering curves were retrieved by solving the system of linear equations C ⋅ SADS = ΔI, where C are the time-dependent concentrations and SADS are the species-associated difference scattering curves. For wild-type DmCry at pH 7, five species were needed, and at pH 9, four states were needed. The measurements on the H378A mutant did not cover nanosecond time scales, and the data could be described using four or three states at pH 7 and 9, respectively. Fewer intermediates resulted in systematic deviations, and additional species did not improve the fit and sometimes resulted in unreasonable values for the rate constants (fig. S1). The rate constants of the model were refined by a least-squares minimization of the residual between the experimental and reconstructed data. The estimated error of the rate constants is retrieved from the Jacobian at the solution
| (1A) |
| (1B) |
| (1C) |
| (1D) |
| (1E) |
By transforming the retrieved basis spectra to a difference in pair distance distribution [ΔP(r)], a structural evaluation of the different intermediates could be performed. The transformation is done according to Eq. 2
| (2) |
ΔP(r) was evaluated from 0 to 90 Å using difference scattering for 0.02 ≤ q ≤ 0.4Å−1, and the regularization parameter α was set to 0.01. The statistical uncertainty of this transformation was estimated by adding white Gaussian noise, with an amplitude similar to that observed in the raw experimental data, to the difference scattering curves associated with each species. One hundred curves with different noise were generated for each basis spectrum and used to estimate the SD of the reconstructed ΔP(r). The radius of gyration (Rg) for each state is calculated according to Eq. 3
| (3) |
Here, P(r) was computed by adding the P(r) of the dark-adapted DmCry from a conventional SAXS measurement to the ΔP(r) for each state, accounting for the photoconversion yield of ca. 6%. The photoconversion yield was estimated by adjusting the absolute scattering curves from the TRXSS experiment to the same scale as the conventional SAXS experiment. The difference scattering of the DmCryε state was then compared to the difference scattering (light-dark) of the conventional SAXS measurement, which, based on UV-vis measurements, is considered to represent complete conversion to the reduced anion radical form. The ΔRg was calculated by subtracting the Rg of the dark-adapted state. The error of ΔRg was estimated by calculating it using 100 ΔP(r) with simulated noise, as described above.
MD simulations
The starting structure used was based on the coordinates of a D. melanogaster cryptochrome crystal structure [Protein Data Bank (PDB) ID: 4GU5] (15, 22). The parameters for the oxidized and anion radical FAD were based on previous work (42). The simulations were run using GROMACS 5.0.4 (43) and the CHARMM27 force field. The simulation box was replicated in all directions with periodic boundary conditions, all bonds were constrained using the linear constraint solver algorithm, and a time step of 2 fs was used. Particle mesh Ewald (PME) electrostatics with fourth-order interpolation and with a grid spacing of 0.12 nm were used. The cutoff scheme was Verlet with a 1.0-nm cutoff, and cutoffs for short-range electrostatic and van der Waals interactions were 1.0 nm as well. During the production run, pressure control was achieved using the Parrinello-Rahman barostat (τP = 2 ps, P = 1 bar), and temperature control was achieved via the modified Berendsen (velocity-rescale) thermostat (τT = 0.1 ps, T = 300 K). The protein was placed in a cubic box, 1 nm larger than the protein in all directions and solvated with transferable interparticle potential with 3 points (TIP3P) water molecules. The system was neutralized by replacing water molecules with sodium and chloride ions to a final concentration of 150 mM. After adding ions, the system underwent initial energy minimization until all forces were below 1000 kJ mol−1 nm−1. Subsequently, the system was minimized for 100 ps in the canonical (NVT) and isothermal-isobaric (NPT) ensembles. During equilibration all nonhydrogen atoms were position restrained with force constants of 1000 kJ mol−1 nm−2. In total, 16 simulations were run for 250 ns each. These simulations used two different force-field parameters for the FAD, and in half of the simulations, histidine 378 was mutated to an alanine. In the simulations with H378 present, it was protonated on Nϵ since this is suggested to be the protonation state of H378 in the dark-adapted DmCry (20). Eight simulations each for the wild type and mutant were using the parameters for oxidized FAD and the anion radical, each with four replicates.
The RMSD of the FFW motif was used as a reporter of the CTT flexibility. Before computing the RMSD, rotational and translational degrees of freedom were removed by aligning the protein on the backbone atoms of residues that displayed the lowest RMSF. The average RMSF for all simulations was computed, and the protein was aligned on the quartile of the residues with the lowest RMSF. After this, the RMSD of the FFW motif was calculated.
Comparison of spectroscopic time constants and those obtained by TRXSS
To find out whether any of the observed structural states correlate with spectroscopic events, we compared the rate constants for deprotonation of W394 and recombination of the radical pair for the two cryptochrome samples (table S5) to the ones obtained from TRXSS (Table 1 and table S4). The deprotonation of the terminal tryptophan (W394) was observed on microsecond time scales, as previously shown for DmCry (19). The time constant of the recombination reaction is mainly dependent on the oxidation potential in the buffer. In the TRXSS experiments, the oxidant was potassium ferricyanide, which was added to prevent FAD•− accumulation during data acquisition. Transient absorption measurements were performed using identical molar ratios of protein and potassium ferricyanide as in scattering experiments. Two transient absorption datasets were recorded per sample, one with a 10-μs time window and one with a 10-ms time window. The 10-μs datasets were fitted by four exponential decays using Glotaran (44) as described in (19). The 10-ms datasets were fitted with two exponential decays (table S5). k3 has previously been assigned as the rate constant for deprotonation of the tryptophanyl cation radical (19), and k5 and k6 describe the recombination reaction of the radical pair. It is obvious from table S5 that the deprotonation rates are similar for DmCry and DmCry(H378A) are comparable to published data (19), and are slightly pH dependent. The recombination of the radical pair required two rate constants in DmCry samples, and their rates are slightly decreased as compared to samples without ferricyanide (19). DmCry(H378A) samples also required two rate constants, and their values were increased with respect to DmCry. A detailed investigation and analysis of this behavior is ongoing.
Supplementary Material
Acknowledgments
We thank I. A. Solov’yov for providing force-field parameters for the FAD chromophore. This research used resources of the APS, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number R24GM111072. The time-resolved x-ray diffraction setup at APS Sector 14 was funded, in part, through a collaboration with P. Anfinrud (NIH/NIDDK). The simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC. Funding: S.Wes. acknowledges funding from the Swedish Foundation for International Cooperation in Research and Higher Education, the European Research Council (725642), and the Foundation of Strategic Research, Sweden. E.S. and S.Web. thank the DFG (235777276/GRK1976) for financial support. J.A.I. acknowledges a grant from the Academy of Finland (296135) and the Jane and Aatos Erkko Foundation. Author contributions: O.B., R.R., S.Web., E.S., and S.Wes. designed the research and planned the experiments. R.R. produced and purified the proteins. C.E. performed the transient absorption measurements. O.B., L.H., M.R.P., A.J.H., J.A.I., R.R., R.H., I.K., and S.Wes. performed the x-ray solution scattering experiments, and O.B. analyzed the data and performed the modeling. O.B. and S.Wes. wrote the paper with input from all other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials or are available through figshare (doi: 10.6084/m9.figshare.7628648). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw1531/DC1
Table S1. TRXSS kinetic modeling.
Table S2. Changes in Rg.
Table S3. SAXS parameters.
Table S4. Rate constants for TRXSS measurements of DmCry at pH 9.
Table S5. Rate constants for TA measurements of DmCry.
Fig. S1. TRXSS data and the reconstructed data using a kinetic model with different numbers of components.
Fig. S2. SAXS scattering profiles for DmCry in the dark and under blue-light illumination.
Fig. S3. The RMSD of the FFW motif at each simulation frame.
Fig. S4. TRXSS data and kinetic modeling for wild-type DmCry and H378A at pH 9.
Fig. S5. Stability analysis of the used samples via SDS-PAGE and SAXS.
REFERENCES AND NOTES
- 1.Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L. O., van der Horst G. T. J., Batschauer A., Ahmad M., The cryptochromes: Blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Kavakli I. H., Baris I., Tardu M., Gül Ş., Öner H., Çal S., Bulut S., Yarparvar D., Berkel Ç., Ustaoğlu P., Aydın C., The photolyase/cryptochrome family of proteins as DNA repair enzymes and transcriptional repressors. Photochem. Photobiol. 93, 93–103 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Emery P., So W. V., Kaneko M., Hall J. C., Rosbash M., CRY, a Drosophila clock and light-regulated cryptochrome, is a major contributor to circadian rhythm resetting and photosensitivity. Cell 95, 669–679 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Stanewsky R., Kaneko M., Emery P., Beretta B., Wager-Smith K., Kay S. A., Rosbash M., Hall J. C., The cryb mutation identifies cryptochrome as a circadian photoreceptor in Drosophila. Cell 95, 681–692 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Crane B. R., Young M. W., Interactive features of proteins composing eukaryotic circadian clocks. Annu. Rev. Biochem. 83, 191–219 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Ritz T., Adem S., Schulten K., A model for photoreceptor-based magnetoreception in birds. Biophys. J. 78, 707–718 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodson C. A., Hore P. J., Wallace M. I., A radical sense of direction: Signalling and mechanism in cryptochrome magnetoreception. Trends Biochem. Sci. 38, 435–446 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Hore P. J., Mouritsen H., The radical-pair mechanism of magnetoreception. Annu. Rev. Biophys. 45, 299–344 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Sheppard D. M. W., Li J., Henbest K. B., Neil S. R. T., Maeda K., Storey J., Schleicher E., Biskup T., Rodriguez R., Weber S., Hore P. J., Timmel C. R., Mackenzie S. R., Millitesla magnetic field effects on the photocycle of an animal cryptochrome. Sci. Rep. 7, 42228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad K. S., Manahan C. C., Crane B. R., Photochemistry of flavoprotein light sensors. Nat. Chem. Biol. 10, 801–809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouly J.-P., Schleicher E., Dionisio-Sese M., Vandenbussche F., van der Straeten D., Bakrim N., Meier S., Batschauer A., Galland P., Bittl R., Ahmad M., Cryptochrome blue light photoreceptors are activated through interconversion of flavin redox states. J. Biol. Chem. 282, 9383–9391 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Peschel N., Chen K. F., Szabo G., Stanewsky R., Light-dependent interactions between the Drosophila circadian clock factors cryptochrome, jetlag, and timeless. Curr. Biol. 19, 241–247 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Dissel S., Codd V., Fedic R., Garner K. J., Costa R., Kyriacou C. P., Rosato E., A constitutively active cryptochrome in Drosophila melanogaster. Nat. Neurosci. 7, 834–840 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Ozturk N., Selby C. P., Annayev Y., Zhong D., Sancar A., Reaction mechanism of Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 516–521 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy C., Zoltowski B. D., Jones A. R., Vaidya A. T., Top D., Widom J., Young M. W., Scrutton N. S., Crane B. R., Leys D., Updated structure of Drosophila cryptochrome. Nature 495, E3–E4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czarna A., Berndt A., Singh H. R., Grudziecki A., Ladurner A. G., Timinszky G., Kramer A., Wolf E., Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 153, 1394–1405 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Nohr D., Franz S., Rodriguez R., Paulus B., Essen L.-O., Weber S., Schleicher E., Extended electron-transfer in animal cryptochromes mediated by a tetrad of aromatic amino acids. Biophys. J. 111, 301–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nohr D., Paulus B., Rodriguez R., Okafuji A., Bittl R., Schleicher E., Weber S., Determination of radical-radical distances in light-active proteins and their implication for biological magnetoreception. Angew. Chem., Int. Ed. Engl. 56, 8550–8554 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Paulus B., Bajzath C., Melin F., Heidinger L., Kromm V., Herkersdorf C., Benz U., Mann L., Stehle P., Hellwig P., Weber S., Schleicher E., Spectroscopic characterization of radicals and radical pairs in fruit fly cryptochrome - protonated and nonprotonated flavin radical-states. FEBS J. 282, 3175–3189 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Ganguly A., Manahan C. C., Top D., Yee E. F., Lin C., Young M. W., Thiel W., Crane B. R., Changes in active site histidine hydrogen bonding trigger cryptochrome activation. Proc. Natl. Acad. Sci. U.S.A. 113, 10073–10078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaidya A. T., Top D., Manahan C. C., Tokuda J. M., Zhang S., Pollack L., Young M. W., Crane B. R., Flavin reduction activates Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 20455–20460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zoltowski B. D., Vaidya A. T., Top D., Widom J., Young M. W., Crane B. R., Structure of full-length Drosophila cryptochrome. Nature 480, 396–399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hitomi K., Nakamura H., Kim S.-T., Mizukoshi T., Ishikawa T., Iwai S., Todo T., Role of two histidines in the (6-4) photolyase reaction. J. Biol. Chem. 276, 10103–10109 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Schleicher E., Hitomi K., Kay C. W. M., Getzoff E. D., Todo T., Weber S., Electron nuclear double resonance differentiates complementary roles for active site histidines in (6-4) photolyase. J. Biol. Chem. 282, 4738–4747 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Yamada D., Iwata T., Yamamoto J., Hitomi K., Todo T., Iwai S., D. Getzoff E., Kandori H., Structural role of two histidines in the (6-4) photolyase reaction. Biophys. Physicobiol. 12, 139–144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berntsson O., Diensthuber R. P., Panman M. R., Björling A., Hughes A. J., Henry L., Niebling S., Newby G., Liebi M., Menzel A., Henning R., Kosheleva I., Möglich A., Westenhoff S., Time-resolved X-ray solution scattering reveals the structural photoactivation of a light-oxygen-voltage photoreceptor. Structure 25, 933–938.e3 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Berntsson O., Diensthuber R. P., Panman M. R., Björling A., Gustavsson E., Hoernke M., Hughes A. J., Henry L., Niebling S., Takala H., Ihalainen J. A., Newby G., Kerruth S., Heberle J., Liebi M., Menzel A., Henning R., Kosheleva I., Möglich A., Westenhoff S., Sequential conformational transitions and α-helical supercoiling regulate a sensor histidine kinase. Nat. Commun. 8, 284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Björling A., Berntsson O., Lehtivuori H., Takala H., Hughes A. J., Panman M., Hoernke M., Niebling S., Henry L., Henning R., Kosheleva I., Chukharev V., Tkachenko N. V., Menzel A., Newby G., Khakhulin D., Wulff M., Ihalainen J. A., Westenhoff S., Structural photoactivation of a full-length bacterial phytochrome. Sci. Adv. 2, e1600920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takala H., Björling A., Berntsson O., Lehtivuori H., Niebling S., Hoernke M., Kosheleva I., Henning R., Menzel A., Ihalainen J. A., Westenhoff S., Signal amplification and transduction in phytochrome photosensors. Nature 509, 245–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson M., Malmerberg E., Westenhoff S., Katona G., Cammarata M., Wöhri A. B., Johansson L. C., Ewald F., Eklund M., Wulff M., Davidsson J., Neutz R., Structural dynamics of light-driven proton pumps. Structure 17, 1265–1275 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Cho H. S., Dashdorj N., Schotte F., Graber T., Henning R., Anfinrud P., Protein structural dynamics in solution unveiled via 100-ps time-resolved x-ray scattering. Proc. Natl. Acad. Sci. U.S.A. 107, 7281–7286 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hub J. S., Interpreting solution X-ray scattering data using molecular simulations. Curr. Opin. Struct. Biol. 49, 18–26 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Levantino M., Schirò G., Lemke H. T., Cottone G., Glownia J. M., Zhu D., Chollet M., Ihee H., Cupane A., Cammarata M., Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun. 6, 6772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brinkmann L. U. L., Hub J. S., Publisher’s Note: “Anisotropic time-resolved solution X-ray scattering patterns from explicit-solvent molecular dynamics” [J. Chem. Phys. 143, 104108 (2015)]. J. Chem. Phys. 143, 129901 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Svergun D., Barberato C., Koch M. H. J., CRYSOL– a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 28, 768–773 (1995). [Google Scholar]
- 36.Ivanović M. T., Bruetzel L. K., Shevchuk R., Lipfert J., Hub J. S., Quantifying the influence of the ion cloud on SAXS profiles of charged proteins. Phys. Chem. Chem. Phys. 20, 26351–26361 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Thöing C., Oldemeyer S., Kottke T., Microsecond deprotonation of aspartic acid and response of the α/β subdomain precede C-terminal signaling in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 137, 5990–5999 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Li C., Wen A., Shen B., Lu J., Huang Y., Chang Y., FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 11, 92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hitomi K., Kim S. T., Iwai S., Harima N., Otoshi E., Ikenaga M., Todo T., Binding and catalytic properties of Xenopus (6-4) photolyase. J. Biol. Chem. 272, 32591–32598 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., Svergun D. I., ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Cryst. 50, 1212–1225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rambo R. P., Tainer J. A., Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solov’yov I. A., Domratcheva T., Moughal Shahi A. R., Schulten K., Decrypting cryptochrome: Revealing the molecular identity of the photoactivation reaction. J. Am. Chem. Soc. 134, 18046–18052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 44.Snellenburg J. J., Laptenok S. P., Seger R., Mullen K. M., van Stokkum I. H. M., Glotaran: AJava-Based graphical user interface for theRPackageTIMP. J. Stat. Softw. 49, 1–22 (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw1531/DC1
Table S1. TRXSS kinetic modeling.
Table S2. Changes in Rg.
Table S3. SAXS parameters.
Table S4. Rate constants for TRXSS measurements of DmCry at pH 9.
Table S5. Rate constants for TA measurements of DmCry.
Fig. S1. TRXSS data and the reconstructed data using a kinetic model with different numbers of components.
Fig. S2. SAXS scattering profiles for DmCry in the dark and under blue-light illumination.
Fig. S3. The RMSD of the FFW motif at each simulation frame.
Fig. S4. TRXSS data and kinetic modeling for wild-type DmCry and H378A at pH 9.
Fig. S5. Stability analysis of the used samples via SDS-PAGE and SAXS.