

Abstract
Background and Purpose
Non‐alcoholic hepatic fatty liver disease (NAFLD) is a manifestation of the metabolic syndrome in the liver and non‐alcoholic steatohepatitis (NASH) represents its advanced stage. R17 derived from bouchardatine, shows benefits in the metabolic syndrome, but has not been tested in the liver. The present study examined the pharmacological effects of R17 in a model of NAFLD/NASH and its mode of action.
Experimental Approach
The effects of R17 were examined in mice fed a high‐fat (HF) diet to induce the pathological characteristics of NAFLD/NASH and in cultures of HuH7 cells. We used histological and immunohistochemical techniques along with western blotting and siRNA. Generation of ROS and apoptosis were measured.
Key Results
Administration of R17 (20 mg·kg−1, i.p. every other day) for 5 weeks reversed HF‐induced hepatic triglyceride content, inflammation (inflammatory cytokines and macrophage numbers), injury (hepatocyte ballooning and apoptosis, plasma levels of alanine aminotransferase and aspartate aminotransferase), and fibrogenesis (collagen deposition and mRNA expression of fibrosis markers). In cultured cells, R17 reduced cell steatosis from both lipogenesis and fatty acid influx. The attenuated inflammation and cell injury were associated with inhibition of both endoplasmic reticulum (ER) stress and oxidative stress. Notably, R17 activated the liver kinase B1‐AMP‐activated protein kinase (AMPK) pathway by inhibiting activity of ATP synthase, rather than direct stimulation of AMPK.
Conclusion and Implications
R17 has therapeutic potential for NAFLD/NASH. Its mode of action involves the elimination of ER and oxidative stresses, possibly via activating the LKB1‐AMPK axis by inhibiting the activity of ATP synthase.
Abbreviations
- ACC
acetyl‐CoA carboxylase
- ALT
alanine aminotransferase
- AMPK
AMP‐activated protein kinase
- AST
aspartate aminotransferase
- CHOP
C/EBP homologous protein
- eIF‐2α
eukaryotic translation initiation factor 2 α
- FA
fatty acid
- FAS
fatty acid synthase
- HF
very high‐fat diet (60% calorie from lard)
- HSCs
hepatic stellate cells
- IRE‐1α
inositol‐requiring enzyme 1 α
- LKB1
liver kinase B1
- MDA
malonaldehyde
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- OA
oleic acid
- PA
palmitic acid
- PERK
PRKR‐like endoplasmic reticulum kinase
- SCD1
stearoyl‐CoA desaturase 1
- SREBP‐1c
sterol regulatory element‐binding protein‐1c
- XBP‐1s
spliced X‐box binding protein 1
What is already known
R17 was synthesized as a derivative of bouchardatine, an alkaloid from the plant Rutaecae.
What this study adds
R17 alleviates all phenotypes of early stage NASH in mice given a high fat diet.
What is the clinical significance
R17 provides a possible new treatment for NASH, through its inhibition of ATP synthase.
1. INTRODUCTION
Non‐alcoholic fatty liver disease (NAFLD) is a major health problem with a prevalence of 20% in the adult population worldwide. Across the wide spectrum of NAFLD, approximately 10–30% of patients progress from the asymptomatic simple steatosis to non‐alcoholic steatohepatitis (NASH) with additional inflammation, injury and various degrees of fibrosis in the liver (Chalasani et al., 2018; Diehl & Day, 2017). NASH is a critical stage in the progression of NAFLD from a reversible and benign stage to the irreversible stage of liver injury which may deteriorate further to hepatic cirrhosis and even hepatic cancer (Drew, 2017; Patton et al., 2018). At present, there is still no pharmacological treatment approved specifically for NASH (Diehl & Day, 2017).
Although a range of different cellular targets have been considered for the identification of potential new drugs for NASH (Musso, Cassader, & Gambino, 2016), a major challenge has been how to obtain therapeutic efficacy for the pathological features of NASH, namely, hepatic steatosis, inflammation, injury, and fibrosis (Diehl & Day, 2017). These therapeutic criteria for the evaluation of a new drug for NASH are fundamental, even though many different mechanisms underlie NASH and their interactions are not well understood (Diehl & Day, 2017; Drew, 2017). Thus, we have based our search for potential therapeutic agents on these pathological characteristics of NASH and identified a low MW derivative of bouchardatine. R17, for investigation in the present study.
This compound, R17, (Figure 1a; MW373.4) was synthesized by us (Chen et al., 2015), based on the structure of bouchardatine, an alkaloid isolated from Bouchardatia neurococca (Rutaecae). Bouchardatine itself ameliorated obesity, insulin resistance, and simple hepatic steatosis (triglyceride [TG] accumulation) in mice on a high‐fat (HF) diet, by activating the SIRT1/liver kinase B1 (LKB1)‐AMP‐activated protein kinase (AMPK) axis (Rao et al., 2017). However, that study did not examine the other characteristics of NAFLD/NASH, such as hepatic inflammation, injury, and fibrosis. As an improved analogue of bouchardatine, R17 would be expected to alleviate hepatic steatosis and possibly the other pathological features of NASH. Indeed, R17 has improved efficacy in decreasing cellular lipid accumulation and much more favourable pharmacokinetic properties than those of bouchardatine (Chen et al., 2015; Rao et al., 2015). Furthermore, R17 decreased cellular stresses including endoplasmic reticulum (ER) and oxidative stresses (Chen et al., 2015), two mechanisms contributing to the progression of simple hepatic steatosis to NASH (Diehl & Day, 2017).
Figure 1.
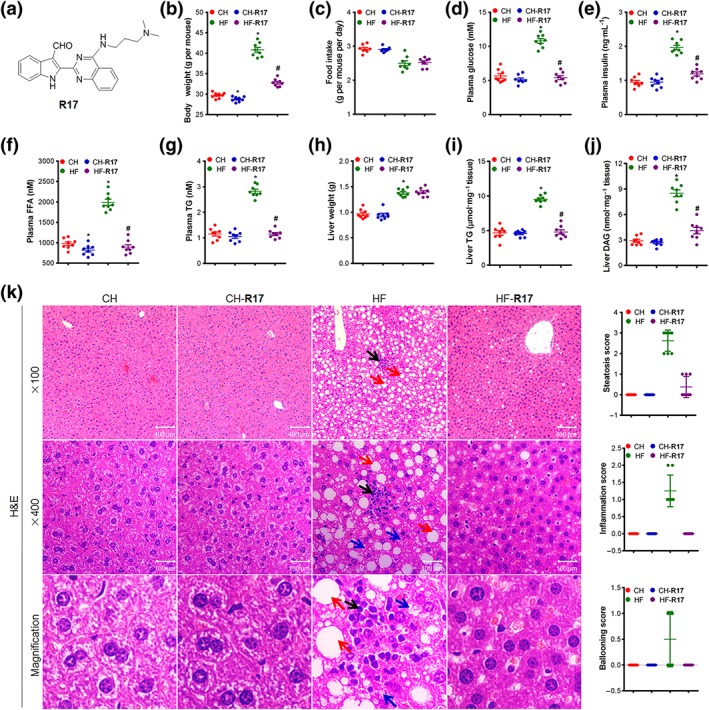
R17 ameliorates phenotypes of metabolic syndrome and NAFLD/NASH in HF‐fed mice. Male adult mice were fed a chow (CH) or high‐fat (HF, 60% calorie as fat) diet for 16 weeks and R17 was administered in the last 5 weeks (20 mg·kg−1, i.p. every other day). Body or tissue weights and parameters of plasma and liver were measured at the end of the experiment after 8 hr of fasting. (a) Chemical structure of R17; (b) body weight; (c) food intake; (d) plasma glucose level; (e) plasma insulin level; (f) plasma level of free fatty acid (FFA); (g) plasma level of triglyceride (TG); (h) liver weight; (i) liver TG level; (j) liver DAG content; and (k) representative images (left) and quantifications (right) of H&E staining of the liver (×100 and ×400 magnifications). Hepatic steatosis (red arrow), inflammation (black arrow), and ballooning (blue arrow) were quantified according to the NAFLD Activity Score (NAS) System, as described. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice
Therefore, the first aim of the present study was to characterize the therapeutic effects of R17 on the pathological characteristics of NASH in a mouse model of NAFLD/NASH generated by prolonged feeding with an HF diet containing a very high level of fat (60% of the calories). Our second aim was to investigate the molecular mode of action of R17 underlying these therapeutic effects. This was carried out by detailed examination of the liver samples from R17‐treated mice, in terms of the important pathological pathways leading to NAFLD/NASH and by relevant intervention studies in cell models.
2. METHODS
2.1. Animal experiments
All animal care and experimental procedures were approved by the Sun Yat‐sen University Committee on Animal Ethics for the Use of Laboratory Animals in accordance with the Animal Welfare Legislation of China. Every effort was made to minimize the use of the animals and their discomfort. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology. Male C57BL/6J mice aged 7–8 weeks (18–20 g, IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664) bred at the Laboratory Animal Centre of Sun Yat‐sen University (Guangzhou, China) were used for the study. The mice were housed under specific pathogen free and reared in line with standardized methods at 22 ± 1°C on a 12‐hr light/dark cycle with free access to food and water. After 1 week of acclimatization to the environment of this study, the mice were fed with either a chow diet (CH, with 70% calories from starch and provided by the Animal Center) or the HF diet ad libitum for up to 16 weeks. Both CH and HF groups were randomly divided into two subgroups at the beginning of Week 12 to receive R17, dissolved in normal saline, or its vehicle (saline ;control group). R17 or vehicle was injected i.p. (20 mg·kg−1 every other day) in the last 5 weeks. The control subgroups were administered the same volume of vehicle. Body weight and food intake were monitored daily.
2.2. Assessment of hepatic de novo lipogenesis
This was performed in a fasted/refeeding model (Chen et al., 2015; Vatner et al., 2015). The mice were fasted overnight and randomly divided into two subgroups, one group (control subgroup) fed a high fructose diet whereas another group of mice (R17‐treated group) were fed a high fructose diet plus R17 treatment (20 mg·kg−1 by i.p. injection once every other day) for 10 days. The control subgroups were administered the same volume of the vehicle. Hepatic de novo lipogenesis was assessed according to the described methods (Vatner et al., 2015).
2.3. Plasma biochemistry analysis
At the end of the study, mice were fasted for 8 hr and anaesthetized by an i.p. injection of 80 mg·kg−1 ketamine and 10 mg·kg−1 xylazine. When the mice were fully anaesthetized, the eyeball was removed to collect blood samples in a tube containing 1‐mM EDTA for the measurement of relevant plasma variables. The mice were then killed by cervical decapitation. The tissues of interest were weighed, photographed, and freeze‐clamped or fixed in 4% formaldehyde solution. Blood samples were centrifuged at 3,000 g for 10 min at 4°C. Collected plasma samples were used for the determination of alkaline phosphatase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), glucose, free fatty acids (FFA), TG, total cholesterol (CHO), HDL cholesterol, and LDL cholesterol using an Olympus AU 600 auto‐analyser (Olympus, Japan). Plasma insulin was determined using an ELISA assay kit (Abnova Cat #KA3812, Wuhan, China).
2.4. Determination of the triglyceride (TG) and diacylglycerol (DAG) contents of liver
Liver TG and DAG were extracted as described previously (Folch, Lees, & Sloane Stanley, 1957; Zeng et al., 2015). Briefly, the liver tissue or cells were homogenized and extracted with equal volumes of chloroform/methanol. The chloroform phase was removed to a new tube and dried and was then re‐suspended in isopropyl alcohol as a total lipid extract sample. The quantities of total TG (Jiancheng Bio Cat# A110‐2, Jiangsu, China) and DAG (Uscn Life Science Inc Cat# CEC038Ge, Shanghai, China) in the livers were then assayed according to the manufacturers' protocols. 100 μl of TG and DAG working reagents were mixed with 10 μl of ddH2O, TG, or DAG standards and the samples respectively. The reaction was incubated at 37°C for 15 min. Absorbance (A) of blank, standards, and samples were recorded at 510 nm. TG and DAG concentrations were calculated as (Asample − Ablank)/(Astandard − Ablank) × concentration of standards and normalized to tissue weight or protein content.
2.5. Histological examination and definition of the scoring system
All antibody‐based procedures used in this study comply with the recommendations made by the British Journal of Pharmacology. The tissues fixed in 4% formaldehyde were embedded in paraffin after dehydration in a graded ethanol series (70–100%). Embedded samples were sectioned (4‐μm thick) with a rotary microtome (Leica, Germany) and subject to haematoxylin and eosin, Masson or Sirius red staining. Both total and monocyte‐derived macrophages were assessed by immunohistochemistry labelled with CD68 antibody (Abcam# ab955, Guangzhou, China; RRID:AB_307338) and MAC387 antibody (Abcam# ab22506, Guangzhou, China; RRID:AB_447111) respectively. Sirius red staining was used to detect collagen and Masson staining was for fibrosis in the liver sections. The sections for histological and immunohistochemical examination were coded before assessment. The scores of steatosis, inflammation, ballooning, and fibrosis in the liver were evaluated by a pathologist, without knowledge of the treatments, using the NAFLD activity scoring system described by Kleiner et al. (2005). All sections were viewed with a light microscope (Olympus, Germany) and photographed at ×100 and/or ×400 magnification. The numbers of CD68‐ and MAC387‐labelled cells in each slide were counted with Image J software (Macbiophotonics, McMaster University, Hamilton, ON, Canada, RRID:SCR_003070) by Servicebio Company (Beijing, China). Quantification analysis was performed in six randomly selected fields per sample in a blinded manner.
2.6. Malonaldehyde level and SOD activity assay
Tissues or cells were homogenized in normal saline on ice and centrifuged at 3,500× g for 10 min at 4°C to remove debris. The collected supernatants containing extracted protein were used to measure the content of malonaldehyde (MDA; Jiancheng Bio Cat# A003, Nanjing, China) and the activity of SOD (Jiancheng Bio Cat# A001, Nanjing, China) according to the manufacturer's instructions. For the determination of SOD activity, 30 μg of extracted protein was incubated with the assay reagents at room temperature (25°C) for 10 min and then measured at an absorbance of 550 nm. For the quantification of MDA, 30 μg of extracted protein was incubated with the assay reagents at 95°C for 40 min and then rapidly cooled to room temperature. After centrifugation at 10,000× g for 10 min, the absorbance of the supernatant was measured at 532 nm. The MDA level and SOD activity in the liver were calculated by normalizing to the protein content.
2.7. TNF‐α and IL‐6 secretion level measurement
The levels of TNF‐α (GenStar Cat# C708, Guangzhou, China) and IL‐6 (GenStar Cat# 704, Guangzhou, China) in plasma (prepared as described before) or cell culture media were measured by ELISA kits according to the manufacturer's instructions. The cell media were collected after a designated treatment and centrifuged at 1,000 g for 10 min at 4°C to remove cell debris. Briefly, 10‐μl plasma or 50‐μl culture medium, saline, and standard was added to the ELISA plate and incubated at 37°C for 90 min. Wells were washed four times with elution buffer and then incubated with biotin‐labelled antibodies at 37°C in the dark for another 30 min followed by reaction with the corresponding substrate. The colour produced was proportional to the concentration of IL‐6 or TNF‐α and was measured at 450 nm and quantified against the standard curve from the known amount of the cytokine.
2.8. Caspase 3/8 activity measurement
The activity of caspase 3 (Biovision Cat# K105‐100, Wuhan, China) and caspase 8 (Promega Cat# G8200, Guangzhou, China) in cultured cells or liver samples was measured by a fluorescence‐based assay. Approximately 10‐μg liver protein or 20‐μg cell lysate was added to the wells and incubated with the fluorogenic substrate rhodamine 110 bis‐(N‐CBZ‐l‐aspartyl‐l‐glutamyl‐l‐valyl‐l‐aspartic acid amide; Z‐DEVD‐R110; 25 μM) at room temperature. Substrate cleavage and rhodamine 110 accumulation was measured fluorometrically at 499 nm (excitation) and at 521 nm (emission) in a multiplate reader (Spectromax Gemini, Molecular Devices). The protein content was determined by BCA Assay Kit (Thermo Cat# 23227, Guangzhou, China), with human serum albumin as a standard.
2.9. TUNEL assay
Liver samples were fixed in 10% formalin and embedded in paraffin. After de‐paraffinization, the liver samples were stripped of protein by incubation with 20 mg·ml−1 proteinase K (Sigma Chemical, Cat# 03115887001, Guangzhou, China) for 15 min at the room temperature. The TUNEL staining was performed using an apoptosis in situ detection kit (Roche, Cat# 11767291190, Guangzhou, China), according to the manufacturer's protocols. The nuclei were stained with DAPI (blue fluorescence, a ratio of 1:200), and TUNEL‐positive cells were visualized by green fluorescence. The sections were viewed with a light microscope (Olympus, Germany) and photographed at ×100 magnification. Samples for immunohistochemistry were coded before analysis. The numbers of TUNEL‐labelled cells in each slide were calculated with Image J software (Macbiophotonics, McMaster University, RRID:SCR_003070) by Servicebio Company (Beijing, China). More than 200 DAPI‐positive cells were examined in six random fields of each slide and TUNEL‐positive cells were counted to calculate the in situ apoptosis, in a blinded manner.
2.10. RNA extraction and qRT‐PCR
Total RNA from cells or mouse livers was isolated using the TRIzol method (Invitrogen, Cat# 15596018, Guangzhou, China). The first‐strand cDNA was synthesized with a cDNA synthesis kit (Takara, Cat# 6210B, Dalian, China). Quantitative real‐time PCR was carried out using 2 × RealStar Green Fast Mixture with ROX (GenStar, Cat# A301, Guangzhou, China). The results were analysed on an ABI StepOnePlus real‐time PCR system (Applied Biosystems, USA, RRID:SCR_015805) using the 2−ΔΔCt method. Primers were synthesized by Generay Biotech (Guangzhou, China), and primers sequences were listed in Table S2. Actin was used as a loading control, and relative mRNA levels were normalized to actin.
2.11. Immunoblotting
This was essentially based on the protocols used in our recent studies (Rao et al., 2017). Liver samples or cells were lysed in ice‐cold RIPA extract buffer (Beyotime, Cat# P0013C, Chengdu, China) supplemented with a mixture of protease inhibitors (Roche, Cat# 4693006001, Guangzhou, China). The extracted proteins were separated by SDS‐PAGE and transferred to a PVDF membrane (Millipore, Guangzhou, China). After blocking with TBS/T (0.1%) containing 5% BSA for 30 min at room temperature, the membrane was incubated with different primary antibodies of acetyl‐CoA carboxylase (ACC; Cell Signaling Technology Cat# 3676, RRID:AB_2219397), fatty acid synthase (FAS; Cell Signaling Technology Cat# 3180, RRID:AB_2100796), stearoyl‐CoA desaturase 1 (SCD1; Cell Signaling Technology Cat# 2794, RRID:AB_2183099), IL‐6 (Cell Signaling Technology Cat# 12153, RRID:AB_2687897), TNF‐α (Cell Signaling Technology Cat# 6945, RRID:AB_26878975), NF‐κB (Cell Signaling Technology Cat# 6956, RRID:AB_10828935), pNF‐κB (Cell Signaling Technology Cat# 3033, RRID:AB_331284), PARP (Cell Signaling Technology Cat# 9542, RRID:AB_2160739), c‐PARP (Cell Signaling Technology Cat# 9548, RRID:AB_2160592), caspase 8 (Cell Signaling Technology Cat# 4927, RRID:AB_2068301), c‐caspase 8 (Cell Signaling Technology Cat# 8592, RRID:AB_10891784), caspase 3 (Cell Signaling Technology Cat# 9665, RRID:AB_2069872), c‐caspase 3 (Cell Signaling Technology Cat# 9664, RRID:AB_2070042), PRKR‐like ER kinase (PERK; Cell Signaling Technology Cat# 5683, RRID:AB_10841299), pPERK (Cell Signaling Technology Cat# 3179, RRID:AB_2095853), inositol‐requiring enzyme 1 α (IRE‐1α; Cell Signaling Technology Cat# 3294, RRID:AB_823545), eukaryotic translation initiation factor 2 α (eIF‐2α; Cell Signaling Technology Cat# 2103, RRID:AB_836874), peIF‐2α (Cell Signaling Technology Cat# 3597, RRID:AB_390740), spliced X‐box binding protein 1 (XBP‐1s Cell Signaling Technology Cat# 12782, RRID:AB_2687943), C/EBP homologous protein (CHOP; Cell Signaling Technology Cat# 2895, RRID:AB_2089254), pAMPKα (Cell Signaling Technology Cat# 2535, RRID:AB_331250), pACC (Cell Signaling Technology Cat# 11818, RRID:AB_2687505), actin (Cell Signaling Technology Cat# 4970, RRID:AB_2223172), ATP synthase (Abcam Cat# 109867, RRID:AB_10866627), pIRE‐1α (Abcam Cat# ab48187, RRID:AB_873899), sterol regulatory element‐binding protein‐1c (SREBP‐1c; Santa Cruz Biotechnology Cat# sc‐367, RRID:AB_2194226), and pLKB1 (Santa Cruz Biotechnology Cat# sc‐28465, RRID:AB_2198347) were used by 1:1,000 dilution in 5% bovine albumin at 4°C overnight. The membrane was washed with TBS/T four times to remove the unbound antibody and then incubated with the HRP‐conjugated secondary antibody (Cell Signaling Technology, Cat# 7076 [mouse] and Cat# 7074 [rabbit], China). Protein bands were visualized with an ECL kit (Millipore, Cat# 64‐201bp, China). Densitometry analysis was performed using Quantity One Software (Bio‐Rad Laboratories, CA, USA) relative to the loading control.
2.12. Cell culture
Human hepatoma HuH7 (CLS Cat# 300156/p7178_HuH7, RRID:CVCL_0336) and HepG‐2 (CLS Cat# 300198/p2277_Hep‐G2, RRID:CVCL_0027) cells were obtained from American Type Culture Collection (China). The cells were cultured in DMEM supplemented with 10% FBS and 10 U·ml−1 penicillin/streptomycin (Thermo, Guangzhou, China) at 37°C in a humidified incubator with 5% CO2. For induction with fatty acids (FAs), cells were seeded and cultured overnight. When the cells reached 70% confluence, they were incubated with 0.5‐mM oleic acid (OA; Sigma, China) or 0.5‐mM palmitic acid (PA; Sigma, China) for 24 hr and then were harvested for subsequent analysis. To examine the effect of R17 on hepatocyte de novo lipogenesis, HuH7 cells cultured in a DMEM containing 25‐mM glucose were primed in a lower glucose (5 mM) DMEM for 8 hr. The de novo lipogenesis was stimulated by switching the cells to a high glucose (30 mM) DMEM for 24 hr, in the presence or absence of R17. To investigate the effect of R17 on fibrogenesis at the cellular level, LX‐2 cells (Millipore Cat# SCC064, Shanghai, China, RRID:CVCL_5792) derived from hepatic stellate cells (HSCs; the main cell type for hepatic fibrosis) were used (Tsuchida & Friedman, 2017). Fibrogenesis was induced by incubation with 20 μg·ml−1 TGF‐β (R&D System, Cat# 240‐B‐002, Beijing, China) for 24 hr, with or without R17.
2.13. Oil red O staining and TG assay
After treatment, cells were fixed by 4% paraformaldehyde for 1 hr at room temperature and then stained with fresh 0.5% Oil red O solution for 30 min in the dark. After the staining, cells were rinsed with 60% isopropanol, washed with distilled water, and analysed under a light microscope (Zeiss, Germany). Cellular TG were extracted and determined as mentioned above, using commercial Peridochrom TG GPO‐PAP kit (Jiancheng Bio, Cat# A110‐2, Nanjing, China).
2.14. Cytotoxicity and cell viability assay
Cytotoxicity was evaluated by using the LDH release assay kit (Beyotime, Cat# C0017, Chengdu, China). Cells were plated at a low density (5,000 cells per well) in a 96‐well plate and separated into four groups: background group (without cells), maximal enzymic group (positive group), control group (without compound R17 treatment), and R17‐treated group. R17 was added at a final concentration of 1 μM. After a period of incubation, the LDH releasing agent (1/10, v/v) was added to the maximal enzymic group. After these treatments, the plates were centrifuged at centrifuged at 3,500× g for 5 min with supernatant collected for the determination of LDH release to assess the cytotoxicity of R17. The supernatant (40 μl) was incubated with detecting reagents in the dark for 30 min and the Absorbance (A) was recorded at 490 nm. The cytotoxicity was calculated as (Acompound − Acontrol)/(Amaximal − Acontrol).
Cell viability was determined by using MTT (Beyotime, Cat# C0009, Chengdu, China) assay. After treatment, cells were incubated with 0.5 mg·ml−1 MTT at 37°C for 4 hr, the supernatant was discarded, and cells were resolved in DMSO solution for the measurement of the absorbance at 490 nm. The relative cell viability was calculated.
For cell counting, treated cells were collected and re‐suspended in 1‐ml PBS. The cell suspension (0.5 ml) was mixed with 0.4% Trypan blue stain (Beyotime, China) at 1:1 (v/v) and incubated for 5 min at room temperature. The haemocytometer was filled with 10 μl of the cell mixture for counting under a light microscope (Zeiss, Germany).
2.15. Apoptosis assay
After incubation with R17, cells were washed with PBS and then stained with Annexin V‐FITC/PI (MultiScience, Cat# 70‐AP101‐100, China) in the dark for 10 min. Each sample was loaded with 15,000 cells for the analysis in a flow cytometer (Beckman, Germany) at the auto‐setting. Data were analysed by Expo 32 Software (Beckman, Germany). According to the instructions of the Annexin V‐FITC/PI, FITC‐labelled cells were regarded as early apoptotic cells whereas those labelled by both PI and FITC were defined as late apoptotic cells. Viable cells were not labelled by either PI or FITC.
2.16. Cellular ROS determination
Intracellular ROS was measured using a DCFH‐DA probe (Beyotine, Cat# S0033, Chengdu, China). Briefly, HuH7 cells were seeded at a density of 100,000 cells per well in a 24‐well plate and cultured overnight. The cells were then incubated with PA or OA (0.5 mM) in the presence or absence of R17 (1 μM) for 24 hr. The treated cells were then incubated with 10‐μM DCFH‐DA at 37°C for 30 min and then immediately analysed by flow cytometry (Beckman, Germany). Data were collected from at least 10,000 cells at a flow rate of 250–300 cells per s. Representative images were captured by confocal microscopy (Zeiss, Germany) under an excitation wavelength of 488 nm.
2.17. ATP level and ATP synthase activity determination
ATP content in cells or liver tissues was quantified by an ATP determination kit following the manufacturer's instructions (Promega, Cat# FF2000, Guangzhou, China). The treated cells or tissues were lysed with an ATP lysis buffer on ice for 30 min and centrifuged at 3,000× g for 15 min. The supernatant was collected for the determination of ATP level; 20‐μl ATP‐free water (background), ATP standard (0, 0.1, 1, 10, 100, 1,000, and 10,000 nM), and samples were incubated with 100‐μl detecting reagents with four replicates in the dark for 2 min, and the RLU values were determined over a 10‐s integration time. The ATP level was calculated according to the obtained ATP standard curve and normalized to the protein content.
The activity of mitochondrial ATP synthase was measured spectrophotometrically at 340 nm. Approximately 50‐μg cellular or liver protein was mixed with the detection reagents and the absorbance (A) was measured immediately and recorded as 0 min (A0). After incubation at 37°C for 5 min, the absorbance was measured again and recorded as 5 min (A5). The lysis buffer was viewed as a blank control. The ATP synthase activity (Jiancheng Bio Cat# A089‐5, Nanjing, China) was calculated according to the manufacturer's instructions and normalized to the protein content of the sample.
2.18. AMPKα siRNA transfection
For the knockdown of AMPKα, AMPKα siRNA was chemically synthesized by RiboBio (Guangzhou, China). The siRNA sequence of AMPKα was listed as follows: sense: 5′‐GAGGAUGCCUCAGGAAAUA‐3′ and anti‐sense: 5′‐AAAGCGUCUGGAAAAGUCG‐3′. HuH7 cells were transfected with control siRNA or AMPKα siRNA using Lipofectamine 3000 according to the manufacturer's instructions (Invitrogen, Guangzhou, China). Briefly, HuH7 cells were seeded at a density of 200,000 cells per well in a six well‐plate and incubated overnight. After this, the cells were transfected with control siRNA or AMPKα siRNA (50 nM) for 24 hr. R17 (1 μM) was added for the last 16 hr. The efficiency of knockdown and related parameters were detected.
2.19. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Data are expressed as the mean ± SEM. Differences between two groups were analysed by Student's t test using Graphpad Prism (Graphpad Software Inc, California, USA, RRID:SCR_002798). Statistical analysis for multiple groups was performed by one‐way ANOVA followed by Tukey's HSD post hoc tests. A P value of ≤.05 was considered statistically significant.
2.20. Materials
R17 (Figure 1a) was synthesized in our laboratory (purity >95%) by introducing a side chain of N,N‐dimethyl‐1,3‐propane diamine, into the structure of bouchardatine, as described previously (Chen et al., 2015). It was dissolved in DMSO to a final concentration of 0.4 and 1 μM for cell culture. 5‐Aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside (AICAR; CID: 17513), LPS (CID: 11970413), compound C (CID: 11524144), SC75741 (CID: 23661638), and oligomycin (CID: 78358496) were purchased from Selleck (Shanghai, China). BSA‐conjugated palmitic acid (PA, CID: 985) and BSA‐conjugated oleic acid (OA, CID: 445639) were purchased from Sigma–Aldrich (USA). AMPKα small interfering RNA was purchased from Ribobio Co., Ltd. (Guangzhou, China). The HF diet (60% of calories from fat, Cat# D12492) and the high‐fructose diet (60% of calories from fructose, Cat# D02022704) were purchased from the Research Diet (New Brunswick, NJ 08901, USA). The formulation of the HF diet and the high‐fructose diet are shown in Table S1.
2.21. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018; Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Fabbro et al., 2017; Alexander, Kelly et al., 2017).
3. RESULTS
3.1. R17 ameliorates hepatic steatosis in HF‐fed mice
As shown in Figure 1, prolonged feeding with the HF diet (with 60% of the calories as fat) produced significant increases in body weight, hyperglycaemia, hyperinsulinaemia, and hyperlipidaemia (measured as FFA and TG levels). Associated with these phenotypes of the metabolic syndrome, the HF mice displayed significant characteristics of NAFLD/NASH indicated by two to threefold increases in hepatic lipid contents (TG and DAG), histological steatosis, inflammation, and hepatocyte ballooning. As expected, treatment with R17 reversed HF‐induced phenotypes of the metabolic syndrome as indicated by reductions in body weight gain, hyperglycaemia, hyperinsulinaemia, and increased plasma level of FFA and TG (Figure 1b–g). These effects occurred without affecting the food intake (Figure 1c). Interestingly, administration of R17 almost corrected all the HF‐induced phenotypes of NAFLD/NASH, namely, hepatic lipid contents of TG and DAG (Figure 1i–j), histological steatosis, inflammation, and hepatocyte ballooning (Figure 1k) to the normal, in mice fed the normal chow (CH mice). These effects of R17 occurred without affecting liver weight (Figure 1h). Similarly, R17 treatment also decreased TG and DAG levels in the liver of fasted/refeeding mice (Figure S1A–C), a model of hepatic steatosis induced by increased de novo lipogenesis (Vatner et al., 2015). There were no detectable abnormalities in tissues of the examined organs after the treatment with R17, as indicated by their weight, H&E staining, and relevant plasma parameters (Figure S2).
3.2. R17 inhibits lipid accumulation within cells
Consistent with the reduced lipid accumulation in the liver, R17 treatment markedly decreased the levels of key lipogenic proteins, ACC, FAS, SCD1 and SREBP‐1c, in both HF‐fed and fasted/refeeding mice (Figures 2a and S1D), indicating a suppression of the rate‐limiting enzymes in hepatic lipogenesis. To elucidate the molecular mode of action at the cellular level, we incubated hepatoma HuH7 cells with different FAs (an unsaturated acid, OA or a saturated acid, PA). As expected, OA significantly increased TG levels with moderate cytotoxicity but only at higher concentrations while PA resulted in less TG accumulation but much greater lipotoxicity (indicated by cell viability; Figure S3). Incubation with R17 dose dependently reduced the accumulation of TG and increased FAS, ACC, and SCD1 induced by OA or PA (Figure 2b–c). Furthermore, R17 markedly inhibited high glucose‐induced accumulation of TG from de novo lipogenesis, in a dose‐dependent manner, as indicated by the decreases in the lipogenic proteins FAS, ACC, and SCD1 (Figure 2d–e).
Figure 2.

R17 decreases lipid synthesis and cellular TG accumulation induced by FAs or glucose. Panel (a), effects of R17 in HF‐fed mice as described for Figure 1. (a) Effects on the expression of key lipogenic proteins in the liver. Animal experiments were described in Section 2 and Figure 1 legend. The extracted protein from the liver samples were immunoblotted with indicated specific antibodies and quantified to the loading control of GAPDH. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (b–e), effects of R17 in cultured HuH7 cells. (b–c) Effects of R17 (0.4, 1 μM) on TG level and expression of lipogenic proteins in cultured HuH7 cells incubated with oleic acid (OA, 0.5 mM) or palmitic acid (PA, 0.5 mM) for 24 hr. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. (d–e) Effect of R17 on the TG level and expression of lipogenic proteins in high glucose stimulated HuH7 cells. Cells cultured at 25‐mM glucose was primed at 5‐mM glucose for 8 hr followed by the stimulation of lipogenesis with 30‐mM glucose for 24 hr. The blank control group was cultured at 25‐mM glucose throughout the experiment. The extracted protein from the cells were immunoblotted with indicated specific antibodies. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. Data shown are individual values with means ± SEM, N = 5 independent experiments. *P < .05, significantly different from blank control; # P < .05, significantly different from the corresponding group of OA, PA, or 30‐mM glucose in the absence of R17
3.3. R17 attenuates hepatic injury and inflammation in HF‐fed mice
To evaluate the therapeutic effects of R17 on hepatic injury and inflammation, we first assayed the markers of liver injury in HF‐fed mice. Administration of R17 reduced plasma levels of ALT, AST, and alkaline phosphatase (ALP) (Figure 3a) in HF mice, indicating an attenuation of the hepatic injury. Further examination showed that R17 treatment significantly inhibited HF‐induced deposition of macrophages, as stained for the specific antigen CD68 (for residual macrophages) and MAC387 (for monocyte‐derived macrophages; Figure 3b). This inhibition was in accompanied by a decrease in CD68 mRNA level of the livers (Figure S4A). These changes were associated with decreased mRNA levels of the inflammatory cytokines TNF‐α, IL‐6, and IFN‐γ in the liver (Figure 3c) and their circulating levels in plasma (Figure 3d–e). To investigate whether the anti‐inflammatory effects of R17 resulted from its effect in lowering cellular accumulation of lipids, we examined its effects on FA‐induced production of these inflammatory cytokines in cultures of hepatoma HuH7 cells. As shown in Figure 3f–h, incubation with OA or PA increased the secretion and mRNA expression of TNF‐α and IL‐6 and activated NF‐κB. R17 treatment reversed these changes, suggesting that the anti‐inflammatory effects of R17 may be due to its effect in reducing cellular lipids. Interestingly, the anti‐inflammatory effects of R17 were additive to those produced by the NF‐κB inhibitor SC75741 (Figure 3i–j). To further test whether R17 may also inhibit inflammation caused by endotoxin (an extra‐hepatic factor of NASH), we examined its efficacy on LPS‐stimulated inflammation and observed similar inhibitory effects in this cell model (Figure S4B–D).
Figure 3.

R17 inhibits lipid‐induced inflammation and macrophage accumulation. Panels (a–e), effects of R17 in HF‐fed mice as described for Figure 1. (a) Plasma levels of enzymes from the liver. (b) CD68 (macrophage marker as illustrated by arrows, top) and MAC387 (monocyte marker as illustrated by arrows, bottom) in the liver and labelled cell were quantified by Image J software as described. (c) mRNA levels of inflammatory cytokines in the liver. (d–e) Plasma levels of inflammatory cytokines determined by ELISA. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (f–h), HuH7 cells were incubated with OA (0.5 mM) or PA (0.5 mM) for 24 hr with or without R17 (1 μM) treatment. (f–g) Secretion level of TNF‐α and IL‐6. Culture medium was collected for the determination of cytokine secretion by an ELISA assay. (h) Protein levels of TNF‐α, IL‐6, and NF‐κB in cells were determined by immunoblotting with specific antibodies. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. (i–j) Additive effects with the NF‐κB inhibitor SC75741 on TNF‐α, IL‐6, and NF‐κB. SC75741 (10 μM) was co‐incubated with R17 (1 μM) for 24 hr, cells were subjected to mRNA level (RT‐PCR) and protein level (immunoblotting) determination. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the Ctrl group. Data shown are individual values with means ± SEM, N = 5 independent experiments. *P < .05, significantly different from blank; # P < .05, significantly different from control (Ctrl); @ P < .05, significantly different from R17‐ or SC75741‐treated cells
3.4. R17 protects against hepatocyte injury
As reported (Osawa et al., 2018), we found chronic HF feeding increased hepatocellular apoptosis in the liver, as indicated by the increased activity of caspases 3 and 8, and the TUNEL assay in situ (Figure 4a–c). These increases in apoptotic markers were markedly inhibited by R17. In cultured HuH7 cells, R17 dose dependently reversed FA‐induced cell death (Figure 4d–e). These inhibitory effects on cell apoptosis were evident as early as 6 hr and sustained to 24 hr (Figure 4f). Addition of R17 to the cultured cells effectively decreased FAs‐induced caspase 3 activity (Figure 4g). Consistent with this, R17 treatment inhibited the activation of apoptosis‐related proteins as indicated by the decreased levels of the cleaved caspase 3 (c‐caspase 3), cleaved caspase 8 (c‐caspase 8), and cleaved PARP (c‐PARP; Figure 4h).
Figure 4.
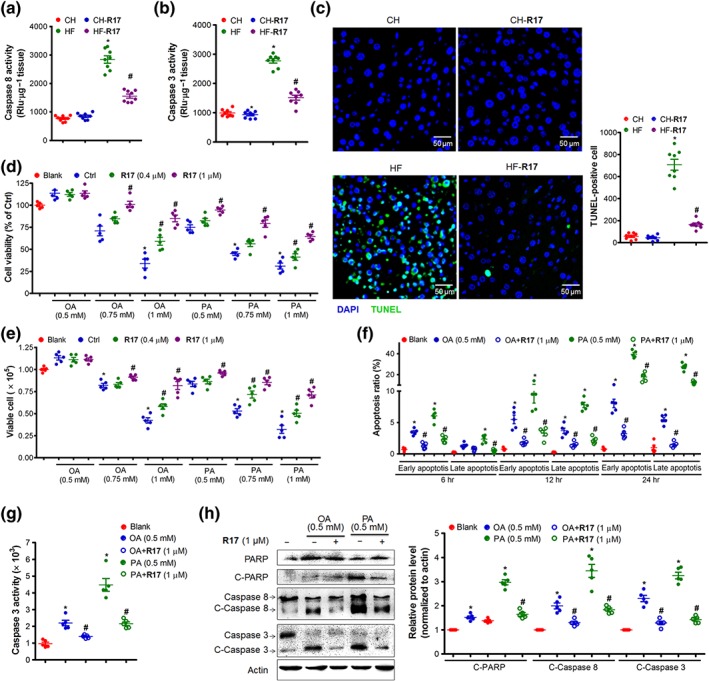
R17 protects against lipid‐induced cell death and apoptosis. Panels (a–c), effects of R17 in HF‐fed mice as described for Figure 1. (a–b) Caspase 8/3 activity in the liver. (c) Apoptosis in the liver determined by a TUNEL assay in situ. Representative images were captured and the foci (green) were quantified by an Image J software. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (d–h), HuH7 cells were incubated with OA (0.5 mM) or PA (0.5 mM) for 24 hr with or without R17 (1 μM). The cell viability, viable cell, cell apoptosis, and protein levels were determined. (d) Effect on cell viability. (e) Effect on cell survival. (f) Effects on the apoptosis over time (6, 12, and 24 hr). (g) Effect on caspase 3 activity. (h) Effects on apoptosis‐related proteins. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. Data shown are individual values with means ± SEM, N = 5 independent experiments. *P < .05, significantly different from blank; # P < .05, significantly different from control (Ctrl)
3.5. R17 inhibits lipid‐induced oxidative stress and ER stress
Increased peroxidation of lipids has been suggested to increase oxidative stress which can drive hepatic steatosis to NASH (Shi et al., 2017). Consistent with this report, there was an increase in oxidative stress in HF‐fed mice as indicated by increased MDA (a key parameter of lipid peroxidation) and decreased SOD (a key enzyme to break down ROS) in the liver. Notably, R17 significantly reversed the HF‐induced increase in MDA and preserved the activity of SOD (Figure 5a–b), suggesting an attenuation of the oxidative stress in the liver. We next examined ER stress because ER stress is increased during obesity and can promote lipid synthesis and inflammation (Jung, Kim, Abd El‐Aty, & Jeong, 2018; Hotamisligil, 2010). The results in Figure 5c show that R17 inhibited HF‐induced ER stress in the liver, as indicated by reduced phosphorylation of eIF‐2α, IRE‐1α and PERK, XBP‐1s, and CHOP content. In cultured HuH7 hepatocytes, R17 also abolished the increased oxidative stress and ER stress induced by FAs (Figure 5d–g). Similar results were also observed for R17 in starved/high glucose cell model (Figure S5A).
Figure 5.

R17 inhibits lipid‐induced oxidative stress and ER stress. Panels (a–c), effects of R17 in HF‐fed mice as described for Figure 1. (a–b) Effects of R17 on the MDA level and SOD activity in the liver of mice. (c) Effects on UPR pathway proteins. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (d–g), HuH7 cells were incubated with OA (0.5 mM) or PA (0.5 mM) in the absence or presence of R17 (1 μM). (d–e) Effect on ROS level. ROS levels were determined by a ROS probe in flow cytometry. Representative images of DAPI (blue) for nucleus and ROS (green) were captured in confocal microscopy at 24 hr. (f) Effect on MDA level. (g) Effects on ER stress. The UPR proteins (PERK, elF‐2α, IRE‐1α, XBP‐1s, and CHOP) were immunoblotted with specific antibodies. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. Panels (h–j), HuH7 cells were incubated with DTT (2 mM, ER stress inducer) in the absence or presence of R17 (0.4, 1, and 2.5 μM) for 2 hr. (h) Effects on UPR proteins; protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. (i) Cell apoptosis. Determination by flow cytometry using an Annexin V‐FITC/PI kit; (j) cell viability. Data shown are individual values with means ± SEM, N = 5 independent experiments.*P < .05, significantly different from blank; # P < .05, significantly different from control (Ctrl)
To investigate the role of inhibited ER stress and ROS in the protective effects of R17 for cells, we examined the effect of R17 on the cell apoptosis and viability in response to DTT, an ER stress inducer not associated to nutrient metabolism. As shown in Figure 5h, incubation with DTT for 2 hr induced a marked increase in ER stress, as indicated by an arrangement of proteins of the UPR pathway (PERK, eIF‐2α, IRE‐1α, XBP‐1s, and CHOP). These changes were dose dependently reversed by R17. Along with this, R17 preserved cell viability and attenuated apoptosis induced by DTT (Figure 5i–j). Similarly, R17 protected the cell viability while dose dependently inhibiting LPS‐induced ER stress and oxidative stress (Figure S5B).
3.6. R17 inhibits lipid‐induced fibrogenesis
We next examined the effect of R17 on hepatic fibrosis as the extent of fibrosis largely determine the prognosis of NASH (Schuppan, Surabattula, & Wang, 2018; Younossi et al., 2018). As illustrated in Figure 6a, Sirius red and Masson staining showed mild aggregations of collagen and fewer fibres in the liver of HF‐fed mice but these fibrogenic changes were abolished by treatment with R17. Consistent with this, HF‐induced increases in the mRNAs of TGF‐β, collagen I, the chemokine CCL2, and SMAD3 were markedly suppressed by R17 (Figure 6b). Because HSCs are the main source of fibrosis within the liver (Tsuchida & Friedman, 2017), we further investigated the effect of R17 in the fibrogenesis induced by TGF‐β in LX‐2 cells derived human HSCs (Kisseleva & Brenner, 2006). These results showed a similar inhibition of TGF‐β‐induced increases in collagen I, TNF‐α, and IL‐6 by R17 (Figure 6c–d).
Figure 6.
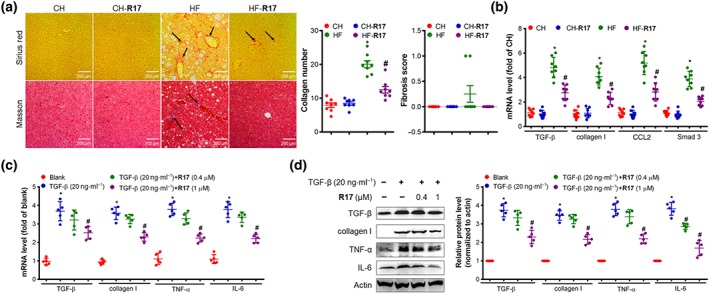
R17 inhibits fibrogenesis in the liver and cultured hepatic stellate cells. Panels (a–b), effects of R17 in HF‐fed mice as described for Figure 1. (a) Liver fibrogenesis. Representative images (×200) of Sirius red for collagen (top, illustrated by arrows) and Masson staining for fibrosis (bottom, illustrated by arrows). Collagen depositions were quantified using an Image J software whereas fibrosis was quantified according to the NAFLD Activity Score System . (b) Expression of liver fibrogenic mRNAs. TGF‐β, collagen I, CCL2, and Smad3. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (c–d), LX‐2 cells (from human stellate cells) were incubated with TGF‐β (fibrogenic inducer, 20 ng·ml−1) for 24 hr in the absence or presence of R17 (0.4, 1 μM) for 24 hr. (c) Fibrogenic and inflammatory mRNAs; (d) fibrogenic and inflammatory proteins, protein levels were quantified and relative folds were determined by comparative with the blank group. Data shown are individual values with means ± SEM, N = 5 independent experiments. *P < .05, significantly different from blank; # P < .05, significantly different from control (Ctrl)
3.7. R17 attenuates mediators of NAFLD/NASH by the AMPK pathway with ATP synthase as an upstream target
In HF‐fed mice, the AMPK pathway in the liver was suppressed as indicated by the reductions in phosphorylation levels of AMPKα, its upstream regulator LKB1, and its downstream substrate ACC (Figure 7a). These reductions were corrected by the treatment with R17 in association with a decrease in ATP content and an increase in the AMP/ATP ratio (Figure 7b). Subsequent studies revealed that the activity of ATP synthase was stimulated, and this increase was reduced to the similar level of normal mice (Figure 7c). We therefore conducted a series of experiments in cultured HuH7 cells to investigate the pathogenic processes involved in the development of NAFLD/NASH. The results in Figure 7d–e show that the HF‐induced changes in AMPK pathways, ATP content, AMP/ATP ratio, and ATP synthase activity were also reproduced in HuH7 cells by incubation with OA or PA, and de novo lipogenesis and LPS‐stimulated cell model (also in Figure S6A–B). The effects of R17 on these changes induced by OA or PA in HuH7 cells were consistent with the results in HF‐fed mice. As shown in Figures 7f–g and S6C, R17 inhibited ATP synthase activity and decreased TG accumulation in HuH7 cells in a similar pattern to the ATP synthase inhibitor oligomycin (also in Figure S6D). Importantly, R17 inhibited the activity of ATP synthase in the absence of changes in the level of this enzyme (Figure S6E) without induction of cytotoxicity (Figure S6F–G). These data together suggest that the restoration of lipid‐induced inhibition of the AMPK pathway by R17 may be due to its increasing the AMP/ATP ratio via inhibition of the activity of ATP synthase.
Figure 7.
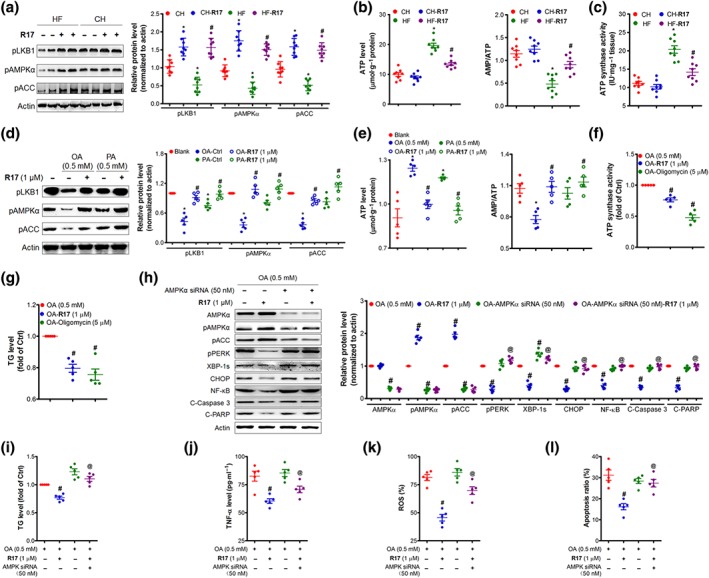
R17 attenuates NAFLD/NASH by activating the AMPK pathway with ATP synthase as an upstream target. Panels (a–c), effects of R17 in HF‐fed mice as described for Figure 1 and the liver samples were used for the assays. (a) Expression of pLKB, pAMPKα, and pACC in the liver. Determined by immunoblotting and quantified to the loading of actin. (b) Liver ATP level and AMP/ATP ratio. Liver ATP and AMP were determined using commercial kits. (c) Liver ATP synthase activity. Data shown are individual values with means ± SEM, N = 8 mice per group. *P < .05, significantly different from CH control mice; # P < .05, significantly different from HF control mice. Panels (d–g), HuH7 cells were incubated with OA (0.5 mM) or PA (0.5 mM) for 24 hr (for d–e) or 12 hr (for f–g) in the presence of indicated treatments. (d) Effects on cellular pLKB, pAMPKα, and pACC. Protein levels were quantified and normalized to actin; and relative fold increases were determined by comparison with the blank group. (e) Cellular ATP and AMP/ATP ratio. (f–g) Cellular ATP synthase activity and TG content. Oligomycin (5 μM) was used as positive control of ATP synthase inhibitor. Panels (h–l), AMPKα was knocked down in HuH7 cells by AMPKα siRNA (50 nM). The cells were then incubated with OA (0.5 mM) in the presence or absence of R17 (1 μM) for 24 hr. (h) Key proteins in AMPK and UPR pathways. Protein levels were quantified and relative fold increases were determined by comparison with the blank group. (i) TG content. (j) TNF‐α secretion. (k) ROS production. (l) Cell apoptosis. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). # P < .05, versus OA‐treated cells (Ctrl); @ P < .05, versus R17 treated alone for all cell‐based experiments
We then examined whether AMPK was required for the therapeutic effects of R17 on NAFLD/NASH by various interventions in HuH7 cells. Indeed, inhibition of the AMPK pathway by AMPKα siRNA diminished the ability of R17 to reduce OA‐induced ER stress (Figure 7h), TG accumulation (Figure 7i), inflammation (Figure 7j), ROS production (Figure 7k), and apoptosis (Figure 7l). Similar results of diminished beneficial effects of R17 were observed when compound C, a direct inhibitor of AMPK, was used to block the ability of R17 to activate the AMPK pathway (Figure S7). These data strongly suggest that activation of the AMPK pathway is likely to be critical for R17 to exert its therapeutic effects on models of NAFLD/NASH.
4. DISCUSSION
The present study investigated the therapeutic effects of the newly synthesized compound R17 on NAFLD/NASH and its molecular mode of action. These effects were tested in a mouse model with changes typical of NAFLD/NASH, as a hepatic manifestation of the metabolic syndrome in humans (Chalasani et al., 2018; Diehl & Day, 2017). As the model did not reach the severity of hepatic fibrosis of full‐blown NASH, we have called it more broadly as NAFLD/NASH. Our results firstly showed that R17 eliminated hepatic steatosis, inflammation, injury, and fibrogenesis in a mouse model of NAFLD/NASH, induced by prolonged feeding with a HF diet. These therapeutic effects appear to largely fulfil the recommended criteria essential for the treatment of NASH (Diehl & Day, 2017). Importantly, R17 exerts these therapeutic effects, at least in part, by activating the AMPK pathway with ATP synthase as an upstream target, without major adverse events. These therapeutic effects together with its safety profile suggest that R17 may be considered for further development for the treatment of NAFLD/NASH.
It is increasingly recognized that the mechanisms underlying NASH involve multiple hits, including hepatic steatosis (first hit) and other additional hits (including oxidative stress and ER stress), which result in inflammation, injury and fibrosis in the liver (Diehl & Day, 2017; Drew, 2017). Regarding the pathogenesis of hepatic steatosis, it usually results from either an increased influx of FAs (such as from HF diet) into the liver or excessive de novo lipogenesis within hepatocytes (such as follows exposure to high glucose or high carbohydrate diet) or both (Suzuki et al., 2017). While FA influx is mainly controlled by FA transporters in the cell membrane (such as CD36), de novo lipogenesis is regulated by a series of lipogenic enzymes, FAS, ACC, and SCD1. The expression of these enzymes is controlled by the master transcription factor SREBP‐1c (Zhao et al., 2012). Our results showed that R17 treatment effectively reversed the up‐regulation of SREBP‐1c and its downstream lipogenic enzymes induced by chronic HF feeding. These results indicate that the molecular mode of the action of R17 in eliminating the first hit of NASH can be attributed to the inhibition of SREBP‐1c and its downstream proteins. Further, the efficacy of R17 in reducing TG accumulation in the liver of HF mice and cultured cells incubated with FAs strongly suggests an increased utilization of accumulated lipids, as another mechanism to eliminate the hepatic steatosis in HF mice.
ER stress has been suggested as an important mechanism in the pathogenesis of NASH because it promotes de novo lipogenesis and elicits inflammation and cell injury (Hotamisligil, 2010). It is of interest to note that R17 reversed the increased hepatic ER stress in HF mice and cultured HuH7 cells exposed to FAs and high concentrations of glucose. Apart from the inhibition of de novo lipogenesis, activation of AMPK also promotes FA oxidation via its downstream enzyme ACC, which abolishes the inhibitory effect of carnitine palmitoyltransferase‐1 on FA entry to mitochondria (Day, Ford, & Steinberg, 2017; Kim, Lee, Ntambi, & Hyun, 2011). Indeed, we found a significant increase in phosphorylation of ACC by R17. Apart from hepatic steatosis, R17 clearly exerted therapeutic effects for other key markers of NASH. This was demonstrated by the decreases in plasma ALT and AST (attenuated hepatocyte injury), infiltration of hepatic macrophages, expression of inflammatory cytokines and NF‐κB (attenuated hepatic inflammation), and hepatic collagen deposition and fibrogenic markers (attenuated hepatic fibrosis) in HF‐fed mice. Also, increased hepatocyte apoptosis is a critical mechanism contributing to inflammation and fibrogenesis of the liver in NASH (Hatting et al., 2013; Jaeschke, 2002). The present study showed that R17 treatment protected against hepatocyte apoptosis, together with inhibiting inflammation.
Cell death by apoptosis can be initiated by various causes including lipid toxicity, unresolved ER stress (Hotamisligil, 2010; Yao et al., 2017), and oxidative stress (Oyadomari & Mori, 2004). Increased ER stress can activate three UPR canonical pathways through three ER transmembrane proteins PERK, eIF‐2α, and ATF6. Prolonged ER stress eventually results in cell death by initiating apoptosis (Hotamisligil, 2010). To examine the mechanism of the therapeutic effects of R17 on hepatocyte apoptosis or death as observed in NASH, we exposed hepatoma cells to OA/PA, high glucose, LPS, and DTT respectively. As expected, R17 added has efficiently reversed the activation of ER stress and the apoptosis activity in DTT‐stimulated cells. These results strongly suggest that R17 protects hepatocytes against apoptosis and inflammation via eliminating ER stress and oxidative stress.
Fibrosis has received increasingly more attention as a critical pathological feature of NASH because the extent of fibrosis is a major factor for the prognosis of NASH (Chalasani et al., 2018; Diehl & Day, 2017; Drew, 2017). Hepatic fibrosis is pathologically characterized by collagen accumulation in the liver produced by activated stellate cells (Li et al., 2018; Tsuchida & Friedman, 2017). In the present study, the prolonged HF feeding markedly increased the collagen content in the liver in parallel with the up‐regulation of the network of genes involved in the development of fibrosis. Treatment with R17 significantly attenuated the HF‐induced collagen deposition in the liver and inhibited the up‐regulation of the fibrogenic genes. These results indicate that R17 is likely to attenuate the fibrogenic pathway, possibly as a result of reduced inflammation and cell death in the liver. However, due to the limitation of the lack of histological fibrosis in the present study, the ultimate effect of R17 on hepatic fibrosis needs to be further evaluated using another NASH animal model with clear fibrosis.
Although AMPK is widely recognized as a therapeutic target for metabolic diseases (Day et al., 2017), its role in ER stress and oxidative stress is less clear. Of particular interest, the effect of R17 to inhibit ER stress and oxidative stress was diminished when AMPK was knocked down or inhibited by its inhibitor, compound C. Consistent with this finding, the effects of R17 in alleviating apoptosis and inflammation were also abolished by knocking down AMPK or by compound C. In parallel with AMPK activation, R17 treatment inhibited the inflammation in the liver of HF mice as well as a decreasing inflammatory cytokine in cells incubated with FA or LPS, along with a robust blockade of the NF‐κB pathway. These protective effects of R17 mediated by AMPK were confirmed by AMPK knockdown and its activator or inhibitor. Such interpretation is also in agreement with the reports showing the beneficial effect of activated AMPK against HF‐induced metabolic syndrome (Jung et al., 2015; Liu et al., 2016). It is possible that the observed dependence on AMPK in cultured cell may also explain the mechanism (at least in part) for the observed effects of R17 in HF mice. Obviously, the dependence on AMPK for the therapeutic effects of R17 for NAFLD/NASH in vivo cannot be confirmed in the present study and remains to be established in relevant models generated in AMPK knockout mice.
Regarding the molecular mechanism by which R17 activates AMPK, we first examined the levels of AMP and ATP because AMPK can be activated by increased AMP and decreased ATP (Day et al., 2017). As expected, the level of ATP was decreased in the liver of R17‐treated HF‐fed mice and cultured HuH7 cells while the ratio of AMP/ATP was increased. We further investigated how ATP was decreased by R17, and the results revealed that the decreased ATP may result from an inhibition (but not complete block) by R17 of the activity of ATP synthase, which is required for the synthesis of ATP. These findings suggest that R17 attenuates NASH by activating the LKB1‐AMPK pathway by blunting the activity of ATP synthase. Inhibition of the synthesis of ATP to activate AMPK is believed to be an important mechanism to exert anti‐ metabolic syndrome effects by compounds such as metformin and berberine (Turner et al., 2008). However, the cellular target sites for R17, metformin or berberine are very different. Although both metformin and berberine target mitochondrial complex I (Turner et al., 2008), the exact target protein is not known because there are more than 40 proteins in mitochondrial complex I (Vinothkumar, Zhu, & Hirst, 2015). In comparison, ATP synthase is a subunit of mitochondrial complex V. Thus, identification of ATP synthase as an upstream target of R17 may provide a potential new cellular mechanism for the treatment of NAFLD/NASH associated with the metabolic syndrome.
Overall, the present study demonstrated that R17 effectively attenuated NAFLD/NASH by reducing lipotoxicity, inflammation, and fibrogenesis in the liver. These therapeutic effects are in accordance with the recommended criteria for a potential new drug to treat NASH (Diehl & Day, 2017). The molecular mode of action involves inhibition of ER stress, oxidative stress, and apoptosis, consistent with the proposed mechanism of the pathogenesis of NASH (Ashraf, & Sheikh, 2015; Koyama et al., 2018). These beneficial effects are driven by its activation of the LKB1‐AMPK pathway triggered by its inhibition of the activity of ATP synthase as the primary cellular target (Figure 8). As R17 exerts these anti‐NAFLD/NASH effects without any detectable adverse effect, it may be further assessed for the treatment of NASH.
Figure 8.
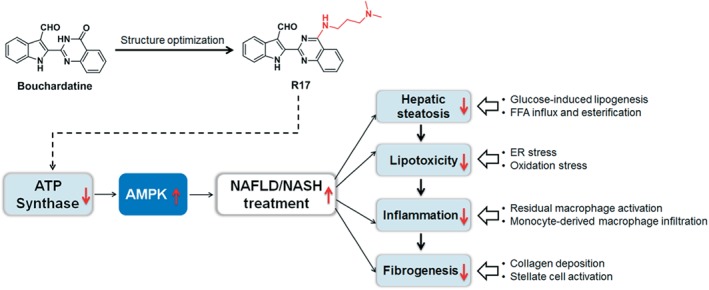
Diagram of the proposed mechanism of the therapeutic effects of R17 on NAFLD/NASH
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
This study was designed by Y.R., J.‐M.Y., and Z.‐S.H. Y.R., Y.‐T.L., C.L., and Q.‐Q.S. performed biological evaluation, animal study, and mechanism study of R17. Z.X., Y.‐T.H., H.Y., and L.G., performed the synthesis and characterization of compound R17. Y.R., Z.‐S.H., and J.‐M.Y. provided reagents, materials, and analysis tools. Data were analysed and interpreted by Y.R. and Z.‐S.H. The manuscript was written by Y.R., J.‐M.Y., and Z.‐S.H., with input from all authors.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1. The formulation of the high fat diet and high fructose diet
Table S2. Primers sequence information
Figure S1. Effects of R17 on TG level in liver of fast/refeding mice. Male adult C57BL/6 J mice were fasted overnight and fed with a high fructose diet with or without the treatment of R17 at the dose of 20 mg kg−1 by i.p. each other day for 10 days. Livers were isolated and weighted, the TG, DAG level and protein related to lipogenesis were determined. (A) Liver weight. (B) Liver TG level. (C) Liver DAG level. (D) Expression of proteins related to lipogenesis and quantification. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 10 mice/group). *p < 0.05, vs. Ctrl group mice.
Figure S2. Evaluation of toxicity of R17 in mice after 5 weeks treatment. Male adult C57BL/6 J mice were fed with chow (CH) or high fat (HF) diet for consecutively 16 weeks, and R17 was administrated at last 5 weeks at the dose of 20 mg/kg by i.p. each other day. After 5 weeks treatment, plasma and interested tissues were isolated and related parameters of plasma and tissues were determined. (A) Hematoxylin & eosin (× 200 magnification) staining of heart, kidney, muscle and pancreas. (B) Ratio of tissue/Body weight of heart, kidney, muscle and pancreas. (C) Determination the level of chemicals in plasma indicative of organ toxicity in mice. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 8 mice/group). *p < 0.05, vs. CH control mice; #p < 0.05, vs. HF control mice.
Figure S3. Change of TG levels in HuH7 hepatocytes after FAs induction. HuH7 cells were incubated with fatty‐free BSA or OA (0.5 mM) and PA (0.5 mM) for 24 hr, TG level and cell viability were determined. (A) Images were captured under a contrasted eyesight or after Oil‐Red O staining, × 200 magnification. (B) Cellular TG level. (C) Cell viability. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. BSA treated group.
Figure S4. Effect of R17 on the inflammation in LPS stimulated cells. (A) CD68 mRNA level in liver of mice. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 8 mice/group). *p < 0.05, vs. CH control mice; #p < 0.05, vs. HF control mice. HepG‐2 cells were exposed to LPS (50 μg/mL) with or without the treatment of R17 (0.4, 1 μM) for 24 hr, the inflammatory cytokines in culture medium and protein level of NF‐κB pathway were determined by ELISA and immunoblotting assay, respectively. (B‐C) Secretion levels of IL‐6 and TNF‐α; (D) Expression of IL‐6, TNF‐α and NF‐κB pathway proteins, and quantification, relative folds were determined by comparative with the Blank group. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S5. Effects of R17 on ER stress in high glucose or LPS stimulated cells. (A) HuH7 cells were starved (cultured in a culture medium containing 5 mM) for 8 hr and then switched to a high glucose (30 mM) culture medium in the presence or absence of R17 (1 μM) treatment for 24 hr. UPR pathway related proteins were determination by immunoblotting and quantification, relative folds were determined by comparative with the Blank group. (B) HepG2 cells were exposed to LPS (50 μg/mL) stimulation with or without the treatment of R17 (0.4, 1 μM) treatment for 24 hr. UPR pathway related proteins were determination by immunoblotting and quantification, relative folds were determined by comparative with the Blank group. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S6. R17 inhibits lipotoxicity in hepatocytes via LKB1‐AMPK axis activation. (A) Effect of R17 on the LKB1AMPK axis in the LPS stimulated HepG‐2 cells. HepG‐2 cells were exposed to LPS (50 μg/mL) stimulation for 24 hr with or without R17 (1 μM) treatment, cells were collected and subjected to western blot assay and quantification, relative folds were determined by comparative with the Blank group. (B) Effect of R17 on the LKB1‐AMPK axis in the high glucose stimulated HuH7 cells as described in Methods. Protein levels were quantified and relative folds were determined by comparative with the Blank group. (C) Effect of R17 (1 μM) on the ATP synthase activity in OA or PA (0.5 mM) stimulated‐HuH7 cells. (D‐G) Effects of ATP synthase inhibitor oligomycin on TG level and AMPK pathway in OA (0.5 mM)‐stimulated HuH7 cells. Cells treated with oligomycin (10 μM) for 12 hr and collected for indicative assay. (D) ATP level and AMP/ATP ratio. (E) Proteins level of AMPK pathway, ATP synthase and quantification, relative folds were determined by comparative with the OA (Ctrl) group. (F) Cell viability. (G) Cell survival. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 S10 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S7. Compound C (CC) abolishes the hepatoprotective effects of R17 in HuH7 cells. Cells were treated with AICAR (0.2 mM), R17 (1 μM) alone or together with compound C (40 μM) in the presence of OA (0.5 mM) for 24 hr. (A) Expression of UPR pathway, inflammation and apoptosis association protein and quantification, relative folds were determined by comparative with the OA (Ctrl) group. (B) TNF‐α secretion level. (C) TG level; (D) Cell apoptosis. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). #p < 0.05, vs. OA (Ctrl) group; @p < 0.05, vs. R17 or AICAR treated group.
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China (Grants 21672265 to Z.S.H. and 81703336 to Y.R.); Special Fund for Science and Technology Development in Guangdong Province (Grant 2016A020217004 to Z.S.H.); the Science and Technology Program of Guangzhou (Grant 201704020104 to Z.S.H.); the Natural Science Foundation of Guangdong Province (Grant 2017A030308003 to Z.S.H.); the 111 Project (Grant B16047); Guangdong Provincial Key Laboratory of Construction Foundation (Grant 2017B030314030); the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (Grant 2017BT01Y093); and Bureau for Foreign Experts (Grant SYSUPCS2014 to J.M.Y.).
Rao Y, Lu Y‐T, Li C, et al. Bouchardatine analogue alleviates non‐alcoholic hepatic fatty liver disease/non‐alcoholic steatohepatitis in high‐fat fed mice by inhibiting ATP synthase activity. Br J Pharmacol. 2019;176:2877–2893. 10.1111/bph.14713
Contributor Information
Yong Rao, Email: raoyong0805@126.com.
Ji‐Ming Ye, Email: jiming.ye@rmit.edu.au.
Zhi‐Shu Huang, Email: ceshzs@mail.sysu.edu.cn.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(Suppl 1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. 10.1111/bph.13883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf, N. U. , & Sheikh, T. A. (2015). Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radical Research, 49(12), 1405–1418. 10.3109/10715762.2015.1078461 [DOI] [PubMed] [Google Scholar]
- Chalasani, N. , Younossi, Z. , Lavine, J. E. , Charlton, M. , Cusi, K. , Rinella, M. , … Sanyal, A. J. (2018). The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology, 67, 328–357. 10.1002/hep.29367 [DOI] [PubMed] [Google Scholar]
- Chen, Y. C. , Zeng, X. Y. , He, Y. , Liu, H. , Wang, B. , Zhou, H. , … Huang, Z. S. (2015). Rutaecarpine analogues reduce lipid accumulation in adipocytes via inhibiting adipogenesis/lipogenesis with AMPK activation and UPR suppression. ACS Chemical Biology, 10, 1570–1571. 10.1021/acschembio.5b00242 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, E. A. , Ford, R. J. , & Steinberg, G. R. (2017). AMPK as a therapeutic target for treating metabolic diseases. Trends in Endocrinology and Metabolism, 28, 545–560. 10.1016/j.tem.2017.05.004 [DOI] [PubMed] [Google Scholar]
- Diehl, A. M. , & Day, C. (2017). Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. The New England Journal of Medicine, 377, 2063–2072. 10.1056/NEJMra1503519 [DOI] [PubMed] [Google Scholar]
- Drew, L. (2017). Fighting the fatty liver. Nature, 550, S102–S103. 10.1038/550S102a [DOI] [PubMed] [Google Scholar]
- Folch, J. , Lees, M. , & Sloane Stanley, G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. The Journal of Biological Chemistry, 226, 497–509. [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatting, M. , Zhao, G. , Schumacher, F. , Sellge, G. , Al Masaoudi, M. , Gaβler, N. , … Trautwein, C. (2013). Hepatocyte caspase‐8 is an essential modulator of steatohepatitis in rodents. Hepatology, 57, 2189–2201. 10.1002/hep.26271 [DOI] [PubMed] [Google Scholar]
- Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell, 140, 900–917. 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke, H. (2002). Inflammation in response to hepatocellular apoptosis. Hepatology, 35, 964–966. 10.1053/jhep.2002.0350964 [DOI] [PubMed] [Google Scholar]
- Jung, T. W. , Hwang, H. J. , Hong, H. C. , Yoo, H. J. , Baik, S. H. , & Choi, K. M. (2015). BAIBA attenuates insulin resistance and inflammation induced by palmitate or a high fat diet via an AMPK‐PPARδ‐dependent pathway in mice. Diabetologia, 58, 2096–2105. 10.1007/s00125-015-3663-z [DOI] [PubMed] [Google Scholar]
- Jung, T. W. , Kim, H. C. , Abd El‐Aty, A. M. , & Jeong, J. H. (2018). Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK‐SERCA2b pathway. The Journal of Biological Chemistry, 293, 3981–3988. 10.1074/jbc.RA117.000885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. J. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E. , Lee, J. H. , Ntambi, J. M. , & Hyun, C. K. (2011). Inhibition of stearoyl‐CoA desaturase1 activates AMPK and exhibits beneficial lipid metabolic effects in vitro. European Journal of Pharmacology, 672, 38–44. 10.1016/j.ejphar.2011.09.172 [DOI] [PubMed] [Google Scholar]
- Kisseleva, T. , & Brenner, D. A. (2006). Hepatic stellate cells and the reversal of fibrosis. Journal of Gastroenterology and Hepatology, 21(Suppl 3), S84–S87. 10.1111/j.1440-1746.2006.04584.x [DOI] [PubMed] [Google Scholar]
- Kleiner, D. E. , Brunt, E. M. , Van Natta, M. , Behling, C. , Contos, M. J. , Cummings, O. W. , … Nonalcoholic Steatohepatitis Clinical Research Network. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology, 41, 1313–1321. 10.1002/hep.20701 [DOI] [PubMed] [Google Scholar]
- Koyama, N. , Yamazaki, T. , Kanetsuki, Y. , Hirota, J. , Asai, T. , Mitsumoto, Y. , … Okanoue, T. (2018). Activation of apoptosis inhibitor of macrophage is a sensitive diagnostic marker for NASH‐associated hepatocellular carcinoma. Journal of Gastroenterology, 53, 770–779. 10.1007/s00535-017-1398-y [DOI] [PubMed] [Google Scholar]
- Li, J. , Zeng, C. , Zheng, B. , Liu, C. , Tang, M. , & Jiang, Y. (2018). HMGB1‐induced autophagy facilitates hepatic stellate cells activation: A new pathway in liver fibrosis. Clinical Science (London, England), 132, 1645–1667. 10.1042/CS20180177 [DOI] [PubMed] [Google Scholar]
- Liu, T. Y. , Xiong, X. Q. , Ren, X. S. , Zhao, M. X. , Shi, C. X. , Wang, J. J. , … Zhu, G. Q. (2016). FNDC5 alleviates hepatosteatosis by restoring AMPK/mTOR‐mediated autophagy, fatty acid oxidation, and lipogenesis in mice. Diabetes, 65, 3262–3275. 10.2337/db16-0356 [DOI] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172, 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso, G. , Cassader, M. , & Gambino, R. (2016). Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nature Reviews. Drug Discovery, 15, 249–274. 10.1038/nrd.2015.3 [DOI] [PubMed] [Google Scholar]
- Osawa, Y. , Kojika, E. , Hayashi, Y. , Kimura, M. , Nishikawa, K. , Yoshio, S. , … Kimura, K. (2018). Tumor necrosis factor‐α‐mediated hepatocyte apoptosis stimulates fibrosis in the steatotic liver in mice. Hepatol Commun, 2, 407–420. 10.1002/hep4.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S. , & Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death and Differentiation, 11, 381–389. 10.1038/sj.cdd.4401373 [DOI] [PubMed] [Google Scholar]
- Patton, H. , Nyberg, A. , Chiang, K. , Yang, S. J. , Caparosa, S. , Stern, J. , & Nyberg, L. , Stern, J. (2018). Clinical characteristicsandepidemiology of patients with non-alcoholic fatty liver diseaseandnon‐alcoholic steatohepatitis in a large community‐based healthcaredeliverysystem in the US. Journal of Hepatology, 68, S839–S840. 10.1016/S0168-8278(18)31956-1S [DOI] [Google Scholar]
- Rao, Y. , Liu, H. , Gao, L. , Yu, H. , Ou, T.‐M. , Tan, J.‐H. , … Huang, Z.‐S. (2015). Synthesis and biological evaluation of novel bouchardatine derivatives as potential adipogenesis/lipogenesis inhibitors for antiobesity treatment. Journal of Medicinal Chemistry, 58(23), 9395–9413. 10.1021/acs.jmedchem.5b01566 [DOI] [PubMed] [Google Scholar]
- Rao, Y. , Yu, H. , Gao, L. , Lu, Y. T. , Xu, Z. , Liu, H. , … Huang, Z. S. (2017). Natural alkaloid bouchardatine ameliorates metabolic disorders in high‐fat diet‐fed mice by stimulating the sirtuin 1/liver kinase B‐1/AMPK axis. British Journal of Pharmacology, 174, 2457–2470. 10.1111/bph.13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan, D. , Surabattula, R. , & Wang, X. Y. (2018). Determinants of fibrosis progression and regression in NASH. Journal of Hepatology, 68, 238–250. 10.1016/j.jhep.2017.11.012 [DOI] [PubMed] [Google Scholar]
- Shi, C. , Wang, Y. , Gao, J. , Chen, S. , Zhao, X. , Cai, C. , … Qiu, L. (2017). Inhibition of aldose reductase ameliorates alcoholic liver disease by activating AMPK and modulating oxidative stress and inflammatory cytokines. Molecular Medicine Reports, 16, 2767–2772. 10.3892/mmr.2017.6895 [DOI] [PubMed] [Google Scholar]
- Southan, C. , Sharman, J. L. , Benson, H. E. , Faccenda, E. , Pawson, A. J. , Alexander, S. P. , … NC‐IUPHAR . (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: Towards curated quantitative interactions between 1300 proteins targets and 6000 ligands. Nucl Acids Res, 44, D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Gao, J. H. , Ishigaki, Y. , Kondo, K. , Sawada, S. , Izumi, T. , … Katagiri, H. (2017). ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Reports, 18, 2045–2057. 10.1016/j.celrep.2017.01.076 [DOI] [PubMed] [Google Scholar]
- Tsuchida, T. , & Friedman, S. L. (2017). Mechanisms of hepatic stellate cell activation. Nature Reviews. Gastroenterology & Hepatology, 14, 397–411. 10.1038/nrgastro.2017.38 [DOI] [PubMed] [Google Scholar]
- Turner, N. , Li, J. Y. , Gosby, A. , To, S. W. , Cheng, Z. , Miyoshi, H. , … Ye, J. M. (2008). Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP‐activated protein kinase and improve insulin action. Diabetes, 57, 1414–1418. 10.2337/db07-1552 [DOI] [PubMed] [Google Scholar]
- Vatner, D. F. , Majumdar, S. K. , Kumashiro, N. , Petersen, M. C. , Rahimi, Y. , Gattu, A. K. , … Shulman, G. I. (2015). Insulin‐independent regulation of hepatic triglyceride synthesis by fatty acids. Proceedings of the National Academy of Sciences of the United States of America, 112, 1143–1148. 10.1073/pnas.1423952112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar, K. R. , Zhu, J. , & Hirst, J. (2015). Architecture of mammalian respiratory complex I. Nature, 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, Y. , Lu, Q. , Hu, Z. , Yu, Y. , Chen, Q. , & Wang, Q. K. (2017). A non‐canonical pathway regulates ER stress signaling and blocks ER stress‐induced apoptosis and heart failure. Nature Communications, 8, 133–147. 10.1038/s41467-017-00171-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi, Z. M. , Loomba, R. , Anstee, Q. M. , Rinella, M. E. , Bugianesi, E. , Marchesini, G. , … Lindor, K. (2018). Diagnostic modalities for non‐alcoholic fatty liver disease (NAFLD), non‐alcoholic steatohepatitis (NASH) and associated fibrosis. Hepatology, 68, 349–360. 10.1002/hep.29721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, X. Y. , Wang, H. , Bai, F. , Zhou, X. , Li, S. P. , Ren, L. P. , … Ye, J. M. (2015). Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. British Journal of Pharmacology, 172, 4303–4318. 10.1111/bph.13209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Feng, D. , Wang, Q. , Abdulla, A. , Xie, X. J. , Zhou, J. , … Yang, F. (2012). Regulation of lipogenesis by cyclin‐dependent kinase 8‐mediated control of SREBP‐1. The Journal of Clinical Investigation, 122, 2417–2427. 10.1172/JCI61462 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The formulation of the high fat diet and high fructose diet
Table S2. Primers sequence information
Figure S1. Effects of R17 on TG level in liver of fast/refeding mice. Male adult C57BL/6 J mice were fasted overnight and fed with a high fructose diet with or without the treatment of R17 at the dose of 20 mg kg−1 by i.p. each other day for 10 days. Livers were isolated and weighted, the TG, DAG level and protein related to lipogenesis were determined. (A) Liver weight. (B) Liver TG level. (C) Liver DAG level. (D) Expression of proteins related to lipogenesis and quantification. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 10 mice/group). *p < 0.05, vs. Ctrl group mice.
Figure S2. Evaluation of toxicity of R17 in mice after 5 weeks treatment. Male adult C57BL/6 J mice were fed with chow (CH) or high fat (HF) diet for consecutively 16 weeks, and R17 was administrated at last 5 weeks at the dose of 20 mg/kg by i.p. each other day. After 5 weeks treatment, plasma and interested tissues were isolated and related parameters of plasma and tissues were determined. (A) Hematoxylin & eosin (× 200 magnification) staining of heart, kidney, muscle and pancreas. (B) Ratio of tissue/Body weight of heart, kidney, muscle and pancreas. (C) Determination the level of chemicals in plasma indicative of organ toxicity in mice. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 8 mice/group). *p < 0.05, vs. CH control mice; #p < 0.05, vs. HF control mice.
Figure S3. Change of TG levels in HuH7 hepatocytes after FAs induction. HuH7 cells were incubated with fatty‐free BSA or OA (0.5 mM) and PA (0.5 mM) for 24 hr, TG level and cell viability were determined. (A) Images were captured under a contrasted eyesight or after Oil‐Red O staining, × 200 magnification. (B) Cellular TG level. (C) Cell viability. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. BSA treated group.
Figure S4. Effect of R17 on the inflammation in LPS stimulated cells. (A) CD68 mRNA level in liver of mice. Data are statistically analysed as means ± SEM, each circle indicated one mice (N = 8 mice/group). *p < 0.05, vs. CH control mice; #p < 0.05, vs. HF control mice. HepG‐2 cells were exposed to LPS (50 μg/mL) with or without the treatment of R17 (0.4, 1 μM) for 24 hr, the inflammatory cytokines in culture medium and protein level of NF‐κB pathway were determined by ELISA and immunoblotting assay, respectively. (B‐C) Secretion levels of IL‐6 and TNF‐α; (D) Expression of IL‐6, TNF‐α and NF‐κB pathway proteins, and quantification, relative folds were determined by comparative with the Blank group. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S5. Effects of R17 on ER stress in high glucose or LPS stimulated cells. (A) HuH7 cells were starved (cultured in a culture medium containing 5 mM) for 8 hr and then switched to a high glucose (30 mM) culture medium in the presence or absence of R17 (1 μM) treatment for 24 hr. UPR pathway related proteins were determination by immunoblotting and quantification, relative folds were determined by comparative with the Blank group. (B) HepG2 cells were exposed to LPS (50 μg/mL) stimulation with or without the treatment of R17 (0.4, 1 μM) treatment for 24 hr. UPR pathway related proteins were determination by immunoblotting and quantification, relative folds were determined by comparative with the Blank group. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S6. R17 inhibits lipotoxicity in hepatocytes via LKB1‐AMPK axis activation. (A) Effect of R17 on the LKB1AMPK axis in the LPS stimulated HepG‐2 cells. HepG‐2 cells were exposed to LPS (50 μg/mL) stimulation for 24 hr with or without R17 (1 μM) treatment, cells were collected and subjected to western blot assay and quantification, relative folds were determined by comparative with the Blank group. (B) Effect of R17 on the LKB1‐AMPK axis in the high glucose stimulated HuH7 cells as described in Methods. Protein levels were quantified and relative folds were determined by comparative with the Blank group. (C) Effect of R17 (1 μM) on the ATP synthase activity in OA or PA (0.5 mM) stimulated‐HuH7 cells. (D‐G) Effects of ATP synthase inhibitor oligomycin on TG level and AMPK pathway in OA (0.5 mM)‐stimulated HuH7 cells. Cells treated with oligomycin (10 μM) for 12 hr and collected for indicative assay. (D) ATP level and AMP/ATP ratio. (E) Proteins level of AMPK pathway, ATP synthase and quantification, relative folds were determined by comparative with the OA (Ctrl) group. (F) Cell viability. (G) Cell survival. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 S10 independent experiments). *p < 0.05, vs. Blank group; #p < 0.05, vs. Ctrl group.
Figure S7. Compound C (CC) abolishes the hepatoprotective effects of R17 in HuH7 cells. Cells were treated with AICAR (0.2 mM), R17 (1 μM) alone or together with compound C (40 μM) in the presence of OA (0.5 mM) for 24 hr. (A) Expression of UPR pathway, inflammation and apoptosis association protein and quantification, relative folds were determined by comparative with the OA (Ctrl) group. (B) TNF‐α secretion level. (C) TG level; (D) Cell apoptosis. Data are statistically analysed as means ± SEM, each circle indicated one independent experiment (N = 5 independent experiments). #p < 0.05, vs. OA (Ctrl) group; @p < 0.05, vs. R17 or AICAR treated group.