

Abstract
Subunit vaccines are composed of pathogen fragments that, on their own, are generally poorly immunogenic. Therefore, the incorporation of an immunostimulating agent, e.g. adjuvant, into vaccine formulation is required. However, there are only a limited number of licenced adjuvants and their im-munostimulating ability is often limited, while their toxicity can be substantial. To overcome these prob-lems, a variety of vaccine delivery systems have been proposed. Most of them are designed to improve the stability of antigen in vivo and its delivery into immune cells. Cell-penetrating peptides (CPPs) are especially attractive component of antigen delivery systems as they have been widely used to enhance drug transport into the cells. Fusing or co-delivery of antigen with CPPs can enhance antigen uptake, pro-cessing and presentation by antigen presenting cells (APCs), which are the fundamental steps in initiating an immune response. This review describes the different mechanisms of CPP intercellular uptake and various CPP-based vaccine delivery strategies.
Keywords: Cell-penetrating peptide, vaccine delivery, antigen presenting cells, adjuvant, antigen uptake, humoral and cellular immunity
1. INTRODUCTION
Vaccination as a preventive immunization approach has been used over two centuries to reduce the burden of human and non-human animal infectious diseases. Indeed, the worldwide mortality and morbidity caused by various infectious diseases declined drastically with the introduction of vaccination. The eradication of smallpox, which started with the first smallpox immunization by Edward Jenner in 1796 [1, 2], is the most impressive example of vaccine success [3]. Since then, the pace of vaccine development has accelerated dramatically. A range of new vaccines have emerged and their success has resulted in a reduced prevalence of infections caused by polio, mumps, rubella, measles, rotavirus, and varicella [4]. Vaccination has gradually become standard practice for the prevention of infectious diseases worldwide. However, despite the successes of conventional whole-organism-based vaccinations, certain limitations still hamper their universal application. For example, pathogens present in the live-attenuated vaccine may revert back to virulent form and trigger disease [5]. In contrast, killed microorganisms have no ability to induce disease and their poor immunogenicity usually means that multiple booster doses are required to achieve a sufficient immune response [6].
Whole-pathogen-based vaccines also often carry reactogenic components (e.g. whole-cell pertussis vaccine) [7] that are associated with undesirable adverse effects with mild to fatal consequences. Furthermore, traditional vaccine designs are not applicable for several pathogens (e.g. hepatitis B, hepatitis C and human papillomavirus) due to the difficulty in, or even inability, to culture them [8]. There is still no vaccine against lethal infectious diseases, such as malaria and rheumatic heart disease caused by group A streptococcus (GAS, Streptococcus pyogenes) [3, 9]. To address and overcome these hurdles, new types of vaccine have emerged over the past several decades.
The current focus of vaccine development has moved from conventional vaccines to subunit vaccines [10]. The greatest advantage of subunit vaccines is that they exhibit an improved safety profile in comparison to conventional vaccines, which is the primary concern in modern vaccine development. Subunit vaccines are designed to contain only particular antigens, which are composed of antigenic peptides, polysaccharides or proteins, allowing for the removal of redundant components and, consequently, reducing the risk of allergic or autoimmune responses [11-13]. Moreover, subunit vaccines can be customized; for example, by being able to carry antigens against different life stages of a pathogen. They can also be produced economically and over a large scale. However, antigenic peptides and proteins are not highly immunogenic, as the removal of redundant pathogen compounds from a vaccine also removes most of the “danger signals”, which naturally trigger activation of the immune system. To overcome this problem, external “danger signals” called “immune stimulators” or adjuvant have been utilized in subunit vaccine design [14-17]. The addition of adjuvants and/or delivery systems are required to enhance vaccine efficiency through stimulating Antigen Presenting Cells (APCs) (e.g. dendritic cells, DCs) and prolonging the vaccines half-life [12, 18, 19]. However, only a limited number of adjuvants have been licensed for specific vaccines and these are approved for use only in certain countries. Most of these adjuvants are oil-based formulations (e.g. liposomes, MF59 and AS21) [14], which are not always effective, especially when used in combination with a poor immunogen, such as peptides. Thus, to improve vaccine performance, the further modification of delivery systems and/or adjuvants is required. The incorporation of Cell-Penetrating Peptides (CPPs) in vaccine delivery systems may improve antigen uptake by APCs and, therefore, can be considered as a safe alternative or additive to classical adjuvant formulations.
CPPs have the ability to overcome the permeability barrier of cell membranes and are able to enter the cell interior in a non-invasive manner without the assistance of membrane proteins [20]. CPPs have been thoroughly explored for the delivery of various cargos, such as peptides, nucleic acids, proteins, nanoparticles and liposomes, into cells [21-23]. CPPs are generally short peptides composed of 5-30 amino acids. They are classified into three major categories: cationic, amphipathic and hydrophobic (Table 1) [24]. In 1988, The first CPP (Tat) was found within the Human Immunodeficiency Virus (HIV) transactivating regulatory protein [25, 26]. A few years later, penetratin (Antp) was discovered in the Drosophila antennapedia homeodomain protein. These two peptides exhibit cationic characteristics and are the most intensively studied of all CPPs. Following their discovery, a large number of protein-derived CPPs have been identified. Subsequently, based on the structure of these natural-derived CPPs, a group of artificial CPPs was designed, including polyarginine [27], polylysine [28] and MPG [29]. Since CPPs are usually nontoxic and can be easily and economically produced, they have been widely applied in drug delivery systems. More recently, CPPs have been suggested as a promising agent for vaccine delivery. CPPs were often fused with antigens to achieve efficient cell membrane translocation, enhancing antigen uptake, processing and presentation by APCs. CPPs have also been incorporated into several DNA vaccine candidates to facilitate the transport of genetic material through nuclear and plasma membranes. Here, we review the mechanisms of CPP action, their interactions with immune cells and their application for vaccine delivery.
Table 1. Examples of different CPP classes.
| Name | Origin | Sequence | Refs. |
|---|---|---|---|
| Cationic | |||
| Polyarginine (R8,R9) | Synthetic | RRRRRRRR, RRRRRRRRR | [30] |
| pAntp, Penetratin | Drosophila antennapedia homeodomain protein | RQIKIWFQNRRMKWKK | [31] |
| Tat | HIV-1 transcriptional activator protein | RKKRRQRRR | [32] |
| PDX-1 | Pancreatic and duodenal homeobox protein | RHIKIWFQNRRMKWKK | [33] |
| KAFAK | Synthetic | KAFAKLAARLYRKALARQLGVAA | [34] |
| Amphipathic | |||
| VP22 | Herpes simplex virus (HSV)-1 protein | DAATATRGRSAASRPTERPRAPARSASRPRRVD | [35] |
| MPG | Conjugate of HIV glycoprotein 41 and nuclear localization sequence (NLS) from simian virus 40 (SV40) | GALFLGWLGAAGSTMGAPKKKRKV | [36] |
| (Model amphipathic peptide) MAP | Synthetic | KLALKLALKALKAALKLA | [29] |
| Pep-1 | Conjugate of SV40 NLS and reverse transcriptase of HIV-1 | KETWWETWWTEWSQPKKKRKV | [37] |
| CADY | PPTG1 peptide | GLWRALWRLLRSLWRLLWRA | [38] |
| pVEC | Murine vascular endothelial cadherin protein | LLIILRRRIRKQAHAHSK | [39] |
| Transportan | Galanin-Lys-mastoparan protein | GWTLNS/AGYLLGKINLKALAALAKKIL | [40] |
| Hydrophobic | |||
| C105Y | HIV glycoprotein 41 | PFVYLI | [41] |
| SG3 | Synthetic | RLSGMNEVLSFRWL | [42] |
2. MECHANISMS OF CELL-PENETRATING PEPTIDE INTERNALIZATION
Although the exact mechanisms of CPP internalization are not fully understood, the uptake pathways of CPPs have been generally classified as non-endocytic (resulting in the delivery of cargo to cytoplasm) and endocytic (passing of cargo to lysosomes). These pathways differ significantly in the internalization processes of CPPs and CPP-cargo. In addition, some CPPs can enter the cytoplasm through endocytic mechanisms [43]. For instance, Tat peptide can be internalized through three endocytic pathways: macropinocytosis, clathrin- and caveolae/lipid-raft-mediated endocytosis [44]. Moreover, different cargo attachments to CPPs influence their uptake pathways. For example, while Tat conjugated with protein used lipid raft-mediated endocytosis, it used clathrin-dependent endocytosis when embedded with fluorophore [45, 46].
2.1. Non-endocytic Pathway
The non-endocytic uptake pathway, also known as the direct-penetration internalization pathway, occurs through pore formation, inverted micelle formation or the carpet-like mechanism; all are energy-independent [47].
In the non-endocytic pathway, CPPs can be taken up by pore formation mechanisms that follow barrel-stave [48] or toroidal models [49] (Fig. 1A and B). In the barrel-stave model, the hydrophobic peptide regions of CPPs align with the lipid core after CPP insertion into the membrane bilayer, while the hydrophilic regions of CPPs form the interior part of the pore. Then, CPP is delivered into the cytoplasm; however, this can damage the cell membrane. In the toroidal model, CPPs attach to the cell surface, then enter the cell membrane. The insertion of CPPs induces continuous bending of the lipid monolayers towards to the pore. Therefore, the inserted peptides and the lipid headgroups both develop transient water pores [50]. CPPs can then enter the cytoplasm through this water pore. The inverted micelle model was first proposed for the penetration of penetratin by Derossi et al. in 1988. In this model [51], charged protein-derived CPP residues interact with phospholipids on the membrane surface. The hydrophobic part of the peptide then interacts with the membrane, which leads to the formation of the inverted micelle (Fig. 1C) and CPPs or CPP-cargo are transferred inside the cell [52, 53]. In the carpet-like model, CPPs cover the surface of the membrane in a carpet-like manner through linkages between charged domains of CPPs and the cell membrane. Subsequently, the hydrophobic part of peptide is flipped by the hydrophobic core of the membrane, contributing to the disruption of the membrane (Fig. 1D), which finally allows the translocation of cargo [54].
Fig. (1).
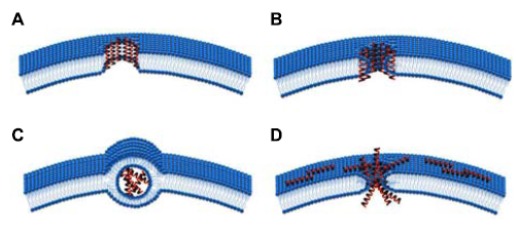
Non-endocytic CPP uptake mechanisms: A) barrel-stave model; B) toroidal model; C) inverted micelle; D) carpet model.
2.2. Endocytic Pathways
In addition to uptake by non-endocytic internalization, CPPs can also be translocated into the cytoplasm through energy-dependent endocytosis, which can be classified into four dominant pathways: micropinocytosis, clathrin-, or caveloae-mediated endocytosis and phagocytosis (Fig. 2) [55, 56].
Fig. (2).
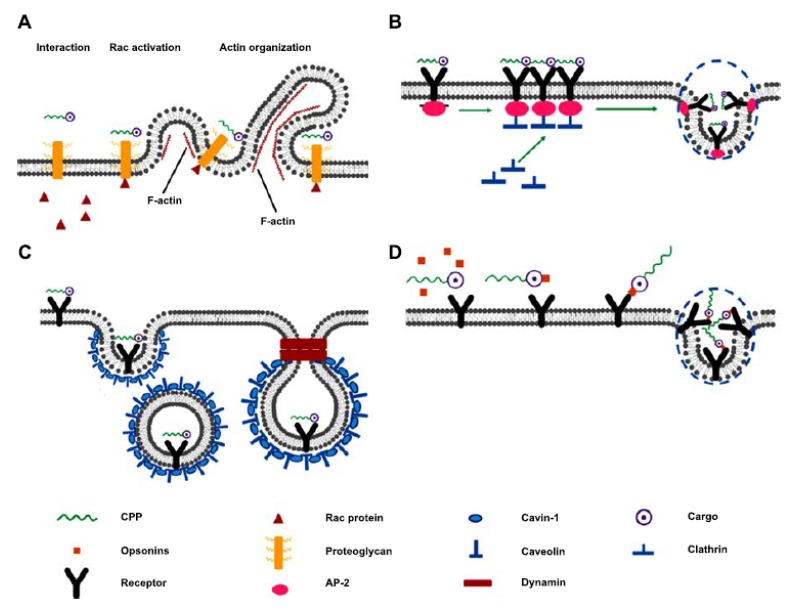
Endocytic CPP-cargo uptake pathways: A) macropinocytosis; B) clathrin-mediated endocytosis; C) caveloae-mediated endocytosis; D) phagocytosis.
Macropinocytosis is one of the major endocytic pathways for CPPs with large cargo attached (Fig. 2A). CPPs carrying the cargo firstly interact with membrane-associated proteoglycans, contributing to the activation of rac protein from the cytoplasm. Subsequently, signals from the rac protein trigger F-actin (filamentous actin) organization. After that, the CPP-cargo complex is absorbed into the cell by macropinocytosis [57]. Arginine-rich CPPs, as a highly positively charged molecule [58], typically undergo clathrin-mediated endocytosis for internalization (Fig. 2B); Arginine-rich CPPs typically undergo clathrin-mediated endocytosis for internalization (Fig. 2B); CPP-cargo is first attached to cell membrane receptors, then epsin protein interact with the cell membrane to generate a curvature and also recruits clathrin and heterotetrameric protein (AP-2) to form a pit beneath the cell membrane. After the invagination of the cell membrane, this pit finally develops into clathrin- and AP-2-coated complex vesicle in the cytoplasm. Ultimately, an uncoating process is activated to release the CPP-cargo, which can also proceed to endosome [59].
Amphipathic Pro-rich or Tat -dominated CPPs were often found to be internalised through caveloae-mediated endocytosis (Fig. 2C) [60, 61]. Caveloae-mediated endocytosis, also known as lipid raft endocytosis, depends on invagination (as with clathrin-mediated endocytosis), but is associated with caveolin and cavin-1 interaction instead of clathrin. In the first stage of this pathway, the CPP-cargo complex binds to specific receptors on a lipid raft domain in the cell membrane that has various chimeric proteins and cholesterol. Then, cavin-1 connects each caveolin. With increased density of caveolin and cavin-1 complexes under the cell membrane, a pit is developed. Finally, a vesicle containing the CPP-cargo complex is invaginated following the formation of a helical tube around the neck of the vesicle by the polymerization of the Gtpase, dynamin [61]. The translocation of CPP through the phagocytic uptake pathway occurs primarily in specialized cells, such as DCs and macrophages (Fig. 2D) [62]. CPP cargo carriers are recognized extracellularly and tagged by opsonins (such as immunoglubulin (Ig) G and complement components), which enables CPPs to be recognized by specialized cells. The CPP complex can then be attached to the Fc receptor in the cell membrane, which stimulates actin assembly and generates a membrane coat for CPPs. Finally, the cargo can be translocated into the cytoplasm [63].
Although receptor-mediated uptake of CPP is widely reported, there is no clear evidence of which receptors CPPs can bind to. The receptors identified as being involved in CPP uptake are actually related to the cargo of the CPP, not the CPP itself. For example, CPPs recruit type A scavenger receptors to the plasma membrane for cellular delivery of nucleic acids [46].
3. CPP AND THE IMMUNE SYSTEM
3.1. The Human Immune System
The immune system identifies threats to its host and eliminates pathogens without damaging host cells [64]. Antigens are molecular substances that are characterized by two major functions: immunogenicity (the ability to induce immune response) and antigenicity (the ability to be recognized by antigen-specific receptors on the cells of the immune system). Human immune responses have been broadly classified into two categories: innate immunity and adaptive/acquired immunity (Fig. 3). As the name suggests, innate immunity consists of the immunological responses that a body is programmed to produce from birth. It is the first line of defence (after the physiological barriers) and it recognizes general molecular patterns that are foreign to the body. Acquired immunity caters to the need to protect the body after encountering an antigen for the first time. It is a more specific immune response that recognizes particular antigens from invading pathogen.
Fig. (3).
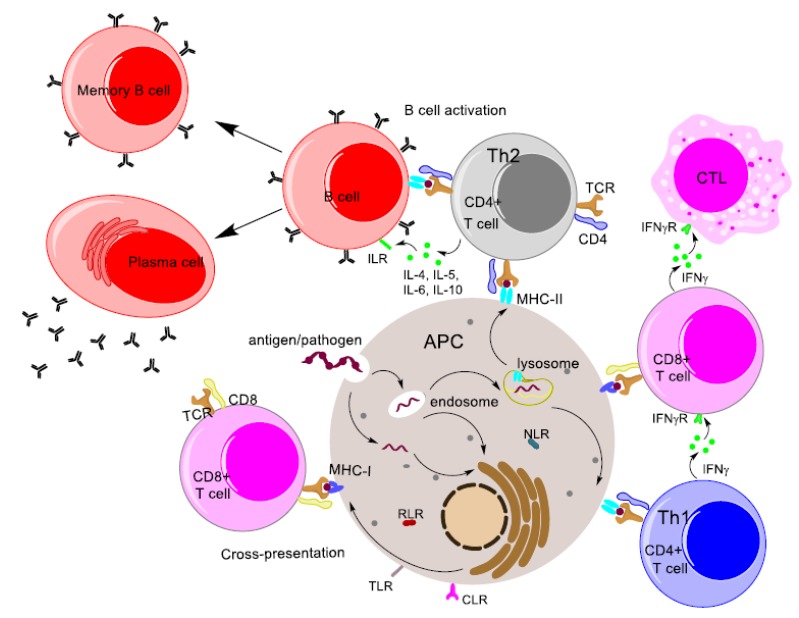
Diagrammatic representation of the major immune response pathways.
Innate immune responses rely on a limited variety of receptors to detect invading pathogens. These receptors, known as Pattern Recognition Receptors (PRR), are expressed by most of the innate immune effector cells and bind to conserved regions, known as pathogen-associated molecular patterns (PAMPs), of a plethora of pathogens [65, 66]. Cells involved in innate immune responses include macrophages, DCs, and granulocytes. These effector cells all act as APCs and bear the major histocompatibility complex (MHC). The function of the MHC receptor is to display peptide fragments derived from digested pathogens/antigens, called epitope [67]. While all nucleated cells express MHC Class I receptors, only APCs express MHC Class II receptors.
Long-term adaptive immunity is the ability of individuals to quickly trigger an immune response against a pathogen that was previously encountered by the immune system [68]. T lymphocytes (T cells) provide cellular immunity and B lymphocytes (B cells) provide humoral immunity. Processed antigen presented on the surface of phagocytic cells is detected by T Cell Receptors (TCRs). Important T cell subsets include CD4+ cells and CD8+ cells. CD4+ and CD8+ cells bind to MHC Class II and MHC Class I, respectively, thereby facilitating the presentation of antigen by MHC to TCRs. Important T cell subsets include CD4+ cells and CD8+ cells. CD4+ and CD8+ cells bind to MHC Class II and MHC Class I, respectively, thereby facilitating the presentation of antigen by MHC to TCRs.
CD4+ cells with CD4 surface molecules are also called T helper (Th) cells as they help to trigger humoral immunity [69]. Th cells are further classified according to the cytokines they produce. Th1 cells secrete interleukin 2 (IL-2), interferon gamma (IFNγ) and tumour necrosis factor beta (TNFβ). Th2 cells release most of the interleukins, such as IL-4, IL-5, IL-6 and IL-10, needed to trigger the activation of B cells, which results in antibody secretion by plasma cells. IFNγ is crucial in activating the Cytotoxic T Lymphocytes (CTL) that induce cell-mediated immunity. Hence, while Th1 cells are associated mainly with cell-mediated immunity, Th2 cells are crucial for humoral immunity [70].
Humoral immunity involves the production of antibodies by plasma cells. Plasma cells are B cells that have been triggered by Th cells for antibody secretion [71]. Antibodies are immunoglobulin molecules that consist of two heavy chains and two light chains. The variable region of the antibody forms the antigen-binding region (Fab). Variability arises from gene recombination, which helps the antibody to bind to a variety of molecular structures [72]. Antibodies exist in two different forms: secreted antibodies are present in the serum, while membrane-bound antibodies are found on the B cell surface as B Cell Receptors (BCRs). Activation of B cells occurs when they encounter an antigen; however, further, activation results from cross-linking by Th cells. Secreted antibodies play an important role in opsonization, the process where antibodies attach to the antigen and, with the help of certain complement proteins, induce the destruction of the antigen through phagocytosis [73].
CD8 surface molecules are found on cytotoxic T cells (CD8+ T cells). They are heterodimers that bind to MHC Class I molecules present on all nucleated cells. CD8+ cells play three important functions in the clearance of antigens: they secrete cytokines for anti-tumor and anti-microbial activity, generate and release cytotoxic granules, and destroy infected cells with the help of Fas receptors on target cells [74].
The fundamental features of adaptive immunity are: immunological memory, immunologic-specificity and self-tolerance [75].
Immunological memory occurs through the generation of memory T cells and memory B cells by multiple pathways. After the elimination of invading pathogens, most of the lymphocytes die; however, a small portion of them get converted to memory cells. Signalling by certain cytokines, such as interleukin 7 (IL-7), is crucial for the survival of memory cells [76]. Important features of memory cells include high sensitivity in case a secondary infection occurs, rapid proliferation in the presence of an antigen, quick differentiation to plasma cells and a long lifespan [77].
Immunologic-specificity arises from pathogen-specific receptors that develop due to somatic gene recombination that occurs during the generation of lymphocytes. The recombination (known as V(D)J recombination) creates variations in B cell and T cell receptors. V(D)J recombination implies that variable (V), diversity (D) and joining (J) gene segments on the antigen-recognition sites of B cell receptors and T cell receptors are involved.
Self-tolerance, or the ability of the immune system to recognise the body’s own cells and not destroy them along with the pathogens, is an important feature of the mammalian immune system. Lymphocytes are deleted when they become specific for self-antigens present in generative lymphoid organs. Certain self-antigens are also “ignored” by immune cells due to the absence of secondary signals for an immune reaction [78]. Failure of proper recognition of self-antigens can lead to multiple autoimmune diseases.
3.2. The Role of CPP in Triggering Immune Responses
A major consideration in the development of subunit vaccines is the components’ ability to stimulate the required level of immune response. To trigger an adaptive immune response, peptide epitope formed from processed antigen needs to be recognized by MHC I and II receptors inside the host cells, principally APCs. Therefore, an antigen must first be recognized and taken up by APCs; hence, the attractiveness of CPP-based vaccine delivery systems that can efficiently deliver antigens into the APCs [79]. Moreover, these delivery systems can deliver antigen directly into the cytoplasm, mimicking viral infections or cancerous cells.
As mentioned, CPPs can transport an antigen into the cytoplasm using direct mechanisms or the phagocytic uptake pathway characteristic to APCs [62]. The delivered antigen is digested by proteasomes in the cytoplasm, recognized and presented by MHC Class I on the cell surface, thereby activating cytotoxic T lymphocytes [80]. Moreover, CPPs can also stimulate the uptake of antigens via endosomes and their presentation by MHC Class II molecules. Therefore, CPPs as a vaccine component should be able to improve both cellular and humoral immune responses.
4. APPLICATIONS OF CPPS IN VACCINE DELIVERY
CPPs have been used extensively over the past decade to improve vaccine formulations. The intracellular delivery of antigens into APCs is a fundamental step in vaccines’ ability to induce immune responses. Although the mechanism(s) of cellular uptake of CPP remains unclear, CPP has been employed to deliver DNA, protein and peptide-based antigens to enhance both cellular immune responses via the delivery of antigen directly to the cytoplasm, and humoral immune responses where antigens are delivered through the endocytic pathway. Cationic CPPs are a class of CPPs that contain a high positive charge and, as the most prominent group of CPPs, they are frequently utilized as carriers to deliver a range of therapeutic targets, including both small molecules and large constructs (e.g. nanoparticles and liposomes) [81, 82].
4.1. Tat
Tat, also known as HIV Tat protein transduction domain (PTD), is a highly positively charged peptide and is the most widely used CPP in drug and gene delivery (Table 1). Not surprisingly, in the vaccine delivery field, Tat is also the most common CPP used for protein and gene delivery. For example, Bahadoran et al. reported an influenza virus (H5N1) vaccine candidate that was constructed based on a DNA plasmid (pBud) and encoded the antigenic H5 gene carried by Tat modified polyamidoamine (PAMAM) dendrimer [83]. This vaccine candidate was prepared as a mixture of Tat-PAMAM/pBud-H5 at N/P ratio 6. A significantly higher expression of the H5 gene in blood was observed in mice transdermally vaccinated with Tat-PAMAM/pBud-H5 compared to the native PAMAM/pBud-H5 immunized mice. Thus, as expected, Tat-enhanced the distribution of the H5 gene in the tissues of immunized mice. Subsequently, interferon-regulatory factor (IRF3) gene as a genetic adjuvant was added to pBud-H5 and encoded by the plasmid. Mice immunized with pBud-H5 carried by Tat-PAMAM dendrimer showed a significant increase in the antigen-specific CD4+ and CD8+ T lymphocyte populations in comparison to the group immunized with pBud-H5/native PAMAM dendrimer, whereas the highest population of antigen-specific CD4+ and CD8+ T cells was observed in mice vaccinated with pBud-H5-IRF3/Tat-PAMAM dendrimer. Moreover, mice treated with Tat-PAMAM/pBud-H5-IRF3 produced a significantly higher level of IFN-γ compared with PAMAM/pBud-H5- and Tat-PAMAM/pBud-H5-immunized mice.
The mucosal surface of the vagina as a primary entry portal of HIV has been considered as an attractive site for HIV vaccination. However, HIV vaccines designed based on recombinant adenovirus (rAd) vectors are not effectively translocated through mucosal layers. Adenoviruses are non-enveloped viruses containing linear double-stranded DNA. They have been widely applied as gene delivery vectors due to the presence of adenovirus receptors on most human cells. Ji et al. designed self-assembled nanocomplexes (Ad-Tat-APS) formulated via electrostatic interaction between rAd, Tat and a polyethylene glycol derivative (APS) [84]. rAd-Tat-APS was able to efficiently cross the mucus layer and enter the vaginal epithelium in in vitro experimentation. In the in vivo validation study, the antigenic HIVgag p24 gene was added to the rAd vector. Mice intravaginally immunized with rAd/HIVgag-Tat-APS nanocomplexes produced enhanced HIVgag-specific CD8+ and CD4+ T cell responses and higher levels of HIVgag-specific IgA and IgG in the vaginal cavity and serum compared to mice immunized with rAd/HIVgag, rAd/HIVgag-Tat.
The ability of Tat to improve DNA delivery to cells has been used not only for DNA-based vaccines but also for DNA adjuvants. As the Tat peptide can be taken up directly into the cytoplasm, it has found application in the delivery of antigens where a cellular immune response is desired. Several Tat fusion protein-based vaccines were incorporated with DNA adjuvant. For instance, Tang et al. created a peptide-based nanoparticle vaccine against cervical cancer [85]. Human papillomavirus (HPV) E7 oncoprotein was chosen as a cellular target, as E7 is responsible for HPV-induced carcinogenesis, cellular immortalization and transformation [86]. The granulocyte-macrophage colony stimulating factor (GM-CSF) gene was encoded by a negative plasmid (pGM-CSF), which self-assembled with a cationic fusion peptide bearing Tat and E7 [49-57] peptide (Tat-E7), a cytotoxic T lymphocyte (CTL) epitope. GM-CSF is a widely used adjuvant in vaccine design, with a potent ability to activate the proliferation and maturation of DCs [87]. Transmission electron microscopy (TEM) showed that the complex (peptide/plasmid) prepared at N/P ratio 2 formed stable, relatively homogeneous and dense nanoparticles with approximate diameters of 20–80 nm. Condensing plasmid GM-CSF into small particles can protect it against degradation in vivo and enhance its uptake by host cells. Mice immunized with nanoparticles Tat-E7/pGM-CSF had higher numbers of E7 specific functional T cells than those immunized with polylysine-E7/pGM-CSF, Tat-E7/empty plasmid, Tat-E7 in IFA, E7 in IFA, and PBS. To determine if the enhanced E7-specific response induced by Tat-E7/pGM-CSF could prevent tumor growth, mice were immunized two times with the nanoparticles and then challenged with TC-1 tumor cells. More than 60 days after the tumor challenge, most of the mice immunized with the nanoparticles (Tat-E7/pGM-CSF) were tumor-free (90%), whereas only 60% of mice immunized with similarly positively charged nanoparticles (polylysine-E7/pGM-CSF) were tumor free. Mice treated with other formulations (Tat-E7/control plasmid, Tat-E7, E7, and PBS) grew tumors rapidly. In addition, TC-1 tumor-bearing mice vaccinated with Tat-E7/pGM-CSF on days 3 and 10 after tumor inoculation exhibited a significant reduction in tumor growth compared with the tumor-bearing mice from other groups (polylysine-E7/pGM-CSF, Tat-E7/plasmid empty, Tat-E7 in IFA, E7 in IFA, PBS). In long-term tumor protection experiments, mice were challenged with TC-1 tumor cells on day 60 after the last vaccination. Four out of the five mice immunized with Tat-E7/pGM-CSF were tumor free, while only two of five mice from the polylysine-E7/pGM-CSF group and one of five mice from the Tat-E7/control plasmid group remained tumor free. In contrast, all mice from the Tat-E7 in IFA, E7 in IFA, and PBS groups progressively grew tumors.
Yang et al. produced an anticancer vaccine based on a virus-sized particle, in which Tat was fused with survivin [85-93] CTL-epitope and self-assembled with a DNA adjuvant [88]. Survivin is involved with the regulation of cell apoptosis, division, migration and metastasis in tumors and is considered to be a potentially relevant target in hampering the progression of tumors. Tat-survivin [85-93] fusion peptide was mixed with the plasmid encoding murine IL-15 gene, which acted as an adjuvant, at the peptide/DNA charge ratio of 2/1 to form Tat-survivin/pIL-15. Heterogeneous and dense particles with diameters of 50-400 nm were observed with TEM. In in vitro trials, Tat-survivin/pIL-15 not only effectively mediated the transfer of material into the cell and the expression of the IL-15 gene, but it also improved survivin [85-93] APC uptake and processing. Mice immunized with Tat-survivin/plasmid without IL-15 induced significantly higher survivin specific IFN-γ levels than the PBS group even five months after the last immunization, but at lower levels than mice vaccinated with Tat-survivin/pIL-15. A rather poor antitumor response from Tat-survivin/pIL-15 was observed in a CT-26 tumor challenge, in which mice were inoculated with tumor cells seven days before immunization. Tumor volume expanded steadily in mice immunized with Tat-survivin/pIL-15, whereas a rapid increase of tumor volume was observed in mice given PBS and the plasmid control group 24 days after tumor injection. The first death occurred on day 40 in tumor-challenged mice treated with Tat-survivin/IL-15, compared to day 35 in the Tat-survivin/ plasmid without IL-15 group and day 30 in the PBS group.
Tat was also often used to deliver protein in DNA-free vaccines. Yu and Yonghua designed a fish vaccine that was constructed by the fusion protein of Tat and Sia10 [89]. Sia10 is a putative secretory antigen identified from the pathogenic Streptococcus iniae (S. iniae) strain, which is capable of infecting a wide range of fish species and causing outbreaks in fish farms [90]. Fish (Japanese flounder) were immunized with recombinant Tat-Sia10 (Tat was fused to the N-terminal of Sia10), Sia10 and PBS, separately. Significantly enhanced macrophage activation and peripheral blood leukocyte proliferation were detected in rTAT-Sia10 vaccinated fish in comparison with fish treated with rSia10 and PBS. Subsequently, fish vaccinated with rTAT-Sia10 were challenged with a lethal dose of S. iniae at different challenge times: one or two months post-vaccination. Significantly lower accumulated mortalities were observed in rTAT-Sia10 vaccinated fish (2% in 1 month challenge and 6% in 2 months challenge) compared to rSia10 (one month: 36%; two month: 34%) and PBS (one month: 84%; two month: 72%) vaccinated fish. Moreover, rTAT-Sia10 vaccinated fish produced significantly higher Sia10-specific serum antibodies than rSia10 vaccinated fish. Transcriptional analysis also showed an up-regulated expression of the genes encoding IL-1β, IL-8, NK, IgD, IgM, TNFα, MHC Iα, MHC IIα, and CD8α in fish administrated with rTAT-Sia10.
The vaccine candidate against the hepatitis B virus has been designed with Tat. Chen et al. concluded that the replication of the hepatitis B virus could be significantly inhibited by fusing Tat with hepatitis B virus core antigen (HBcAg) [91]. HBcAg, Tat and maltose binding protein (MBP) were cloned into a plasmid and then transcribed in Escherichia coli (E. coli) to produce MBP-HBcAg-Tat fusion protein. MBP as a protein expression tag can improve the solubility of fusion protein and protect against proteolysis. In an in vivo study, anti-HBc IgM antibody production was strongly enhanced in mice immunized with MBP-Tat-HBcAg compared to MBP-HBcAg, while no specific anti-HBc antibodies were detected in Tat and PBS immunized mice. Furthermore, mice vaccinated with 40 µg MBP-Tat-HBcAg not only produced high levels of cytokine (IFN-γ, IL-2, IL-4, IL-10) in splenocytes, they also exhibited significantly higher percentages of HBcAg-specific IFN-γ+CD8+ T cells compared to mice treated with MBP-Tat, MBP-HBcAg and PBS. An immunization trial was then performed on HBV transgenic mice, which, as a model of chronic HBV infection, have immune tolerance to HBV encoded antigens. HBV transgenic mice immunized with 50 µg MBP-Tat-HBcAg produced a higher level of CD8+ IFN-γ T cells and significantly reduced titers of serum HBsAg and HBV DNA levels in comparison to MBP-HBcAg and PBS immunized mice. This effect was even more significant when the higher dose of MBP-Tat-HBcAg (100 µg) was applied.
White spot syndrome virus (WSSV) is a highly pathogenic virus that can infect crayfish and crabs. VP28 is one of WSSV’s major envelope proteins that play a crucial role in WSSV infection. A recombinant protein based-oral subunit vaccine [92] was constructed by the expression of recombinant plasmids encoding antigen VP28 gene and Tat sequence in E.coli host. Hemolymph supernatants of orally immunized crayfish (Cambarus clarkia) were collected to detect immune-related enzyme activities. Crayfish fed with Tat-VP28 showed higher phenoloxidase and superoxide dismutase activity than VP28 treated crayfish. The vaccination’s effectiveness was demonstrated when crayfish were challenged with 100 µL of a WSSV solution after the last day of feeding. As a consequence, the highest relative percent survival (RPS) was observed in the Tat-VP28 immunized group (63% at 7 days, and 68% at 14 days) compared to the RPS of crayfish fed with VP28 (44% at 7 days, and 54% at 14 days). The vaccination’s effectiveness was demonstrated when two sets of crayfish were challenged with 100 µL of a WSSV solution after feeding them with vaccine for 7 days and 14 days, respectively. As a consequence, the highest RPS was observed in the Tat-VP28 immunized group (63% at 7 days feeding, and 68% at 14 days feeding) compared to the RPS of crayfish fed with VP28 (44% at 7 days feeding, and 54% at 14 days feeding). Subsequently, another two sets of crayfish were vaccinated by feeding them for 14 days and then challenged with WSSV on days 3 and 7 after last day of feeding. Tat-VP28 immunized group resulted the highest RPS (59% and 47%) compared to VP28 (41% and 33%).
Kronenberg et al. reported a Tat-fusion protein vaccine against Leishmania major [93]. LACK (Leishmania homolog of receptors for activated C kinase) was demonstrated as an antigen against Leishmania, which is a T cell epitope, and showed a protective effect against Leishmania challenge. DCs were pulsed with Tat-LACK protein (Tat–LACK-pulsed DCs) to primarily induce strong CD4-dependent immunity. In both in vivo and in vitro studies, more efficient proliferation of Leishmania-specific CD8+ T cells was achieved with Tat–LACK-pulsed DCs, when compared with DCs incubated with LACK alone (LACK-pulsed DCs). Moreover, Tat–LACK-pulsed IL-12p40-deficient DCs failed to protect vaccinated mice from L. major infection, which indicated that the release of IL-12p40 from DCs is crucial for Tat–LACK mediated immunity against L. major.
In an attempt to develop an anti-tuberculosis vaccine, Dong et al. constructed plasmid expressing the recombinant fusion protein of antigen Ag85B gene and Tat, which was subsequently transferred into E. coli to produce fusion proteins (Tat-Ag85B) [94]. Ag85B is a major protein secreted by all Mycobacterium species and is responsible for inducing protective responses against Mycobacterium tuberculosis (MTB). Mice immunised with Tat-Ag85B produced higher levels of Ag85B specific IgG, IgG2a and cytokine (IFN-γ and TNFα) than mice immunised with Ag85B and PBS five months after the final vaccination. Moreover, two months after mice were challenged with MTB H37Rv (a virulent MTB strain), MTB loads in the lungs and spleen were significantly reduced in Tat-Ag85B-treated mice compared to those in Ag85B vaccinated mice. Similarly, Huang et al. prepared a recombinant construct, Tat-HDAg (Hepatitis delta antigen) as a fusion protein expressed by E. coli [95]. Immunisation with Tat-HDAg inhibited Hepatitis D virus assembly and secretion in in vitro liver cancer cell lines and in vivo mice model.
4.2. Polyarginine
Polyarginine (Table 1) is an artificial cationic CPP designed based on the Tat sequence and exhibits translocation properties similar to Tat [63]. Polyarginine is a popular drug carrier; however, it was found to also have application in vaccine delivery. For instance, Sakuma et al. indicated that polyarginine-modified polymer (N-vinylacetamide-co-acrylic acid, PNVA-co-AA) is a potential antigen carrier that induces humoral immunity following intranasal administration [96]. Immunization with polyarginine-PNVA-co-AA containing OVA antigen showed a significant enhancement in the production of OVA-specific IgG and IgA in the serum and the nasal cavity, respectively, compared to PBS and OVA. However, OVA adjuvanted with CTB was superior to polyarginine-PNVA-co-AA-OVA in enhancing antibody production. In contrast, mice immunized with polyarginine-PNVA-co-AA containing antigenic influenza virus hemagglutinin (HA) protein induced both mucosal and systemic immune responses against influenza virus, similar to those of HA protein administered with CTB. Upon introduction of linker between polyarginine and PNVA-co-AA further, increase of HA protein-specific IgA titers was observed [97].
Similarly, Nakamura et al. reported the antigen delivery potential of polyarginine-modified liposome (polyarginine-Lip) using OVA as an antigen. polyarginine-Lip-OVA induced significantly higher MHC-I antigen presentation than OVA, conventional pH-sensitive Lip-OVA and cationic Lip-OVA [98]. To evaluate the effectiveness of polyarginine-Lip-OVA in vivo, immunised mice were inoculated with OVA tumors. polyarginine-Lip-OVA produced a significant tumor inhibitory effect in comparison to no treatment, OVA, and polyarginine-Lip treated groups. Subsequently, α-galactosylceramide (αGC) as an adjuvant was added to polyarginine-Lip, which was further modified with polyethylene glycol (PEG) to improve stability and suppress non-specific interactions [99]. αGC is a synthetic glycolipid that can activate natural killer (NK) T cells to produce large amounts of IFN-γ. Liposomes with 2 mol% PEG content and a size of around 250 nm stimulated the most efficient IFN-γ production in vitro. Furthermore, the antitumor effect of optimized αGC/polyarginine/PEG-Lip was evaluated by treating mice with a highly malignant and metastatic type of melanoma (B16 melanoma cells) four days before immunization. Lung metastasis was significantly inhibited in αGC/polyarginine/ PEG-Lip-treated mice, while no significant inhibition was exhibited in mice that received αGC only or no treatment. However, vaccine candidates were not compared to antigen delivered with standard adjuvants, e.g. CTB, in above studies.
Wang et al. have recently developed a novel vaccine carrier peptide Cys-Trp-Trp-(Arg)8-Cys-(Arg)8-Cys-(Arg)8-Cys, which bound to an antigen protein (OVA) via electrostatic self-assembly to form nanocomposites (peptide/OVA) [100]. The nanocomposite was stabilized by the spontaneous oxidization of the cysteine thiol moieties, which formed disulfide bond cross-links. The construct was more efficiently uptaken by DC than OVA, and induced stronger humoral and cellular immune responses than OVA alone, however, weaker in comparison to OVA adjuvanted with Complete Freund’s Adjuvant (CFA). Similarly, Zhang et al. demonstrated that polyarginine containing nanoparticles loaded with recombination urease subunit B (rUreB) are more efficient in induction humoral immune responses against Helicobacter pylori infection than rUreB antigen alone upon oral delivery [101].
4.3. Penetratin
Penetratin peptide, also known as antennapedia transduction sequence (Antp), is natural cationic CPP derived from the homeodomain protein of Antennapedia. One of the earliest uses of penetratin in peptide-based vaccine design was reported by Marie-Paule et al. to enhance tumor antigen percutaneous delivery [102]. Biotinylated penetratin-linked antigenic OVA peptide was applied on one side of a mouse’s ear. Skin biopsies indicated that OVA peptide was distributed uniformly on the skin surface of mice receiving this peptide, and no obvious penetration into deeper layers of the epidermis or the dermis was observed. In comparison, penetratin-OVA peptide stimulated penetration of OVA across the skin surface. Mice epicutaneous immunization with penetratin-OVA elicited a high level of OVA-specific CD8+ T cell response compared to mice treated by an equimolar amount of OVA alone. The addition of adjuvant (cholera toxin) to OVA slightly increased the level of antigen-specific CD8 T cells, but this level was still lower than those observed in mice immunized with penetratin-OVA. Different routes of administration of penetratin-associated peptide were subsequently examined. Epicutaneous immunization with penetratin-OVA efficiently induced OVA-reactive CD8+ T cells producing IFN-γ, whereas OVA-reactive CD8+ T cells were not detected in mice subcutaneously or intraperitoneally immunized with penetratin-OVA. The efficacy of the delivery system was improved with the addition of CpG-ODN, a DNA-based adjuvant. Almost all mice immunized epicutaneously with penetratin-OVA-CpG-ODN remained tumor free for 30 days after subcutaneous injection of OVA tumors to mice on day 16 after the last immunization. In contrast, mice epicutaneously immunized with penetratin-OVA and OVA-CpG ODN and challenged with tumors exhibited only very limited tumor-protective effect. Also, Dodie et al. demonstrated that penetratin in a nasal vaccination system could significantly delay the growth of OVA tumors [103] and Muto et al. reported that penetratin can improve humoral immune responses. Intranasal coadministration of penetratin with influenza antigen enhanced the production of systemic IgG and nasal mucosal IgA in comparison to immunization with influenza antigen only [104].
Brooks et al. chemically synthesized tripartite peptide (AntpMUC1tet), which consists of a penetratin, a single variable number of tandem repeat of the mucin 1 (MUC1) antigen and a tetanus toxoid universal CD4 T helper epitope peptide (tetCD4) [105]. MUC1 is a type I transmembrane glycoprotein, and have been widely used as targets for cancer vaccines [106]. In in vivo challenge study, mice were immunized with PBS, AntpMUC1tet and AntpMUC1tet/CpG and then challenged with B16-MUC1 cells seven days after last immunization. AntpMUC1tet/CpG immunized group showed a significant delay in tumor growth. However, no apparent inhibition of tumor growth was observed in mice immunized with AntpMUC1tet. Besides, AntpMUC1tet/ CpG induced a higher level of IFN-γ and IL-4 production and long term immune responses in mice compared to non-adjuvanted AntpMUC1tet.
4.4. VP22
Amphipathic CPPs contain both polar (hydrophilic) and nonpolar (hydrophobic) regions. They are often chimeric peptides that are formed through the conjugation of a hydrophilic domain with a hydrophobic domain. For instance, the sequence of MPG (Table 1) (GALFLGWLGAAGSTMGAPKKKRKV) was devised based on the highly hydrophilic simian virus 40 nuclear localization sequence (CGYGPKKKRKVGG) and a hydrophobic sequence that originated from the fusion sequence of HIV gp 41 [36]. There are also some natural amphipathic CPPs, such as pVEC (Table 1), which are derived from vascular endothelial cadherin protein [39]. Some amphipathic CPPs have a unique tridimensional structure, e.g. helical conformation, formed by the presence of hydrophilic and hydrophobic residues on different sides of the helix (e.g. model amphipathic peptide, MAP). Currently, VP22 (Table 1), derived from HIV-1, is the most commonly used amphiphilic CPP in vaccine delivery. When fused to a protein of interest, VP22 was able to spread antigenic protein to cells neighboring the transfected cells [107]. This unique property of spreading protein to many surrounding cells makes VP22 ideal for overcoming the poor spreading limitation of DNA vaccines.
The vaccine application of VP22 is mainly focused on DNA vaccine delivery, with some of the earliest investigation carried out by Cheng et al. The capability of VP22 to enhance linked protein spreading was successfully demonstrated by the generation of several DNA constructs (E7/GFP, VP22/GFP, VP22/E7/GFP, and VP221–267/E7/GFP) with a mammalian cell expression vector (pcDNA3) [108]. VP221–267 is a mutant version of VP22 that lacks 34 C-terminal residues and is unable to trigger intercellular spread. A significant spread of green fluorescent protein (GFP, which is used as a marker to track intercellular spreading) in cells transfected with VP22/E7/GFP DNA was exhibited compared to cells transfected with E7/GFP or VP221–267/E7/GFP DNA. The correlation between significant spreading and antigen-specific T cell activities was examined in vivo. Mice vaccinated with pcDNA3-VP22/E7 exhibited a dramatic increase (around 50-fold) in the number of E7-specific IFN-γ+ CD8+ T cell precursors compared to mice that received pcDNA3-wild-type E7 only. Mice vaccinated with pcDNA3-VP22/E7 had increased CTL activity compared to mice vaccinated with pcDNA3 (no insert), pcDNA3-VP22, and pcDNA3-E7 only. In an in vivo tumor protection study, mice were challenged with TC-1 tumor cells seven days after the last vaccination. No tumor cells were detected in mice vaccinated with pcDNA3-VP22/E7 63 days after the tumor challenge. In contrast, all of the mice from unvaccinated, pcDNA3 (no insert), pcDNA3-VP22 and pcDNA3-wild-type E7 groups developed tumors within 14 days. The therapeutic potential of VP22/E7 in treating TC-1 tumor metastases in the lungs has also been investigated. Mice were immunized three days after challenge with TC-1 tumor cells. Mice receiving pcDNA-VP22/E7 showed the lowest number of metastatic pulmonary nodules in comparison to mice that received pcDNA3-E7, pcDNA3-VP22 and the mixture of VP22 and E7 DNA. In an in vivo antibody depletion study, the depletion of lymphocyte subsets (CD4, CD8, and NK 1.1) was started one week before tumor challenge, which was performed after immunization. All mice without depletion and depleted of CD4+ T cells remained tumor-free 63 days after tumor injection. In contrast, parts of mice (40%) depleted of NK 1.1 exhibited tumor growth six weeks after the tumor challenge, whereas all mice vaccinated with pcDNA-VP22/E7 and depleted of CD8+ T cells and mice without immunization showed tumor growth within 14 days after tumor injection, indicating that the induced antitumor effect was CD8 dependent.
Cheng et al. explored Sindbis virus (SIN)-based replicon particle encoding VP22 linked to antigen E7 (SIN-VP22/E7) [109]. Sindbis virus is a promising alphavirus with vectors widely used in the development of gene and vaccine therapy. Mice injected intramuscularly with SIN-VP22/E7 produced more E7-specific CD81 T cells than those immunized via intraperitoneal and subcutaneous administration. When a TC-1 tumor challenge was performed one week after vaccination, all mice immunized with SIN-VP22/E7 remained tumor-free 60 days post tumor challenge. In comparison, all mice treated with SIN-E7, SIN-VP22 and control SIN exhibited tumor growth within 20 days. Mice immunized with SIN-VP22/E7 three days after tumor challenge showed a significantly lower mean number of pulmonary nodules than tumor-bearing mice treated with SIN-E7 and SIN-VP22. Depletion of CD8+ T cells, CD4+ T cells, and NK cells showed that SIN-VP22/E7-induced antitumor effects were more reliant on CD8+ T cells than CD4+ T cells or NK cells. Kim et al. extended this strategy and demonstrated that mice immunized with VP22/E7 DNA elicited more E7-specific CD8+ memory T cells and showed enhanced long-term protective antitumor immunity [110].
Resistance is a major problem in developing antimicrobial therapy against Pseudomonas aeruginosa (P. aeruginosa). Thus, Yu et al. designed a recombinant DNA vaccine [111] that incorporated VP22 against P. aeruginosa infection. A DNA vaccine vector, pVAX1, was designed to encode the antigen OprF gene that was fused to either the carboxyl terminal or the amino terminal of VP22. OprF is the antigenic surface protein of P. aeruginosa and is considered to be a selective immunogen against P. aeruginosa infection. Cells transfected with pVAX1-OprF-VP22 expressed a higher level of OprF protein than those with pVAX1-VP22-OprF, pVAX1-OprF or pVAX1-VP22. Furthermore, mice vaccinated with pVAX1-OprF-VP22 produced significantly higher OprF-specific antibody titers than those elicited by pVAX1-VP22-OprF and pVAX1-OprF. Four weeks after DNA vaccine administration, mice were challenged with a lethal dose of P. aeruginosa. Mice that received pVAX1-OprF-VP22 showed better survival rates (75% on day 8, 40% on day 10) than mice injected pVAX1-OprF (50% on day 7) and pVAX1-VP22-OprF (50% on day 8). Similarly, promising immune responses were also observed in mice immunized with plasmid DNA encoding VP22 and influenza virus nucleoprotein (NP) gene, a widely used antigen for influenza vaccines. Mice immunized with pVP22/NP exhibited the highest survival rate following intranasal challenge with H1N1 and H3N2 strains of influenza virus two weeks after final immunization compared to plasmid only, plasmid encoding NP and plasmid encoding VP221–267/NP treated mice [111]. Other infection studies also demonstrated the promising potential of VP22/DNA vaccine strategies against bovine herpesvirus 1 [112, 113] and porcine reproductive and respiratory syndrome virus [114].
4.5. Other CPPs
Pep-1 peptide (Table 1) is a 21-residue amphipathic CPP consisting of a hydrophilic lysine-rich domain originated from SV40 NLS, a hydrophobic tryptophan-rich domain, and a spacer domain that improves the integrity and flexibility between the hydrophilic and hydrophobic domains. Pep-1 is used as a carrier to deliver various macromolecules in drug delivery. In vaccine delivery, Pep-1 improved the delivery and desired intracellular localization of antigenic proteins. For example, Mardani et al. developed a nanoparticle-based HPV vaccine incorporating Pep-1 peptide [115]. E7 protein/Pep-1 peptide complex was mixed at a ratio of 1/20 to form nanoparticles with average diameters ranging from 120 to 250 nm. Mice that received E7/Pep-1 produced higher levels of E7 specific-IgG, IgG1and IgG2a compared to mice immunized with E7, Pep-1 and PBS. Mice were then challenged with tumors three weeks after the last vaccination. Mice immunized with E7 alone remained partially tumor-free (40%) on day 40 post-challenge, while most mice immunized with E7/Pep-1 remained tumor free (80%). Tumor growth was observed in all mice immunized with Pep-1 and PBS on day 12 post-challenge. Importantly, mice immunized first with E7/complete Freund adjuvant (50:50 v/v) followed by two boosters with E7/incomplete Freund adjuvant (50:50 v/v) exhibited a similar protective effect against tumor growth.
MPG is an amphipathic CPP designed based on the sequence of SV40 NLS and fusion sequence of the HIV glycoprotein 41. Saleh et al. demonstrated an efficient MPG-based gene delivery system against HPV infection [116]. The complex of MPG peptide/plasmid encoding antigen E7 gene at the ratio of 10/1 was produced. In a cell transfection assay, PEI/pE7 as the positive control showed similar E7 transfection efficacy with MPG/pE7. Two weeks after the final immunization, mice were challenged subcutaneously with tumors; a complete regression of tumor growth was observed in mice that received MPG/pE7. In comparison, all mice immunized with PBS, MPG, and pE7 developed tumors on days 21, 25 and 35, respectively. When the tumor challenge was performed one week before administration, mice immunized with MPG/pE7 exhibited complete tumor regression and were tumor-free 70 days after challenge. In contrast, all mice developed tumors by day 45 post-challenge when treated with MPG, pE7, and PBS. However, no commercial adjuvant was used in any of the control groups.
CONCLUSION
Subunit vaccines require potent adjuvants to overcome their poor immunogenicity, improve antigen uptake by APCs and stimulate antigen-specific cellular and humoral immune responses. Currently licensed adjuvants are often toxic and have limited effectiveness. CPPs are typically short peptides that have the capacity to facilitate antigen translocation through the cell membrane and, therefore, enhance antigen uptake, processing and presentation by APCs, which is the initial step of evoking an immune response. Since CPPs are cheap, easy to manufacture and usually non-toxic, they are attractive components of vaccine delivery systems. Among them, Tat has been most intensively studied. CPPs VP22 polyarginine and penetratin were also able to significantly improve vaccine efficacy. In general, CPPs facilitated the delivery of protein-based antigens and DNA material in vitro and in vivo in murine studies. Moreover, CPPs have also been used to formulate and deliver adjuvants. However, many studies reviewed here did not use positive controls- antigen administered with a known adjuvant- and, therefore, the real efficacy of these CPPs was not determined. When adjuvant was used in vaccine formulation, CPPs were applied rather as a co-stimulant than adjuvant replacement. Nevertheless, the incorporation of CPPs in vaccine formulation usually resulted in improved immune responses. Thus, even if CPPs do not replace classical adjuvants, they can still improve adjuvant effectiveness and/or reduce the quantity of adjuvant needed to achieve desired immune responses.
The other limitation in wider CPP application for vaccine delivery is the mechanism of how CPP-cargo is internalized, which still remains unclear. The types of CPPs, carried cargos, cell types and CPP concentration all affect the mechanism of cellular uptake and subsequent processing. Though, it is widely recognised that the uptake pathways of CPPs are generally possible by both non-endocytic and endocytic pathways, and, therefore, CPPs can stimulate both cellular and humoral immune responses. Considering this, a more detailed investigation is required to determine the factors that influence the CPP penetration process.
Importantly, CPPs demonstrated promising efficacy in clinical trials when used for drug delivery purposes. Therefore, we can expect that vaccine formulation bearing CPPs will reach the clinical testing phase soon. The unique penetrating ability of CPPs and their ability to deliver various antigens provides reliability for a wide range of vaccine applications; however, there are still many knowledge gaps, which first need to be filled.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the support of The University of Queensland.
LIST OF ABBREVIATIONS
- Antp
Penetratin
- APC
Antigen-presenting cells
- B cells
B lymphocytes
- BCRs
B cell receptors
- CLRs
C-type lectin receptors
- CPPs
Cell-penetrating peptides
- CTL
Cytotoxic T lymphocytes
- DCs
Dendritic cells
- Fab
Antigen binding fragment
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- HBcAg
Hepatitis B virus core antigen
- HIV
Human immunodeficiency virus
- HPVs
Human papillomavirus
- HSV
Herpes simplex virus
- IFNγ
Interferon gamma
- IgG
Immunoglubulin G
- IL-2
Interleukin 2
- IL-7
Interleukin 7
- IRF3
Interferon-regulatory factor 3
- LACK
Leishmania homolog of receptors for activated C kinase
- MHC
Major histocompatibility complex
- MTB
Mycobacterium tuberculosis
- NK
Natural killer
- NLRs
Nucleotide-oligomerisation receptors
- NLS
Nuclear Localization Sequence
- PAMAM
Polyamidoamine
- PAMPs
Pathogen-associated molecular patterns
- PRR
Pattern recognition receptors
- R8,R9
Polyarginine
- RLRs
RIG-1-like receptors
- RPS
Relative percent survival
- SV40
Simian virus 40
- T cells
T lymphocytes
- TCRs
T cell receptors
- TEM
Transmission electron microscopy
- TLRs
Toll-like receptors
- TNFβ
Tumour necrosis factor beta
- WSSV
White spot syndrome virus
- αGC
α-galactosyl ceramide
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by the National Health and Medical Research Council [NHMRC Program Grant APP1132975].
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Maurer D.M., Harrington B., Lane J.M. Smallpox vaccine: Contraindications, administration, and adverse reactions. Am. Fam. Physician. 2003;68:889–906. [PubMed] [Google Scholar]
- 2.Lakhani S. Early clinical pathologists: Edward Jenner (1749-1823). J. Clin. Pathol. 1992;45:756. doi: 10.1136/jcp.45.9.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alonso P.L., Tanner M. Public health challenges and prospects for malaria control and elimination. Nat. Med. 2013;19:150. doi: 10.1038/nm.3077. [DOI] [PubMed] [Google Scholar]
- 4.Alm J.S., Lilja G., Pershagen G., Scheynius A. Early BCG vaccination and development of atopy. Lancet. 1997;350:400–403. doi: 10.1016/S0140-6736(97)02207-1. [DOI] [PubMed] [Google Scholar]
- 5.Bull J.J. Evolutionary reversion of live viral vaccines: Can genetic engineering subdue it? Virus Evol. 2015;1:vev005. doi: 10.1093/ve/vev005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baxter D. Active and passive immunity, vaccine types, excipients and licensing. Occup. Med. (Lond.) 2007;57:552–556. doi: 10.1093/occmed/kqm110. [DOI] [PubMed] [Google Scholar]
- 7.Koj S., Ługowski C., Niedziela T. Bordetella pertussis lipooligosaccharide-derived neoglycoconjugates-new components of pertussis vaccine. Postepy Hig. Med. Dosw. 2015;69:1013–1030. [PubMed] [Google Scholar]
- 8.Singh M., O’Hagan D. Advances in vaccine adjuvants. Nat. Biotechnol. 1999;17:1075. doi: 10.1038/15058. [DOI] [PubMed] [Google Scholar]
- 9.Steer A.C., Law I., Matatolu L., Beall B.W., Carapetis J.R. Global emm type distribution of group A streptococci: Systematic review and implications for vaccine development. Lancet Infect. Dis. 2009;9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 10.Skwarczynski M., Toth I. Peptide-based synthetic vaccines. Chem. Sci. (Camb.) 2016;7:842–854. doi: 10.1039/c5sc03892h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaberolansar N., Toth I., Young P.R., Skwarczynski M. Recent advances in the development of subunit-based RSV vaccines. Expert Rev. Vaccines. 2016;15:53–68. doi: 10.1586/14760584.2016.1105134. [DOI] [PubMed] [Google Scholar]
- 12.Skwarczynski M., Toth I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine (Lond.) 2014;9:2657–2669. doi: 10.2217/nnm.14.187. [DOI] [PubMed] [Google Scholar]
- 13.Moyle P.M., Toth I. Modern subunit vaccines: Development, components, and research opportunities. ChemMedChem. 2013;8:360–376. doi: 10.1002/cmdc.201200487. [DOI] [PubMed] [Google Scholar]
- 14.Nevagi R.J., Toth I., Skwarczynski M. Peptide-based vaccines. In: Koutsopoulos S., editor. Peptide Applications in Biomedicine, Biotechnology and Bioengineering. Cambridge, United Kingdom: Woodhead Publishing; 2018. pp. 327–358. [Google Scholar]
- 15.Azmi F., Ahmad Fuaad A.A., Skwarczynski M., Toth I. Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccin. Immunother. 2014;10:778–796. doi: 10.4161/hv.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reed S.G., Orr M.T., Fox C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 17.Montomoli E., Piccirella S., Khadang B., Mennitto E., Camerini R., De Rosa A. Current adjuvants and new perspectives in vaccine formulation. Expert Rev. Vaccines. 2011;10:1053–1061. doi: 10.1586/erv.11.48. [DOI] [PubMed] [Google Scholar]
- 18.Zhao G., Chandrudu S., Skwarczynski M., Toth I. The application of self-assembled nanostructures in peptide-based subunit vaccine development. Eur. Polym. J. 2017;93:670–681. doi: 10.1016/j.eurpolymj.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussein W.M., Liu T.Y., Skwarczynski M., Toth I. Toll-like receptor agonists: A patent review (2011-2013). Expert Opin. Ther. Pat. 2014;24:453–470. doi: 10.1517/13543776.2014.880691. [DOI] [PubMed] [Google Scholar]
- 20.Deshayes S., Morris M.C., Divita G., Heitz F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 2005;62:1839–1849. doi: 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X. Current and future technological advances in transdermal gene delivery. Adv. Drug Deliv. Rev. 2018;127:85–105. doi: 10.1016/j.addr.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Rádis-Baptista G., Campelo I.S., Morlighem J-É.R., Melo L.M., Freitas V.J. Cell-penetrating peptides (CPPs): From delivery of nucleic acids and antigens to transduction of engineered nucleases for application in transgenesis. J. Biotechnol. 2017;252:15–26. doi: 10.1016/j.jbiotec.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Chen J., Guan X., Hu Y., Tian H., Chen X. Peptide-Based and Polypeptide-Based Gene Delivery Systems. Top. Curr. Chem. (Cham) 2017;375:32. doi: 10.1007/s41061-017-0115-x. [DOI] [PubMed] [Google Scholar]
- 24.Lindgren M., Hällbrink M., Prochiantz A., Langel Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 25.Green M., Loewenstein P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 26.Frankel A.D., Pabo C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 27.Wender P.A., Mitchell D.J., Pattabiraman K., Pelkey E.T., Steinman L., Rothbard J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heitz F., Morris M.C., Divita G. Twenty years of cell‐penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris M., Vidal P., Chaloin L., Heitz F., Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997;25:2730–2736. doi: 10.1093/nar/25.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tünnemann G., Ter‐Avetisyan G., Martin R.M., Stöckl M., Herrmann A., Cardoso M.C. Live‐cell analysis of cell penetration ability and toxicity of oligo‐arginines. J. Pept. Sci. 2008;14:469–476. doi: 10.1002/psc.968. [DOI] [PubMed] [Google Scholar]
- 31.Derossi D., Joliot A.H., Chassaing G., Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 32.Vives E., Brodin P., Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 33.Noguchi H., Matsushita M., Matsumoto S., Lu Y-F., Matsui H., Bonner-Weir S. Mechanism of PDX-1 protein transduction. Biochem. Biophys. Res. Commun. 2005;332:68–74. doi: 10.1016/j.bbrc.2005.04.092. [DOI] [PubMed] [Google Scholar]
- 34.Bartlett R.L., Panitch A. Thermosensitive nanoparticles with pH-triggered degradation and release of anti-inflammatory cell-penetrating peptides. Biomacromolecules. 2012;13:2578–2584. doi: 10.1021/bm300826v. [DOI] [PubMed] [Google Scholar]
- 35.Elliott G., O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 36.Oehlke J., Scheller A., Wiesner B., Krause E., Beyermann M., Klauschenz E., Melzig M., Bienert M. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta Biomembr. 1998;1414:127–139. doi: 10.1016/s0005-2736(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 37.Morris M.C., Depollier J., Mery J., Heitz F., Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001;19:1173. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- 38.Crombez L., Aldrian-Herrada G., Konate K., Nguyen Q.N., McMaster G.K., Brasseur R., Heitz F., Divita G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009;17:95–103. doi: 10.1038/mt.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nan Y.H., Park I.S., Hahm K.S., Shin S.Y. Antimicrobial activity, bactericidal mechanism and LPS‐neutralizing activity of the cell‐penetrating peptide pVEC and its analogs. J. Pept. Sci. 2011;17:812–817. doi: 10.1002/psc.1408. [DOI] [PubMed] [Google Scholar]
- 40.Pooga M., Hällbrink M., Zorko M. Cell penetration by transportan. FASEB J. 1998;12:67–77. doi: 10.1096/fasebj.12.1.67. [DOI] [PubMed] [Google Scholar]
- 41.Rhee M., Davis P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J. Biol. Chem. 2006;281:1233–1240. doi: 10.1074/jbc.M509813200. [DOI] [PubMed] [Google Scholar]
- 42.Gao S., Simon M.J., Hue C.D., Morrison B., III, Banta S. An unusual cell penetrating peptide identified using a plasmid display-based functional selection platform. ACS Chem. Biol. 2011;6:484–491. doi: 10.1021/cb100423u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peptides C.P. Intracellular Pathways and Pharmaceutical Perspectives Patel, Leena N.; Zaro, Jennica L.; Shen, Wei-Chiang. Pharm. Res. 2007;24:1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 44.Duchardt F., Fotin‐Mleczek M., Schwarz H., Fischer R., Brock R. A comprehensive model for the cellular uptake of cationic cell‐penetrating peptides. Traffic. 2007;8:848–866. doi: 10.1111/j.1600-0854.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 45.Fittipaldi A., Ferrari A., Zoppé M., Arcangeli C., Pellegrini V., Beltram F., Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 46.Tali C. In: Scavenger receptors as a target for nucleic acid delivery with peptide vectors, Pooga, M. Padarik K., editor. University of Tarita Press; 2017. [Google Scholar]
- 47.Madani F., Lindberg S., Langel Ü., Futaki S., Gräslund A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011;2011:414729. doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khandia R., Munjal A., Kumar A., Singh G., Karthik K., Dhama K. Cell penetrating peptides: Biomedical/therapeutic applications with emphasis as promising futuristic hope for treating cancer. Int. J. Pharm. 2017;13:677–689. [Google Scholar]
- 49.Derossi D., Chassaing G., Prochiantz A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 50.Elmquist A. Cell-penetrating peptides: Cellular uptake and biological activities. Stockholm University; 2003. [Google Scholar]
- 51.Pouny Y., Rapaport D., Mor A., Nicolas P., Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry. 1992;31:12416–12423. doi: 10.1021/bi00164a017. [DOI] [PubMed] [Google Scholar]
- 52.Wang F., Wang Y., Zhang X., Zhang W., Guo S., Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release. 2014;174:126–136. doi: 10.1016/j.jconrel.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 53.Islam M.Z., Sharmin S., Moniruzzaman M., Yamazaki M. Elementary processes for the entry of cell-penetrating peptides into lipid bilayer vesicles and bacterial cells. Appl. Microbiol. Biotechnol. 2018;102:3879–3892. doi: 10.1007/s00253-018-8889-5. [DOI] [PubMed] [Google Scholar]
- 54.Futaki S., Nakase I., Tadokoro A., Takeuchi T., Jones A.T. Arginine-rich peptides and their internalization mechanisms. England, United Kingdom: In Portland Press Limited; 2007. [DOI] [PubMed] [Google Scholar]
- 55.Mousavi S.A., Malerød L., Trond B., Kjeken R. Clathrin-dependent endocytosis. Biochem. J. 2004;377:1–16. doi: 10.1042/BJ20031000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conner S.D., Schmid S.L. Regulated portals of entry into the cell. Nature. 2003;422:37. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 57.Pujals S., Giralt E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008;60:473–484. doi: 10.1016/j.addr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 58.Futaki S., Nakase I. Cell-surface interactions on arginine-rich cell-penetrating peptides allow for multiplex modes of internalization. Acc. Chem. Res. 2017;50:2449–2456. doi: 10.1021/acs.accounts.7b00221. [DOI] [PubMed] [Google Scholar]
- 59.El-Sayed A., Harashima H. Endocytosis of gene delivery vectors: From clathrin-dependent to lipid raft-mediated endocytosis. Mol. Ther. 2013;21:1118–1130. doi: 10.1038/mt.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 61.Hillaireau H., Couvreur P. Nanocarriers’ entry into the cell: Relevance to drug delivery. Cell. Mol. Life Sci. 2009;66:2873–2896. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaro J.L., Shen W-C. Quantitative comparison of membrane transduction and endocytosis of oligopeptides. Biochem. Biophys. Res. Commun. 2003;307:241–247. doi: 10.1016/s0006-291x(03)01167-7. [DOI] [PubMed] [Google Scholar]
- 63.Futaki S., Suzuki T., Ohashi W., Yagami T., Tanaka S., Ueda K., Sugiura Y. Arginine-rich peptides An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 64.Chaplin D.D. Overview of the immune response. J. Allergy Clin. Immunol. Pract. 2010;125:S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turvey S.E., Broide D.H. Innate immunity. J. Allergy Clin. Immunol. 2010;125:S24–S32. doi: 10.1016/j.jaci.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janeway C.A., Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 67.Janeway C.A., Jr, Travers P., Walport M., Shlomchik M.J. The major histocompatibility complex and its functions. New York, USA: Garland Science; 2001. [Google Scholar]
- 68.Delves P.J., Martin S.J., Burton D.R., Roitt I.M. Essential immunology. New Jersey, United States: John Wiley & Sons; 2017. [Google Scholar]
- 69.Bottomly K. A functional dichotomy in CD4+ T lymphocytes. Immunol. Today. 1988;9:268–274. doi: 10.1016/0167-5699(88)91308-4. [DOI] [PubMed] [Google Scholar]
- 70.Romagnani S. Type 1 T helper and type 2 T helper cells: Functions, regulation and role in protection and disease. Int. J. Clin. Lab. Res. 1992;21:152–158. doi: 10.1007/BF02591635. [DOI] [PubMed] [Google Scholar]
- 71.Bonilla F.A., Oettgen H.C. Adaptive immunity. J. Allergy Clin. Immunol. Pract. 2010;125:S33–S40. doi: 10.1016/j.jaci.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 72.Saper C.B. A guide to the perplexed on the specificity of antibodies. J. Histochem. Cytochem. 2009;57:1–5. doi: 10.1369/jhc.2008.952770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Borghesi L., Milcarek C. From B cell to plasma cell. Immunol. Res. 2006;36:27–32. doi: 10.1385/IR:36:1:27. [DOI] [PubMed] [Google Scholar]
- 74.Gulzar N., Copeland K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004;2:23–37. doi: 10.2174/1570162043485077. [DOI] [PubMed] [Google Scholar]
- 75.Moticka E.J. A historical perspective on evidence-based immunology. Oxford, USA: Newnes; 2015. [Google Scholar]
- 76.MacLeod M.K., Kappler J.W., Marrack P. Memory CD4 T cells: Generation, reactivation and re‐assignment. Immunology. 2010;130:10–15. doi: 10.1111/j.1365-2567.2010.03260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Y-H., Lin K-I. Advances in immunology. Vol. 131. Oxford, USA: Elsevier; 2016. Factors that regulate the generation of antibody-secreting plasma cells. pp. 61–99. [DOI] [PubMed] [Google Scholar]
- 78.Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune system: Turning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- 79.Trombetta E.S., Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 80.Jiang Y., Li M., Zhang Z., Gong T., Sun X. Cell-penetrating peptides as delivery enhancers for vaccine. Curr. Pharm. Biotechnol. 2014;15:256–266. doi: 10.2174/1389201015666140813130114. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y., Røise J.J., Lee K., Li J., Murthy N. Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol. 2018;52:25–31. doi: 10.1016/j.copbio.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharfstein S.T. Non-protein biologic therapeutics. Curr. Opin. Biotechnol. 2018;53:65–75. doi: 10.1016/j.copbio.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 83.Bahadoran A., Ebrahimi M., Yeap S.K., Safi N., Moeini H., Hair-Bejo M., Hussein M.Z., Omar A.R. Induction of a robust immune response against avian influenza virus following transdermal inoculation with H5-DNA vaccine formulated in modified dendrimer-based delivery system in mouse model. Int. J. Nanomedicine. 2017;12:8573. doi: 10.2147/IJN.S139126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ji Z., Xie Z., Zhang Z., Gong T., Sun X. Engineering intravaginal vaccines to overcome mucosal and epithelial barriers. Biomaterials. 2017;128:8–18. doi: 10.1016/j.biomaterials.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 85.Tang J., Yin R., Tian Y., Huang Z., Shi J., Fu X., Wang L., Wu Y., Hao F., Ni B. A novel self-assembled nanoparticle vaccine with HIV-1 Tat49-57/HPV16 E749-57 fusion peptide and GM-CSF DNA elicits potent and prolonged CD8+ T cell-dependent anti-tumor immunity in mice. Vaccine. 2012;30:1071–1082. doi: 10.1016/j.vaccine.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 86.MuÈnger. K.; Basile, J.R.; Duensing, S.; Eichten, A.; Gonzalez, S.L.; Grace, M.; Zacny, V.L. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 87.Bonnem E.M., Chaudry I.A., Stupak E. Use of GM-CSF as a vaccine adjuvant. In Google Patents: US5679356A; 1997. [Google Scholar]
- 88.Yang Z., Wang L., Wang H., Shang X., Niu W., Li J., Wu Y. A novel mimovirus vaccine containing survivin epitope with adjuvant IL-15 induces long-lasting cellular immunity and high antitumor efficiency. Mol. Immunol. 2008;45:1674–1681. doi: 10.1016/j.molimm.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 89.Sun Y., Hu Y-h. Cell-penetrating peptide-mediated subunit vaccine generates a potent immune response and protection against Streptococcus iniae in Japanese flounder (Paralichthys olivaceus). Vet. Immunol. Immunopathol. 2015;167:96–103. doi: 10.1016/j.vetimm.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 90.Weinstein M.R., Litt M., Kertesz D.A., Wyper P., Rose D., Coulter M., McGeer A., Facklam R., Ostach C., Willey B.M. Invasive infections due to a fish pathogen, Streptococcus iniae. N. Engl. J. Med. 1997;337:589–594. doi: 10.1056/NEJM199708283370902. [DOI] [PubMed] [Google Scholar]
- 91.Chen X., Lai J., Pan Q., Tang Z., Yu Y., Zang G. The delivery of HBcAg via Tat-PTD enhances specific immune response and inhibits Hepatitis B virus replication in transgenic mice. Vaccine. 2010;28:3913–3919. doi: 10.1016/j.vaccine.2010.03.070. [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y., Ning J-F., Qu X-q., Meng X-L., Xu J-P. TAT-mediated oral subunit vaccine against white spot syndrome virus in crayfish. J. Virol. Methods. 2012;181:59–67. doi: 10.1016/j.jviromet.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 93.Kronenberg K., Brosch S., Butsch F., Tada Y., Shibagaki N., Udey M.C., Von Stebut E. Vaccination with TAT-antigen fusion protein induces protective, CD8+ T cell-mediated immunity against Leishmania major. J. Invest. Dermatol. 2010;130:2602–2610. doi: 10.1038/jid.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dong H., Jing W., Yingru X., Wenyang W., Ru C., Shengfa N., Congjing X., Jingjing D., Wan W., Jiang H. Enhanced anti-tuberculosis immunity by a TAT-Ag85B protein vaccine in a murine tuberculosis model. Pathog. Glob. Health. 2015;109:363–368. doi: 10.1080/20477724.2015.1111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang H-C., Lu H-F., Lai Y-H., Lee C-P., Liu H-K., Huang C. Tat-enhanced delivery of the C terminus of HDAg-L inhibits assembly and secretion of hepatitis D virus. Antiviral Res. 2018;150:69–78. doi: 10.1016/j.antiviral.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 96.Sakuma S., Suita M., Inoue S., Marui Y., Nishida K., Masaoka Y., Kataoka M., Yamashita S., Nakajima N., Shinkai N. Cell-penetrating peptide-linked polymers as carriers for mucosal vaccine delivery. Mol. Pharm. 2012;9:2933–2941. doi: 10.1021/mp300329r. [DOI] [PubMed] [Google Scholar]
- 97.Mohri K., Miyata K., Egawa T., Tanishita S., Endo R., Yagi H., Ukawa M., Ochiai K., Hiwatari K-I., Tsubaki K. Effects of the chemical structures of oligoarginines conjugated to biocompatible polymers as a mucosal adjuvant on antibody induction in nasal cavities. Chem. Pharm. Bull. (Tokyo) 2018;66:375–381. doi: 10.1248/cpb.c17-00834. [DOI] [PubMed] [Google Scholar]
- 98.Nakamura T., Moriguchi R., Kogure K., Shastri N., Harashima H. Efficient MHC class I presentation by controlled intracellular trafficking of antigens in octaarginine-modified liposomes. Mol. Ther. 2008;16:1507–1514. doi: 10.1038/mt.2008.122. [DOI] [PubMed] [Google Scholar]
- 99.Nakamura T., Yamazaki D., Yamauchi J., Harashima H. The nanoparticulation by octaarginine-modified liposome improves α-galactosylceramide-mediated antitumor therapy via systemic administration. J. Control. Release. 2013;171:216–224. doi: 10.1016/j.jconrel.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 100.Wang K., Yang Y., Xue W., Liu Z. Cell penetrating peptide-based redox-sensitive vaccine delivery system for subcutaneous vaccination. Mol. Pharm. 2018;15:975–984. doi: 10.1021/acs.molpharmaceut.7b00905. [DOI] [PubMed] [Google Scholar]
- 101.Zhang Y., Li H., Wang Q., Hao X., Li H., Sun H., Han L., Zhang Z., Zou Q., Sun X. Rationally designed self‐assembling nanoparticles to overcome mucus and epithelium transport barriers for oral vaccines against Helicobacter pylori. Adv. Funct. Mater. 2018;28(33):1802675 [Google Scholar]
- 102.Schutze-Redelmeier M-P.M., Kong S., Bally M.B., Dutz J.P. Antennapedia transduction sequence promotes anti tumour immunity to epicutaneously administered CTL epitopes. Vaccine. 2004;22:1985–1991. doi: 10.1016/j.vaccine.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 103.Pouniotis D.S., Esparon S., Apostolopoulos V., Pietersz G.A. Whole protein and defined CD8+ and CD4+ peptides linked to penetratin targets both MHC class I and II antigen presentation pathways. Immunol. Cell Biol. 2011;89:904. doi: 10.1038/icb.2011.13. [DOI] [PubMed] [Google Scholar]
- 104.Muto K., Kamei N., Yoshida M., Takayama K., Takeda-Morishita M. Cell-penetrating peptide penetratin as a potential tool for developing effective nasal vaccination systems. J. Pharm. Sci. 2016;105:2014–2017. doi: 10.1016/j.xphs.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 105.Brooks N., Hsu J., Esparon S., Pouniotis D., Pietersz G. Immunogenicity of a tripartite cell penetrating peptide containing a MUC1 variable number of tandem repeat (VNTR) and AT helper epitope. Molecules. 2018;23:2233. doi: 10.3390/molecules23092233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taylor-Papadimitriou J., Burchell J.M., Graham R., Beatson R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018;46:659–668. doi: 10.1042/BST20170400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Elliott G., O’Hare P. Intercellular trafficking of VP22-GFP fusion proteins. Gene Ther. 1999;6:149. doi: 10.1038/sj.gt.3300850. [DOI] [PubMed] [Google Scholar]
- 108.Hung C-F., Cheng W-F., Chai C-Y., Hsu K-F., He L., Ling M., Wu T-C. Improving vaccine potency through intercellular spreading and enhanced MHC class I presentation of antigen. J. Immunol. 2001;166:5733–5740. doi: 10.4049/jimmunol.166.9.5733. [DOI] [PubMed] [Google Scholar]
- 109.Cheng W-F., Hung C-F., Hsu K-F., Chai C-Y., He L., Polo J.M., Slater L.A., Ling M., Wu T-C. Cancer immunotherapy using Sindbis virus replicon particles encoding a VP22–antigen fusion. Hum. Gene Ther. 2002;13:553–568. doi: 10.1089/10430340252809847. [DOI] [PubMed] [Google Scholar]
- 110.Kim T.W., Hung C-F., Kim J.W., Juang J., Chen P-J., He L., Boyd D.A., Wu T-C. Vaccination with a DNA vaccine encoding herpes simplex virus type 1 VP22 linked to antigen generates long-term antigen-specific CD8-positive memory T cells and protective immunity. Hum. Gene Ther. 2004;15:167–177. doi: 10.1089/104303404772679977. [DOI] [PubMed] [Google Scholar]
- 111.Yu X., Wang Y., Xia Y., Zhang L., Yang Q., Lei J. A DNA vaccine encoding VP22 of herpes simplex virus type I (HSV-1) and OprF confers enhanced protection from Pseudomonas aeruginosa in mice. Vaccine. 2016;34:4399–4405. doi: 10.1016/j.vaccine.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 112.Zheng C., Babiuk L.A. Bovine herpesvirus 1 VP22 enhances the efficacy of a DNA vaccine in cattle. J. Virol. 2005;79:1948–1953. doi: 10.1128/JVI.79.3.1948-1953.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng C., Brownlie R., Huang D., Babiuk L. Intercellular trafficking of the major tegument protein VP22 of bovine herpesvirus-1 and its application to improve a DNA vaccine. Arch. Virol. 2006;151:985–993. doi: 10.1007/s00705-005-0694-7. [DOI] [PubMed] [Google Scholar]
- 114.Zhao W., Xiao S-B., Fang L-R., Jiang Y., Song Y., Yan L., Yu X., Chen H. Immunogenicity of DNA vaccine expressing GP5 of porcine reproductive and respiratory syndrome virus fused with VP22 of bovine herpesvirus 1. Sheng Wu Gong Cheng Xue Bao Chin. J. Biotechnol. 2005;21:725–730. [PubMed] [Google Scholar]
- 115.Mardani G., Bolhassani A., Agi E., Shahbazi S., Mehdi Sadat S. Protein vaccination with HPV16 E7/Pep‐1 nanoparticles elicits a protective T‐helper cell‐mediated immune response. IUBMB Life. 2016;68:459–467. doi: 10.1002/iub.1503. [DOI] [PubMed] [Google Scholar]
- 116.Saleh T., Bolhassani A., Shojaosadati S.A., Aghasadeghi M.R. MPG-based nanoparticle: An efficient delivery system for enhancing the potency of DNA vaccine expressing HPV16E7. Vaccine. 2015;33:3164–3170. doi: 10.1016/j.vaccine.2015.05.015. [DOI] [PubMed] [Google Scholar]