

Abstract
Background and Purpose
Ursodeoxycholic acid (UDCA) is the first‐line treatment for primary biliary cholangitis, but its effects on the enterohepatic circulation of bile acid (BA) have been under‐investigated. Therefore, we studied the influence of UDCA on BA enterohepatic circulation in vivo and the mechanisms by which UDCA affects the BA kinetics.
Experimental Approach
Mice were treated with UDCA and other BAs to observe changes in BA pool and BA transporters involved in enterohepatic circulation. Isotope dilution techniques and biochemical analyses were applied to study BA kinetics after oral administration of UDCA, and the mechanism involved.
Key Results
Oral administration of UDCA in mice reduced the overall BA pool and produced a unique BA profile with high‐abundance conjugated UDCA species, including tauroursodeoxycholic acid (TUDCA) and GUDCA. We found increased expression of several main BA transporters in the ileum and liver. BA kinetic experiment showed that feeding UDCA shortened cycling time of BA and accelerated BA enterohepatic circulation. Additionally, we found evidence that the effect of UDCA administration on accelerating BA enterohepatic circulation was due to the inhibition of farnesoid X receptor (FXR) signalling in the ileum and FGF15/19 in the liver.
Conclusion and Implications
Oral administration of UDCA produced a unique BA profile with high‐abundance TUDCA and GUDCA and significantly accelerated BA enterohepatic circulation through the inhibition of intestinal FXR signalling and reduced level of FGF15/19, which in turn, induced the expression of BA transporters in the liver. These findings highlight a critical role for UDCA in maintaining the homeostasis of BA enterohepatic circulation in vivo.
Abbreviations
- ASBT
apical sodium‐dependent bile acid transporter
- BSEP
canalicular bile salt export pump
- CA
cholic acid
- CCK
cholecystokinin
- CDCA
chenodeoxycholic acid
- CYP7A1
cholesterol 7α‐hydroxylase
- CYP7B1
oxysterol 7α‐hydroxylase
- CYP8B1
sterol 12α‐hydroxylase
- DCA
deoxycholic acid
- FXR
farnesoid X receptor
- IBABP
ileal bile acid binding protein
- MRP
multidrug resistance‐associated protein
- NTCP
sodium taurocholate cotransporting polypeptide
- OATP
organic anion transporting polypeptide
- OSTα/β
organic solute transporter α/β
- SHP
small heterodimer partner
- UDCA
ursodesoxycholic acid
What is already known
Ursodeoxycholic acid (UDCA) is a bile acid (BA) with choleretic, anti‐inflammatory, and cytoprotective properties.
What this study adds
UDCA accelerates the enterohepatic circulation of BAs by inhibiting intestinal farnesoid X receptor (FXR)‐SHP and FXR‐FGF15/19 signalling.
What is the clinical significance
UDCA affects the BA kinetics, which has profound effects on BA homeostasis and drug metabolism.
1. INTRODUCTION
Bile acids (BAs) have long been recognized as signalling molecules that help regulate lipid, glucose, and energy metabolism and maintain liver and gut metabolic homeostasis via activation of BA sensitive nuclear receptors such as the farnesoid X receptor (FXR) and G protein‐coupled BA receptor‐1 (Chiang, 2013). Ursodeoxycholic acid (UDCA) is a BA with choleretic, anti‐inflammatory, and cytoprotective properties (Paumgartner & Beuers, 2004; Ward & Lajczak, 2017) and is a first‐line treatment approved by the Food and Drug Administration for primary biliary cholangitis (PBC; Marschall et al., 2005). UDCA has been shown to enhance the expression of hepatobiliary secretion transporters (Marschall et al., 2005), which are positively regulated by the FXR (Trauner & Boyer, 2003; Halilbasic, Claudel, & Trauner, 2013). However, UDCA has also been reported to be an FXR antagonist in humans (Mueller et al., 2015), leaving some questions as to the mechanism by which UDCA facilitates hepatic BA secretion and exerts protective effects on the liver.
The enterohepatic circulation of BAs from the liver to the intestine and back to the liver plays a central role in nutrient absorption and distribution, as well as metabolic regulation and homeostasis (Chiang, 2013). A dysfunction in BA homeostasis and a compromised enterohepatic circulation may lead to cholestasis, non‐alcoholic fatty liver disease, microbiota dysbiosis, or non‐hepatobiliary disease (Chow, Lee, & Guo, 2017; Dermadi et al., 2017; Jovanovich et al., 2018; Li, Tang, Leung, Gershwin, & Ma, 2017). Thus, BA homeostasis is important for an optimal enterohepatic circulation and it depends on adequate hepatic synthesis, biliary secretion, intestinal reabsorption, and hepatic uptake of BAs. The de novo synthesis and transport of BAs are both directly and indirectly regulated by the nuclear receptor FXR (Halilbasic et al., 2013). As endogenous ligands of FXR, BAs have been shown to regulate their own homeostasis by activation of the FXR (Makishima et al., 1999; Parks et al., 1999). The ability of individual BAs to activate FXRs and influence the expression of transporters has been studied, but there is limited consensus among the results (Liu et al., 2014; Song, Rockwell, Cui, & Klaassen, 2015). The frequency of BA enterohepatic recycling is another key factor for BA homeostasis. The kinetics of the enterohepatic circulation of BAs may be closely associated with an individual's liver pathophysiology and has been under‐investigated in recent years.
In the present study, we conducted comprehensive studies in male C57BL/6 mice to compare the effects of administering UDCA and other individual BAs on the enterohepatic circulation, including BA pool size, composition, distribution, and excretion. Based on the detection of BA levels in various tissues and BA transporters in the ileum and liver, we found that UDCA decreased the BA pool size relative to the other BAs administered. Furthermore, an investigation of BA enterohepatic circulation kinetics demonstrated that UDCA accelerates BA circulation frequency in vivo. Intestinal FXR and FGF15/19 were subsequently found to be altered and may play an important role in the mechanism by which UDCA and its taurine‐ and glycine‐conjugated derivatives affect these kinetics.
2. METHODS
2.1. Animal experiments
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and the editorial on reporting animal studies (Mcgrath & Lilley, 2015) with the recommendations made by the British Journal of Pharmacology.
2.2. Animal experiment 1
Pathogen‐free male C57BL/6 mice (18.97 ± 0.84 g; MGI Cat# 5656552, RRID:MGI:5656552), at 5 weeks of age, were purchased from SLAC Laboratory (SH, China) and were housed under specific pathogen‐free condition (12 hr on/off; lights on at 7:00 a.m.) and temperature‐controlled (22–25°C) environment. Mice were acclimatized to the housing facility for 1 week with mouse chow and water available ad libitum.
The mice were divided into six groups, three of cage companions, six in each group. One group was given control chow, and the others were supplied with a diet containing various BAs: CA group (cholic acid, J&K 977474, 0.1% w/w), CDCA group (chenodeoxycholic acid, J&K 951898, 0.1% w/w), DCA group (deoxycholic acid, J&K 393388, 0.1% w/w), UDCA group (ursodeoxycholic acid, J&K 970735, 0.1% w/w), and LCA group (lithocholic acid, J&K 241301, 0.01% w/w; Trophic Animal Feed High‐Tech Co., Ltd, JS, China). All the mice were given diet and water ad libitum. After 8 weeks, mice were fasted for 12 hr and then serum and faeces were collected and livers, gall bladders, intestines, and intestinal contents were removed immediately after mice had been killed. All samples and tissues collected were then frozen in liquid nitrogen and stored at −80°C prior to testing.
2.3. Animal experiment 2
In a second experiment, pathogen‐free male C57BL/6 mice (18.96 ± 0.56 g), at 5 weeks of age, were purchased from SLAC Laboratory (SH, China) and acclimatized to the housing facility as described above. The mice were then divided into three groups (n = 12 per group, three in each cage), one was supplied with control chow, one with a diet containing UDCA (0.1% w/w), and one with a diet containing DCA (0.1% w/w). All the mice were housed under specific pathogen‐free conditions (12 hr on/off; lights on at 7:00 a.m.) and temperature‐controlled (22–25°C) environment, with food and water available ad libitum. After 8 weeks, 240 μg of [2H4]cholate (CDN, D‐2452) in a solution of PBS (pH = 7.4) was administered i.v. to half of the mice in each group (n = 6 per group). Blood samples (100 μl) were subsequently collected at 12, 24, 36, 48, and 60 hr after administration of the [2H4]cholate. Serum was obtained from the blood samples by centrifugation at 6000 rpm (4065× g) for 10 min and stored at −80°C until analysed. The other half of the three groups of mice (n = 6 per group) were fasted for 12 hr at 8 weeks and anaesthetized with pentobarbital sodium (60 mg·kg−1) by intraperitoneal injection and subjected to bile duct cannulation. To ensure accurate measurements, the initial 5 min of bile collected after cannulation was discarded (Kok et al., 2003). Bile was sampled for 30 min thereafter and stored at −80°C until analysis.
2.4. Animal experiment 3
In the third experiment, pathogen‐free male C57BL/6 mice (18.75 ± 0.91 g), at 5 weeks of age, were acclimatized to the housing facility as described above. The mice were then divided into two groups (n = 6 per group, three in one cage), one was given FGF19 (100 μg·kg−1 BW; 969‐FG‐025/CF, R&D Systems, Inc.) by i.p. injection and the other was given normal saline i.p. as the control. After 2 hr, blood was collected, the mice were then killed and both liver and gall bladder were removed. Livers were frozen in liquid nitrogen and stored at −80°C before testing. Gall bladders were weighed by precision electronic autobalance and then stored at −80°C. Serum was obtained by centrifugation of blood samples at 6000 rpm (4065× g) for 10 min and stored at −80°C until analysis was performed.
2.5. Animal experiment 4
Pathogen‐free male C57BL/6 mice (18.93 ± 0.55 g), at 5 weeks of age, were purchased from SLAC Laboratory (SH, China) and acclimatized to the housing facility as described in the first experiment. The mice then were divided into three groups, five in each group (five in one cage), and were given control chow, UDCA diet, and DCA diet respectively. The diet, housing conditions, and environment were the same as the first animal experiment. After 8 weeks, the mice were fasted for 12 hr, venous blood was collected and aprotinin (Sigma A1153) was added immediately. Then next, mice were administered a test meal (20% dextrose, 5% casein, and 1% corn oil; 10 μl·g−1 BW) by oral gavage. Venous blood was collected 30 min after the test, and aprotinin was added immediately. Serum was obtained from the blood samples by centrifugation at 6000 rpm (4065× g) for 10 min and stored at −80°C until analysed. Two days later, mice were killed and livers, gall bladders, intestines, and intestinal contents were removed immediately. All samples and tissues collected were then frozen in liquid nitrogen and stored at −80°C prior to testing.
2.6. Cell culture and treatment
Human liver L‐02 (RRID:CVCL_6926) and intestine NCI‐H716 (ATCC Cat# CCL‐251, RRID:CVCL_1581) cell lines were cultured in DMEM supplemented with 10% charcoal‐stripped FBS (Omega Scientific, Tarzana, CA) and were incubated at 37°C in a humidified atmosphere containing 5% CO2 in air. The 2D and 3D cultures of L‐02 and CRL‐251 cell lines, respectively, were exposed to DMSO (control), UDCAs (TUDCA, GUDCA, UDCA, TLCA, and LCA at a ratio of 100:100:25:5:0.5, 50 μM:50 μM:12.5 μM:2.5 μM:0.25 μM, termed as UDCAs) or DCAs (TCA, TDCA, GDCA, and DCA at a ratio of 100:25:25:1, 50 μM:12.5 μM:12.5 μM:0.5 μM, termed as DCAs). In addition, FGF19 (20, 80, and 160 ng·ml−1) was added to 2D cultures of liver L‐02 cells. After 5 days of each treatment, cells were harvested for western blotting and immunofluorescence staining. Targeted protein levels were determined by integrated densities or mean fluorescence intensity.
2.7. Real‐time quantitative PCR
Total RNA was isolated from frozen mouse liver and intestinal tissue using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. RNA concentration was measured using the Nano Drop 2000C (Thermofisher). Reverse transcription was performed using the RT reagent kit (Takara, Otsu, Japan) according to the manufacturer's instructions. Real‐time quantitative PCR was performed on cDNA samples to quantify mRNA levels by using ABI 7900 (Applied Biosystems, Foster City, CA). Primer sequences were synthesized by Sangon Biotech (Shanghai, China). The target gene mRNA relative to GAPDH mRNA was shown as fold change relative to those of the control group.
2.8. Measurement of BAs
The BA levels in plasma, liver, gall bladder, intestine, intestinal contents, and faeces were quantitatively measured by ultra‐performance liquid chromatography triple quadrupole mass spectrometry according to our previously reported protocol (Xie et al., 2013; Wang et al., 2016). All separations were performed with an ACQUITY BEH C18 column (1.7 mm, 100 mm 3 2.1 mm internal dimensions; Waters, Milford, MA). Data acquisition was performed using MassLynx version 4.1 and BA levels were quantified using the TargetLynx applications manager version 4.1 (Waters, Milford, MA). The bile acid standards are shown in Table 1.
Table 1.
Standards list
| Bile acid standard | Full name | From | No. | |
|---|---|---|---|---|
| 1 | CA | cholic acid | SIGMA | C1129 |
| 2 | TCA | taurocholate acid | FLUKA | 86339 |
| 3 | GCA | glycocholic acid | SIGMA | G7132 |
| 4 | CDCA | chenodeoxycholic acid | SIGMA | C9377 |
| 5 | TCDCA | Taurochenodeoxycholic acid | SIGMA | T6260 |
| 6 | GCDCA | glycochenodeoxycholic acid | FLUKA | 50534 |
| 7 | aMCA | α‐muricholic acid | Steraloids | C1890‐000 |
| 8 | TaMCA | tauro α‐muricholic acid | Steraloids | C1893‐000 |
| 9 | bMCA | β‐muricholic acid | Steraloids | C1895‐000 |
| 10 | TbMCA | tauro β‐muricholic acid | Steraloids | C1899‐000 |
| 11 | UDCA | ursodeoxycholic acid | SIGMA | U5127 |
| 12 | TUDCA | tauroursodeoxycholic acid | SIGMA | S1755898 |
| 13 | GUDCA | glycoursodeoxycholic acid | SIGMA | 06863 |
| 14 | LCA | lithocholic acid | SIGMA | L6250 |
| 15 | TLCA | taurolithocholic acid | SIGMA | T7515 |
| 16 | GLCA | glycolithocholic acid | Steraloids | C1437‐000 |
| 17 | DCA | deoxycholic acid | FLUKA | 30960 |
| 18 | TDCA | taurodeoxycholate acid | SIGMA | T0875 |
| 19 | GDCA | glycodeoxycholic acid | SIGMA | G9910 |
| 20 | wMCA | ω‐muricholic acid | Steraloids | C1888‐000 |
| 21 | TwMCA | tauro ω‐muricholic acid | Steraloids | C1889‐000 |
| 22 | HDCA | hyodeoxycholic acid | Steraloids | C0860‐000 |
| 23 | THDCA | taurohyodeoxycholic acid | SIGMA | T0682 |
| 24 | GHDCA | glycohyodeoxycholic acid | Steraloids | C0867‐000 |
| 25 | HCA | hyocholic acid | Steraloids | C1850‐000 |
| 26 | THCA | taurohyocholic acid | Steraloids | C1887‐000 |
| 27 | GHCA | glycohyocholic acid | Steraloids | C1860‐000 |
| 28 | 7‐ketoDCA | 7‐ketodeoxycholic acid | Steraloids | C1250‐000 |
| 29 | ACA | allocholic acid | TRC | A545000 |
| 30 | dehydroLCA | dehydrocholic acid | SIGMA | 30830 |
| 31 | alloLCA | allolithocholic acid | Steraloids | C0700‐000 |
| 32 | isoLCA | isolithocholic acid | Steraloids | C1475‐000 |
| 33 | 23‐norDCA | 23‐nordeoxycholic acid | Steraloids | N2000‐000 |
| 34 | 6‐ketoLCA | 6‐ketolithocholic acid | Steraloids | C1560‐000 |
| 35 | 7‐ketoLCA | 7‐ketolithocholic acid | Steraloids | C1600‐000 |
| 36 | 12‐ketoLCA | 12‐ketolithocholic acid | Steraloids | C1650‐000 |
| 37 | apoCA | apocholic acid | Steraloids | C2500‐000 |
| 38 | muroCA | Murocholic acid | Steraloids | C0910‐000 |
| 39 | beta‐UDCA | 3β‐Ursodeoxycholic Acid | TRC | U850005 |
| 40 | 6, 7‐diketoLCA | 6,7‐diketolithocholic acid | Steraloids | C1485‐000 |
| 41 | 3‐ketoCA | 3‐dehydrocholic acid | Steraloids | C1272‐000 |
| 42 | 7, 12‐diketoLCA | 7,12‐diketolithocholic acid | Steraloids | C1500‐000 |
| 43 | 12‐ketoCA | 12‐dehydrocholic acid | Steraloids | C1200‐000 |
| 44 | UCA | ursocholic acid | TRC | U849900 |
| 45 | betaCA | 3β‐Cholic Acid | TRC | C432605 |
| 46 | isoDCA | isodeoxycholic acid | Steraloids | C1170‐000 |
| 47 | DHLCA | dehydrolithocholic acid | Steraloids | C1750‐000 |
| 48 | TDHCA | taurodehydrocholic acid | Steraloids | C2045‐000 |
| 49 | GDHCA | glycodehydrocholic acid | Steraloids | C2022‐000 |
2.9. Western blotting
Protein was extracted from liver, intestine tissue, or cultured cells using RIPA lysis buffer (with PMSF) according to the manufacturer's instructions. Protein concentrations were determined using a bicinchoninic acid protein assay kit. Each sample was diluted to 5 μg·μl−1 and mixed with an equal volume of loading buffer (Beyotime Biotechnology). Protein was loaded and electrophoresed through 12% or 8% SDS‐PAGE and electrotransferred onto PVDF membranes (Millipore, Bedford, MA). After being blocked with 5% non‐fat milk for 1 hr at room temperature, membranes were incubated overnight at 4°C with primary antibodies and then washed six times for 5 min each; the membranes were subsequently incubated with HRP‐conjugated secondary antibodies for 1 hr. Then, membranes were again washed six times in TBST buffer for 5 min each. Protein–antibody complexes were detected using an enhanced chemiluminescent kit (Thermo) and exposure to film. After the film had been developed, integrated densities were quantified using ImageJ software (ImageJ, RRID:SCR_003070). Antibodies used in this procedure include ASBT, ileal bile acid binding protein (IBABP), NTCP, OATP1, BSEP (ABCB11), MRP2 (ABCC2), CYP7A1, FXR, SHP, FGF15, FGF19, β‐actin, goat anti‐rabbit IgG HRP‐linked antibody (1:5,000), and horse anti‐mouse IgG HRP‐linked antibody (1:3,000). Data were normalized to β‐actin protein levels and expressed as fold change to those of the control group. The catalogue number and RRIDs of antibodies are provided in the table below. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
| ASBT | (Abcam Cat# ab174610, RRID:AB_2801375) |
| IBABP | (Abcam Cat# ab91184, RRID:AB_10563324) |
| NTCP | (Abcam Cat# ab131084, RRID:AB_11155311) |
| OATP1 | (Abcam Cat# ab203036, RRID:AB_2801377) |
| BSEP | (Abcam Cat# ab112494, RRID:AB_10860892) |
| MRP2 | (Abcam Cat# ab203397, RRID:AB_2801378) |
| CYP7A1 | (Abcam Cat# ab65596, RRID:AB_1566114) |
| FXR | (Abcam Cat# ab56902, RRID:AB_942089) |
| SHP | (Abcam Cat# ab186874, RRID:AB_2797389) |
| FGF15 | (Santa Cruz Biotechnology Cat# sc‐27177, RRID:AB_2104062) |
| FGF19 | (Abcam Cat# ab85042, RRID:AB_10562610) |
| β‐actin | (Cell Signaling Technology Cat# 4970, RRID:AB_2223172) |
| goat anti‐rabbit IgG | (Cell Signaling Technology Cat# 7074, RRID:AB_2099233) |
| horse anti‐mouse IgG | (Cell Signaling Technology Cat# 7076, RRID:AB_330924) |
2.10. Immunohistochemistry and immunofluorescence
Liver and intestine tissue were fixed in 4% paraformaldehyde solution or frozen in optimal cutting temperature compound (OCT; Sakura Finetek USA). Paraffin‐embedded or OCT frozen liver and intestine tissue were processed for immunohistochemistry and immunofluorescence staining. The 2D or 3D cultured cells were processed for immunofluorescence staining. Primary antibodies used include those against ASBT, IBABP, NTCP, BSEP, MRP2, OSTα, FXR, SHP, FGF15, and FGF19. Secondary antibodies used included Alexa Fluor 488‐conjugated anti‐mouse IgG (1:500) and goat anti‐rabbit IgG (H+L) Superclonal (1:1,000). The slides were photographed using a digitalized microscope camera (Zeiss, Oberkochen, Germany). The images from both the immunohistochemistry and immunofluorescence samples were quantified using Image‐Pro Plus software (Image‐Pro Plus, RRID:SCR_007369). The catalogue number and RRIDs of antibodies used are provided in the table below.
| ASBT | (Abcam Cat# ab203205, RRID:AB_2801376) |
| IBABP | (Abcam Cat# ab91184, RRID:AB_10563324) |
| NTCP | (Abcam Cat# ab131084, RRID:AB_11155311) |
| BSEP | (Abcam Cat# ab112494, RRID:AB_10860892) |
| MRP2 | (Abcam Cat# ab203397, RRID:AB_2801378) |
| OSTα | (Santa Cruz Biotechnology Cat# sc‐100078, RRID:AB_2092072) |
| FXR | (Santa Cruz Biotechnology Cat# sc‐1205, RRID:AB_2155054) |
| SHP | (Abcam Cat# ab186874, RRID:AB_2797389) |
| FGF15 | (Santa Cruz Biotechnology Cat# sc‐27177, RRID:AB_2104062) |
| FGF19 | (Abcam Cat# ab85042, RRID:AB_10562610) |
| Alexa Fluor 488‐conjugated anti‐mouse IgG | (Abcam Cat# ab150117, RRID:AB_2688012) |
| Goat anti‐rabbit IgG (H+L) superclonal | (Thermo Fisher Scientific Cat# A27033, RRID:AB_2536096) |
2.11. ELISA
Cholecystokinin (CCK) concentration were measured by ELISA kits (Phoenix Pharmaceuticals, Inc.) according to the manufacturer's instructions. Aprotinin was added to blood samples for CCK measurement immediately after they had been collected. The kits were read at 450 nm by using SpectraMax i3 (Molecular Devices).
2.12. Calculations
2.12.1. Isotope dilution technique
The isotope dilution technique has previously been described in detail by Hulzebos et al. (2001; Kok et al., 2003; Koopman et al., 1988; Stellaard et al., 1984). Enrichment was defined as the increase in M4‐cholate/M0‐cholate relative to baseline measurements after administration of [2H4]cholate and is expressed as the natural logarithm of the atom percent excess (ln APE) value. The decay of ln APE (atom per cent excess) over time was calculated by linear regression analysis. From the linear decay curve, the fractional turnover rate and pool size of cholate were calculated. The fractional turnover rate per day equals the slope of the regression line multiplied by 24.
The pool size (nmol·g−1 BW) was determined according to the following formula:
Pool size = D × b × 100/ea ‐ D, where D is the amount of label administered, b is the isotope purity, and a is the intercept on the y axis of ln APE versus time curve.
2.12.2. Enterohepatic cycling time
The cholate cycling time, that is, the time it takes the cholate pool to circulate once in the enterohepatic circulation, was calculated by dividing the cholate pool size (nmol·g−1 BW) by the biliary secretion rate of cholate (nmol·g−1 BW/h). The cholate biliary secretion rate was calculated by multiplying the bile flow (μl·g−1 BW/h) by the cholate concentration (nmol·L−1) in a single 30‐min fraction, obtained from 5 to 35 min after cannulation of the gall bladder. The cholate cycling frequency is equal to the cholate cycling time divided by 24.
2.13. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015; Curtis et al., 2018). Results are expressed as mean ± SEM for the indicated number of independent experiments. The PLS‐DA was used in multivariate analysis of BA profile. Student's unpaired two‐tailed t test was used for comparison between two groups of mice. P values less than .05 were considered statistically significant.
2.14. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski, et al., 2017; Alexander, Fabbro, et al., 2017; Alexander, Kelly, et al., 2017).
3. RESULTS
3.1. UDCA has a distinct effect on BA pool size and composition
To explore the effects of UDCA on BA enterohepatic circulation and its difference between other BAs in vivo, we measured the BAs in the serum, liver, gall bladder, small intestine, caecum, colon, and the contents of various intestinal segments from mice that were given either UDCA or other BAs in their food. The total BAs in these tissues comprised the BA pool. The results of a multivariate analysis (PLS‐DA), groups of mice that were administered different BAs exhibited distinctly different BA profiles especially when comparing the groups fed UDCA and DCA (Figure 1a). To further characterize the difference, we compared the BA pool size, composition, distribution, and excretion after BA supplementation. The BA pool size was sharply decreased after UDCA supplementation (Figure 1b), while other BAs did not cause a significant change. In the control group, the proportion of primary BAs was slightly higher than that of secondary BAs (Figure 1c), and the ratio of 12‐OH‐BAs (cholic acid [CA] and its derivatives) to non‐12‐OH‐BAs ( CDCA and its derivatives) was almost 3:7 (Figure S1A). After dietary supplementation with UDCA, the composition of the BA pools was significantly altered, which was demonstrated by the proportion of TUDCA, which increased to 44.1%, while that of taurocholic acid (TCA) decreased to 2.99% (Figure 1c), and secondary BAs became more predominant than primary BAs (Figure 1c) and non‐12‐OH‐BAs occupied 95% of the BA pool (Figure S1A). In contrast, the primary BA proportion increased to 73.7% in the CA group and 76.2% in the DCA group (Figure 1c), in which taurocholic acid was detected with 43.4% and 52.0% in the CA and DCA groups respectively (Figure 1c). The proportions of primary and secondary BAs showed a little change in the CDCA supplemented group (Figure 1c), but non‐12‐OH‐BAs were significantly increased (Figure S1A).
Figure 1.
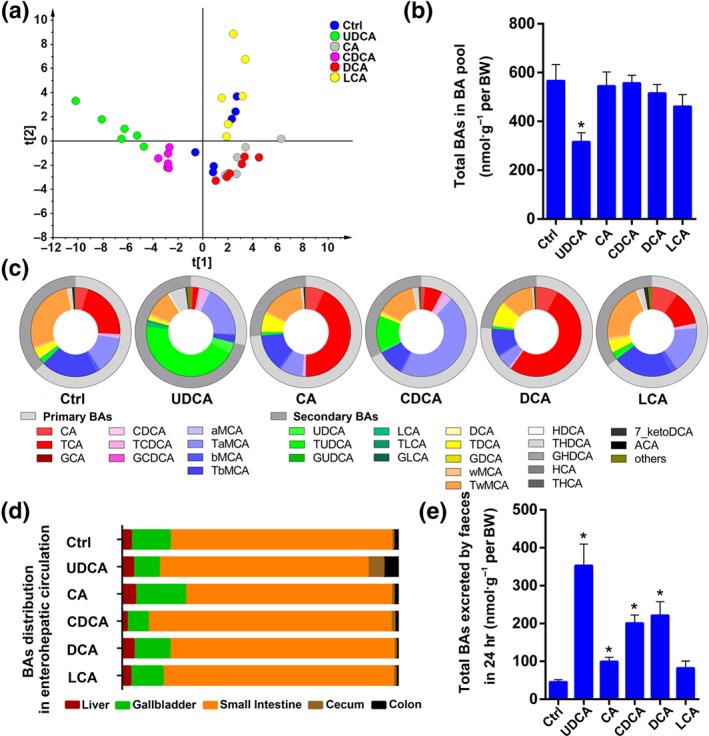
UDCA has a distinct effect on bile acid pool. (a) The multivariate analysis model (PLS‐DA) based on the bile acid profile of the bile acid pool shows that groups of mice that were administered different bile acids were clearly separated, especially the UDCA and DCA group (n = 6 per group). (b) The total concentration of BAs per body weight in the BA pool was shown to be different depending on specific BA supplementation; 44 bile acids were detected and quantitated in different organs and tissues. The BA pool is equal to the sum of the total bile acids in liver, gall bladder, small intestine, caecum, colon, and serum (n = 6 per group). (c) The bile acid composition of bile acid pool. The outside circle shows the proportion of primary and secondary bile acids in the bile acid pool. The inner circle shows 28 individual bile acid proportions and the sum of 16 low content bile acid proportions of the bile acid pool (n = 6 per group). (d) The bile acid distribution of the bile acid pool in the enterohepatic circulation (n = 6 per group). (e) The amount of BA excreted in faeces within 24 hr (n = 6 per group). Mean values ± SEM are expressed for the bar chart. *P < .05 versus normal control group
Aside from BA pool composition, we also calculated the total BAs in different organs involved in enterohepatic circulation and compared their distribution (Figure 1d). In all groups, most BAs were distributed in the small intestine and stored in gall bladder. However, the proportion (Figure 1d) and amount of total BAs distributed in the caecum and colon were significantly increased in the UDCA supplemented group. In addition, the amount of total BAs excreted through faeces within 24 hr was dramatically elevated after UDCA supplementation (Figure 1e). In summary, UDCA intervention caused unique effects on the BA pool including changes in size, composition, distribution, and excretion.
3.2. UDCA induces the expression of BA synthetase in the liver and BA transporters in the ileum
In order to explore the reason why the BA pool was different after UDCA intervention compared to other BAs, we performed mRNA detection of BA synthetic enzymes in the liver and BA transporters in the ileum along with the relative protein expression of the transporters, apical sodium‐dependent BA transporter (ASBT), and the carrier protein, ileal BA binding protein (IBABP). Samples from a second BA group, DCA, were also examined as an intervention control as its BA pool composition displayed the greatest variance to that of the UDCA group in the prior study.
The mRNAs of enzymes involved in the classic BA‐synthetic pathway as well as the alternative BA‐biosynthetic pathway were quantified. Cholesterol degradation to BAs in the liver can be initiated by cholesterol 7α‐hydroxylase (CYP7A1) and sterol 12α‐hydroxylase (CYP8B1) of the classic (neutral) pathway or by mitochondrial sterol 27‐hydroxylase (CYP27A1) and oxysterol 7α‐hydroxylase (CYP7B1) of the alternative (acidic) pathway (Chiang, 2004; Chiang, 2013), synthesizing two primary BAs, CA and CDCA, in humans. In the classic pathway, CYP8B1 is required for synthesis of CA, but without 12α‐hydroxylase transformation, the product is CDCA, and initiation through the alternative pathway also results in the production of CDCA (Chiang, 2013).
After BA supplementation, CYP7A1 and CYP8B1 mRNA expression had almost no change in the UDCA experimental group, while in the DCA group exhibited significant enzyme suppression (Figure 2a). However, the UDCA experiment group did show a significant induction of CYP7B1, the enzyme involved in the alternative BA‐biosynthetic pathway (Figure 2a), which was consistent with its BA pool composition in which non‐12‐OH‐BAs were increased (Figure S1A).
Figure 2.
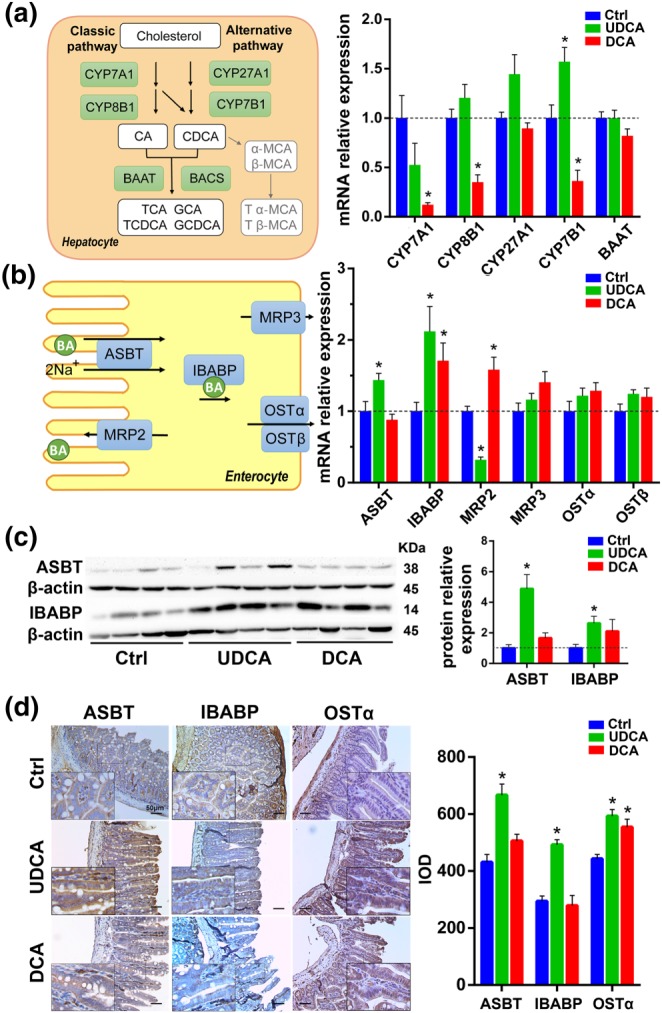
UDCA promotes bile acid synthesis, especially in the alternative pathway and induces the expression of bile acid transporter genes and proteins in the ileum. (a) Diagram and mRNA analysis of bile acid biosynthesis markers in the liver (n = 6 per group). (b) Diagram and mRNA analysis of bile acid transporters in the ileum (n = 6 per group). (c) Protein expression levels of bile acid transporters in the ileum and densities of protein levels relative to β‐actin shown as fold change relative to the control group (n = 6 per group). (d) Representative pictures of immunohistochemistry of bile acid transporter proteins in the ileum and integrated option density shown as a bar chart (n = 6 per group). Mean values ± SEM are expressed for the bar chart. *P < .05 versus normal control group
In enterohepatic circulation, transporters play a critical role in maintaining BA homeostasis, so next we quantified the BA transporters in the ileum and liver. The gene of the main transporter involved in BA active reabsorption in the terminal ileum, ASBT (Hagenbuch & Dawson, 2004), was up‐regulated in the UDCA group (Figure 2b). Multidrug resistance‐associated protein 2 (MRP2), another transporter presenting at the apical membrane of enterocytes that ensures apical excretion of BA (Thomas, Pellicciari, Pruzanski, Auwerx, & Schoonjans, 2008), was down‐regulated in the UDCA group (Figure 2b). IBABP, the carrier protein which binds BAs and promotes BA flux (Dawson, Lan, & Rao, 2009), was also induced by UDCA supplementation (Figure 2b). The protein level of heteromeric organic solute transporter α (OSTα), a major BA efflux pump located in the ileal basolateral membrane (Dawson et al., 2005), was up‐regulated as measured by immunohistochemistry (IHC) after UDCA supplementation (Figure 2d). The protein expression of other BA transporters in the ileum was further examined by western blot (WB) and IHC (Figure 2c,d), confirming that protein levels were consistent with changes seen in gene expression for the UDCA group. For example, WB and IHC showed a significant elevation in ASBT and IBABP levels relative to control in UDCA group. In the DCA group, ASBT showed no changes in mRNA expression by RT‐PCR (Figure 2b), WB (Figure 2c), or IHC (Figure 2d). However, the mRNA expression of MRP2 in the DCA group was significantly up‐regulated in contrast to the UDCA supplemented group (Figure 2b). Together, these results indicated that UDCA induced the expression of BAs transporters in the ileum and had not repressed the BAs synthesis.
3.3. UDCA induces the expression of BA transporters in the liver
BAs returning to the liver via the portal vein are taken up by the Na+‐taurocholate cotransporting polypeptide (NTCP) and to a lesser extent by organic anion transporter 1(OATP1, Slco1a1; Thomas et al., 2008). Both NTCP and OATP1 transporters were up‐regulated by UDCA supplementation both in mRNA expression (Figure 3a) and protein levels (Figure 3b,c). The canalicular bile salt export pump (BSEP) is the predominant transporter responsible for the excretion of monovalent BAs into the bile through the canalicular membrane, and MRP2 mediates canalicular membrane transport of divalent BAs (Trauner & Boyer, 2003). After UDCA intervention, both BSEP and MRP2 mRNA (Figure 3a) and protein (Figure 3b,c) expression were induced. In the DCA group, OATP1 and BSEP showed almost no changes in mRNA or protein level (Figure 3a–c). However, NTCP and MRP2 results were not consistent between different detection methods. Meanwhile, we found that total BA concentration in the liver tended to increase in the DCA group (Figure S1B) while BA synthesis was repressed (Figure 2a); therefore, we hypothesized that BA levels in the liver resulted from the observed changes in hepatic transporter protein levels. When there is hepatocellular BA overload, alternative basolateral BA transporters, such as multidrug resistance‐associated protein 3 (MRP3), multidrug resistance‐associated protein 4 (MRP4), organic anion transporting polypeptide 2 (OATP2, Slco1a4), and OSTα/β, can transport excess BAs to the systemic circulation (Halilbasic et al., 2013; Thomas et al., 2008). DCA did in fact induce the mRNA expression of OSTβ (Figure 3a), which was consistent with the finding of significantly increased total BAs concentration in serum (Figure S1C). However, an increased level of MRP4 was induced in UDCA group (Figure 3a) but the total BA concentration in the serum and liver was unchanged (Figure S1C), which may be due to the up‐regulated BSEP and MRP2.
Figure 3.
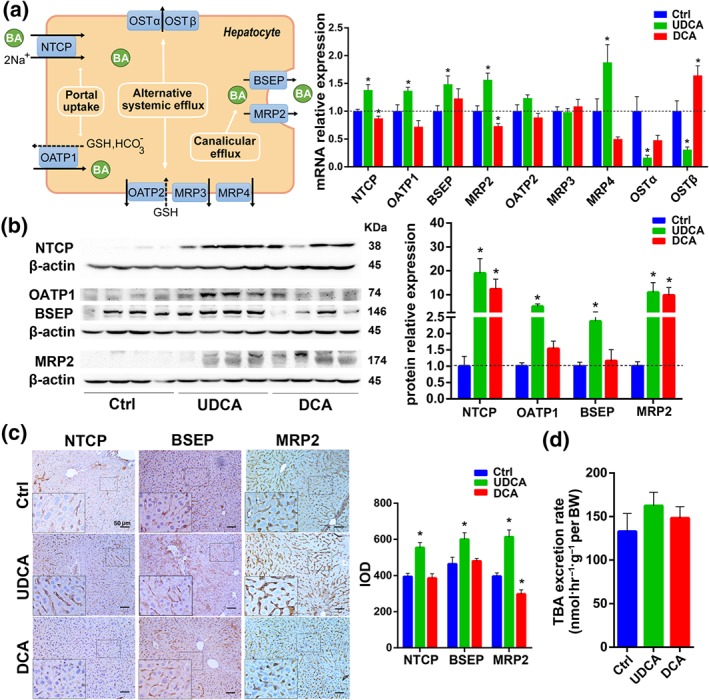
UDCA induces the expression of bile acid transporter genes and protein in the liver. (a) Diagram and mRNA analysis of bile acid transporters in the liver, OATP1 (Slco1a1), OATP2 (Slco1a4; n = 6 per group). (b) Protein expression levels of bile acid transporters in the liver and densities of protein levels relative to β‐actin shown as fold change relative to the control group (n = 6 per group). (c) Representative pictures of immunohistochemistry of bile acid transporter proteins in liver and integrated option density shown as a bar chart (n = 6 per group). (d) Total bile acid secretion rates by bile flow (n = 6 per group). Mean values ± SEM are expressed for the bar chart. *P < .05 versus normal control group
In conclusion, we found that (a) after UDCA intervention, BA transporters in the ileum such as ASBT, IBABP, and OSTα/β were up‐regulated (Figure 2), which enhanced BA reabsorption from the ileum, while in the liver, administration of UDCA induced NTCP, OATP1, BSEP, and MRP2 (Figure 3), which led to both increased BA uptake from the portal vein to the liver and increased BA efflux from the liver to the cholangiocytes. (b) In addition, CYP27A1 and CYP7B1 up‐regulation was induced after UDCA feeding (Figure 2a), implying that the synthesis of bile acids was not inhibited but rather enhanced through the alternative synthetic pathway. Considering the size of the BA pool was decreased in the UDCA group (Figure 1b), the results seem to indicate an increased BA output that is even greater than their increased synthesis. Indeed, BAs excreted in the faeces were increased (Figure 1d) and could not be attributed to reduced reabsorption of BAs due to the higher expression of ASBT. The smaller BA pool size may therefore be the result of a highly enhanced bile efflux. (c) The total BA secretion rate through bile was somewhat higher than control (Figure 3d), consistent with the up‐regulated expression of BSEP and MRP2 in the liver (Figure 3a–c), but the difference was not statistically significant (Figure 3d). From the above results, we hypothesized that UDCA administration accelerates the BA enterohepatic circulation frequency and that this is manifested by increased BA output and decreased BA pool size. We therefore employed an isotope dilution technique in a mouse model to test this hypothesis.
3.4. UDCA accelerates the BA enterohepatic circulation frequency
Isotope dilution techniques applying radioactive or stable isotopes in vivo have been accepted as the preferred method to study BA kinetics (Hulzebos et al., 2001; Kok et al., 2003; Koopman et al., 1988; Stellaard, Sackmann, Sauerbruch, & Paumgartner, 1984). CA decay curves measured in serum exhibited first‐order kinetics (Figure 4a). The average linear regression correlation coefficients were 0.9393 (P < .05) in the control group, 0.8119 (P < .05) in the UDCA group, and 0.7674 (P < .05) in the DCA group. Mean values of the slopes of the ln APE‐versus‐time curves determined in individual mice were represented as the fractional turnover rate (Figure 4b), among which UDCA administration induced the highest fractional turnover rate. Calculated from the y‐intercept of the linear regression line, CA pool size was shown (Figure 4c). In agreement with the previous data that administration of UDCA led to a smaller BA pool size (Figure 1b), it resulted in a much smaller CA pool size for this experiment (Figure 4c), as well. UDCA supplementation also resulted in less CA cycling time and faster CA cycling frequency (Figure 4d,e). Under the assumption that all bile salt species would display a similar cycling frequency to CA, we can use these calculations to compare BA cycling kinetics (Hulzebos et al., 2001; Kok et al., 2003; Koopman et al., 1988; Stellaard et al., 1984). These results, therefore, demonstrate that UDCA does indeed accelerate the BA enterohepatic circulation frequency in vivo.
Figure 4.
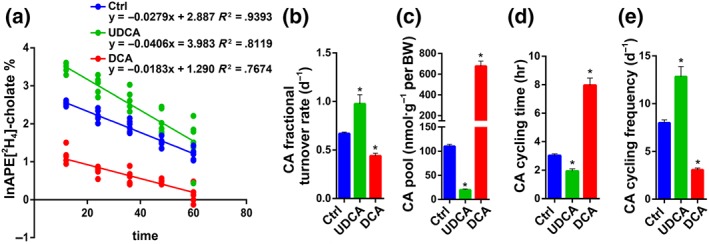
UDCA accelerates the bile acids enterohepatic circulation. (a) Decay of i.v. administered [2H4]‐cholate in the mice (n = 6 per group). Fractional turnover rate (b) (n = 6 per group), pool size (c) (n = 6 per group), cycling time (d) (n = 6 per group), and cycling frequency for 24 hr (e) (n = 6 per group) of cholate as derived from [2H4]‐cholate isotope enrichment measurements in serum of the mice were shown. Data are mean values ± SEM. *P < .05 versus normal control group
3.5. Conjugated UDCAs are antagonists of intestinal FXR in vivo and in vitro
To explore the mechanism by which UDCA accelerates BA enterohepatic circulation in healthy mice, we measured both mRNA and protein levels for the BA nuclear receptor, FXR, considered the master transcriptional regulator of BA homeostasis. FXR acts as a transcription factor for BA synthetic enzymes and transporter proteins thereby maintaining homeostasis through BA synthesis, influx, and efflux (Halilbasic et al., 2013).
In vivo experiments revealed that intestinal FXR mRNA expression was repressed significantly by UDCA administration (Figure 5a). Similarly, intestinal FXR protein expression was also reduced after UDCA feeding (Figure 5b). In the IHC staining of ileal tissue sections, we observed that the downstream partners of FXR, small heterodimer partner (SHP), and FGF15 protein levels were decreased (Figure 5c), which was consistent with the change of FXR (Figure 5a,b). On the contrary, for the DCA group, intestinal FXR, SHP, and FGF15 were up‐regulated, either in mRNA or protein expression (Figure 5a–c). In the liver, FXR and SHP mRNA expression and protein level were unchanged in UDCA group (Figure 5a,b). In summary, the intestinal FXR signalling pathway was inhibited after UDCA feeding, while liver FXR signalling was not.
Figure 5.
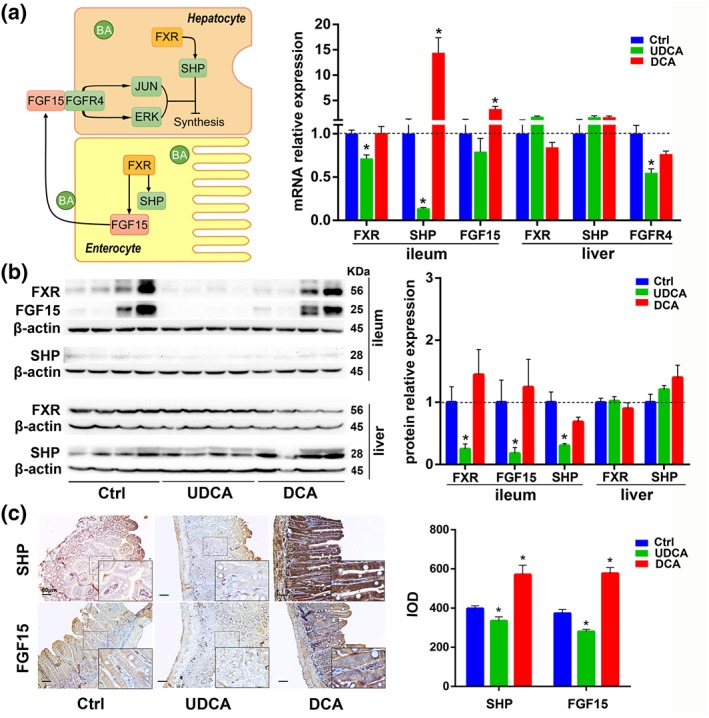
The influence of UDCA on the FXR signalling pathway in vivo. (a) Diagram and mRNA analysis of FXR signalling pathway components in the ileum and liver (n = 6 per group). (b) Protein expression levels of FXR signal pathway components in the ileum and liver and densities of protein levels relative to β‐actin shown as fold change relative to control group (n = 6 per group). (c) Representative pictures of immunohistochemistry staining of FXR signal pathway components in the ileum and integrated option density shown as a bar chart (n = 6 per group). Data are mean values ± SEM. *P < .05 versus normal control group
To further verify the effect of BAs on FXR and BA transporters, we added UDCAs and DCAs, mimicking the BA profiles derived from UDCA or DCA administration in vivo (Figure S2A–C), to 3D and 2D cultures of enterocytes and hepatocytes. We found that treatment with UDCAs which contained predominantly TUDCA and GUDCA inhibited FXR significantly in intestinal cell cultures, with a simultaneous down‐regulation of FGF19 (Figure 6a,b). In the same cells, the main BA transporter, ASBT, was up‐regulated (Figure 6a). Next, we used UDCA and its derivatives (50 μmol·L−1) to treat 2D and 3D cultures of enterocytes respectively and found that TUDCA was the most potent inhibitor of FXR expression (Figure S3A–C). In liver cell culture, FXR was also found to be inhibited by UDCAs (Figure S4A,B), and further, we found that the transporter NTCP was up‐regulated but BSEP was down‐regulated (Figure S4A). The change in transporters we observed in liver cell culture was consistent with previously findings observed when liver FXR is repressed (Alrefai & Gill, 2007; Thomas et al., 2008; Halilbasic et al., 2013) but was not in accordance with what we saw for BSEP in vivo (Figure 3a–c). From the above, it may be inferred that some other factors participate in controlling some of the BA transporters in liver cells.
Figure 6.
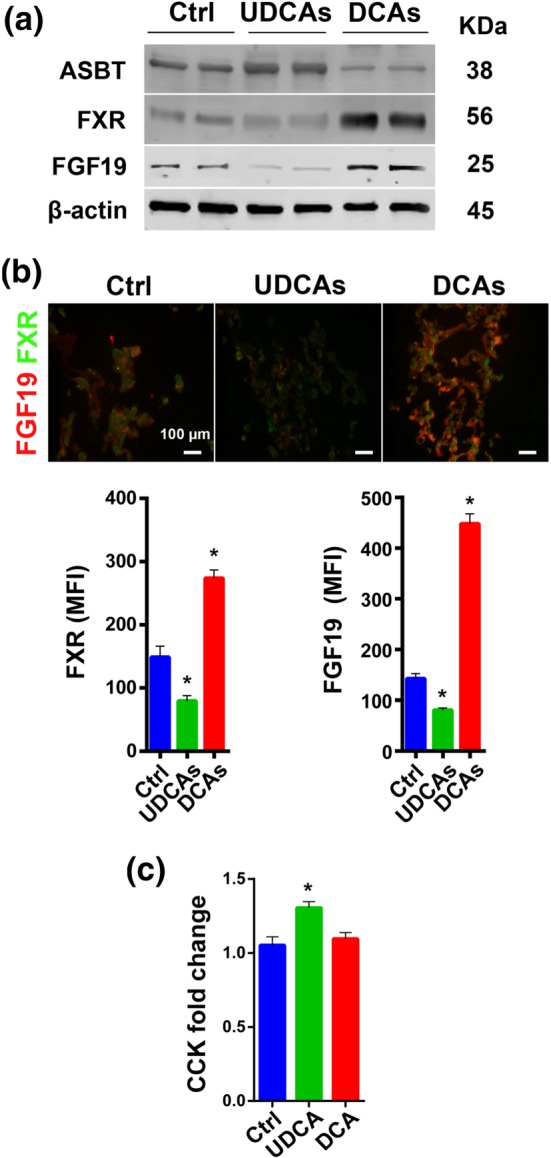
The influence of UDCAs on the FXR signalling pathway in vitro and CCK fold change in vivo. (a) Representative pictures of protein expression of the FXR signalling pathway components and the bile acid transporter ASBT from six samples each group in 3D cultured intestine cells. (b) Immunofluorescent staining of FXR signalling pathway components in 3D cultured intestine cells and mean fluorescence intensity shown as a bar chart (FGF19 n = 15 per group, FXR n = 6 per group). (c) Fold change in CCK postprandial versus fasting condition (n = 5 per group). (UDCAs means the mixture of TUDCA, GUDCA, UDCA, TLCA, and LCA at a concentration of 50, 50, 12.5, 2.5, and 0.25 μM; DCAs means the mixture of TCA, TDCA, GDCA, and DCA at a concentration of 50, 12.5, 12.5, and 0.5 μM.) Data are mean values ± SEM. *P < .05 versus normal control group
3.6. FGF15/19 regulates liver BA transporters in vivo and in vitro and represses cholecystokinin level
FGF15/19 (FGF15 in mouse and FGF19 in human), is a gut‐derived hormone which upon binding to its receptor FGF receptor‐4 (FGFR4) in the liver acts to down‐regulate BA synthesis (Inagaki et al., 2005). Our results showed that level of FGF15 was down‐regulated in the UDCA group from the in vivo study (Figure 5c), so we treated cultured liver cells with FGF19 at different concentrations (20, 80, and 160 ng·ml−1) to observe its influence on BA transporters. As a result, liver BA transporters MRP2 and BSEP were inhibited by FGF19 in a dose‐dependent manner (Figure 7a). Quantification of the mean fluorescence intensity, using a 160‐ng·ml−1 FGF19 cell treatment, also indicated that both MRP2 and BSEP were inhibited (Figure 7b). We also investigated liver BA transporters in vivo after intraperitoneal injection of FGF19 in mice and observed its influence. We found that the mRNA of BA synthetic enzyme CYP7A1 was inhibited 2 hr after the FGF19 injection, and at the same time, several BA transporters were also inhibited (Figure 7c). Evidence was shown that FGF15/19 inhibited expression of hepatic BA uptake transporters NTCP and OATPs both in vivo and in vitro (Slijepcevic, Roscam Abbing, Katafuchi, & Blank, 2017). In our study, we observed that FGF15/19 repressed MRP2 and BSEP both in vivo and in vitro (Figure 7a–c). Based on these results and that level of FGF15 decreased significantly in the UDCA fed mice, we conclude that FGF15 is a factor in the regulation of BA transporters in the liver. Moreover, FGF15 has been reported to be required for gall bladder filling via its function of inhibiting cholecystokinin (CCK) levels (Choi et al., 2006). In the mouse study, we found that the size of the gall bladder in the UDCA group was smaller, especially when compared with the DCA group (Figure S5A,B). Quantification of the CCK level in different treatment groups revealed that UDCA intervention led to a much higher fold change in CCK levels between the postprandial and fasted status (Figure 6c) which corresponded to the decreased FGF15 levels (Figure 5c). After intraperitoneal injection of FGF19 in vivo, the weight of gall bladder increased (Figure 7d).
Figure 7.
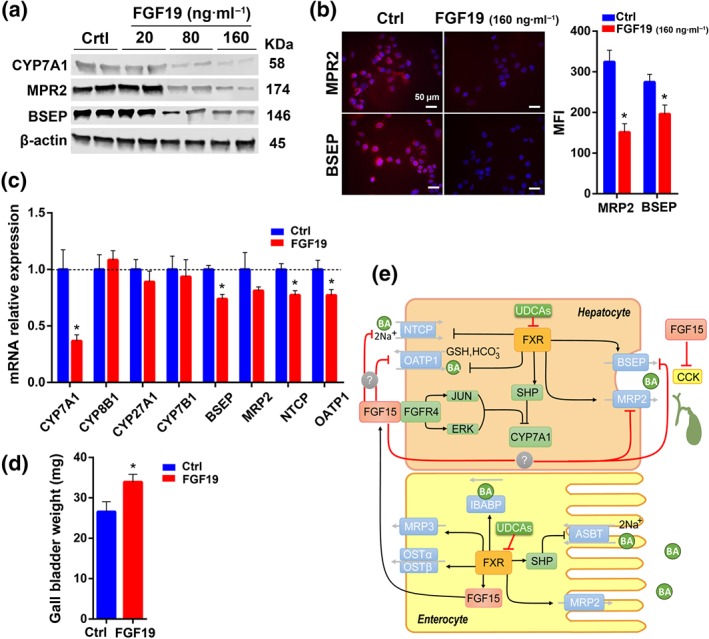
The influence of FGF15/19 in regulating BA transporters in vivo and in vitro. (a) Representative pictures of protein expression of BA transporters in 2D cultured liver cells from six samples each group treated with different concentrations of FGF19. (b) Immunofluorescent staining of BA transporters in 2D cultured liver cells after treatment with FGF19 and mean fluorescence intensity shown as a bar chart (n = 6 per group). (c) mRNA analysis of BA synthetic enzymes, and bile acid transporters in liver after treatment with FGF19 (100 μg·kg−1 per BW) (n = 6 per group). (d) Gall bladder weight after i.p. injection of FGF19 (n = 6 per group). (e) A diagram of the proposed mechanism for UDCA‐induced acceleration of enterohepatic circulation. Data are mean values ± SEM. *P < .05 versus normal control group
In conclusion, oral administration of UDCA, or taurine‐/glycine‐conjugated UDCA derivatives in vitro, were shown to inhibit intestinal FXR‐SHP and FXR‐FGF15 (Figures 5 and 6). Reduced SHP and FGF15, in turn, induced more expression of ASBT and promoted increased BA reabsorption from the ileum (Li et al., 2005). With the reduction of FGF15 level, the liver transporters, NTCP, BSEP, and MRP2 were induced (Figure 3), although liver FXR signalling showed no change (Figure 5b). The increased expression of NTCP, BSEP, and MRP2 thereby facilitated BA influx and efflux in the liver. Furthermore, decreased level of FGF15 led to enhanced gall bladder contraction along with increased CCK levels (Figure 6c) which promoted bile release from the gall bladder to the small intestine. In summary, FGF15/19 plays an important role in regulating BA synthesis and transport, and intestinal FXR is important for overall BA homeostasis.
4. DISCUSSION
In this study, we showed that UDCA supplementation led to a smaller BA pool and enhanced the expression of BA transporters involved in the enterohepatic circulation, including ASBT for reabsorption of BAs in the distal ileum and NTCP, MRP2, and BSEP for uptake and excretion of BAs in the liver. Several years ago, there were a number of studies in the field that were focused on hepatic BA transporters after oral UDCA administration (Marschall et al., 2005; Paumgartner & Beuers, 2004). However, in the present study, we evaluated the effect of oral UDCA administration on BA transporters in the ileum in conjunction with the liver, in addition to an examination of the kinetics associated with BA enterohepatic circulation. We found that reabsorption and excretion of BAs were both enhanced upon UDCA administration. A calculation of the BA circulation frequency using widely accepted isotope dilution techniques demonstrated that UDCA actually accelerates BA circulation in vivo.
In order to examine how UDCA might accelerate BA enterohepatic circulation, we examined the expression of various FXR transcriptional targets in both the ileum and liver, as BA transporters are both directly and indirectly regulated by the FXR nuclear receptor (Halilbasic et al., 2013). We verified that oral administration of UDCA antagonized intestinal FXR signalling. In vitro studies showed that FXR was not inhibited by UDCA alone, but by a mixture of UDCA derivatives that mimicked the BA composition derived from UDCA administration in vivo. After oral administration of UDCA, TUDCA increased dramatically, while MCAs remained unchanged or slightly decreased (Figure S7). Conjugated MCAs are thought to be FXR antagonists (Jiang et al., 2015). Our results suggest that UDCA conversion to MCAs was minimal in this study, and the suppression of FXR signalling was attributed to conjugated UDCA species such as TUDCA. ASBT was found to be negatively regulated by ileal FXR‐SHP (Li et al., 2005; Neimark, Chen, Li, & Shneider, 2004), which was consistent with our finding that ASBT was induced after UDCA suppression of ileal FXR‐SHP expression. The inconsistency in the results for hepatic FXR signalling in vivo and in vitro brought our attention to the gut‐derived hormone FGF15/19 that linked the crosstalk between the gut and liver (Inagaki et al., 2005). Evidence was shown that FGF15/19 inhibits expression of hepatic BA uptake transporters NTCP and OATPs both in vivo and in vitro (Slijepcevic et al., 2017). In our study, we observed that the hepatic BA transporters, including NTCP, OATP1, BSEP, and MRP2, were down‐regulated after FGF19 administration both in vivo and in vitro. This implied that FGF15/19 was involved in regulating BA transporters in the liver. The use of FGF19 to replace FGF15 was a limitation in this study, although FGF19 was previously used to mimic FGF15 in mouse (Slijepcevic et al., 2017). In addition, it was previously reported that FGF15/19 causes gall bladder filling via inhibition of CCK (Choi et al., 2006). In this study, UDCA administration led to lower level of FGF15, and higher level of serum CCK, which corresponded to smaller gall bladders and increased bile flow in UDCA‐treated mice. The lower level of FGF15 and higher level of CCK in vivo would strengthen gall bladder contraction and contribute to the acceleration of BA enterohepatic circulation.
UDCA, the first Food and Drug Administration approved treatment for PBC, has also been used for non‐cholestatic as well as non‐hepatobiliary diseases (Roma et al., 2011). It has also been shown to attenuate colon carcinogenesis in both humans and in animal models (Krishna‐Subramanian et al., 2012). Additionally, it is thought to play a protective role in bile‐induced pancreatic ductal injury (Katona et al., 2016) and has been evaluated for the treatment of non‐alcoholic steatohepatitis, although the therapeutic effect remains controversial (Dufour et al., 2006; Laurin et al., 1996; Leuschner et al., 2010; Lindor et al., 2004; Mueller et al., 2015; Ratziu et al., 2011). Even though UDCA is widely used for PBC, and also for more common ailments, such as gallstones or obstetric cholestasis, very little study has been done regarding its mechanism of action, especially in organisms without prior liver disease. In the present investigation, we found that at least one of these mechanisms is to accelerate enterohepatic circulation via an intestinal FXR signalling pathway. This is a critical finding as enterohepatic circulation has profound effects on the pharmacology and toxicology of everything that is processed by the liver and excreted into the bile. In addition, the involvement of the FXR receptor is important as its impact is seen not only in BA homeostasis but as an important regulator of hepatic triglycerides and intestinal fluid equilibrium. In addition to our finding of UDCA's influence on FXR, we showed that the UDCA derived TUDCA was an even more potent inhibitor of this pathway in intestinal cells in vitro, which is consistent with a study recently published (Sun et al., 2018). There have been some reports which have indicated that TUDCA has the ability to decrease ER stress, act as a leptin‐sensitizing agent, and thus may provide a novel treatment for obesity (Ozcan et al., 2009). Along this line, Nor‐UDCA is another derivative of UDCA, which has been approved for the attenuation of non‐alcoholic steatohepatitis progression (Beraza et al., 2011). Considering the impact on critical enterohepatic processes and signalling pathways, UDCA derivatives may yet have untapped therapeutic potential for the treatment of other still unidentified diseases in the gut–liver axis. The fact that this study used male mice only is a limitation. Future studies should include both male and female mice and compare the metabolic differences between the two genders. In the meantime, an important next step will be to further characterize human serum and other samples to test that our results are indeed translational.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Y.Z. drafted the manuscript, conducted animal experiments and measurements of BAs and other protein and metabolite markers, and analysed the data. R.J. performed the cell experiments, immunohistochemistry and immunofluorescence experiments, and analysed the data. X.Z., S.L., and F.H. participated in animal experiments and measurement of BAs. G.X., S.K., H.Y., C.F., B.S., A.Z., and W.J. revised the manuscript. W.J. was the project leader and designed the study.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
Bile acids in different tissues.
Figure S2. Bile acids composition and fold change in small intestine content and in liver.
Figure S3. The influence of individual bile acid on FXR signal pathway in enterocyte.
Figure S4. The change of hepatic FXR signal pathway in vitro.
Figure S5. The gallbladder picture and gallbladder weight in different bile acids feeding group.
Figure S6. The body weight, food intake, and liver weight in each animal experiment.
Figure S7. The concentration of bile acids.
Figure S8. The cytotoxicity assays of UDCAs and DCAs in liver and intestine cell by Cell Counting Kit‐8.
Figure S9. The mRNA expression of BA synthetase and transporters in liver at different time point after pentobarbital sodium intraperitoneal injection.
Figure S10. Supplementary pictures of protein expressions of main BA transporters (A, B) involved in enterohepatic circulation and FXR signaling pathway components in liver and in ileum (C).
ACKNOWLEDGEMENTS
This work was supported by the National Key R&D Program of China (Grant 2017YFC0906800), National Natural Science Foundation of China (Grants 81772530, 31500954, 81772569, and 81572370), and International Science and Technology Cooperation Program of China (Grant 2014DFA31870).
Zhang Y, Jiang R, Zheng X, et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. Br J Pharmacol. 2019;176:2848–2863. 10.1111/bph.14705
Yunjing Zhang and Runqiu Jiang share first authorship.
REFERENCES
- Alexander, S. P. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174(Suppl 1), S208–s224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174(Suppl 1), S360–s446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alrefai, W. A. , & Gill, R. K. (2007). Bile acid transporters: Structure, function, regulation and pathophysiological implications. Pharmaceutical Research, 24(10), 1803–1823. 10.1007/s11095-007-9289-1 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals andpracticalities of immunoblotting and immunohistochemistry: a guide for submission to the british journal of pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraza, N. , Ofner‐Ziegenfuss, L. , Ehedego, H. , Boekschoten, M. , Bischoff, S. C. , Mueller, M. , … Trautwein, C. (2011). Nor‐ursodeoxycholic acid reverses hepatocyte‐specific nemo‐dependent steatohepatitis. Gut, 60(3), 387–396. 10.1136/gut.2010.223834 [DOI] [PubMed] [Google Scholar]
- Chiang, J. Y. (2004). Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. Journal of Hepatology, 40(3), 539–551. 10.1016/j.jhep.2003.11.006 [DOI] [PubMed] [Google Scholar]
- Chiang, J. Y. (2013). Bile acid metabolism and signaling. Comprehensive Physiology. 10.1002/cphy.c120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, M. , Moschetta, A. , Bookout, A. L. , Peng, L. , Umetani, M. , Holmstrom, S. R. , … Kliewer, S. A. (2006). Identification of a hormonal basis for gallbladder filling. Nature Medicine, 12(11), 1253–1255. 10.1038/nm1501 [DOI] [PubMed] [Google Scholar]
- Chow, M. D. , Lee, Y. H. , & Guo, G. L. (2017). The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Molecular Aspects of Medicine, 56, 34–44. 10.1016/j.mam.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … McGrath, J. C. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP. British Journal of Pharmacology, 172(14), 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P. A. , Hubbert, M. , Haywood, J. , Craddock, A. L. , Zerangue, N. , Christian, W. V. , & Ballatori, N. (2005). The heteromeric organic solute transporter α‐β, Ostα‐Ostβ, is an ileal basolateral bile acid transporter. Journal of Biological Chemistry, 280(8), 6960–6968. 10.1074/jbc.M412752200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, P. A. , Lan, T. , & Rao, A. (2009). Bile acid transporters. Journal of Lipid Research, 50(12), 2340–2357. 10.1194/jlr.R900012-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermadi, D. , Valo, S. , Ollila, S. , Soliymani, R. , Sipari, N. , Pussila, M. , … Nyström, M. (2017). Western diet deregulates bile acid homeostasis, cell proliferation, and tumorigenesis in colon. Cancer Research, 77(12), 3352–3363. 10.1158/0008-5472.CAN-16-2860 [DOI] [PubMed] [Google Scholar]
- Dufour, J. F. , Oneta, C. M. , Gonvers, J. J. , Bihl, F. , Cerny, A. , Cereda, J. M. , … Swiss Association for the Study of the Liver (2006). Randomized placebo‐controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clinical Gastroenterology and Hepatology: The Official Clinical Practice Journal of the American Gastroenterological Association, 4(12), 1537–1543. 10.1016/j.cgh.2006.09.025 [DOI] [PubMed] [Google Scholar]
- Hagenbuch, B. , & Dawson, P. (2004). The sodium bile salt cotransport family SLC10. Pflügers Archiv, 447(5), 566–570. 10.1007/s00424-003-1130-z [DOI] [PubMed] [Google Scholar]
- Halilbasic, E. , Claudel, T. , & Trauner, M. (2013). Bile acid transporters and regulatory nuclear receptors in the liver and beyond. Journal of Hepatology, 58(1), 155–168. 10.1016/j.jhep.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–d1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulzebos, C. V. , Renfurm, L. , Bandsma, R. H. , Verkade, H. J. , Boer, T. , Boverhof, R. , … Stellaard, F. (2001). Measurement of parameters of cholic acid kinetics in plasma using a microscale stable isotope dilution technique: Application to rodents and humans. Journal of Lipid Research, 42(11), 1923–1929. [PubMed] [Google Scholar]
- Inagaki, T. , Choi, M. , Moschetta, A. , Peng, L. , Cummins, C. L. , McDonald, J. G. , … Kliewer, S. A. (2005). Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metabolism, 2(4), 217–225. 10.1016/j.cmet.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Jiang, C. , Xie, C. , Lv, Y. , Li, J. , Krausz, K. W. , Shi, J. , … Gonzalez, F. J. (2015). Intestine‐selective farnesoid X receptor inhibition improves obesity‐related metabolic dysfunction. Nature Communications, 6, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovich, A. , Isakova, T. , Block, G. , Stubbs, J. , Smits, G. , Chonchol, M. , & Miyazaki, M. (2018). Deoxycholic acid, a metabolite of circulating bile acids, and coronary artery vascular calcification in CKD. American Journal of Kidney Diseases, 71(1), 27–34. 10.1053/j.ajkd.2017.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona, M. , Hegyi, P. , Kui, B. , Balla, Z. , Rakonczay, Z. Jr. , Razga, Z. , … Venglovecz, V. (2016). A novel, protective role of ursodeoxycholate in bile‐induced pancreatic ductal injury. American Journal of Physiology. Gastrointestinal and Liver Physiology, 310(3), G193–G204. 10.1152/ajpgi.00317.2015 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok, T. , Hulzebos, C. V. , Wolters, H. , Havinga, R. , Agellon, L. B. , Stellaard, F. , … Kuipers, F. (2003). Enterohepatic circulation of bile salts in farnesoid X receptor‐deficient mice efficient intestinal bile salt absorption in the absence of ileal bile acid‐binding protein. Journal of Biological Chemistry, 278(43), 41930–41937. 10.1074/jbc.M306309200 [DOI] [PubMed] [Google Scholar]
- Koopman, B. , Kuipers, F. , Bijleveld, C. , Van Der Molen, J. , Nagel, G. , Vonk, R. , & Wolthers, B. G. (1988). Determination of cholic acid and chenodeoxycholic acid pool sizes and fractional turnover rates by means of stable isotope dilution technique, making use of deuterated cholic acid and chenodeoxycholic acid. Clinica Chimica Acta, 175(2), 143–155. 10.1016/0009-8981(88)90004-6 [DOI] [PubMed] [Google Scholar]
- Krishna‐Subramanian, S. , Hanski, M. L. , Loddenkemper, C. , Choudhary, B. , Pages, G. , Zeitz, M. , & Hanski, C. (2012). UDCA slows down intestinal cell proliferation by inducing high and sustained ERK phosphorylation. International Journal of Cancer, 130(12), 2771–2782. 10.1002/ijc.26336 [DOI] [PubMed] [Google Scholar]
- Laurin, J. , Lindor, K. D. , Crippin, J. S. , Gossard, A. , Gores, G. J. , Ludwig, J. , … McGill, D. B. (1996). Ursodeoxycholic acid or clofibrate in the treatment of non‐alcohol‐induced steatohepatitis: A pilot study. Hepatology, 23(6), 1464–1467. 10.1002/hep.510230624 [DOI] [PubMed] [Google Scholar]
- Leuschner, U. F. , Lindenthal, B. , Herrmann, G. , Arnold, J. C. , Rossle, M. , Cordes, H. J. , … NASH Study Group (2010). High‐dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: A double‐blind, randomized, placebo‐controlled trial. Hepatology, 52(2), 472–479. 10.1002/hep.23727 [DOI] [PubMed] [Google Scholar]
- Li, H. , Chen, F. , Shang, Q. , Pan, L. , Shneider, B. L. , Chiang, J. Y. , … Xu, G. (2005). FXR‐activating ligands inhibit rabbit ASBT expression via FXR‐SHP‐FTF cascade. American Journal of Physiology. Gastrointestinal and Liver Physiology, 288(1), G60–G66. 10.1152/ajpgi.00170.2004 [DOI] [PubMed] [Google Scholar]
- Li, Y. , Tang, R. , Leung, P. S. C. , Gershwin, M. E. , & Ma, X. (2017). Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmunity Reviews, 16(9), 885–896. 10.1016/j.autrev.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Lindor, K. D. , Kowdley, K. V. , Heathcote, E. J. , Harrison, M. E. , Jorgensen, R. , Angulo, P. , … Colin, P. (2004). Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: Results of a randomized trial. Hepatology, 39(3), 770–778. 10.1002/hep.20092 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Lu, H. , Lu, Y.‐F. , Lei, X. , Cui, J. Y. , Ellis, E. , … Klaassen, C. D. (2014). Potency of individual bile acids to regulate bile acid synthesis and transport genes in primary human hepatocyte cultures. Toxicological Sciences, 141(2), 538–546. 10.1093/toxsci/kfu151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima, M. , Okamoto, A. Y. , Repa, J. J. , Tu, H. , Learned, R. M. , Luk, A. , … Shan, B. (1999). Identification of a nuclear receptor for bile acids. Science, 284(5418), 1362–1365. 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- Marschall, H. U. , Wagner, M. , Zollner, G. , Fickert, P. , Diczfalusy, U. , Gumhold, J. , … Trauner, M. (2005). Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology, 129(2), 476–485. 10.1016/j.gastro.2005.05.009 [DOI] [PubMed] [Google Scholar]
- Mcgrath, J. C. & Lilley, E. (2015). Implementing guidelines on reportingresearch using animals (arrive etc.): new requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, M. , Thorell, A. , Claudel, T. , Jha, P. , Koefeler, H. , Lackner, C. , … Trauner, M. (2015). Ursodeoxycholic acid exerts farnesoid X receptor‐antagonistic effects on bile acid and lipid metabolism in morbid obesity. Journal of Hepatology, 62(6), 1398–1404. 10.1016/j.jhep.2014.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neimark, E. , Chen, F. , Li, X. , & Shneider, B. L. (2004). Bile acid‐induced negative feedback regulation of the human ileal bile acid transporter. Hepatology, 40(1), 149–156. 10.1002/hep.20295 [DOI] [PubMed] [Google Scholar]
- Ozcan, L. , Ergin, A. S. , Lu, A. , Chung, J. , Sarkar, S. , Nie, D. , … Ozcan, U. (2009). Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metabolism, 9(1), 35–51. 10.1016/j.cmet.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Parks, D. J. , Blanchard, S. G. , Bledsoe, R. K. , Chandra, G. , Consler, T. G. , Kliewer, S. A. , … Lehmann, J. M. (1999). Bile acids: Natural ligands for an orphan nuclear receptor. Science, 284(5418), 1365–1368. 10.1126/science.284.5418.1365 [DOI] [PubMed] [Google Scholar]
- Paumgartner, G. , & Beuers, U. (2004). Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clinics in Liver Disease, 8(1), 67–81, vi. 10.1016/S1089-3261(03)00135-1 [DOI] [PubMed] [Google Scholar]
- Ratziu, V. , de Ledinghen, V. , Oberti, F. , Mathurin, P. , Wartelle‐Bladou, C. , Renou, C. , … Spénard, J. (2011). A randomized controlled trial of high‐dose ursodesoxycholic acid for nonalcoholic steatohepatitis. Journal of Hepatology, 54(5), 1011–1019. 10.1016/j.jhep.2010.08.030 [DOI] [PubMed] [Google Scholar]
- Roma, M. G. , Toledo, F. D. , Boaglio, A. C. , Basiglio, C. L. , Crocenzi, F. A. , & Pozzi, E. J. S. (2011). Ursodeoxycholic acid in cholestasis: Linking action mechanisms to therapeutic applications. Clinical Science, 121(12), 523–544. 10.1042/CS20110184 [DOI] [PubMed] [Google Scholar]
- Slijepcevic, D. , Roscam Abbing, R. L. P. , Katafuchi, T. , & Blank, A. (2017). Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology, 66(5), 1631–1643. https://doi.org/10.1002/hep.29251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, P. , Rockwell, C. E. , Cui, J. Y. , & Klaassen, C. D. (2015). Individual bile acids have differential effects on bile acid signaling in mice. Toxicology and Applied Pharmacology, 283(1), 57–64. 10.1016/j.taap.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellaard, F. , Sackmann, M. , Sauerbruch, T. , & Paumgartner, G. (1984). Simultaneous determination of cholic acid and chenodeoxycholic acid pool sizes and fractional turnover rates in human serum using 13C‐labeled bile acids. Journal of Lipid Research, 25(12), 1313–1319. [PubMed] [Google Scholar]
- Sun, L. , Xie, C. , Wang, G. , Wu, Y. , Wu, Q. , Wang, X. , … Jiang, C. (2018). Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nature Medicine, 24(12), 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. , Pellicciari, R. , Pruzanski, M. , Auwerx, J. , & Schoonjans, K. (2008). Targeting bile‐acid signalling for metabolic diseases. Nature Reviews Drug Discovery, 7(8), 678–693. 10.1038/nrd2619 [DOI] [PubMed] [Google Scholar]
- Trauner, M. , & Boyer, J. L. (2003). Bile salt transporters: Molecular characterization, function, and regulation. Physiological Reviews, 83(2), 633–671. 10.1152/physrev.00027.2002 [DOI] [PubMed] [Google Scholar]
- Xie, G. , Zhong, W. , Li, H. , Li, Q. , Qiu, Y. , Zheng, X. , … Jia, W. (2013). Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. The FASEB Journal, 27(9), 3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Xie, G. , Zhao, A. , Zheng, X. , Huang, F. , Wang, Y. , … Liu, P. (2016). Serum bile acids are associated with pathological progression of hepatitis B‐induced cirrhosis. Journal of proteome research, 15(4), 1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, J. B. J. , Lajczak, N. K. , Kelly, O. B. , O'Dwyer, A. M. , Giddam, A. K. , Ní Gabhann, J. , … Keely, S. J. (2017). Ursodeoxycholic acid and lithocholic acid exert anti‐inflammatory actions in the colon. American Journal of Physiology – Gastrointestinal and Liver Physiology, 312(6), G550–g558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Bile acids in different tissues.
Figure S2. Bile acids composition and fold change in small intestine content and in liver.
Figure S3. The influence of individual bile acid on FXR signal pathway in enterocyte.
Figure S4. The change of hepatic FXR signal pathway in vitro.
Figure S5. The gallbladder picture and gallbladder weight in different bile acids feeding group.
Figure S6. The body weight, food intake, and liver weight in each animal experiment.
Figure S7. The concentration of bile acids.
Figure S8. The cytotoxicity assays of UDCAs and DCAs in liver and intestine cell by Cell Counting Kit‐8.
Figure S9. The mRNA expression of BA synthetase and transporters in liver at different time point after pentobarbital sodium intraperitoneal injection.
Figure S10. Supplementary pictures of protein expressions of main BA transporters (A, B) involved in enterohepatic circulation and FXR signaling pathway components in liver and in ileum (C).