

Abstract
Pseudohypoparathyroidism 1B (PHP1B) is caused by maternal epigenetic defects in the imprinted GNAS cluster. PHP1B can follow an autosomal dominant mode of inheritance or occur sporadically (spor-PHP1B). These latter patients present broad methylation changes of two or more differentially methylated regions (DMR) that, when mimicking the paternal allele, raises the suspicious of the occurrence of paternal uniparental disomy of chromosome 20 (upd(20)pat).
A cohort of 33 spor-PHP1B patients was screened for upd(20)pat using comparative genomic hybridization with SNP-chip. Methylation analyses were assessed by methylation specific-multiplex ligation-dependent probe amplification. Upd(20)pat was identified in 6 patients, all exhibiting typical paternal methylation pattern compared to normal controls, namely a complete loss of methylation of GNAS A/B:TSS-DMR, negligible methylation at GNAS-AS1 :TSS-DMR and GNAS-XL:Ex1-DMR and complete gain of methylation at GNAS-NESP:TSS-DMR. The overall frequency of upd(20) is 18% in our cohort when searched considering both severe and partial loss of imprinting. However, twenty five patients displayed severe methylation pattern and the upd(20)pat frequency reaches 24% when searching in this group. Consequently, up to day, upd(20)pat is the most common anomaly than other genetic alterations in spor-PHP1B patients.
Upd(20)pat occurrence is not linked to the parental age in contrast to upd(20)mat, strongly associated with an advanced maternal childbearing age.
This study provides criteria to guide further investigations for upd(20)pat needed for an adequate genetic counseling.
Keywords: PHP1B, Pseudohypoparathyroidism 1B, Paternal uniparental disomy of chromosome 20, Upd(20)pat, Upd(20)mat, GNAS, Methylation
1. Introduction
GNAS gene encodes the stimulatory guanine nucleotide-binding protein (Gas) and maps to 20q13.2–13.3, a complex locus subjected to parent-specific methylation. Molecular variant at GNAS lead to a wide spectrum of phenotypes depending on the parental localization of the genetic defect [1,2]. Heterozygous inactivating GNAS mutations are the cause of pseudohypoparathyroidism type 1a (PHP1A: OMIM N°103580), a syndrome characterized by multi-hormonal resistance and Albright Hereditary Osteodystrophy (AHO), when maternally inherited or PseudoPseudoHypoParathyroidism (PPHP: OMIM N°300800) or Progressive Osseous Heteroplasia (POH: OMIM N°166350) [3,4] when paternally inherited. As with PHP1A, patients affected by pseudohypoparathyroidism type 1B (PHP1B) develop resistance to PTH leading to hypocalcemia and hyperphosphatemia, which is often associated with resistance to TSH and occasionally with features of AHO. PHP1B patients don’t carry variations in the GNAS exons coding Gas, but methylation changes at one or several differential methylated regions (DMRs) including at least GNAS A/B:TSS-DMR.
The GNAS locus gives rise to several different transcripts expressed from upstream promoters/first exons that show, with the exception of the Gas promoter, parent-specific methylation and are thus derived only from the non-methylated parental allele [5]. All alternative first exons are spliced onto the shared exons 2–13, giving rise to either coding (Gαs, XLαs, and NESP55), non-coding (AS), or presumably non-translated mRNAs (exon A/B).
The promoter region for the A/B transcript (GNAS A/B:TSS-DMR) is methylated on the maternal allele and its expression occurs only from the paternal allele. Likewise, the sense exon XL (GNAS-XL:Ex1-DMR) and the antisense exon AS (GNAS-AS1 :TSS-DMR) are methylated on the maternal allele and transcribed only from the paternal allele. Conversely, the exon coding for NESP55 shows methylation on the paternal allele (GNAS-NESP:TSS-DMR) and its transcript is derived from the maternal allele.
PHP1B may follow an autosomal dominant pattern of inheritance (AD-PHP1B) or it can occur as a disorder that appears sporadically (spor-PHP1B). Loss-of-methylation (LoM) at GNAS A/B:TSS-DMR alone is found in AD-PHP1B cases caused either by microdeletions on the maternal allele comprising the gene encoding syntaxin 16 (STX16) [6–8], or by inversion of the region comprising exon A/B through exon 13 [9]. Copy numbers abnormalities encompassing one or several DMRs can affect the methylation patterns. For example maternal intragenic GNAS deletions including exon A/B to NESP55 DMR result in a paternal methylation pattern [10]. The four GNAS DMRs were also affected by deletions of AS exon 3–4 and/or NESP and AS 3–4 [6,8,11–13].
Genomic DNA from spor-PHP1B patients show broad GNAS methylation changes including LoM at GNAS A/B:TSS-DMR, GNAS-XL:Ex1-DMR and GNAS-AS1 :TSS-DMR promoter as well as gain of methylation (GoM) at GNAS-NESP:TSS-DMR [14]. So far, no specific genetic mutation has been identified as a cause of these changes.
Uniparental disomy (UPD) is a condition in which individual inherited both copies of one chromosome from only a single parent. Consequently, the patient harbors the parent-specific methylation pattern. Since the first description by Bastepe et al. [15], thirteen patients have been defined at the molecular level revealing a paternal disomy of chromosome 20 (upd(20)pat) affecting either the entire chromosome or only the long arm (upd(20q)pat) [15–22].
We now describe six new patients harboring upd(20)pat from a cohort of 33 patients affected by spor-PHP1B. One patient presented with a particularly mixed pattern of paternal iso- and heterodisomy at chromosome 20. Taken previous reports into consideration, we estimated the frequency of upd(20)pat in spor-PHP1B patients and defined a specific methylation signature. Last, we evaluated the influence of the parents’ childbearing ages on the occurrence of these epigenetic changes.
2. Subjects and methods
2.1. Patients
We studied 33 cases from Caucasian origin (17 males and 16 females) with the diagnosis of spor-PHP1B, who had been evaluated by our laboratory over the past 5 years. Data describing clinical symptoms were collected retrospectively using records from hospitals or primary care physicians. Routine biochemical assays were performed at the time of diagnosis, or during the follow up, in a variety of different clinical laboratories. This study analysis was conducted according to the Declaration of Helsinki with written consent for collection of DNA, clinical and laboratory data to conduct molecular studies obtained from the patient and/or their parents.
2.2. Molecular investigations
2.2.1. Methylation studies
Leukocyte genomic DNA was screened for GNAS mutations and STX16 deletions using procedures routinely used in the laboratory [13]. Copy number variants (CNV, such as deletions and duplications) and methylation profiling were assessed by methylation specific-multiplex ligation-dependent probe amplification (MS-MLPA) assay using the ME031-A1 kit (MRC-Holland, Amsterdam, the Netherlands) [13,23] according to the manufacturer’s instruction. PCR products were analyzed on an ABI3130 sequencer using GeneMapper (Applied Biosystems) and Coffalyser (MRC-Holland) softwares. The reproducibility of the assay was assessed by measuring the methylation at each DMR in a same control individual in eleven independent assays (Table S1), even if the reproducibility and reliability of the MS-MLPA assay were previously described by Elli et al. [24].
The diagnosis of spor-PHP1B was established on the basis of lack of a positive familial history, absence of CNV within STX16 or NESP, LoM at GNAS A/B:TSS-DMR, associated to epigenetic changes at two or more DMRs including GNAS-XL:Ex1-DMR, GNAS-AS1 :TSS-DMR as well as GoM at GNAS-NESP:TSS-DMR.
2.2.2. Uniparental disomy
Uniparental disomy was searched on DNA using two different procedures: array CGH and SNP genotyping. CGH/SNP array analysis (Agilent SurePrint G3 Cancer CGH + SNP 4×180K) was processed according to the manufacturer’s instruction (Agilent). One patient was also studied using a 300K SNP chip (CytoSNP-12 Illumina®) which included 7614 markers (7598 SNPs and 16 CNVs) located on chromosome 20.
Thirty six specific SNP primers (listed in Fig. S2) were designed across chromosome 20 to build the chromosome 20 haplotypes. Briefly, the amplification refractory mutation system (ARMS)-PCR technique [25] was used to amplify the 36 SNPs using 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) which allows single-plex SNP interrogation through fluorescence detection. Analysis of microsatellite markers across the 20q13.2 region was performed by the center of Human Genetic Research of the Massachusetts General Hospital as previously described [26]. Loss of heterozygosity (LoH) at each SNP indicates inheritance of two copies of the chromosome from one parent (isodisomy). Heterodisomy of chromosome 20 (hUPD) correspond to the inheritance of the two chromosomal homologs from one parent. It was searched in trio including the patient and both parental DNAs or in duo with only one parental DNA. Heterodisomy is inferred from allelic discordance between the patient and his/her mother or allelic concordance with his/her father. All genomic positions were based upon hg19/GRCh37 assembly.
2.3. Statistical analysis
For each biochemical parameter, differences between groups were calculated using the Mann and Whitney nonparametric U test for unpaired samples using GraphPadPrism software. A two-tailed p value < .05 was considered statistically significant.
3. Results
Clinical and biochemical data of the 33 patients diagnosed as spor-PHP1B are shown in Table S3. Depending of the degree of methylation defect at GNAS-XL:Ex1-DMR, patients can be subdivided in two major groups, one with a severe methylation defect (n = 25; < 10%) and another with a partial methylation defect (n = 8; > 10%). The main clinical findings at presentation were convulsions associated with hypocalcemia and an elevated PTH level. Ten patients also had elevated TSH levels. Fifteen patients presented with mild AHO features as suggested by round face and slight metacarpal shortening. One patient (patient 6) for whom data were available exhibited a significant early-onset weight gain; the BMI had returned to normal before puberty (Fig. S4).
3.1. Upd(20)pat was identified in 6 patients (Figs. 1, 2 and Table S3, patients 1–6)
Fig. 1.
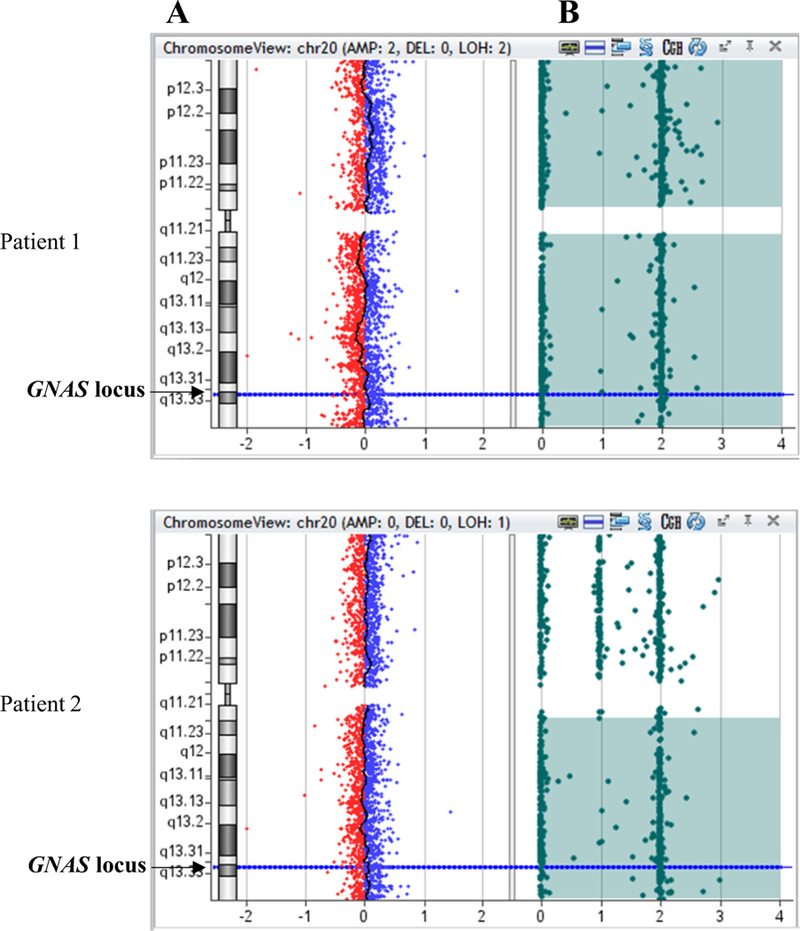
CGH/SNP-array in two patients presenting upd(20)pat (patient 1) or upd(20q)pat (patient 2). A: copy number, the X-axis represents the normalized log2 ratio Cy5(patient)/Cy3(healthy control) fluorescence intensity thresholds – 1 (loss) and 1 (gain), while the Y-axis represents the chromosome 20; B: SNP array; each SNP probe (represented by a filled circle) at a given locus can exist either in homozygous state (a single allele at 0 or 2) or in heterozygous state (at 1). Homozygosity is demonstrated by probes present at either 0 or 2 and the absence of probes at 1.
Fig. 2.
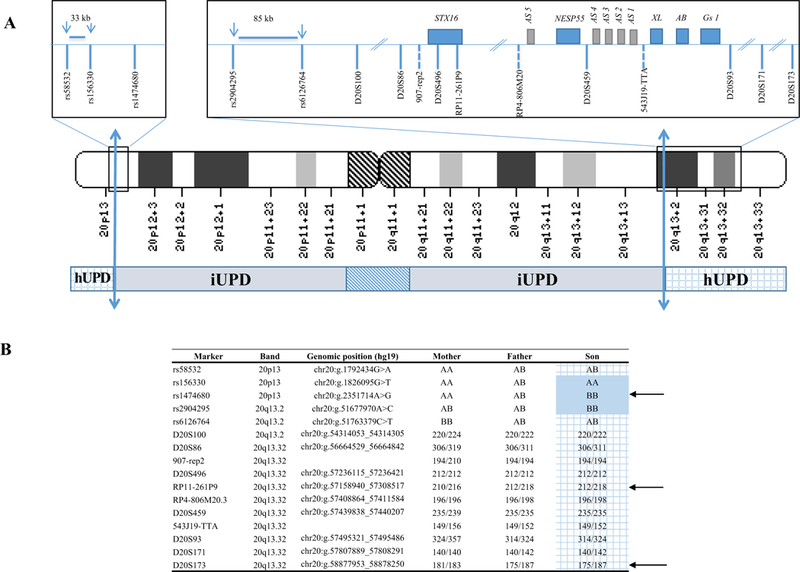
Mixed rearrangement in chromosome 20 (patient 6).
upd(20)pat.arr[GRCh37] 20p13(63244_1792434)x2 htz,20p13q13.2(1800783_51758074)x2 hmz, 20q13.2q13.33(51763379_62909908)x2 htz. A. The inferred breakpoints between paternal isodisomy (iUPD) and paternal heterodisomy (hUPD) are indicated by the two-way arrows: in the short arm, they are located between the last heterozygous SNP (rs58532) (chr20:g.1792434) and the first homozygous one rs156330 (chr20:g.1826095) and in the long arm between rs2904295 (chr20:g.51677970) and rs6126764 (chr20:g.51763379).
B. SNP (single nucleotide polymorphism) and microsatellite markers analysis in patient 6 confirmed mixed rearrangement with paternal isodisomy (iUPD) extending from 20p13 to 20q13.2 with telomeric paternal heterodisomy (hUPD)(grid). Arrows indicate informative SNPs or microsatellite markers. The hUPD overlaps the GNAS locus at position 20q13.3.
Four patients displayed complete upd(20)pat, one patient a upd (20q)pat (Fig. 1, patients 1 and 2). Patient 6 harbors two paternal chromosomes 20, one of which had undergone mixed upd(20) rearrangement (Fig. 2). Analysis of DNA from this patient and his parents using a 300K SNP chip showed a LoH (paternal isodisomy, iUPD) extending from 20p13 to 20q13.2 with telomeric paternal heterodisomy (hUPD). The SNP analysis indicated that the hUPD overlaps the GNAS locus at position 20q13.3. Finally, on 6 patients with upd(20) identified in this study, origin of parental disomy was determined only for two patients (paternal DNAs were available). Others upd(20)pat were inferred on the phenotype patient.
3.2. Percent of methylation at each GNAS DMRs was compared between spor-PHP1B patients with or without upd(20)pat (Fig. 3)
Fig. 3.

Methylation pattern (%) at GNAS locus in patients with spor-PHP1B with or without upd(20)pat.
Dosage and differential methylation analyses were assessed by multiplex ligation-dependent probe amplification and methylation specific-MLPA (MS-MLPA). The relative peak area of each target probe from the ligation-only reaction of the patient DNA was compared to that obtained from the controls. Data are expressed as percentage of methylation (mean ± SD) for each DMRs. The percentage was 50 in the control for the DMRs in which allele-specific methylation was observed. A percentage closed to 100 suggests biallelic methylation. In contrast, a value closed to 0 suggests biallelic demethylation at these DMRs. p values for comparison between groups are indicated. ns: no significance.
All patients with upd(20)pat display a complete loss of methylation of GNAS A/B:TSS-DMR (0 ± 0%), negligible methylation at GNAS-AS1:TSS-DMR and GNAS-XL:Ex1-DMR (1.1 ± 1.6% and 0.77 ± 0.9%, respectively) and complete methylation at GNAS-NESP:TSS-DMR (97 ± 3.3%) compare to normal control.
No statistical differences in the degree of methylation at GNAS-AS1 :TSS-DMR and GNAS A/B:TSS-DMR were observed between patients with or without upd(20)pat. Interestingly, we observed that spor-PHP1B patients without upd(20)pat displayed a broader range in the degree of methylation at GNAS-XL:Ex1-DMR [0–44%] compared to spor-PHP1B with upd(20)pat [0–2%], even if the threshold of significance is not reached (p = .11).
3.3. Parental age at conception in upd(20) (Fig. 4)
Fig. 4.
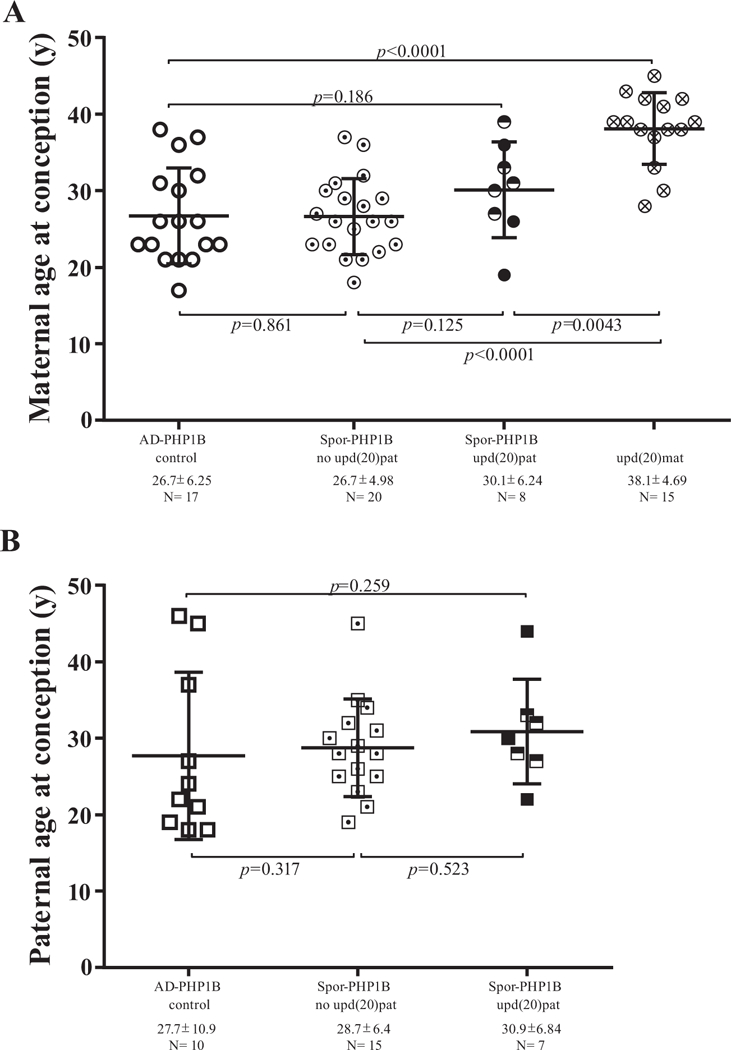
Maternal and paternal age at conception in patients with spor-PHP1B with and without UPD.
Circles display maternal age (A) and squares paternal age (B) respectively. Empty form: control patient (AD-PHP1B); form with a point: spor-PHP1B without upd(20) pat; half full form (literature [15,18,21]) and full form (this study): spor-PHP1B due to upd(20)pat; form with a cross: upd(20)mat (literature [24,27]). N = number of subjects per group. The horizontal and vertical bars for each group represent the mean ± SD. p values for comparison between groups are indicated; y: years.
We compared parental ages at conception between spor-PHP1B patients with or without upd(20)pat and used as control AD-PHP1B patients from our laboratory (n = 17). We added to our data patients previously described with paternal and maternal upd(20) [15,18,21,27–29]. The maternal age at conception was similar for AD-PHP1B and spor-PHP1B patients without upd(20)pat (26.7 ± 6.25 years, n = 17 vs. 26.7 ± 4.98, n = 20; p = .861). We observed a slightly higher in maternal age of conception in upd(20)pat patients (30.1 ± 6.24 years, n = 8) even it is not statistically different from the other two groups (spor-PHP1B, p = .125 or AD-PHP1B, p = .186). By contrast, in upd(20)mat patients, maternal age at conception is statistically advanced compared to others groups and particularly to AD-PHP1B group (38.1 ± 4.69 years; n = 15 vs. 26.7 ± 6.25, n = 17, p < .0001) [27–29]. The paternal age at conception was higher in upd(20)pat patients, but the difference is not statistically significant (30.9 ± 6.84 years, n = 7 vs. 28.7 ± 6.4, n= 15).
4. Discussion
This report presents clinical and biochemical data on a large cohort of patients with an imprinted disorder known as spor-PHP1B caused by methylation abnormalities at GNAS locus.
With respect to clinical implications, no significant differences were found in upd(20)pat patients compared with patients with methylation defect at GNAS locus of unknown origin. Clinical symptoms, mainly convulsion or seizure, develop at an age similar to other PHP1B patients [30]. Out of 17 patients with upd(20)pat in whom the data were available, six (35%) presented advanced weight (> +2 SD) for age at the time of diagnosis [15–18]. An intriguing feature remains the normalization of the weight after six years and indeed, Takatani et al. [21] reported that none other patients with upd(20)pat are obese at the age of 7 and 12, respectively (Table 1). Patients with AD-PHP1B due to STX16 deletions and spor-PHP1B display very similar early overgrowth [31]. Recently, Gruters-Kieslich et al. [32] found obesity during the first year of life being the first clinical evidence for PHP1B suggesting that epigenetic GNAS analyses should be considered for unexplained obesity. Patients with upd(20) are therefore a good model to better understand the pathological mechanism responsible for the much-enhanced growth. With the exception of A/B transcript, our study brings arguments suggesting that the paternal transcripts which are biallelically expressed in spor-PHP1B with upd(20)pat and mono-allelically expressed in AD-PHP1B, cannot be responsible for the overgrowth in all forms of PHP1B patients. That restrains further investigations to the role of the defect of Gas expression and/or the biallelic expression of A/B.
Table 1.
Clinical and epigenetics features with parental age at conception of patients with spor-PHP1B due to upd(20)pat reported in the literature and comparison with our patients. upd(20)pat: paternal disomy at chromosome 20; ND: not determinated.
| Authors | Bastepe et al. [15] | Lecumberri et al. [22] | Bastepe et al. [16] | Fernandez-Rebollo et al. [18] | Dixit et al. [17] | |||||
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 1 | Patient 2 | |||||
| Frequency | Case report | Case report | 1/22 (4.5%) | 4/20 (20%) | Cases reports | |||||
| Upd(20)pat type | Upd(20q) | Upd(20q) | Upd(20) | Upd (20q13.13-qter) | iUPD(20q) | HUPD(20p), iUPD +putative HUPD (20) (q13.33q23) | Upd(20q13.13-qter) | Upd(20q12-q13.33-qter) | Upd(20q13.31-q13.32) | |
| Method | STR | STR | SNPs + 100K SNP array |
STR, SNP array | STR analysis; SNP array |
|||||
| GNAS TSS-DMRs methylation analysis | Methylation-sensitive restriction enzyme | MS-MLPA | ND | MS-MLPA | Methylation-specific PCR | |||||
| GNAS-NESP: TSS-DMR | GoM | |||||||||
| GNAS-AS 1: TSS-DMR | LoM | |||||||||
| GNAS-XL: Ex1-DMR | LoM | |||||||||
| GNAS A/B:TSS-DMR | LoM | |||||||||
| Sex | M | M | F | F | M | M | M | M | M | |
| Age at diagnosis (years) | 5 | 9 | 3.5 | 26 | 9 | 5 | 46 | 14.5 | 5.5 | |
| Weight at birth (percentile) | ND | 50–75 | 25–50 | |||||||
| Weight at diagnosis (SD) | + 3.4 | −1 | + 2 | −0.1 | −0.92 | + 5.5 | + 1.5 | + 0.75 | + 2 | |
| Height at diagnosis (SD) | + 3.6 | −2 | > + 2 | −1 | −1.6 | + 2 | −2 | 0 | −2 | |
| AHO signs | − | + | − | − | + | − | ND | − | ND | |
| Maternal age at conception (years) | 39 | ND | 27 | 31 | 33 | ND | ||||
| Paternal age at conception (years) | ND | 27 | 32 | 33 | ND | |||||
| Authors | Takatani et al. [21] | Park et al. [20] | Jin et al. [19] | This paper | ||||||
| Patient 1 | Patient 2 | 1 | 2 | 3 | 4 | 5 | 6 | |||
| Frequency | 2/23 (8,7%) | Case report osteosarcoma | 1/7 (14%) | 6/33 (18.2%) | ||||||
| Upd(20)pat type | Upd(20q) | Upd(20q) | Upd(20q) | Upd(20) | Upd(20q) | Upd(20) | Upd(20) | Upd (20) | Mixed HUPD/iUPD | |
| Method | SNPs GNAS + STR | STR | STR | CGH/SNP array | STR + SNP array | |||||
| GNAS TSS DMRs methylation analysis | MS-MLPA | |||||||||
| GNAS-NESP: TSS-DMR | GoM | 97% | 91% | 101% | 97% | 98% | 95% | |||
| GNAS-AS 1: TSS-DMR | LoM | 0% | 0% | 0% | 3% | 3% | 0% | |||
| GNAS-XL: Ex1-DMR | LoM | 0% | 0% | 1% | 2% | 2% | 0% | |||
| GNAS A/B:TSS-DMR | LoM | 0% | 0% | 0% | 0% | 0% | 0% | |||
| Sex | M | F | M | M | F | M | M | F | F | M |
| Age at diagnosis (years) | 12 | 7 | 21 | 8 | 33 | 42 | ND | 27.5 | 10 | 4 |
| Weight at birth (percentile) | ND | > 90 | ND | 75−90 | 10 | 50–75 | ||||
| Weight at diagnosis (SD) | + 0.6 | + 1.2 | −1 | + 1 | + 0.5 | > +3 | ND | 0 | + 3 | |
| Height at diagnosis (SD) | + 1.5 | + 1.2 | −1.5 | + 0.45 | −1 | + 0.5 | ND | −1.5 | + 1 | 0 |
| Symptoms of AHO | − | − | − | − | − | − | ND | + | − | − |
| Maternal age at conception (years) | 30 | ND | 19 | ND | 36 | 26 | ||||
| Paternal age at conception (years) | 28 | ND | 22 | ND | 44 | 30 | ||||
UPD can involve either the whole chromosome and reflects complete isodisomy or affect the long arm of the chromosome 20 or be distal, from 20q13.13-qter [16,18]. It can be segmental, interspersed with regions of heterodisomy. In this study, we described a rare mixed chromosomal rearrangement associating paternal telomeric heterodisomy and paternal central isodisomy different from that previously described with different disomy breakpoint in the long arm [18]. The co-occurrence of meiotic recombination, abnormal segregation and subsequent correction are implicated [33].
We found that 18% of spor-PHP1B had inherited two copies of the paternal alleles at GNAS locus, a frequency closed to that reported by Fernandez-Rebollo et al. [18]. When combined with data from previous cohort reports [16,18,19,21], the frequency decreased to 13% (14 with upd(20)pat among 105 spor-PHP1B, Table 1). In this study, we used SNP genotyping with high density arrays that detect blocks of homozygosity which offers a more reliable procedure than microsatellite markers with high informativity but limited density that can miss a small segmental UPD [17,18]. We have however to point out that the number of reported UPD cases could be a slight underestimate because heterodisomy could be missed in absence of parental samples.
Different technologies have been used to study the methylation at GNAS locus. Maupetit-Mehouas et al. [34], Elli et al. [35] and Elli et al. [24] described complex LoM and GoM associations with complete and partial defects (pLoM) in spor-PHP1B. The overall frequency of upd(20) was low (8%) when searched considering both severe and partial loss of imprinting (pLoI) [35]. In the present paper we studied a cohort with the same MS-MLPA protocol which is currently used as a routine procedure for the quantification of the methylation defect at GNAS locus. In our cohort, severe methylation defects at all GNAS TSS-DMRs were only detected in upd(20)pat. Indeed, a broader range in the degree of methylation at GNAS-XL:Ex1-DMR is not in favour of upd(20). UPD might only be suspected in the presence of severe LoI especially GNAS-XL:Ex1-DMR. So, twenty five patients displayed severe methylation pattern (Table S3) and the upd(20)pat frequency reaches 24% when searching in this group.
The variation of methylation at GNAS-XL:Ex1-DMR remains without underlying genetic cause. A de novo mutation somewhere, within or not the GNAS domain that impacts on methylation establishment would not be excluded. In fact, mixed pattern of methylation defects and pLoM at GNAS-XL:Ex1-DMR appear to be the most prevalent methylation pattern defect in spor-PHP1B [35]. Susceptibility to methylation defects has been described in multilocus imprinted disorders (MLID) [36,37] leading to the hypothesis of an imprinted gene network. Methylation defects in GNAS have been found in a subset of Beckwith-Wiedemann syndrome (BWS) cases [38], and the presence of PHP1B in BWS patients has been described, at least, once [39]. Finally, sporadic cases of PHP1B with pLoI could represent true stochastic errors in early post-zygotic phases and embryonic maintenance of methylation [40] and might correspond to mosaicism [24].
The identification of a genetic cause allows adapted genetic counseling for the affected patient and their families. With a prevalence estimated to be approximately 1.65/10000 [41], UPD is a rare condition resulting from meiotic nondisjunction followed by compensatory mechanisms including trisomic rescue, monosomic rescue, compensatory UPD and/or somatic recombination errors [42]. No case of recurrence has been reported until now. A reassuring genetic counseling can be brought to the family for prenatal diagnosis.
Last, our study does not show a correlation between paternal or maternal ages at the conception and the occurrence of upd(20)pat in their child. In contrast an advanced maternal age is clearly associated to upd(20)mat [27–29,43,44]. Incidence of aneuploidie in oocytes increased with maternal age at childbirth and would contribute to the development of trisomy rescue type maternal UPD or monosomy rescue type paternal UPD by replication of a single paternally derived chromosome [45]. The lack of connection with parental age suggests upd (pat) to be a random event during the formation of reproductive cells (eggs and sperm).
In conclusion, our study emphasizes the high frequency of upd(20) pat in spor-PHP1B patients. Methylation profiling using MS-MLPA is a convenient tool for evaluating the methylation pattern at GNAS locus to better define the phenotype of spor-PHP1B patients who should benefit of genetic screening for upd(20)pat. While upd(20)mat is strongly associated with an advanced maternal childbearing age, the upd(20)pat occurrence is not linked to the parental age at the conception.
Supplementary Material
Acknowledgments
The project was supported by a grant from the Direction de la recherche Clinique, CHU de Caen Normandie to NR: 2013 APRI Michropuces 13-031. We are grateful to the patients and their parents for their cooperation in this study; We thank the physicians who have contributed to collect DNA samples and clinical and laboratory data. In particular, we are indebted to Dr Henry (intercomunal hospital, Vesoul 70014) and Dr Coombes (70000 Vesoul), for their extensive help. We thank Dr S Chantot-Bastaraud (CHU Paris Est - Children Hospital Armand-Trousseau, 75571 Paris) for performing SNP-array.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bone.2019.03.023.
References
- [1].Weinstein LS, Yu S, Warner DR, Liu J, Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting, Endocr. Rev. 22 (2001) 675–705. [DOI] [PubMed] [Google Scholar]
- [2].Juppner H, The genetic basis of progressive osseous heteroplasia, N. Engl. J. Med. 346 (2002) 128–130. [DOI] [PubMed] [Google Scholar]
- [3].Lebrun M, Richard N, Abeguile G et al. : Progressive osseous heteroplasia: a model for the imprinting effects of GNAS inactivating mutations in humans. J. Clin. Endocrinol. Metab. 2010; 95: 3028–3038. [DOI] [PubMed] [Google Scholar]
- [4].Shore EM, Ahn J, Jan de Beur S et al. : Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia, N. Engl. J. Med. 346 (2002) 99–106. [DOI] [PubMed] [Google Scholar]
- [5].Hayward BE, Kamiya M, Strain L et al. : The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc. Natl. Acad. Sci. U. S. A. 1998; 95: 10038–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Linglart A, Gensure RC, Olney RC, Juppner H, Bastepe M, A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS, Am. J. Hum. Genet. 76 (2005) 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Elli FM, de Sanctis L, Peverelli E et al. : Autosomal dominant pseudohypoparathyroidism type Ib: a novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J. Clin. Endocrinol. Metab. 2014; 99: E724–728. [DOI] [PubMed] [Google Scholar]
- [8].Bastepe M, Frohlich LF, Hendy GN et al. : Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J. Clin. Invest. 2003; 112: 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Grigelioniene G, Nevalainen PI, Reyes M et al. : A large inversion involving GNAS exon A/B and all exons encoding Gsalpha is associated with autosomal dominant Pseudohypoparathyroidism Type Ib (PHP1B). Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research 2017; 32: 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fernandez-Rebollo E, Garcia-Cuartero B, Garin I et al. : Intragenic GNAS deletion involving exon A/B in pseudohypoparathyroidism type 1A resulting in an apparent loss of exon A/B methylation: potential for misdiagnosis of pseudohypoparathyroidism type 1B. J. Clin. Endocrinol. Metab. 2010; 95: 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bastepe M, Frohlich LF, Linglart A et al. : Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat. Genet. 2005; 37: 25–27. [DOI] [PubMed] [Google Scholar]
- [12].Chillambhi S, Turan S, Hwang DY, Chen HC, Juppner H, Bastepe M, Deletion of the noncoding GNAS antisense transcript causes pseudohypoparathyroidism type Ib and biparental defects of GNAS methylation in cis, J. Clin. Endocrinol. Metab. 95 (2010) 3993–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Richard N, Abeguile G, Coudray N et al. : A new deletion ablating NESP55 causes loss of maternal imprint of A/B GNAS and autosomal dominant pseudohypoparathyroidism type Ib. J. Clin. Endocrinol. Metab. 2012; 97: E863–867. [DOI] [PubMed] [Google Scholar]
- [14].Fernandez-Rebollo E, Perez de Nanclares G, Lecumberri B et al. : Exclusion of the GNAS locus in PHP-Ib patients with broad GNAS methylation changes: evidence for an autosomal recessive form of PHP-Ib? Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research 2011; 26: 1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bastepe M, Lane AH, Juppner H, Paternal uniparental isodisomy of chromosome 20q—and the resulting changes in GNAS1 methylation-as a plausible cause of pseudohypoparathyroidism, Am. J. Hum. Genet. 68 (2001) 1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Juppner H, Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypoparathyroidism type Ib (PHP-Ib), Bone 48 (2011) 659–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dixit A, Chandler KE, Lever M et al. : Pseudohypoparathyroidism type 1b due to paternal uniparental disomy of chromosome 20q. J. Clin. Endocrinol. Metab. 2013; 98: E103–08. [DOI] [PubMed] [Google Scholar]
- [18].Fernandez-Rebollo E, Lecumberri B, Garin I et al. : New mechanisms involved in paternal 20q disomy associated with pseudohypoparathyroidism. European Journal of Endocrinology/European Federation of Endocrine Societies 2010; 163: 953–962. [DOI] [PubMed] [Google Scholar]
- [19].Jin HY, Lee BH, Choi JH et al. : Clinical characterization and identification of two novel mutations of the GNAS gene in patients with pseudohypoparathyroidism and pseudopseudohypoparathyroidism. Clin. Endocrinol. 2011; 75: 207–213. [DOI] [PubMed] [Google Scholar]
- [20].Park HS, Kim CG, Hong N, Lee SJ, Seo DH, Rhee Y, Osteosarcoma in a patient with pseudohypoparathyroidism type 1b due to paternal uniparental disomy of chromosome 20q, Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research 32 (2017) 770–775. [DOI] [PubMed] [Google Scholar]
- [21].Takatani R, Minagawa M, Molinaro A et al. : Similar frequency of paternal uniparental disomy involving chromosome 20q (patUPD20q) in Japanese and Caucasian patients affected by sporadic pseudohypoparathyroidism type Ib (sporPHP1B). Bone 2015; 79: 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lecumberri B, Fernandez-Rebollo E, Sentchordi L et al. : Coexistence of two different pseudohypoparathyroidism subtypes (Ia and Ib) in the same kindred with independent Gs{alpha} coding mutations and GNAS imprinting defects. J. Med. Genet. 2010; 47: 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alsum Z, Abu Safieh L, Nygren AO, Al-Hamed MA, Alkuraya FS, Methylation-specific multiplex-ligation-dependent probe amplification as a rapid molecular diagnostic tool for pseudohypoparathyroidism type 1b, Genetic Testing and Molecular Biomarkers 14 (2010) 135–139. [DOI] [PubMed] [Google Scholar]
- [24].Elli FM, Bordogna P, Arosio M, Spada A, Mantovani G, Mosaicism for GNAS methylation defects associated with pseudohypoparathyroidism type 1B arose in early post-zygotic phases, Clin. Epigenetics 10 (2018) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Newton CR, Graham A, Heptinstall LE et al. : Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989; 17: 2503–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Juppner H, Schipani E, Bastepe M et al. : The gene responsible for pseudohypoparathyroidism type Ib is paternally imprinted and maps in four unrelated kindreds to chromosome 20q13.3. Proc. Natl. Acad. Sci. U. S. A. 1998; 95: 11798–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mulchandani S, Bhoj EJ, Luo M et al. : Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genetics in Medicine: Official Journal of the American College of Medical Genetics 2016; 18: 309–15. [DOI] [PubMed] [Google Scholar]
- [28].Kawashima S, Nakamura A, Inoue T et al. : Maternal uniparental disomy for chromosome 20: physical and endocrinological characteristics of five patients. J. Clin. Endocrinol. Metab. 2018; 103: 2083–2088. [DOI] [PubMed] [Google Scholar]
- [29].Eggermann T, Mergenthaler S, Eggermann K et al. : Identification of interstitial maternal uniparental disomy (UPD) (14) and complete maternal UPD(20) in a cohort of growth retarded patients. J. Med. Genet. 2001; 38: 86–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Linglart A, Bastepe M, Juppner H, Similar clinical and laboratory findings in patients with symptomatic autosomal dominant and sporadic pseudohypoparathyroidism type Ib despite different epigenetic changes at the GNAS locus, Clin. Endocrinol. 67 (2007) 822–831. [DOI] [PubMed] [Google Scholar]
- [31].Hanna P, Grybek V, Perez de Nanclares G et al. : Genetic and epigenetic defects at the GNAS locus lead to distinct patterns of skeletal growth but similar early-onset obesity. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gruters-Kieslich A, Reyes M, Sharma A et al. : Early-onset obesity: unrecognized first evidence for GNAS mutations and methylation changes. J. Clin. Endocrinol. Metab. 2017; 102: 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kotzot D, Complex and segmental uniparental disomy updated, J. Med. Genet. 45 (2008) 545–556. [DOI] [PubMed] [Google Scholar]
- [34].Maupetit-Mehouas S, Mariot V, Reynes C et al. : Quantification of the methylation at the GNAS locus identifies subtypes of sporadic pseudohypoparathyroidism type Ib. J. Med. Genet. 2011; 48: 55–63. [DOI] [PubMed] [Google Scholar]
- [35].Elli FM, Linglart A, Garin I et al. : The prevalence of GNAS deficiency-related diseases in a large cohort of patients characterized by the EuroPHP network. J. Clin. Endocrinol. Metab. 2016; 101: 3657–3668. [DOI] [PubMed] [Google Scholar]
- [36].Court F, Martin-Trujillo A, Romanelli V et al. : Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum. Mutat. 2013; 34: 595–602. [DOI] [PubMed] [Google Scholar]
- [37].Eggermann T, Elbracht M, Schroder C et al. : Congenital imprinting disorders: a novel mechanism linking seemingly unrelated disorders. J. Pediatr. 2013; 163: 1202–1207. [DOI] [PubMed] [Google Scholar]
- [38].Bliek J, Verde G, Callaway J et al. : Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. European Journal of Human Genetics: EJHG 2009; 17: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bakker B, Sonneveld LJ, Woltering MC, Bikker H, Kant SG, A girl with Beckwith-Wiedemann syndrome and pseudohypoparathyroidism type 1B due to multiple imprinting defects, J. Clin. Endocrinol. Metab. 100 (2015) 3963–3966. [DOI] [PubMed] [Google Scholar]
- [40].Kelsey G, Imprinting on chromosome 20: tissue-specific imprinting and imprinting mutations in the GNAS locus, Am. J. Med. Genet. C: Semin. Med. Genet. 154C (2010) 377–386. [DOI] [PubMed] [Google Scholar]
- [41].Engel E, DeLozier-Blanchet CD, Uniparental disomy, isodisomy, and imprinting: probable effects in man and strategies for their detection, Am. J. Med. Genet. 40 (1991) 432–439. [DOI] [PubMed] [Google Scholar]
- [42].Tucker T, Schlade-Bartusiak K, Eydoux P, Nelson TN, Brown L, Uniparental disomy: can SNP array data be used for diagnosis? Genetics in Medicine: Official Journal of the American College of, Medical Genetics 14 (2012) 753–756. [DOI] [PubMed] [Google Scholar]
- [43].Chudoba I, Franke Y, Senger G et al. : Maternal UPD 20 in a hyperactive child with severe growth retardation. European Journal of Human Genetics: EJHG 1999; 7: 533–540. [DOI] [PubMed] [Google Scholar]
- [44].Velissariou V, Antoniadi T, Gyftodimou J et al. : Maternal uniparental isodisomy 20 in a foetus with trisomy 20 mosaicism: clinical, cytogenetic and molecular analysis. European Journal of Human Genetics: EJHG 2002; 10: 694–698. [DOI] [PubMed] [Google Scholar]
- [45].Matsubara K, Ogata T, Advanced maternal age at childbirth and the development of uniparental disomy. A commentary on the proportion of uniparental disomy is increased in Prader-Willi syndrome due to an advanced maternal childbearing age in Korea, J. Hum. Genet. 58 (2013) 118–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.