

Abstract
We herein present the design, synthesis, and biological evaluation of potent and highly selective β-secretase 2 (memapsin 1, beta-site amyloid precursor protein cleaving enzyme 2 or BACE 2) inhibitors. BACE2 has been recognized as an exciting new target for type 2 diabetes. The X-ray structure of BACE1 bound to inhibitor 2a containing a hydroxyethylamine isostere was determined and based on this structure, a computational docking study was performed which led to inhibitor 2a-bound BACE2 models which were used to optimize the potency and selectivity of inhibitors. A systematic structure-activity relationship study led to identification of determinants of inhibitors’ potency and selectivity towards the BACE2 enzyme. Inhibitors 2d (Ki = 0.031 nM, selectivity over BACE1 ≈174,000-fold) and 3l (Ki = 1.6 nM, selectivity over BACE1 over 500-fold) displayed outstanding potency and selectivity. Inhibitor 3l is non-peptide in nature and may pave the way to the development of a new class of potent and selective BACE2 inhibitors with clinical potential.
Keywords: BACE2 inhibitors, β-secretase 2, Tmem27, type 2 diabetes, structure-based design
Design of Selective BACE2 Inhibitors
Selective BACE2 inhibitors have been designed and synthesized as a potential new treatment for type 2 diabetes. Utilizing the X-ray structural studies with BACE1 along with computational docking approaches with BACE2, a series of potent and highly selective BACE2 inhibitors containing hydroxyethylamine isostere have been designed. The present investigation led to the identification of structural determinants and development for achieving potency and selectivity towards BACE2 enzyme. Inhibitor 3l exhibited excellent BACE2 potency and high selectivity over BACE1.
Graphical Abstract
Introduction
Type 2 diabetes mellitus is a major medical problem that seriously threatens human health worldwide, especially in developing countries.1, 2 International Diabetes Federation estimated that worldwide, there were 415 million adults between the age of 20–70 living with diabetes and 5 million deaths due to diabetes in 2015.3 The global burden for diabetes patient care exceeded over 673 billion dollars in 2015.3 The prevalence of diabetes has been increasing steadily, particularly in developing countries, with three quarters of patients being from low and middle income families. In some countries, the number of patients affected with diabetes is expected to increase even more rapidly due to the aging population and adding further to the healthcare burden.4, 5
Diabetes mellitus is a chronic, progressive metabolic disorder. It is characterized by hyperglycemia resulting from reduced insulin secretion from pancreatic beta-cells.6 The disease also originates from an increase in insulin demand associated with tissue insulin resistance. There is no cure for type 2 diabetes, however, treatment modalities include lifestyle modifications, treatment of obesity, administration of oral hypoglycemic agents and insulin sensitizers like metformin, a biguanide which reduces insulin resistance, a recommended first-line medication particularly for obese patients. Other effective antidiabetic medications include sulfonylureas, meglitinide, thiazolidinediones, sodium-glucose cotransporter 2 (SGLT2) inhibitors, and α-glucosidase inhibitors.7–9 Moreover, enhancing incretin action has been, in recent years, a widely utilized and effective approach for the treatment of type 2 diabetes. Strategies have been developed to circumvent dipeptidyl peptidase 4 (DPP-4) action, leading to the development of distinct glucagon-like peptide 1 (GLP-1) receptor agonists with longer half-lives (such as exenatide) as well as compounds blocking DPP-4 action and leading to increased endogenous GLP-1 levels (eg. sitagliptin).10 Despite these currently available options for lowering blood glucose, there are severe liabilities associated with these existing antidiabetic drugs, especially related to cardiovascular complications.11, 12 Although examples of recent drugs with reduced risk of cardiovascular death have been reported (eg. the SGLT2 inhibitor empagliflozin),13 new therapies based upon innovative mechanisms are still urgently needed to address improvement of treatment and current unmet needs.
A recent study demonstrated that inhibition of BACE2 in insulin-resistant mice resulted in an increase of beta-cell mass and improvement of insulin levels in mice.14 Importantly, another recent study has shown that BACE2 suppression promotes beta-cell survival and function in a model of type 2 diabetes induced by human islet amyloid polypeptide over-expression.15 Tmem27 (collectrin) is a type 1 transmembrane protein and its over expression in pancreatic beta-cells leads to cellular proliferation, increased cell mass and increased insulin production. The excess Tmem27 in beta-cells is regulated by BACE2 cleavage of Tmem27, resulting in the release of the Tmem27 ectodomain outside the cell.16,17 These studies suggested that BACE2 inhibitors may have the potential for novel treatments of type 2 diabetes.18
BACE2 (memapsin 1) is a type 1 membrane-bound aspartic acid protease that belongs to the pepsin family.19 It is a close homologue of BACE1 which is highly expressed in the brain.20,21 BACE2 is expressed in the alpha-cells of pancreatic islets and it is found in other peripheral tissues, the colon and kidney.22 It has been demonstrated that BACE2 inhibition results in beta-cell survival through a potential substrate, islet amyloid polypeptide (IAPP). Overexpression of IAPP leads to defects in glucose tolerance.23 BACE2 also cleaves plasma membrane protein Tmem27 and regulates beta-cell mass and function.14 Taken together, BACE2 inhibition is potentially a viable new path for the treatment of diabetes.11,14,15 However, BACE2 inhibitors may need to be selective for a number of reasons. First of all, its closest homologue, BACE1, has a distinct physiological function in down-regulating brain neuronal function by cleaving APP and overproducing β-amyloid peptide (Aβ) in the brain.24 While BACE1 has been mechanistically an exciting target for possible treatment of Alzheimer’s disease, BACE1 gene deletion resulted in behavioral abnormalities in mice.25 Furthermore, BACE1 is involved in the formation of myelin sheath in central and peripheral nerves.26,27 BACE1 may also have other important protein substrates.26,28
Diabetes is a chronic disease and for effective chronic treatment, any off-target toxicities need to be minimized, as collective toxicity burden over time could be severe. Therefore, development of selective BACE2 inhibitors over cathepsin D is also critically important. Cathepsin D is abundant in cells and it plays an important role in protein catabolism.29,30 In addition, it has an important role in retinal function of the eyes.31,32 The majority of current BACE1 inhibitors lack selectivity and as a result, clinical toxicity has become a major issue. Thus far, there have been few selective BACE1 inhibitors that have been taken through clinical development.33,34 For the development of BACE2 inhibitor drugs for diabetes, inhibitor selectivity is critically important and there are only a few limited reports of BACE2 inhibitors with marginal selectivity.14,35 We recently reported our preliminary work on the design and synthesis of selective BACE2 inhibitors based upon transition-state isosteres.36 We have now expanded structure-activity relationship studies, determined a high-resolution X-ray structure of a potent and selective inhibitor complexed with BACE1 and created a structural model of inhibitor and BACE2 active site interactions. We herein report the full account of our studies detailing the design, synthesis, biological evaluation, X-ray crystallography and computational docking studies leading to potent and selective BACE2 inhibitors.
Results and Discussion
In our preliminary efforts towards the design of potent and selective BACE2 inhibitors, we focused our inhibitor design based upon hydroxyethylamine isosteres as these dipeptide mimics are inherent to several FDA approved HIV-1 protease inhibitor drugs.37,38 Our starting point for the design of selective BACE2 inhibitors is compound 1 (Figure 1, Ki = 1.8 nM, BACE1) which was designed by us as a potent BACE1 inhibitor.39 This inhibitor exhibits many drug-like properties including low toxicity and lipophilicity. Inhibitor 1 is quite potent against BACE1, but it also displayed inhibition towards BACE2 with a Ki value of 137 nM which is 76-fold less potent than BACE1.40 Ki values for these compounds and all other compounds described herein were determined from kinetic data determined under tight-binding inhibition conditions and using the Morrison equation as described previously.36
Figure 1.

Structures of BACE2 inhibitors 1, 2a, 2d and 3l.
We next determined a high-resolution X-ray structure of inhibitor 1-bound BACE1 complex.39 The X-ray structure provided critical ligand-binding site interactions in the BACE1 active site as shown in Figure 2A. We have made a number of attempts to crystallize inhibitor 1 in complex with BACE2 but so far we have been unable to grow crystals suitable for structural studies. In the absence of an X-ray structure of a compound 1-bound BACE2 complex, we performed computational docking to create a model of inhibitor 1 bound in the BACE2 active site. The model was constructed using the available low-resolution X-ray structure of an inhibitor-bound BACE2 complex.41,42 The resulting model of inhibitor 1 bound in the active site of BACE2 is shown in Figure 2B. Our analysis of a compound 1-bound BACE2 computational model and the X-ray structure of 1-bound BACE1, suggested that certain BACE2 active site residues can be specifically targeted in order to achieve potency and selectivity (e.g. Gly50 and Thr88). Also, we hypothesized that the incorporation of polar substituents on the prime side would reduce BACE1 activity thus leading to more potent and selective BACE2 inhibitors. In particular, we observed that the methoxybenzyl moiety in inhibitor 1 is mostly engaged in non-polar interactions with residues in the hydrophobic S1’-S2’ subsites of BACE1. Therefore, replacement of the methoxybenzyl group with polar functionalities could improve selectivity towards BACE2. We therefore sought to simultaneously insert alcohol and amide polar functionalities by replacing the methoxybenzyl group with an allothreonine isobutylamide and other derivatives.36 Thus, replacement of the P1’-methoxybenzylamine of inhibitor 1 with an allothreonine derivative resulted in inhibitor 2a. Further structural modification led to the very potent and selective inhibitors 2d and 3l.
Figure 2.

(A) X-ray structure of inhibitor 1 (carbon chain, orange) bound to BACE1 (PDB code: 2VKM). (B) An energy-minimized model of inhibitor 1 (carbon chain, green) bound in the BACE2 active site. All strong hydrogen bonding interactions are shown as dotted lines. The docking was performed with AutoDock Vina (see Supplementary Material for details).
Chemistry
Our synthesis of BACE2 inhibitors 2f–j is shown in Scheme 1. Various isophthalamide derivatives 4a–7b were prepared using previously described procedures.43–46 For the synthesis of allothreonine isobutylamide derivatives, the Boc group of known43 isobutylamide derivatives 8a,b was deprotected in the presence of trifluoroacetic acid (TFA) in CH2Cl2 at 0 °C to 23 °C for 1 h. Reaction of the resulting free amine with commercially available tert-butyl ((S)-1-((S)-oxiran-2-yl)-2-phenylethyl) carbamate 9 in 2-propanol heated at 80 °C for 18 h, provided the corresponding epoxide open product. Exposure of the resulting Boc derivative to TFA in CH2Cl2 afforded amine derivatives 10a,b. Coupling of acids 5a,b and 6a,b with amines 10a,b using EDCI and HOBt in the presence of N,N-diisopropylethylamine (DIPEA) in CH2Cl2 at 23 °C led to compounds 2f–j with yields ranging from 39% to 78%.
Scheme 1.

Reagents and conditions (a) TFA, dry CH2Cl2, 1 h, from 0 °C to 25 °C; (b). 9, i-PrOH, 80 °C, 18 h; (c) TFA, dry CH2Cl2, 1 h, from 0 °C to 25 °C, 66–86% (3 steps) (d) Suitable acids 5a,b and 6a,b and appropriate amines 10a or 10b, EDCI·HCl, HOBt·H2O, DIPEA, dry CH2Cl2, 15 h, rt, 39–78%.
The synthesis of aminoalcohol derivatives containing the substituted aromatic rings is shown in Scheme 2. Commercially available aldehydes 11a,b were subjected to Wittig reaction with the appropriate alkyl triphenylphosphonium bromide leading to the corresponding olefins as E/Z mixture. Isomerization of the olefin mixture in the presence of bis(acetonitrile)dichloropalladium(II) resulted in the E-olefins 12a–c in nearly quantitative yields.47 Asymmetric dihydroxylation of olefins 12a–c with AD-mix-α provided stereochemically defined diols 13a–c. These diols were converted into the corresponding diastereoisomeric mixtures (1:1) of cyclic sulfites 14a–c by treatment with thionyl chloride in the presence of triethylamine.48 Treatment of sulfites 14a–c with sodium azide in DMF at 80 °C, afforded azidoalcohols 15a–c in excellent yields.
Scheme 2.

Reagents and conditions. a) R2CH2P+Ph3Br−, nBuLi, dry THF, 1 h, −78 °C; b) (CH3CN)2Cl2Pd, CHCl3, 72 h, 25 °C, 69–84% (2 steps); c) AD-mix-α t-BuOH/H20, 24 h, 25 °C, 68–73%; d) TEA, thionyl chloride, 0 °C, 45 min, 66–79%; e) NaN3, DMF, 10 h, 80 °C, 74–88%; f) (Boc)2O, H2, Pd/C, EtOAc, 1 atm, 25 °C, 15 h; g) TFA, dry CH2Cl2, 1 h, from 0 °C to 25 °C, 66–86% (2 steps).
Catalytic hydrogenation of these azides in the presence of di-t-butyldicarbonate provided the Boc-protected aminoalcohols. Exposure of these Boc-derivatives to TFA in CH2Cl2 at 0 °C to 23 °C for 1 h furnished free aminoalcohols 16a–c.49
The synthesis of BACE2 inhibitors 3a–h (Table 2) with varying substituents on the aromatic rings and on the side chain is shown in Scheme 3. Opening of epoxide 9 carried on with amine derivatives 16a–c in isopropanol at 80 °C afforded aminoalcohol derivatives 17a–c. Deprotection of Boc-group by exposure to TFA followed by coupling of the resulting amines with the suitable carboxylic acids 4a–d36 using EDCI and HOBt in the presence of DIPEA led to final compounds 3a–h (Table 2).
Table 2.
Structure and activity on BACE1 and BACE2 enzymes for title inhibitors 3a–h.
Scheme 3.

Reagents and conditions, a) 16a, 16b or 16c, i-PrOH, 18 h, 80 °C, 65–71%; b) TFA, dry CH2Cl2, 2 h, from 0 °C to 25 °C; c) Acids 4a–d, EDCI·HCl, HOBt·H2O, DIPEA, dry CH2Cl2, 15 h, 25 °C, 55–71% (2 steps).
The syntheses of BACE2 inhibitors 3i–n (Table 3) with variation of P3 and P2’-ligands of inhibitor 1 is shown in Scheme 4. Opening of epoxide 9 with various benzyl amine derivatives provided amines 18a,b.39,50 These amines were coupled with acids 4a, 4b,36 7a or 7b,51 using EDCI and HOBt in the presence of DIPEA to provide the final compounds 3i-n (Table 3). Inhibitor 20 was synthesized starting from acetophenone derivative 19 following reported procedures.35
Table 3.
Structure and activity on BACE1 and BACE2 enzymes for title inhibitors 3i–o.
Scheme 4.

Reagents and conditions, a) Acids 4a, 4b, 7a or 7b, EDCI·HCl, HOB·tH2O, DIPEA, dry CH2Cl2, 15 h, 25 °C, 55–71% (2 steps).
The synthesis of inhibitor 3o with a substituted oxazole derivative as the P3-ligand is shown in Scheme 5. Alkylation of commercially available oxazole 21 with methyl iodide in the presence of sec-butillithium and TMEDA led to alkylated derivative 22. Deprotection of the Boc group by exposure to TFA followed by coupling of the resulting amine with acid 23 in the presence of EDCI, HOBt and DIPEA furnished the amide derivative. Hydrolysis of ester with aqueous LiOH led to acid derivative 24. Coupling of acid 24 with amine 18b, provided final inhibitor 3o in 48% yield.
Scheme 5.

Reagents and conditions, a) s-BuLi, TMEDA, Et2O, −78 °C, 30 min, then Mel, −78 °C to 25 °C, 8 h, 86%; b) TFA:CH2Cl2 (3:1), 25 °C, 1 h; c) 23, EDCI, HOBt, DIPEA, CH2Cl2, 0 °C to 25 °C, 12 h, 66% (2 steps); d) LiOH·H2O, THF:H2O (2:1), 25 °C, 6 h, 88%; e) 18b, EDCI, HOBt, DIPEA, CH2Cl2, 0 °C to 25 °C, 12 h, 48%.
Structure-Activity Relationships
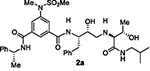
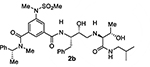
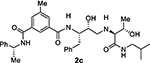
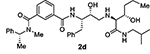
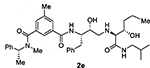
At the outset, we replaced the methoxybenzylamine P1’-ligand of inhibitor 1 with an allothreonine isobutyl amide group. The resulting inhibitor 2a showed improvement of BACE2 potency with a Ki value of 14.6 nM and demonstrated nearly 3-fold selectivity over BACE1. We next determined the X-ray structure of inhibitor 2a-bound BACE1 and created an active site model of 2a-bound to BACE2. The X-ray structure of 2a in complex with BACE1 is shown in Figure 3 and the computational model of 2a bound to the BACE2 active site is shown in Figure 4. We gained critical molecular insights on specific active site interactions that could improve potency and selectivity against the BACE2 enzyme. In particular, from the model of compound 2a bound to the BACE2 enzyme, it appeared that the P2 amide NH and the sulfonamide group on the P2-isophthalamide were not involved in significant polar interactions, in contrast to interactions of 2a in BACE1 active site. Based on this speculation, we first assessed compound 2b as the amide N-methylated counterpart of compound 2a. The resulting inhibitor 2b displayed reduction of BACE2 potency, but improved selectivity over BACE1 (Ki = 80 nM for BACE2 and Ki = 1812 nM for BACE1).36 We then designed compound 2c in which the sulfonamide moiety of 2a was replaced by a methyl group (Compound 2c displayed a BACE1 Ki value of 731 nM and a BACE2 Ki value of 183 nM). Our analysis of binding properties of inhibitor 2c with respect to the model of compound 2a within the BACE2 active site (Figure 4) suggested that a larger P1’ alkyl chain replacing the methyl group of allothreonine could result more effective in establishing van der Waals interactions with the surrounding hydrophobic residues.36 We also planned to alkylate the amide NH and remove the P2-sulfonamide functionality. This prompted us to design compounds 2d and 2e (Table 1), which differ by the presence of an additional methyl on the isophthalic moiety benzene ring. Both compounds displayed excellent inhibitory activity on BACE2 (Ki = 0.031 nM for 2d and Ki = 0.038 nM for 2e) with inhibitor 2d showing outstanding selectivity over BACE1 (174,000 fold).36 To date, the only other relevant work on the design of selective BACE2 inhibitors is related to a patent which reported a series of 2-aminodihydro[1,3]-thiazines, as exemplified by inhibitor 20 (Scheme 4).33
Figure 3.

A Stereoview of the X-ray structure of inhibitor 2a (carbon chain, magenta color) in complex with BACE1 (memapsin 2) (PDB code: 5DQC). All strong hydrogen bonding interactions are shown as dotted lines.
Figure 4.

A stereoview of an energy-minimized model of 2a (carbon chain, green) bound within the BACE2 active site. All strong hydrogen bonding interactions are shown as dotted lines. The docking was performed with AutoDock Vina (see SupplementaryInformation for details).
Table 1.
Structure and activity on BACE1 and BACE2 enzymes for title inhibitors 2a–j.
Based on the high potency of 2a against the purified BACE2 enzyme, we next investigated whether inhibitor 2a could prevent processing of Tmem27 in the pancreatic beta-cell line MIN6.52,53 Tmem27 behaves like a canonical BACE½ substrate, being subjected to subsequent intramembrane cleavage by the γ-secretase complex. This was confirmed by incubation of MIN6 cells with increasing concentrations of γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) which led to the stabilization of a 22 kDa C-terminal fragment (CTF) in the cell lysate.14 We tested our inhibitor 2a following the protocol employed by Esterhazy.14 Accordingly, the C-terminal fragment of Tmem27 was preserved from degradation in the presence of DAPT as shown in lane 1 (Figure 5). Without the treatment of DAPT, most of the CTF of Tmem27 was degraded as shown in lane 5. Hence, DAPT was added in the cellular assay for studying the in cellulo potency of potent inhibitors. For these studies, MIN6 cells were grown in the presence of various inhibitors, lysed and subjected to Western blot using a monoclonal antibody vs. Tmem27 C-terminal region. The results are shown in Figure 5.14 As can be seen, the 22-kDa C-terminal fragment of Tmem27 was formed only in small extent in the presence of 0.4 μM of inhibitor 2a (less than 5%, lane 3). Then, in the presence of 0.9 μM inhibitor 2a, the processing of Tmem27 was completely abolished as shown in lane 4. We used inhibitor 20, also known as compound J (BACE2 Ki = 6 nM; BACE1 Ki = 18 nM) as a positive control and the results are shown in lane 2.14
Figure 5.

Inhibition of BACE2 processing of Tmem27 by CpdJ and compound 2a (at two different concentrations) in MIN6 cells. Lanes 1 and 5 represent vehicle (DMSO) controls with and without DAPT, respectively.
With the assumption that BACE2 processes its protein substrate PMEL in endosomes54 these results indicated that inhibitor 2a penetrated MIN6 cells and blocked the processing of Tmem27 by BACE2. We now have greatly extended our structure-activity relationships and have further investigated determinants of inhibitors’ potency and selectivity towards BACE2 through the design, synthesis and biological evaluation of a series of new inhibitors, namely 2f–j and 3a–o. First of all, we synthesised a focused subset of derivatives (compounds 2f–j, Table 1) in search of an effective P2-P3 ligand. Accordingly, we incorporated the isophthalamide terminal nitrogen in a substituted pyrrolidyl moiety (compounds 2f,g) and replaced the P2 benzylamine portion with an isosteric thiazolylamine moiety (compounds 2h–j). We speculated that suitable heterocyclic moieties and their substituents could form favorable hydrogen bonds with backbone atoms as well as stronger van der Waals interactions in the active site of BACE2. In particular, we selected a 3-(R)-2-(methoxymethyl)pyrrolidinylisophtalamide and a ((4-methylthiazol-2-yl)methyl)-isophthalamide moiety in combination with a methyl or a longer propyl chain at the P1’ alkylamide portion. In compounds 2f,g bearing a 3-((R)-2-(methoxymethyl)pyrrolidinylisophtalamide moiety, the insertion of a P1’ propyl chain and a methyl group on 5-position of the isophthalamide benzene ring led to a significant increase of BACE1 and BACE2 activity. Accordingly, inhibitor 2g with a propyl P1’-side chain and a 3-methyl group on the P2-isophthalamide ligand led to improvement of both BACE1 and BACE2 inhibitory activity. However, inhibitor 2g is more potent against BACE1 (Ki= 6.6 nM) than BACE2 (Ki = 27.9 nM). Compound 2h with a ((4-methylthiazol-2-yl)methyl)isophthalamide moiety as the P3-ligand showed a BACE2 Ki of 97 nM and displayed 2-fold selectivity over BACE1. Inhibitor 2i with a longer P1’ alkyl chain in combination with no substitution on the isophthalamide phenyl ring showed improvement of BACE1 and BACE2 potency. Compound 2j with a methyl group on the isophthalamide group, displayed comparable BACE1 and BACE2 activity, however, no improvement of selectivity over BACE1 was observed.
Based on our structural insights, we then examined a small series on inhibitors combining the ((R)-1–1phenylethyl) isophthalamide as the P2-P3 ligand and a P1’ ligand containing a reduced peptide character. Thus, we incorporated a stereochemically defined aminoalcohol moiety as a convenient handle to insert an alkyl chain of variable length similar to inhibitors 2d and 2e coupled with a substituted phenyl ring in place of isobutylamide. Beyond improving hydrophobic interactions, such an aryl moiety could also serve to mimic the P2’ isobutylamide interaction with a suitably positioned polar group. Accordingly, we synthesized compounds 3a–h (Table 2). In general, this set of compounds showed decreased affinity towards BACE2 compared to their terminal isobutylamide counterparts such as inhibitors 2d and 2e. Inhibitor 3a, containing a 3-sulfonamide group on the isophthalic moiety displayed higher affinity towards the BACE1 enzyme (Ki = 205 nM for BACE1 and Ki = 731 nM for BACE2). The amide N-methylation in inhibitor 3b led to higher affinity for BACE2 with slight improvement of potency and selectivity toward BACE2 over BACE1 (Ki = 4852 nM for BACE1 and Ki = 479 nM for BACE2).
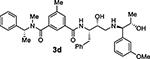
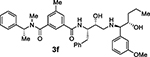
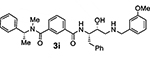
The removal of the sulfonamide functionality on the isophthalamide phenyl ring (compounds 3c and 3d), did not improve potency but did increase BACE2 selectivity (54-fold for compound 3c and 34-fold for compound 3d). In agreement with the previous series of compounds, a longer P1’ alkyl chain had a beneficial effect for BACE2 inhibitory activity. Compound 3f, bearing a methyl substituent on the isophthalic phenyl ring and a terminal (1R,2S)-1-amino-1-(3-methoxyphenyl)pentan-2-ol displayed BACE2 inhibitory potency with a Ki value of 141 nM. Interestingly, compound 3e without a methyl group on the isophthalamide group, showed a more marked selectivity on BACE2 over BACE1 (159-fold for 3e and 62-fold for 3f). The removal of the 3-methoxy substituent from the P1’-ligand led to compound 3g. However, this compound displayed a significant drop in BACE2 inhibitory activity. Similarly, removal of the 3-methoxy group from the P1’-ligand of inhibitor 3f resulted in compound 3h which also showed reduction of BACE2 Ki value of 905 nM over 3f (Ki value of 141 nM). These results indicated that the 3-methoxy substituent on the phenyl ring moiety may be involved in significant interactions in the active site.
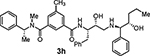
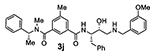
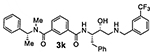
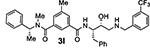
Bearing in mind the data on prime and non-prime site preferences, we investigated a series of compounds (3i–o, Table 3) where we removed the 3-sulfonamide functionality from the P2 isophthalamide ligand and N-methylated the amide NH of our initial lead inhibitor 1. Compound 3i with a 3-methoxylbenzylamine moiety as the P1’-ligand improved BACE2 potency with a Ki value of 39.4 nM. This compound showed over 52-fold selectivity for BACE2. Incorporation of a 3-methyl group on the isophthalamide group resulted in inhibitor 3j which showed nearly 3-fold improvement of BACE2 Ki. Also, it showed slight improvement of BACE2 selectivity (nearly 60-fold). Replacement of the 3-methoxy group on the P1’-ligand with a lipophilic 3-trifluoromethyl group resulted in inhibitor 3k. This inhibitor (3k) displayed slight improvement of BACE2 activity (Ki value of 35.7 nM). However, compound 3k showed reduction of BACE2 selectivity compared to inhibitor 3i (50-fold for 3i versus 37-fold for 3k). Interestingly, incorporation of a 3-methyl group on the P2-isophthalamide group of inhibitor 3k resulted in a very potent and selective inhibitor 3l. Inhibitor 3l exhibited a BACE2 Ki value of 1.6 nM. Furthermore, inhibitor 3l showed over 500-fold selectivity for BACE2. The combination of a P2 ((4-methyloxazol-2-yl)methyl)isophthalamide derivative and a P1’ 3-methoxybenzylamino moiety provided compound 3m. This inhibitor showed a dramatic loss of BACE2 potency but very potent BACE1 activity (3m, Ki = 69 nM for BACE1 and Ki = 7.3 μM for BACE2) with selectivity over 100-fold against BACE2.
Compound 3n, with a methyl substituent on the isophthalamide ring, further improved BACE1 activity with a Ki of 25 nM and a selectivity >75-fold against BACE2. Based on the efficacy of the α-methyl group on the benzylisophthalamide moiety, we sought to explore the outcome of an α-methyl functionality on the oxazole-based inhibitors. Accordingly, compound 3o was synthesized as a mixture of diastereomers (1:1) on the methyl bearing center on the oxazolylmethyl group. This compound exhibited comparable BACE1 and BACE2 activity with no appreciable selectivity.
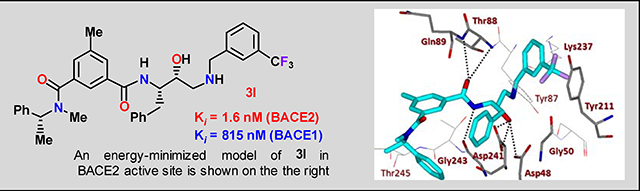
To obtain insight into the molecular binding properties responsible for potency and selectivity of inhibitors 2d and 3l, we created energy-minimized active models for these inhibitors as shown in Figure 6A and 7A. The resulting models were selected based upon the X-ray structure of the protein-ligand complex of BACE2 (Figure 6B and 7B). Figure 6B depicts an overlay of the inhibitor 2d model and the X-ray crystal structure of a known hydroxyethylamine transition state inhibitor in the BACE2 active site. Inhibitor 2d shows a similar binding orientation as the hydroxyethylamine transition state inhibitor in the crystal structure (also applied to 3l in Figure 7B). The detailed docking procedures are shown in the supporting information. As can be seen in Figure 6A, inhibitor 2d makes extensive contacts in the S2′ and S3′ subsites. The P1′-NH is within proximity to form hydrogen bonds with the Gly50 backbone NH. The P2′-carbonyl as well as P2′-NH are also within proximity to form hydrogen bonds with Thr88 backbone NH and side chain hydroxyl groups, respectively. Furthermore, the P3′-hydroxyl group is oriented toward Tyr211 hydroxyl group to form a hydrogen bond. The (S)-propyl group occupies a hydrophobic pocket surrounding Lys86, Ile239, and Tyr211. The P2 carboxamide carbonyl functionality is within proximity to form a hydrogen bond with Gln89 and the amide NH is also within hydrogen bonding distance with Thr244 side chain hydroxyl group. The P3-phenylmethyl group forms extensive hydrophobic interactions with Trp131, Thr245, and Phe125. Overall, the number of polar interactions of P2 and P3-ligands are significantly less compared to inhibitor 2a interactions with the BACE1 active site. The major difference between inhibitors 2d and 3l is that the inhibitor 3l contains a 3-trifluoromethylbenzyl group as the P2’-ligand in place of (2S,3S)-2-amino-3-hydroxy-N-isobutylhexanamide group as the P2-P3 ligand for 2d. The P2’-ligand appears to make extensive hydrophobic interactions in the S2’-subsite. The interactions of P2 and P3-ligands are similar to inhibitor 2d. The combination of active site interactions of inhibitors 2d and 3l with BACE2 may be responsible for their high BACE2 potency and selectivity.
Figure 6.

(A) An energy-minimized model of inhibitor 2d (carbon chain, green) bound in the BACE2 active site. All strong hydrogen bonding interactions are shown as dotted lines. (B) Overlay of inhibitor 2d model with the X-ray crystal structure of hydroxyethylamine transition state inhibitor (carbon chain, orange) in BACE2 active site (PDB : 2EWY). The docking was performed with AutoDock Vina (see SupplementaryInformation for details).
Figure 7.

(A) An energy-minimized model of inhibitor 3l (carbon chain, turquoise) bound in the BACE2 active site. All strong hydrogen bonding interactions are shown as dotted lines. (B) Overlay of inhibitor 3l model with the X-ray crystal structure of hydroxyethylamine transition state inhibitor (carbon chain, orange) in BACE2 active site (PDB : 2EWY). The docking was performed with AutoDock Vina (see SupplementaryInformation for details).
Conclusions
Selective BACE2 inhibitors represent a potentially new treatment for type 2 diabetes. We present here the design, synthesis, and evaluation of a series of potent and highly selective BACE2 inhibitors. We have utilized the X-ray structural studies with BACE1 along with computational docking approaches with BACE2 to optimize the potency and selectivity. Furthermore, we have carried out an extensive SAR analysis to investigate determinants of inhibitors’ potency and selectivity towards BACE2 enzyme using inhibitors 1 and 2a as the starting points. We have shown that inhibitor 2a blocked the processing of Tmem27 in the pancreatic beta-cell line MIN6. The X-ray structure of 2a-BACE1 complex was determined and then used to computationally generate a working model of 2a bound in the BACE2 active site. Based upon our SAR studies and structural requirements for BACE2 selectivity, we have designed selective inhibitors 2d and 3l by removing the P2-sulfonamide, while incorporating an N-methyl amide on the non-prime side. For the most selective inhibitor 2d, we incorporated a propyl chain on the prime side in inhibitor 2a. On the other hand, for inhibitor 3l, we incorporated a highly lipophilic 3-trifluoromethylphenyl ring. Inhibitor 3l has shown excellent BACE2 potency and high selectivity over BACE1. Further studies including the design of more selective BACE2 inhibitors are in progress.
Experimental Section
General
Reactions were carried out under an argon atmosphere in either flame- or oven-dried (120 °C) glassware. All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Anhydrous solvents were obtained as follows: dichloromethane from calcium hydride, diethyl ether and tetrahydrofuran from sodium benzophenone, methanol and ethanol from activated magnesium under argon. All purification procedures were carried out with reagent-grade solvents (purchased form VWR) in air. TLC analysis was conducted using glass-backed thin-layer silica gel chromatography plates (60 a, 250 mm thickness, F254 indicator). Column chromatography was performed using 230–400 mesh, 60 a pore diameter silica gel. 1H and 13C NMR spectra were recorded at room temperature on a Bruker AV800, DRX-500 and ARX-400 or a Varian INOVA300. Chemical shifts (δ values) are reported in parts per million and are referenced to the deuterated residual solvent peak. NMR data are reported as: δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet). Low resolution mass analyses were performed on an Agilent 1290 Infinity II spectrometer. High Resolution mass analyses were performed at the Purdue University Campus-wide Mass Spectrometry Center. HPLC analysis was performed on an Agilent 1260 Infinity instrument. All test inhibitors showed purity >95% by HPLC analysis.
N-((2S,3R)-3-Hydroxy-4-(((2S,3S)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl)amino)-1-phenylbutan-2-yl)-3-((R)-2-(methoxymethyl)pyrrolidine-1-carbonyl)benzamide (2f)
Carboxylic acid 5a (16.5 mg, 0.063 mmol) was dissolved in dry CH2Cl2 (3.0 mL), then EDCI (22 mg, 0.114 mmol), HOBt (15.4 mg, 0.114 mmol) and N,N-DIPEA (20 μL, 0.114 mmol) were sequentially added. Isosteric amine 10a (0.057 mmol) dissolved in dry CH2Cl2 (2 mL) was then added and the reaction mixture was stirred at 25 °C for 14 h. Saturated aqueous sodium bicarbonate was added and the reaction mixture was extracted three times with CH2Cl2. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (8% MeOH in CH2Cl2) affording the title compound as a white amorphous solid (45% yield). δ 1H NMR (400 MHz, CDCl3) δ 8.08 – 7.04 (m, 11H), 4.41 (br, 1H), 4.15 (br, 1H), 3.83 – 3.50 (m, 2H), 3.56 – 3.21 (m, 4H), 3.21 – 2.63 (m, 6H), 2.26 – 1.63 (m, J = 79.7 Hz, 6H), 1.65 – 1.02 (m, 10H), 0.86 (s, 6H); LRMS-ESI (m/z): 583.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C32H47N4O6, 583.3490; found 583.3496.
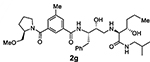
N-((2S,3R)-3-Hydroxy-4-(((2S,3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl)amino)-1-phenylbutan-2-yl)-3-((R)-2-(methoxymethyl)pyrrolidine-1-carbonyl)-5-methylbenzamide (2g)
Title compound was obtained from acid 5b and isosteric amine 10b following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.84 (s, 1H), 7.49 (s, 1H), 7.40 (s, 2H), 7.37 – 7.10 (m, 7H), 4.48 – 4.33 (m, 2H), 3.90 – 3.78 (m, 1H), 3.78 – 3.70 (m, 1H), 3.69 – 3.53 (m, 3H), 3.36 (s, 3H), 3.31 – 3.21 (m, 1H), 3.17 (s, 1H), 3.13 – 2.91 (m, 6H), 2.89 – 2.73 (m, 2H), 2.30 (s, 2H), 2.12 – 1.84 (m, 4H), 1.82 – 1.64 (m, 2H), 1.57 – 1.28 (m, 4H), 0.98 – 0.74 (m, 9H); 13C NMR (200 MHz, CDCl3) δ 172.8), 169.5, 168.0, 138.3, 138.0, 136.9, 134.9, 130.3, 129.7, 129.3, 128.5, 126.5, 123.1, 72.5, 72.0, 71.3, 67.2, 59.2, 57.0, 55.1, 50.7, 50.3, 46.6, 36.5, 35.3, 29.7, 28.5, 27.6, 25.1, 21.2, 20.2, 19.1, 14.0. LRMS-ESI (m/z): 625.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C35H53N4O6, 625.3960; found 625.3972.
N1-((2S,3R)-3-Hydroxy-4-(((2S,3S)-3-hydroxy-1-(isobutylamino)-1-oxobutan-2-yl)amino)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((4-methylthiazol-2-yl)methyl)isophthalamide (2h)
Title compound was obtained from acid 6b and isosteric amine 10a following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.99 – 7.69 (m, 1H), 7.69 – 7.45 (m, 2H), 7.46 – 7.01 (m, 7H), 6.96 – 6.79 (m, 1H), 5.06 – 4.70 (m, 2H), 4.50 – 4.34 (m, 1H), 4.26 – 3.96 (m, 3H), 3.97 – 3.74 (m, 2H), 3.67 (s, 1H), 3.48 – 3.24 (m, 1H), 3.24 – 2.98 (m, 6H), 2.91 (s, 2H), 2.55 – 2.42 (m, 3H), 2.34 (s, 3H), 1.88 – 1.73 (m, 1H), 1.24 (d, J = 6.0 Hz, 3H), 1.02 – 0.83 (m, 6H); 13C NMR (200 MHz, CDCl3) δ 172.6, 171.0, 167.6, 165.0, 138.9, 137.6, 134.7, 130.4, 129.7, 129.2, 128.7, 126.7, 122.8, 114.8, 71.5, 68.3, 67.7, 54.7, 50.5, 48.8, 46.6, 37.7, 36.5, 29.7, 28.5, 21.2, 20.2, 19.1, 17.0. LRMS-ESI (m/z): 624.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C33H46N5O5S, 624.3214; found 624.3220.
N1-((2S,3R)-3-Hydroxy-4-(((2S,3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl)amino)-1-phenylbutan-2-yl)-N3-methyl-N3-((4-methylthiazol-2-yl)methyl)isophthalamide (2i)
Title compound was obtained from acid 6a and isosteric amine 10b following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.80 – 7.68 (m, 2H), 7.56 (d, J = 6.8 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.32 – 7.01 (m, 7H), 6.89 (s, 1H), 4.89 (br, 2H), 4.54 – 4.36 (m, 1H), 3.86 (dd, J = 11.4, 5.1 Hz, 1H), 3.76 (d, J = 4.1 Hz, 1H), 3.18 – 2.93 (m, 8H), 2.91 – 2.73 (m, 2H), 2.46 (s, 3H), 1.77 (dt, J = 13.4, 6.7 Hz, 1H), 1.60 – 1.19 (m, 6H), 1.01 – 0.83 (m, 9H); 13C NMR (200 MHz, CDCl3) δ 172.6, 170.8, 167.2, 164.9, 152.5, 137.7, 135.4, 134.9, 129.9, 129.6, 129.5, 129.2, 128.6, 127.8, 126.7, 125.9, 114.8, 113.9, 109.8, 72.1, 71.1, 67.0, 54.8, 50.3, 48.9, 46.6, 37.8, 36.6, 35.5, 29.7, 28.4, 20.2, 19.1, 17.0, 14.0. LRMS-ESI (m/z): 638.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C34H48N5O5S, 638.3371; found 638.3380.
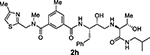
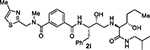
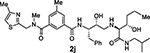
N1-((2S,3R)-3-Hydroxy-4-(((2S,3S)-3-hydroxy-1-(isobutylamino)-1-oxohexan-2-yl)amino)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((4-methylthiazol-2-yl)methyl)isophthalamide (2j)
Title compound was obtained from acid 6b and isosteric amine 10b following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.64 – 7.46 (m, 2H), 7.44 – 7.17 (m, 6H), 7.10 (s, 2H), 6.89 (s, 1H), 4.91 (s, 2H), 4.43 (s, 1H), 3.87 (s, 1H), 3.74 (s, 1H), 3.33 – 2.94 (m, 9H), 2.84 (s, 2H), 2.46 (s, 3H), 2.37 (s, 3H), 1.86 – 1.70 (m, 1H), 1.55 – 1.18 (m, 6H), 1.03 – 0.81 (m, J = 6.5 Hz, 9H); 13C NMR (200 MHz, CDCl3) δ 172.7, 171.0, 167.6, 165.0, 152.5, 138.9, 137.7, 135.4, 134.8, 130.1, 129.6, 129.5, 129.3, 128.6, 126.7, 122.8, 114.8, 113.9, 72.1, 71.2, 67.0, 54.7, 48.9, 46.6, 37.8, 36.6, 35.5, 29.7, 28.5, 21.2, 20.2, 19.1, 17.0, 14.1, 13.9. LRMS-ESI (m/z): 652.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C35H50N5O5S, 652.3527; found 652.3531.
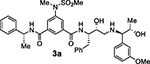
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)propyl)ammo)-1-phenylbutan-2-yl)-5-(N-methylmethylsulfonamido)-N3-((R)-1-phenylethyl)isophthalamide (3a)
Title compound was obtained from acid 4d and the free amine of Boc compound 17a following the procedure described for compound 2f. 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 8.0 Hz, 1H), 8.15 (s, 1H), 7.94 – 7.83 (m, 2H), 7.42 – 7.12 (m, 13H), 7.04 (br, 1H), 6.87 – 6.79 (m, 3H), 5.37 – 5.23 (m, 1H), 4.56 – 4.40 (m, 1H), 4.15 (dd, J = 6.4, 3.7 Hz, 1H), 3.84 – 3.77 (m, 3H), 3.75 – 3.65 (m, 1H), 3.51 (d, J = 3.5 Hz, 1H), 3.33 – 3.27 (m, 3H), 3.05 (dd, J = 13.9, 7.5 Hz, 1H), 2.88 – 2.75 (m, 5H), 2.75 – 2.64 (m, 1H), 1.58 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.4 Hz, 5H).
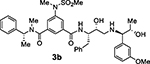
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)propyl)ammo)-1-phenylbutan-2-yl)-N3-methyl-5-(N-methylmethylsulfonamido)-N3-((R)-1-phenylethyl)isophthalamide (3b)
Title compound was obtained from acid 4c and the free amine of Boc compound 17a following the procedure described for compound 2f. 1H NMR (300 MHz, CDCl3) δ 7.93 – 7.72 (m, 2H), 7.59 (s, 1H), 7.48 – 7.03 (m, 12H), 6.99 – 6.72 (m, 3H), 6.11 (s, 0.5H), 5.01 (s, 0.5H), 4.49 (s, 1H), 4.20 (s, 1H), 3.79 (s, 3H), 3.77 – 3.63 (m, 2H), 3.59 (s, 1H), 3.40 – 3.20 (m, 3H), 3.17 – 2.97 (m, 2H), 2.97 – 2.69 (m, 7H), 2.70 – 2.53 (m, 2H), 1.61 (d, J = 6.8 Hz, 3H), 1.02 (d, J = 5.9 Hz, 3H). LRMS-ESI (m/z): 717.8 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C39H49N4O7S, 717.3317; found 717.3310.
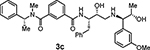
N1- ((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)propyl)ammo)-1-phenylbutan-2-yl)-N3-methyl-N3-((R)-1-phenylethyl)isophthalamide (3c)
Title compound was obtained from acid 4a and the free amine of Boc compound 17a following the procedure described for compound 2f. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 7.5 Hz, 2H), 7.54 (br, 1H), 7.51 – 7.15 (m, 13H), 6.95 (d, J = 8.5 Hz, 2H), 6.92 – 6.83 (m, 1H), 6.21 (s, 0.5H), 5.03 (s, 0.5H), 4.52 (s, 1H), 4.19 (s, 1H), 3.94 – 3.81 (m, 3H), 3.81 – 3.68 (m, 1H), 3.67 – 3.57 (m, 1H), 3.11 (s, 2H), 2.99 (dd, J = 14.1,6.3 Hz, 2H), 2.95 – 2.70 (m, 4H), 2.63 (s, 2H), 1.77 – 1.52 (m, 3H), 1.14 – 1.05 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 171.3, 167.7, 160.1, 140.9, 138.2, 137.1, 129.8, 130.0, 128.8, 128.1, 127.8, 127.0, 125.9, 121.1, 114.6, 113.2, 71.0, 70.4, 68.9, 55.6, 55.4, 49.6, 37.4, 20.1, 15.8. LRMS-ESI (m/z): 610.0 [M+H]+, 632.2 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C37H44N3O5, 610.3276; found 610.3270.
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)propyl)amino)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((R)-1-phenylethyl)isophthalamide (3d)
Title compound was obtained from acid 4b and the free amine of Boc compound 17a following the procedure described for compound 2f. 1H NMR (500 MHz, CDCl3) δ 7.70 – 7.57 (m, 2H), 7.54 – 7.17 (m, 13H), 7.08 – 6.84 (m, 3H), 6.21 (s, 0.5H), 5.03 (s, 0.5H), 4.51 (s, 1H), 4.19 (s, 1H), 3.85 (s, 3H), 3.80 – 3.68 (m, 1H), 3.62 (s, 1H), 3.10 (s, 1H), 3.07 – 2.86 (m, 3H), 2.86 – 2.70 (m, 3H), 2.62 (s, 2H), 2.44 (s, 3H), 1.75 – 1.58 (m, 3H), 1.11 (d, J = 6.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.0, 160.1, 140.7, 138.1, 137.2, 135.4, 130.2, 129.6, 129.4, 129.0, 128.1, 127.8, 127.1, 122.9, 121.2, 114.6, 113.3, 71.1,70.4, 68.9, 55.6, 55.3, 49.5, 37.3, 30.2, 21.8, 20.2. LRMS-ESI (m/z): 624.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C38H46N3O5, 624.3432; found 624.3437.
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)pentyl)amino)-1-phenylbutan-2-yl)-N3-methyl-N3-((R)-1-phenylethyl)isophthalamide (3e)
Title compound was obtained from acid 4a and the free amine of Boc compound 17b following the procedure described for compound 2f. 1H NMR (800 MHz, CDCl3) δ 7.96 – 7.73 (m, 2H), 7.51 (d, J = 7.5 Hz, 1H), 7.46 – 7.37 (m, 4H), 7.34 (dd, J = 15.2, 7.5 Hz, 1H), 7.28 – 7.22 (m, 5H), 7.22 – 7.14 (m, 2H), 6.92 (d, J = 7.9 Hz, 2H), 6.85 – 6.82 (m, 1H), 6.16 (s, 0.5H), 5.01 (s, 0.5H), 4.48 (s, 1H), 3.97 (s, 1H), 3.82 (s, 3H), 3.73 (dd, J = 12.6, 3.2 Hz, 1H), 3.61 (d, J = 1.9 Hz, 1H), 3.09 (s, 1H), 2.97 (dd, J = 14.1, 6.3 Hz, 1H), 2.92 – 2.81 (m, 1H), 2.84 – 2.69 (m, 2H), 2.59 (s, 1H), 1.70 – 1.56 (m, 3H), 1.40 – 1.25 (m, 4H), 1.19 (td, J = 9.1, 4.7 Hz, 1H), 0.89 – 0.80 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 159.9, 140.8, 138.0, 136.9, 129.8, 129.6, 129.8, 128.7, 127.9, 127.7, 126.9, 125.8, 121.0, 114.5, 113.0, 70.9, 70.3, 68.8, 55.5, 55.30, 53.8, 49.5, 37.2, 32.2, 20.0, 15.7. LRMS-ESI (m/z): 638.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C39H48N3O5, 638.3589; found 638.3596.
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)pentyl)ammo)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((R)-1-phenylethyl)isophthalamide (3f)
Title compound was obtained from acid 4b and the free amine of Boc compound 17b following the procedure described for compound 2f. 1H NMR (300 MHz, CDCl3) δ 7.82 (s, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.50 – 7.07 (m, 12H), 6.90 (d, J = 6.6 Hz, 2H), 6.81 (d, J = 8.1 Hz, 1H), 6.13 (s, 0.5H), 4.98 (s, 0.5H), 4.44 (s, 1H), 3.95 (s, 1H), 3.77 (s, 3H), 3.75 – 3.62 (m, 1H), 3.58 (s, 1H), 3.17 – 2.90 (m, 3H), 2.92 – 2.48 (m, 6H), 2.36 (s, 3H), 1.70 – 1.42 (m, 4H), 1.44 – 1.09 (m, 4H), 0.83 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.1, 160.2, 140.8, 138.5, 137.9, 137.3, 135.5, 130.7, 130.0, 129.1, 129.3, 128.7, 128.4, 128.1, 127.98, 127.6, 127.4, 126.8, 123.0, 121.7, 121.0, 115.1, 114.3, 113.7, 113.0, 71.2, 70.4, 69.0, 55.7, 55.4, 49.6, 37.4, 32.4, 30.2, 21.8, 20.2, 18.1, 15.9. LRMS-ESI (m/z): 652.4 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C40H50N3O5, 652.3745; found 652.3749.
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-phenylpentyl)ammo)-1-phenylbutan-2-yl)-N3-methyl-N3-((R)-1-phenylethyl)isophthalamide (3g)
Title compound was obtained from acid 4a and the free amine of Boc compound 17c following the procedure described for compound 2f. 1H NMR (300 MHz, CDCI3) δ 8.03 (s, 1H), 7.80 (s, 2H), 7.62 – 7.10 (m, 17H), 6.13 (s, 1H), 4.98 (s, 1H), 4.56 – 4.39 (m, 1H), 3.95 (s, 1H), 3.68 (s, 1H), 3.60 (d, J = 3.3 Hz, 1H), 3.08 (dd, J = 13.8, 7.6 Hz, 2H), 2.92 (dd, J = 14.0, 6.4 Hz, 2H), 2.88 – 2.65 (m, 4H), 2.57 (s, 2H), 1.68 – 1.42 (m, 3H), 1.37 – 1.02 (m, 4H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (200 MHz, CDCI3) δ 167.4, 137.7, 136.7, 129.3, 128.8, 128.4, 127.5, 126.6, 126.5, 73.5, 70.5, 67.5, 55.1, 49.0, 36.9, 36.3, 29.7, 19.2. LRMS-ESI (m/z): 608.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C38H46N3O4, 608.3483; found 608.3493.
N1-((2S,3R)-3-Hydroxy-4-(((1R,2S)-2-hydroxy-1-phenylpentyl)amino)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((R)-1-phenylethyl)isophthalamide (3h)
Title compound was obtained from acid 4b and the free amine of Boc compound 17c following the procedure described for compound 2f. 1H NMR (500 MHz, CDCl3) δ 8.49 – 8.26 (m, J = 24.0 Hz, 2H), 8.15 (d, J = 19.2 Hz, 2H), 8.01 – 7.61 (m, 15H), 6.67 (s, 1H), 5.02 (s, 1H), 4.64 (s, 2H), 4.24 (s, 2H), 3.80 –– 3.60 (m, 2H), 3.58 – 3.17 (m, 2H), 3.12 (s, 2H), 2.91 (s, 2H), 2.14 (s, 3H), 2.01 (s, 2H), 1.88 – 1.57 (m, 5H), 1.42 – 1.28 (m, 3H); 13C NMR (200 MHz, CDCl3) δ 167.4, 137.6, 136.8, 130.8, 129.6, 129.4, 128.5, 127.8, 127.5, 126.6, 70.6, 67.4, 54.9, 48.8, 36.7, 36.3, 29.7, 22.7, 21.3, 19.1, 14.0. LRMS-ESI (m/z): 622.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C39H47N3O4, 622.3640; found 622.3635.
N1-((2S,3R)-3-Hydroxy-4-((3-methoxybenzyl)ammo)-1-phenylbutan-2-yl)-N3-methyl-N3-((R)-1-phenylethyl)isophthalamide (3i)
Title compound was obtained from acid 4a and isosteric amine 18b following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.52 (d, J = 7.4 Hz, 1H), 7.47 – 7.12 (m, 12H), 7.05 (br, 1H), 6.99 – 6.89 (m, 2H), 6.84 (d, J = 7.6 Hz, 1H), 4.49 – 4.34 (m, 1H), 3.96 – 3.74 (m, 6H), 3.72 – 3.45 (m, 2H), 3.13 – 2.97 (m, 2H), 2.93 – 2.77 (m, 2H), 2.78 – 2.57 (m, 2H), 1.62 (d, J = 6.8 Hz, 3H); 13C NMR (200 MHz, CDCl3) δ 170.7, 167.2, 159.9, 137.8, 129.7, 129.3, 128.8, 128.7, 127.7, 127.5, 126.6, 120.9, 114.2, 113.3, 70.4, 55.3, 54.2, 53.4, 50.5, 36.4, 29.7. LRMS-ESI (m/z): 566.3 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C35H40N3O4, 566.3013; found 566.3009.
N1-((2S,3R)-3-Hydroxy-4-((3-methoxybenzyl)ammo)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((R)-1-phenylethyl)isophthalamide (3j)
Compound was synthesized as reported.36 1H, 13C, LRMS and HRMS are identical to those reported in literature.36
N1-((2S,3R)-3-Hydroxy-1-phenyl-4-((3-(trifluoromethyl)benzyl)amino)butan-2-yl)-N3-methyl-N3-((R)-1-phenylethyl)isophthalamide (3k)
Title compound was obtained from acid 4a and isosteric amine 18a following the procedure described for compound 2f. 1H NMR (400 MHz, CDCl3) δ 7.85–- 7.08 (m, 18H), 6.94 (br, 1H), 6.14 (br, 1H), 4.92 (s, 1H), 4.50 – 4.23 (m, 1H), 3.86 (q, J = 13.5 Hz, 2H), 3.75 – 3.57 (m, 1H), 3.17 – 2.90 (m, 3H), 2.91 – 2.65 (m, 4H), 2.66 –– 2.43 (m, 1H), 1.72 – 1.47 (m, 3H); 13C NMR (200 MHz, CDCl3) δ 170.6, 167.2, 140.1, 137.6, 137.2, 131.7, 131.4, 131.3, 130.9, 129.3, 128.8, 129.0, 127.9, 127.7, 127.4, 126.7, 126.5, 126.3, 126.4, 126.42, 125.44, 124.2, 123.4, 113.9, 70.7, 54.0, 53.3, 50.7, 36.4, 29.7, 22.70. LRMS-ESI (m/z): 604.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C35H37F3N3O3, 604.2782; found 604.2785.
N1-((2S,3R)-3-Hydroxy-1-phenyl-4-((3-(trifluoromethyl)benzyl)ammo)butan-2-yl)-N3,5-dimethyl-N3-((R)-1-phenylethyl)isophthalamide (3l)
Title compound was obtained from acid 4b and isosteric amine 18a following the procedure described for compound 2f. 1H NMR (300 MHz, CDCl3) 1H NMR (800 MHz, CDCl3) δ 7.62 (s, 1H), 7.56 (dd, J = 21.7, 7.5 Hz, 2H), 7.52 – 7.38 (m, 5H), 7.39 – 7.30 (m, 3H), 7.27 – 7.12 (m, 5H), 6.85 – 6.69 (m, 1H), 6.16 (br, 1H), 4.94 (br, 1H), 4.43 – 4.30 (m, 1H), 3.92 (dd, J = 37.4, 13.4 Hz, 2H), 3.79 – 3.64 (m, 1H), 3.10 (dd, J = 14.2, 5.0 Hz, 2H), 2.99 (s, 1H), 2.91 – 2.69 (m, 4H), 2.56 (s, 2H), 2.36 (s, 3H), 1.68 – 1.49 (m, 3H); 13C NMR (200 MHz, CDCl3) δ 170.8, 167.5, 139.9, 137.6, 131.8, 131.2, 130.2, 130.4, 130.2, 130.0, 130.0, 129.3. 127.5, 127.2, 126.9, 126.6, 125.2, 124.8, 124.4, 123.4, 70.8, 53.8, 53.2, 50.6, 36.4, 29.7, 21.2. LRMS-ESI (m/z): 618.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C36H39F3N3O3, 618.2938; found 618.2945.
N1-((2S,3R)-3-Hydroxy-4-((3-methoxybenzyl)amino)-1-phenylbutan-2-yl)-N3-methyl-N3-((4-methyloxazol-2-yl)methyl)isophthalamide (3m)
Title compound was obtained from acid 7a and isosteric amine 18b following the procedure described for compound 2f. 1H NMR (800 MHz, CDCl3) δ 7.87 (d, J = 47.0 Hz, 1H), 7.80 – 7.62 (m, 2H), 7.63 – 7.46 (m, 1H), 7.36 (dd, J = 20.3, 12.6 Hz, 5H), 7.24 – 7.15 (m, 1H), 7.16 – 7.10 (m, 1H), 7.01 (s, 1H), 6.94 (d, J = 7.3 Hz, 1H), 6.82 (dd, J = 8.3, 2.2 Hz, 1H), 4.85 – 4.77 (m, 1H), 4.41 (s, 1H), 4.35 (d, J = 5.3 Hz, 1H), 4.13 – 4.04 (m, 2H), 4.01 – 3.94 (m, 1H), 3.72 (s, 3H), 3.15 – 3.03 (m, 3H), 2.98 – 2.86 (m, 3H), 2.22 – 2.10 (m, 3H); 13C NMR (200 MHz, CDCl3) δ 167.4, 159.9, 159.3, 137.8, 136.9, 136.2, 135.8, 135.5, 135.2, 134.2, 130.0, 129.4, 128.6, 128.5, 127.8, 126.5, 126.1, 124.5, 121.7, 114.8, 114.7, 114.6, 113.9, 70.0, 55.3, 54.1, 52.3, 50.4, 35.9, 29.7. LRMS-ESI (m/z): 557.2 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C32H37N4O5, 557.2759; found 557.2755.
N1-((2S,3R)-3-Hydroxy-4-((3-methoxybenzyl)amino)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-((4-methyloxazol-2-yl)methyl)isophthalamide (3n)
Title compound was obtained from acid 7b and isosteric amine 18b following the procedure described for compound 2f. 1H NMR (800 MHz, CDCl3) δ 7.67 – 7.47 (m, 1H), 7.46 – 7.34 (m, 1H), 7.34 – 7.08 (m, 8H), 7.06 – 6.87 (m, 2H), 6.84 (d, J = 8.3 Hz, 1H), 4.82 (s, 1H), 4.44 (s, 1H), 4.41 – 4.32 (m, 1H), 3.96 (s, 1H), 3.88 (d, J = 12.0 Hz, 2H), 3.78 (s, 3H), 3.17 – 3.05 (m, 3H), 3.05 – 2.89 (m, 5H), 2.89 – 2.73 (m, 2H), 2.35 (s, 3H), 2.19 (s, 3H); 13C NMR (200 MHz, CDCl3) δ 171.1, 167.5, 159.9, 159.3, 139.0, 137.8, 136.6, 135.6, 135.1, 134.3, 130.7, 129.8, 129.5, 129.3, 128.5, 126.6, 122.9, 121.2, 114.4, 113.9, 70.3, 55.3, 54.0, 52.9, 50.5, 36.2, 29.7, 21.2, 11.5. LRMS-ESI (m/z): 571.3 [M+H]+; HRMS-APCI (m/z): [M+H]+ calcd for C33H39N4O5, 571.2915; found 571.2921.
N1-((2S,3R)-3-Hydroxy-4-((3-methoxybenzyl)ammo)-1-phenylbutan-2-yl)-N3,5-dimethyl-N3-(1-(4-methyloxazol-2-yl)ethyl)isophthalamide (3o)
To a solution of carboxylic acid 24 (42 mg, 0.13 mmol) in CH2Cl2 (2 mL) were added EDCI HCl (32 mg, 0.16 mmol) and HOBt·H2O (22 mg, 0.16 mmol) at 0 °C and the mixture was stirred for 10 min. Amine 18b (50 mg, 0.16 mmol) in CH2Cl2 (1 mL) and DIPEA (30 μL, 0.16 mmol) were added at 0 °C and the resulting mixture was stirred at 23 °C for 12 h. After this period, the reaction was quenched with saturated aqueous NH4Cl solution. The mixture was extracted with CH2Cl2, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography over silica gel (3% MeOH/CH2Cl2) to afford inhibitor 3o (39 mg, 48%). 1H NMR (800 MHz, CD3OD): δ 7.70–7.61 (m, 1H), 7.53–7.37 (m, 3H), 7.34 (t, J = 7.3 Hz, 1H), 7.30–7.21 (m, 4H), 7.17 (m, 1H), 7.10 (m, 1H), 7.07 (d, J = 6.5 Hz, 1H), 6.98 (d, J = 7.6 Hz, 1H), 5.95 (m, 0.6H), 4.96 (m, 0.4H), 4.28–4.19 (m, 3H), 3.96 (m, 1H), 3.81 (s, 3H), 3.38 (d, J = 12.5 Hz, 1H), 3.19 (d, J = 12.0 Hz, 1H), 3.06 (m, 1H), 2.89 (m, 1H), 2.86–2.73 (m, 3H), 2.42 (s, 3H), 2.23–2.14 (m, 3H), 1.74–1.56 (m, 3H); 13C NMR (200 MHz, CD3OD): δ 171.4, 168.8, 160.3, 139.1, 138.2, 136.3, 135.9, 135.7, 135.5, 134.2, 132.0, 130.3, 129.9, 129.1, 129.0, 128.7, 128.1, 126.2, 122.6, 121.8, 115.2, 114.9, 69.2, 54.6, 54.4, 50.8, 49.6, 48.1, 48.0, 35.8, 32.3, 19.8,14.9, 13.8, 9.8; LRMS-ESI (m/z): 585 [M+H]+; HRMS-ESI (m/z): [M+H]+ calcd for C34H41N4O5, 585.3072; found 585.3068.
(2S,3S)-2-(((2R,3S)-3-Amino-2-hydroxy-4-phenylbutyl)amino)-3-hydroxy-N-isobutylbutanamide (10a)
Compound was synthesized from derivative 8a and epoxide 9 as reported. 1H data are identical to those reported in literature.36
(2S,3S)-2-(((2R,3S)-3-Amino-2-hydroxy-4-phenylbutyl)amino)-3-hydroxy-N-isobutylhexanamide (10b)
Compound was synthesized from derivative 8b and epoxide 9 as reported. 1H data are identical to those reported in literature.36
(E)-1-Methoxy-3-(prop-1-en-1-yl)benzene (12a)
n-Butyllithium (1.6 M solution in n-hexane, 6.5 mL, 10.43 mmol) was added dropwise to a stirred suspension of ethyltriphenylphosphonium bromide (3.8 g, 10.28 mmol) in dry THF (37 mL) at −78 °C under argon atmosphere. The temperature of the reaction mixture was allowed to rise to 0 °C for 30 min and then the solution was cooled again to −78 °C. Subsequently, a solution of 3-methoxybenzaldehyde 11a (1.0 g, 7.35 mmol) in dry THF (10 mL) was added over 30 min. The reaction mixture was stirred for an additional 20 min at −78 °C and then for 3 h at 25 °C. Water was added and the reaction mixture was extracted three times with EtOAc. The organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) providing 1-methoxy-3-(prop-1-en-1-yl)benzene as E/Z mixture (88% yield). The E/Z mixture of alkenes (959 mg, 6.41 mmol) was dissolved in chloroform (40 mL). Bis(acetonitrile)dichloropalladium (II) (30 mg) was added and the reaction mixture was stirred at 25 °C for 72 h. Then, solvent was evaporated, and the residue was purified by using a short silica gel path eluted with n-hexane-EtOAc 9:1 to afford 12a as a pale yellow oil (95% yield). 1H NMR (300 MHz, CDCl3) δ 7.24 (t, J = 7.9 Hz, 1H), 7.01 – 6.85 (m, 2H), 6.78 (dd, J = 8.2, 2.5 Hz, 1H), 6.50 – 6.35 (m, 1H), 6.36 – 6.16 (m, 1H), 3.83 (s, 3H), 1.91 (d, J = 6.3 Hz, 3H).
(E)-1-Methoxy-3-(pent-1-en-1-yl)benzene (12b)
Title compound was obtained following the procedure described for 12a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to afford title compound as a dark yellow oil (69% yield over two steps). 1H NMR (300 MHz, CDCl3) δ 7.33 – 7.18 (m, 1H), 7.05 – 6.92 (m, 2H), 6.88 – 6.74 (m, 1H), 6.51 – 6.34 (m, 1H), 6.35 – 6.18 (m, 1H), 3.84 (s, 3H), 2.32 – 2.13 (m, 2H), 1.63 – 1.43 (m, 2H), 1.00 (t, J = 7.3 Hz, 3H).
(E)-Pent-1-en-1-ylbenzene (12c)
Title compound was obtained following the procedure described for 12a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 15:1) to afford title compound as a dark yellow oil (81% yield over 2 steps). 1H NMR (300 MHz, CDCl3) δ 7.45 – 7.26 (m, 4H), 7.25 – 7.14 (m, 1H), 6.46 – 6.34 (m, 1H), 6.33 – 6.16 (m, 1H), 2.20 (td, J = 7.6, 0.8 Hz, 2H), 1.61 – 1.40 (m, 2H), 1.08 – 0.92 (m, 3H).
(1S,2S)-1-(3-Methoxyphenyl)propane-1,2-diol (13a)
Compound 12a (200 mg, 1.35 mmol) was dissolved in a 1:1 mixture of t-BuOH and water (20 mL). Methanesulfonamide (193 mg, 2.02 mmol) and AD-mix-α (1.9 g) were added in sequence and the reaction mixture was stirred at 25 °C for 36 h. The reaction was quenched with Na2SO3 and extracted three times with EtOAc. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel flash chromatography (n-hexane/EtOAc 1:1) providing title compound as a white amorphous solid (73% yield). 1H NMR (300 MHz, CDCl3) δ 7.32 – 7.16 (m, 1H), 6.96 – 6.75 (m, 3H), 4.40 – 4.23 (m, 1H), 3.89 – 3.73 (m, 4H), 3.11 – 2.96 (m, 2H), 2.80 (br, 1H), 1.04 (d, J = 6.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 159.6, 142.6, 129.4, 119.1, 113.3, 112.3, 79.2, 72.0, 55.1, 18.6.
(1S,2S)-1-(3-Methoxyphenyl)pentane-1,2-diol (13b)
Title compound was obtained following the procedure described for 13a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 1:1) to afford title compound as a dark yellow oil (68% yield). 1H NMR (300 MHz, CDCl3) δ 7.29 – 7.12 (m, 1H), 6.91 – 6.70 (m, 3H), 4.42 – 4.18 (m, 1H), 3.78 – 3.65 (m, 4H), 3.11 – 2.96 (m, 2H), 2.87 (br, 1H), 1.71 – 1.23 (m, 4H), 0.98 – 0.75 (m, 3H).
(1S,2S)-1-Phenylpentane-1,2-diol (13c)
Title compound was obtained following the procedure described for 13a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 1:1) to afford title compound as a dark yellow oil (68% yield). 1H NMR (300 MHz, CDCl3) δ 7.49 – 7.24 (m, 5H), 4.44 (d, J = 6.8 Hz, 1H), 3.70 (ddd, J = 8.3, 6.9, 3.5 Hz, 1H), 2.50 (br, 2H), 1.62 – 1.19 (m, 4H), 0.96 – 0.78 (m, 3H).
(4S,5S)-4-Methyl-5-(3-methoxyphenyl)-1,3,2-dioxathiolane-2-oxide (14a)
Diol 13a (300 mg, 1.65 mmol) was dissolved in triethylamine (5 mL) and cooled to 0 °C in an ice bath under an argon atmosphere. Thionyl chloride (144 μL, 1.98 mmol) was added dropwise and the reaction mixture stirred at 0 °C for 50 min (monitored by TLC). After completion, ice-cold water (20 mL) was added and the mixture was extracted with Et2O. The ethereal layer was washed with 10% HCl, saturated aqueous NaHCO3 and brine. The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) providing the compound as a dark yellow oil (79% yield, mixture of diastereoisomers). 1H NMR (300 MHz, CDCl3) δ (1:1 diastereomeric mixture) 7.33 (td, J = 7.9, 3.1 Hz, 1H), 7.09 – 6.88 (m, 3H), 5.41 (d, J = 9.1 Hz, 0.5H), 4.93 – 4.77 (m, 1H), 4.39 (dq, J = 9.0, 6.2 Hz, 0.5H), 3.83 (s, 3H), 1.60 (t, J = 10.4 Hz, 1.5H), 1.52 – 1.43 (m, 1.5H).
(4S,5S)-4-Propyl-5-(3-methoxyphenyl)-1,3,2-dioxathiolane-2-oxide (14b)
Title compound was obtained following the procedure described for 14a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to afford title compound as a brown oil (66% yield). 1H NMR (300 MHz, CDCl3) δ (1:1 diastereomeric mixture) 7.32 (td, J = 7.8, 3.0 Hz, 1H), 6.96 (ddd, J = 13.3, 9.2, 5.9 Hz, 3H), 5.45 (d, J = 9.0 Hz, 0.5H), 4.88 (d, J = 9.6 Hz, 0.5H), 4.74 (dt, J = 9.6, 6.0 Hz, 0.5H), 4.31 (td, J = 8.8, 3.7 Hz, 0.5H), 3.81 (s, 3H), 2.00 – 1.32 (m, 4H), 0.92 (td, J = 7.2, 2.1 Hz, 3H).
(4S,5S)-4-Propyl-5-phenyl-1,3,2-dioxathiolane-2-oxide (14c)
Title compound was obtained following the procedure described for 14a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to afford title compound as a brown oil (54% yield). 1H NMR (300 MHz, CDCl3) δ (1:1 diastereomeric mixture) 7.63 – 7.33 (m, 5H), 5.49 (d, J = 9.1 Hz, 0.5H), 4.91 (d, J = 9.6 Hz, 0.5H), 4.85 – 4.68 (m, 0.5H), 4.44 – 4.22 (m, 0.5H), 2.01 – 1.29 (m, 4H), 1.08 – 0.82 (m, 3H).
(1R,2S)-1-Azido-1-(3-methoxyphenyl)propan-2-ol (15a)
To a stirred solution of cyclic sulfite 14a (150 mg, 0.66 mmol) in DMF (5 mL) was added sodium azide (171 mg, 263 mmol). The reaction flask was then heated to 80 C for 8 h. After completion, the reaction mixture was poured in 25 mL of water, and then extracted three times with ether. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude azidoalcohol was then purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to provide the title compound as a light yellow oil (88% yield). 1H NMR (300 MHz, CDCl3) δ 7.41 – 7.23 (m, 1H), 6.98 – 6.81 (m, 3H), 4.43 (d, J = 5.7 Hz, 1H), 4.06 – 3.85 (m, 1H), 3.81 (s, 3H), 2.03 (br, 1H), 1.17 (d, J = 6.3 Hz, 3H).
(1R,2S)-1-Azido-1-(3-methoxyphenyl)pentan-2-ol (15b)
Title compound was obtained following the procedure described for 15a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to afford title compound as a brown oil (74% yield). 1H NMR (300 MHz, CDCl3) δ 7.42 – 7.23 (m, 2H), 7.04 – 6.83 (m, 2H), 4.45 (d, J = 5.8 Hz, 1H), 3.93 – 3.71 (m, 4H), 1.77 – 1.20 (m, 5H), 0.91 (t, J = 6.5 Hz, 3H).
(1R,2S)-1-Azido-1-phenylpentan-2-ol (15c)
Title compound was obtained following the procedure described for 15a. The residue was purified by silica gel flash chromatography (n-hexane-EtOAc 9:1) to afford title compound as a brown oil (74% yield). 1H NMR (300 MHz, CDCl3) δ 7.58 – 7.29 (m, 4H), 4.48 (d, J = 5.7 Hz, 1H), 3.79 (d, J = 5.7 Hz, 1H), 1.76 – 1.17 (m, 5H), 1.06 – 0.83 (m, 3H).
(1R,2S)-1-Ammo-1-(3-methoxyphenyl)propan-2-ol (16a)
To a solution of 15a (120 mg, 0.58 mmol) and di-tert-butyldicarbonate (164 mg, 0.75 mmol) in EtOAc (5 mL), 10% Pd/C (15 mg) was added and the reaction mixture was stirred at 25 °C under hydrogen atmosphere for 16 h. The reaction mixture was then filtered through a celite plug. The residue containing tert-butyl ((1R,2S)-2-hydroxy-1-(4-methoxyphenyl)propyl)carbamate 1H NMR (300 MHz, CDCl3) δ 7.26–7.20 (m, 1H), 6.85–6.75 (m, 3H), 5.51 (br, 1H), 4.56 (br, 1H), 4.10–3.96 (m, 1H), 3.76 (s, 3H), 2.31 (d, J = 6.6 Hz, 1H), 1.39 (s, 9H), 1.05 (d, J = 6.6 Hz, 3H) was submitted to the following step without further purification. The compound was dissolved in CH2Cl2 (2 mL) and TFA (300 μL) was added. The reaction mixture was stirred at 25 °C for 2 h, then solvents evaporated. CH2Cl2 (3 mL) was added to the residue and evaporated under reduced pressure. This operation was repeated additional two times. The residue was then taken up in CH2Cl2 and saturated aqueous NaHCO3 (1 mL) was added. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford desired aminoalcohol as a white amorphous solid (86% yield, 2 steps).
(1R,2S)-1-Ammo-1-(3-methoxyphenyl)pentan-2-ol (16b)
Title compound was obtained following the procedure described for 16a to afford title compound as a white amorphous solid (66% yield, 2 steps). tert-Butyl ((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)pentyl)carbamate 1H NMR (300 MHz, CDCl3) δ 7.23 (t, J = 7.8 Hz, 1H), 7.01 – 6.74 (m, 3H), 5.56 (br, 1H), 4.60 (br, 1H), 3.95 – 3.63 (m, 4H), 2.11 – 1.85 (m, 1H), 1.67 – 1.02 (m, 13H), 0.86 (t, J = 7.0 Hz, 3H).
(1R,2S)-1-Amino-1-phenylpentan-2-ol (16c)
Title compound was obtained following the procedure described for 16a. to afford title compound as a white amorphous solid (66% yield over two steps). tert-Butyl ((1R,2S)-2-hydroxy-1-phenylpentyl)carbamate 1H NMR (300 MHz, CDCl3) δ 7.51 – 7.19 (m, 5H), 5.44 (br, 1H), 4.63 (br, 1H), 3.88 (d, J = 8.8 Hz, 1H), 1.72 – 1.02 (m, 14H), 0.88 (t, J = 7.1 Hz, 3H).
(tert-Butyl ((2S,3R)-3-hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)propyl)amino)-1-phenylbutan-2-yl)carbamate (17a)
Compound 16a (0.565 mmol) was dissolved in i-PrOH (7 mL) and 9 (74 mg, 0.283 mmol) was added. The reaction mixture was stirred at 80 °C for 14 h. The solvent was evaporated under reduced pressure and the residue was purified by silica gel flash chromatography (CH2Cl2/MeOH 100:0 to 98:2) providing title compound as an amorphous white solid (65% yield). 1H NMR (300 MHz, CDCl3) δ 7.43 – 7.10 (m, 5H), 7.00 – 6.74 (m, 3H), 4.85 (d, J = 8.9 Hz, 1H), 4.38 – 4.23 (m, 1H), 4.12 – 3.95 (m, 2H), 3.93 – 3.75 (m, 4H), 3.75 – 3.61 (m, 5H), 3.62 – 3.48 (m, 2H), 3.47 – 3.15 (m, 1H), 3.04 – 2.48 (m, 6H), 1.48 – 1.12 (m, 9H), 1.05 (d, J = 6.2 Hz, 3H).
tert-Butyl ((2S,3R)-3-hydroxy-4-(((1R,2S)-2-hydroxy-1-(3-methoxyphenyl)pentyl)amino)-1-phenylbutan-2-yl)carbamate (17b)
Title compound was obtained following the procedure described for 17a. The residue was purified by silica gel flash chromatography (CH2Cl2/MeOH 100:0 to 98:2) to afford title compound as a pale yellow oil (71% yield). 1H NMR 1H NMR (300 MHz, CDCl3) δ 7.39 – 7.07 (m, 5H), 6.95 – 6.72 (m, 3H), 4.74 (d, J = 8.8 Hz, 1H), 3.90 – 3.74 (m, 5H), 3.66 – 3.36 (m, 3H), 3.13 – 2.74 (m, 6H), 2.74 – 2.51 (m, 2H), 1.55 – 1.12 (m, 13H), 0.87 (t, J = 7.0 Hz, 3H).
tert-Butyl ((2S,3R)-3-hydroxy-4-(((1R,2S)-2-hydroxy-1-phenylpentyl)amino)-1-phenylbutan-2-yl)carbamate (17c)
Title compound was obtained following the procedure described for 17a. The residue was purified by silica gel flash chromatography (CH2Cl2/MeOH 100:0 to 98:2) to afford title compound as a pale yellow oil (71% yield). 1H NMR (300 MHz, CDCl3) δ 7.50 – 7.09 (m, 10H), 4.77 (d, J = 8.8 Hz, 1H), 3.95 – 3.78 (m, 2H), 3.78 – 3.33 (m, 2H), 3.31 – 3.00 (m, 2H), 2.98 – 2.74 (m, 2H), 2.76 – 2.51 (m, 2H), 1.65 – 1.06 (m, 13H), 0.86 (t, J = 7.0 Hz, 3H).
tert-Butyl methyl(1-(4-methyloxazol-2-yl)ethyl)carbamate (22)
A solution of 21 (110 mg, 0.486 mmol) in diethyl ether (5 mL) was cooled to −78 °C and treated with TMEDA (95 μL, 0.63 mmol), followed by s-BuLi (45 μL, 0.63 mmol) dropwise. The mixture was stirred for 30 min at −78 °C and treated with a solution of iodomethane (60 μL, 0.97 mmol) in 1 mL of diethyl ether and then slowly warmed to room temperature and stirred for 8 h. The mixture was diluted with 6 mL of water and extracted with ether (2 × 20 mL) and washed with saturated aqueous NH4Cl solution. The combined extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to give a crude product which was purified by column chromatography over silica gel to afford 22 (64 mg, 55%) as a colorless oil and 28 mg of starting material was recovered. 1H NMR (400 MHz, CDCl3): δ 7.30 (q, J = 4.0 Hz, 1H), 5.54 (m, 0.6H), 5.20 (m, 0.4H), 2.67 (brs, 3H), 2.15 (d, J = 1.2 Hz, 3H), 1.54 (d, J = 7.0 Hz, 3H), 1.46 (s, 9H). LRMS-ESI (m/z): 241 [M+H]+.
3-Methyl-5-(methyl(1-(4-methyloxazol-2-yl)ethyl)carbamoyl)benzoic acid (24)
Methyl 3-methyl-5-(methyl(1-(4-methyloxazol-2-yl)ethyl)carbamoyl)benzoate. To a stirred solution of 22 (64 mg, 0.26 mmol) in dichloromethane (3 mL) was added TFA (1 mL) at 0 °C under argon atmosphere and the mixture was stirred at 23 °C for 2 h. Upon completion, trifluoroacetic acid and CH2Cl2 were removed under reduced pressure, the resulting residue was dissolved in ethyl acetate and washed with saturated aqueous NaHCO3 solution. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford amine derivative (40 mg) which used for next step without further purification.
To a solution above N-methyl-1-(4-methyloxazol-2-yl)ethan-1-amine (40 mg, 0.28 mmol) and 3-(methoxycarbonyl)-5-methylbenzoic acid 23 (66 mg, 0.34 mmol) in CH2Cl2 (4 mL) at 23 °C was added EDCI. HCl (109 mg, 0.57 mmol), HOBt·H2O (74 mg, 0.57 mmol) and N,N-DIPEA (150 μL, 0.85 mmol). The resulting mixture was stirred at 23 °C for 12 h. After this period, the reaction was quenched with saturated aqueous NaHCO3 solution. The mixture was extracted with EtOAc, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (40% EtOAc/hexanes) to provide the corresponding amide derivative Methyl 3-methyl-5-(methyl(1-(4-methyloxazol-2-yl)ethyl)carbamoyl)benzoate (60 mg, 66% over 2 steps). 1H NMR (400 MHz, CDCl3) δ 8.02–7.84 (m, 2H), 7.47 (m, 1H), 7.36 (m, 1H), 6.05 (m, 0.6H), 4.96 (m, 0.4H), 3.88 (s, 3H), 2.86 (s, 1.4H), 2.73 (1.6H), 2.39 (s, 3H), 2.14 (s, 3H), 1.68–1.54 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 171.0, 170.5, 166.4, 162.3, 161.7, 139.0, 138.8, 136.2, 136.1, 135.0, 134.9, 132.1, 131.7, 131.3, 130.3, 130.1, 125.1, 124.8, 52.2, 46.9, 36.5, 32.5, 28.0, 24.6, 21.1, 16.2, 15.0, 11.3. LRMS-ESI (m/z): 317 [M+H] +.
To a solution of this latter (48 mg, 0.15 mmol) in a mixture of water (1 mL) and THF (2 mL) at 0 °C was added LiOH•H2O (19 mg, 0.45 mmol). The resulting mixture was stirred for at 23 °C for 8 h. Upon completion, the solvent was removed under reduced pressure, and the resulting mixture was diluted with H2O and washed with diethyl ether. The aqueous layer was acidified with aqueous 1 N HCl and extracted with ethyl acetate (3 × 10 mL). The combined organic fractions were dried over anhydrous Na2SO4 and concentrated in vacuo to furnish carboxylic acid 24 (42 mg, 92%). 1H NMR (400 MHz, CDCl3): δ 10.17 (brs, 1H), 8.05–7.89 (m, 2H), 7.51 (m, 1H), 7.38 (s, 1H), 6.07 (m, 0.6H), 5.04 (m, 0.4H), 2.91 (s, 1.3H), 2.79 (s, 1.7H), 2.40 (s, 3H), 2.17 (s, 3H), 1.73–1.57 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 176.6, 171.2, 170.8, 169.9, 162.5, 161.8, 139.1, 138.8, 136.3, 136.2, 135.9, 135.2, 135.0, 132.6, 132.2, 131.2, 130.1, 129.9, 125.7, 125.3, 52.4, 47.2, 32.8, 29.58, 28.2, 21.1, 16.1, 14.9, 11.1. LRMS-ESI (m/z): 303 [M+H] +.
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health and Purdue University is gratefully acknowledged. A.D.M also wishes to acknowledge partial support from the Walther Cancer Foundation. NMR, mass spectrometry and protein crystallization were supported in part by the Purdue Center for Cancer Research Shared Resources, which are supported by NIH grant (P30 CA023168). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Abbreviations
- BACE1
beta-site amyloid precursor protein cleaving enzyme 1
- BACE2
beta-site amyloid precursor protein cleaving enzyme 2
- DPP-4
dipeptidyl peptidase 4
- FDA
Food and Drug Administration
- SAR
structure-activity relationships
- SGLT2
sodium-glucose linked transporter 2
Footnotes
Supporting information for this article is given via a link at the end of the document
References:
- [1].Kahn SE, Cooper ME, Del Prato S, Lancet 2014, 383, 1068–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zimmet P, Alberti KG, Shaw J, Nature 2001, 414, 782–787. [DOI] [PubMed] [Google Scholar]
- [3].Stumvoll M, Goldstein BJ, van Haeften TW, Lancet 2005, 365, 1333–1346. [DOI] [PubMed] [Google Scholar]
- [4].IDF Diabetes Atlas Seventh Edition. Available at: http://www.diabetesatlas.org/kev-messages.html. (Accessed: 1th April 2016).
- [5].Li MZ, Su L, Liang BY, Tan JJ, Chen Q, Long JX, Xie JJ, Wu GL, Yan Y, Guo XJ, Gu L, int. J. Endocrinol 2013, 2013, 753150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liu L, Zhou C, Du H, Zhang K, Huang D, Wu J, Sci. Rep 2014, 4, 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chaudhury A, Duvoor C, Reddy Dendi VS, Kraleti S, Chada A, Ravilla R, Marco A, Shekhawat NS, Montales MT, Kuriakose K, Sasapu A, Beebe A, Patil N, Musham CK, Lohani GP, Mirza W, Front. Endocrinol. (Lausanne) 2017, 8, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Standards of medical care in diabetes−−2014. Diabetes Care 2014, 37 Suppl 1, S14–80. [DOI] [PubMed] [Google Scholar]
- [9].Standards of Medical Care in Diabetes-2016: Summary of Revisions. Diabetes Care 2016, 39 Suppl 1, S4–5. [DOI] [PubMed] [Google Scholar]
- [10].Eckerle Mize DL, Salehi M, Curr. Diabetes Rep 2013, 13, 307–318. [DOI] [PubMed] [Google Scholar]
- [11].Ferrannini E, DeFronzo RA, Eur. Heart J 2015, 36, 2288–2296. [DOI] [PubMed] [Google Scholar]
- [12].Chamberlain JJ, Rhinehart AS, Shaefer CF Jr., Neuman A, Ann. intern. Med 2016, 164, 542–552. [DOI] [PubMed] [Google Scholar]
- [13].Verma S, Mazer CD, Fitchett D, Inzucchi SE, Pfarr E, George JT, Zinman B, Diabetologia 2018, 61, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Esterhazy D, Stutzer I, Wang H, Rechsteiner MP, Beauchamp J, Dobeli H, Hilpert H, Matile H, Prummer M, Schmidt A, Lieske N, Boehm B, Marselli L, Bosco D, Kerr-Conte J, Aebersold R, Spinas GA, Moch H, Migliorini C, Stoffel M, Cell Metab. 2011, 14, 365–377. [DOI] [PubMed] [Google Scholar]
- [15].Alcarraz-Vizan G, Castano C, Visa M, Montane J, Servitja JM, Novials A, Cell Mol. Life Sci 2017, 74, 2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Altirriba J, Gasa R, Casas S, Ramirez-Bajo MJ, Ros S, Gutierrez-Dalmau A, de Villa MCR, Barbera A, Gomis R, Diabetologia 2010, 53, 1406–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Akpinar P, Kuwajima S, Krutzfeldt J, Stoffel M, Cell Metab. 2005, 2, 385–397. [DOI] [PubMed] [Google Scholar]
- [18].Southan C, Expert Opin. Ther Pat 2013, 23, 649–663. [DOI] [PubMed] [Google Scholar]
- [19].Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J, Proc. Natl. Acad. Sci. U. S. A 2000, 97, 1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Turner RT 3rd, Loy JA, Nguyen C, Devasamudram T, Ghosh AK, Koelsch G, Tang J, Biochemistry 2002, 41, 8742–8746. [DOI] [PubMed] [Google Scholar]
- [21].Turner RT 3rd, Koelsch G, Hong L, Castanheira P, Ermolieff J, Ghosh AK, Tang J, Biochemistry 2001, 40, 10001–10006. [DOI] [PubMed] [Google Scholar]
- [22].Voytyuk I, Mueller SA, Herber J, Snellinx A, Moechars D, van Loo G, Lichtenthaler SF, De Strooper B, Life Sci. All 2018, 1, e201800026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rulifson IC, Cao P, Miao L, Kopecky D, Huang L, White RD, Samayoa K, Gardner J, Wu X, Chen K, Tsuruda T, Homann O, Baribault H, Yamane H, Carlson T, Wiltzius J, Li Y, PLoS One 2016, 11, e0147254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R, Neuron 2003, 37, 925–937. [DOI] [PubMed] [Google Scholar]
- [25].Kobayashi D, Zeller M, Cole T, Buttini M, McConlogue L, Sinha S, Freedman S, Morris RG, Chen KS, Neurobiol. Aging 2008, 29, 861–873. [DOI] [PubMed] [Google Scholar]
- [26].Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C, Science 2006, 314, 664–666. [DOI] [PubMed] [Google Scholar]
- [27].Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R, Neurosci J 2010, 30, 8819–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Harrison SM, Harper AJ, Hawkins J, Duddy G, Grau E, Pugh PL, Winter PH, Shilliam CS, Hughes ZA, Dawson LA, Gonzalez MI, Upton N, Pangalos MN, Dingwall C, Mol. Cell. Neurosci 2003, 24, 646–655. [DOI] [PubMed] [Google Scholar]
- [29].Diment S, Leech MS, Stahl PD, J. Biol. Chem 1988, 263, 6901–6907. [PubMed] [Google Scholar]
- [30].Liaudet-Coopman E, Beaujouin M, Derocq D, Garcia M, Glondu-Lassis M, Laurent-Matha V, Prebois C, Rochefort H, Vignon F, Cancer Lett. 2006, 237, 167–179. [DOI] [PubMed] [Google Scholar]
- [31].Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W, Saftig P, Gartner J, Am. J. Hum. Genet 2006, 78, 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Benes P, Vetvicka V, Fusek M, Crit. Rev. Oncol. Hematol 2008, 68, 12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, Jhee S, Kleijn HJ, Kuvelkar R, Li W, Mattson BA, Mei H, Palcza J, Scott JD, Tanen M, Troyer MD, Tseng JL, Stone JA, Parker EM, Forman MS, Sci. Transl. Med 2016, 8, 363ra150. [DOI] [PubMed] [Google Scholar]
- [34].Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot-Blomley M, Laue G, Beltz K, Vogg B, Schmid P, Frieauff W, Shimshek DR, Staufenbiel M, Jacobson LH, Mol. Neurodegener 2015, 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Beauchamp J; Bernardeau A;, Hilpert H;, Migliorini C;, Riboulet W;, Wang H, 2- Aminodihydro [1,3]-thiazines as BACE2 inhibitors for the treatment of diabetes, WO2011029803A1. 2011.
- [36].Ghosh AK, Reddy BS, Yen YC, Cardenas E, Rao KV, Downs D, Huang X, Tang J, Mesecar AD, Chem. Sci 2016, 7, 3117–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ghosh AK; Anderson DD; Mitsuya H, Burger’s Medicinal Chemistry, Drug Discovery and Development 2010; Vol. 7. [Google Scholar]
- [38].Ghosh AK, Chapsal BD, Mitsuya H, Methods Prins. Med. Chem 2010; Vol. 45. [Google Scholar]
- [39].Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J, Bioorg. Med. Chem. Lett 2008, 18, 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, Tang J, F.A.S.E.B. J 2011, 25, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ostermann N, Eder J, Eidhoff U, Zink F, Hassiepen U, Worpenberg S, Maibaum J, Simic O, Hommel U, Gerhartz B, J. Mol. Biol 2006, 355, 249–261. [DOI] [PubMed] [Google Scholar]
- [42].Stutzer I, Selevsek N, Esterhazy D, Schmidt A, Aebersold R, Stoffel M, J. Biol. Chem 2013, 288, 10536–10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ghosh AK, Venkateswara Rao K, Yadav ND, Anderson DD, Gavande N, Huang X, Terzyan S, Tang J, J. Med. Chem 2012, 55, 9195–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang W, Cary DR, Jacobs JW, Lu W, Lu Y, Sun J, Zhong M, Preparation of amino carboxamide derivatives as aspartyl protease inhibitors. 2003; Vol. WO 2003106405, A1 20031224. [Google Scholar]
- [45].Mauser H, Nettekoven M, Schmitt S, Isophthalamide derivatives as BACE2 inhibitors and their preparation and use for the treatment of diabetes. 2012; Vol. US 20120053200 A1 20120301. [Google Scholar]
- [46].Ghosh AK, Kumaragurubaran N, Liu C, Devasamudram T, Lei H, Swanson L, Ankala S, Tang J, Bilcer G, Preparation of benzene-1,3-dicarboxamides which inhibit β-secretase activity. 2006; Vol. WO 2006110668 A1 20061019. [Google Scholar]
- [47].Green IR, October N, Arkivoc. 2010, 2, 71–96. [Google Scholar]
- [48].Sayyed IA, Sudalai A, Tetrahedron Asymm. 2004, 15, 3111–3116. [Google Scholar]
- [49].Prevost S, Gauthier S, de Andrade MCC, Mordant C, Touati AR, Lesot P, Savignac P, Ayad T, Phansavath P, Ratovelomanana-Vidal V, Genet JP, Tetrahedron Asymm. 2010, 21, 1436–1446. [Google Scholar]
- [50].Maillaird M, Hom C, Gailunas A, Jagodzinska B, Fang LY, John V, Freskos JN, Pulley SR, Beck JP, Tenbrink RE, Preparation of substituted amines to treat Alzheimer’s disease. 2002; Vol. WO 2002002512 A2 20020110. [Google Scholar]
- [51].Ghosh AK, Brindisi M, Nyalapatla PR, Takayama J, Ella-Menye JR, Yashchuk S, Agniswamy J, Wang YF, Aoki M, Amano M, Weber IT, Mitsuya H, Bioorg. Med. Chem 2017, 25, 5114–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Skelin M, Rupnik M, Cencic A, ALTEX 2010, 27, 105–113. [DOI] [PubMed] [Google Scholar]
- [53].Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y, Diabetologia 1993, 36, 1139–1145. [DOI] [PubMed] [Google Scholar]
- 54.[] Rochin L, Hurbain I, Serneels L, Fort C, Watt B, Leblanc P, Marks MS, De Strooper B, Raposo G, van Niel G, Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10658–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.