

Abstract
Non-alcoholic fatty liver disease (NAFLD) is estimated to affect 24% of the global adult population. NAFLD is a major risk factor for the development of cirrhosis and hepatocellular carcinoma, as well as being strongly associated with type 2 diabetes and cardiovascular disease. It has been proposed that up to 88% of obese adults have NAFLD, and with global obesity rates increasing, this disease is set to become even more prevalent. Despite intense research in this field, the molecular processes underlying the pathology of NAFLD remain poorly understood. Hepatic intracellular lipid accumulation may lead to dysregulated tricarboxylic acid (TCA) cycle activity and associated alterations in metabolite levels. The TCA cycle metabolites alpha-ketoglutarate, succinate and fumarate are allosteric regulators of the alpha-ketoglutarate-dependent dioxygenase family of enzymes. The enzymes within this family have multiple targets, including DNA and chromatin, and thus may be capable of modulating gene transcription in response to intracellular lipid accumulation through alteration of the epigenome. In this review, we discuss what is currently understood in the field and suggest areas for future research which may lead to the development of novel preventative or therapeutic interventions for NAFLD.
Keywords: Non-alcoholic fatty liver disease, Mitochondria, TCA cycle, Alpha-ketoglutarate-dependent dioxygenases, Metabolite
Background
Globally, non-alcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease [1], and comprises a spectrum of pathologies, from benign hepatic steatosis to inflammatory non-alcoholic steatohepatitis (NASH), liver cirrhosis, and hepatocellular carcinoma (HCC) [2]. Benign hepatic steatosis is the most common form of NAFLD and is characterised by a hepatic fat content of greater than 5% of total liver weight [3]. Approximately 37% of adult patients with benign steatosis will develop NASH, which is strongly associated with hepatic inflammation, insulin resistance (IR), and type 2 diabetes (T2D) [4]. Despite much research aimed at developing therapeutics to target the disease, currently, the only way to reverse the development of benign steatosis or NASH is through lifestyle modifications associated with weight loss, including diet and exercise [5], or gastric banding [6], for example, in a 5-year human study gastric banding was shown to be effective in reversing hepatic steatosis, NASH, and IR [6]. However this remains an invasive surgical procedure, the risks of which are exacerbated in overweight individuals [7]; indeed a systematic review of the outcomes found that a subset of patients experienced new or worsening features of NAFLD, including fibrosis, demonstrating that this surgery is not suitable in all cases [8]. A major hurdle in the development of therapeutics for NAFLD is that the cause is multi-factorial [9]. Early studies advanced a two-hit model, in which benign steatosis, characterised by the accumulation of intracellular triglycerides, represents the first ‘hit’. The second hit involves the additional presence of oxidative stress, the production of inflammatory cytokines and mitochondrial dysfunction [10]. This has now been further developed into a ‘multiple-hit’ model, which considers that numerous other factors may drive pathology, including diet and genetic predisposition [11].
Obesity, type 2 diabetes, and NAFLD
The relationship between NAFLD, IR, and T2D is complex, and although NAFLD and its progression are strongly associated with the metabolic syndrome, it also occurs in the absence of overt IR and T2D. Indeed, epidemiological studies suggest that the presence of NAFLD may be predictive of the development of IR and T2D [3], but the precise molecular bases of these associations are not fully understood.
Hepatic steatosis is a consequence of an imbalance between hepatic lipid uptake, de novo lipogenesis, and lipid clearance. In obesity, the excess accumulation of lipids in visceral adipose tissue results in an increased release of free fatty acids (FFAs) into the circulation, with FFAs derived from adipose lipolysis being the principal source of lipids supplied to the liver [12]. In the steady state, stimulation of hepatic insulin receptors activates the PKB/AKT pathway, inducing phosphorylation and nuclear exclusion of the fork-head box protein (FOXO1) transcription factor [13]. In the presence of excess FFAs, insulin receptor activation is blunted, leading to a disruption of the insulin signalling pathway [14]. This disruption prevents the phosphorylation of FOXO1, allowing it to translocate to the nucleus, where it stimulates the expression of genes associated with gluconeogenesis and de novo lipogenesis [15]. This can stimulate hepatic intracellular lipid accumulation and the development of large lipid droplets within the cytoplasm [16]. Overnutrition, accompanied by hyperinsulinemia and hyperglycemia, drives steatosis by promoting de novo lipogenesis in the liver which contributes substantially to the accumulation of multiple lipid species [17]. Studies in rodents have shown that obesity is associated with an increase in the expression of lipogenic transcription factors, including carbohydrate response element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c). This leads to a downstream increase in the expression of lipogenic genes, increased flux through acetyl-CoA carboxylase, and increased hepatic malonyl-CoA. These changes result in increased hepatic de novo lipogenesis, suppressed hepatic fatty acid oxidation, and the development of steatosis [18]. Further, the increase in hepatic diacylglycerol, which binds to and activates protein kinase C [19], can in turn inhibit the hepatic insulin signalling cascade [20]. FFAs also repress the expression of glucose transporter 4 (GLUT4), and studies in immortalised primary human hepatocytes suggest that the subsequent increase in intracellular glucose levels can stimulate glucokinase activity, providing more substrates for glycolysis through conversion of glucose to glucose-6-phosphate [21] and further contributing to de novo lipogenesis [22]. Increased rates of glycolysis may also provide pyruvate, which can be used as a substrate for the TCA cycle.
Mitochondria and the TCA cycle in NAFLD
Studies in humans and mice indicate that alterations in mitochondrial TCA cycle activity may play a central role in the pathogenesis of NAFLD [23, 24]. The TCA cycle occurs predominantly in the mitochondrial matrix and is a central pathway for the metabolism of amino acids, fatty acids, and carbohydrates, in order to generate cellular energy [25]. The cycle utilises the substrate acetyl coenzyme A (acetyl-CoA), which is generated by glycolysis via pyruvate, through the action of pyruvate dehydrogenase (PDH) [26] and through amino acid degradation and β-oxidation. Acetyl-CoA is oxidised through a series of steps, producing NADH and FADH2, which transfer electrons to the electron transport chain to fuel the production of ATP. Pyruvate can also be carboxylated by pyruvate carboxylase (PC) to form oxaloacetate (OAA), which then undergoes a condensation reaction with acetyl-CoA to form citrate (Fig. 1). Additionally, the TCA cycle can be fuelled by the deamination of glutamate by glutamate dehydrogenase, to produce α-ketoglutarate (αKG) [27]. Increased influx of substrates into the TCA cycle can increase cycle flux, as well as the abundance of the intermediates αKG, succinate, and fumarate [28].
Fig. 1.

The TCA cycle occurs predominantly in the mitochondrial matrix, although some reactions can occur beyond the mitochondria, including in the cytoplasm and nucleus. Solid arrows represent the reaction direction (enzymes omitted for simplicity), whilst dashed arrows represent the transport of metabolites into the cytoplasm
Whilst the TCA cycle is typically a feature of the mitochondria, specific TCA cycle reactions can also occur in the cytoplasm [29] and the nucleus [30] (Fig. 1). For example, αKG can be generated in the cytoplasm by glutamate dehydrogenase, before being transported into the mitochondria. The malate/aspartate shuttle exports aspartate to the cytoplasm, where it is converted to oxaloacetate, and then to malate, whilst also oxidising NADH to NAD+. Malate and NAD+ are then transported back into the mitochondria, and αKG is simultaneously exported to the cytoplasm. Data from studies in PC12 cells suggest that alterations in the balance of NADH and NAD+ may play a role in the development of fatty liver, through increased oxidative stress [31] and disruption of β-oxidation, leading to increased lipid accumulation [32]. There are mitochondrial and cytoplasmic forms of the TCA cycle enzymes isocitrate dehydrogenase (IDH) and malate dehydrogenase (MDH). Fumarate hydratase, which only exists in one form, is most commonly located in the mitochondria but can be found in the cytoplasm and can translocate to the nucleus upon DNA damage [33]. Furthermore, several mitochondrial TCA cycle enzymes, including PDH, PC, and isocitrate dehydrogenase 3 (IDH3), can translocate to the nucleus, where they ultimately generate αKG [30], although to date this has only been demonstrated during a time-limited phase of zygotic genome activation. The generation and presence of TCA cycle metabolites in multiple cellular compartments increase the chance of them being able to interact with a number of enzymes, and this will be discussed later in this review.
Whilst the standard model of the TCA cycle describes a unidirectional process, the landscape is more complex than this. Instead of OAA condensing with acetyl-CoA to form citrate, it can instead be converted to malate, then fumarate, and back to malate and OAA, in a process termed ‘backflux’ [34]. Truncation of the TCA cycle is also possible in certain disease states, including hypoglycaemia and in various cancers, in which specific steps are no longer functional and are bypassed. During hypoglycaemia, for example, glutamate can become the primary source of carbon, allowing OAA to be generated from αKG instead of citrate [35]. Loss of FH function, as found in some cancers, results in the accumulation of fumarate and a corresponding decrease in NADH generation [36]. However, despite these metabolic alterations fumarate hydratase-deficient cells (UOK262 cell line) are viable, and resist hypoxia, perhaps through stabilisation of hypoxia-inducible factors [36], which will be discussed later in this review. The plasticity of the TCA cycle serves to illustrate that it can undergo complex adaptations in pathological states and promote cell survival.
Hepatic lipid accumulation is associated with mitochondrial dysfunction and oxidative stress, through disruption of mitochondrial energy metabolism pathways [17, 37–39]. Studies in human and animal models show that the transport of excess FFAs into the mitochondria leads to compensatory adaptations, which increase the rates of β-oxidation, TCA cycle flux, and oxidative phosphorylation [23, 40, 41]. These alterations are associated with increased reactive oxygen species (ROS) generation, lipid peroxidation, and apoptosis [42], which may contribute to the inflammatory phenotype observed in patients with NASH. Humans with NASH demonstrate increased expression of mitochondrial uncoupling protein 2 (UCP2), which may occur in response to increased ROS production [43]. UCP2 uncouples the electron transport chain, by increasing proton leakage and lowering redox stress through decreased reactive ROS production [44]. Whilst this may initially be beneficial, it may eventually become detrimental in chronic conditions such as NAFLD, leading to mitochondrial dysfunction, with loss of electron transport chain efficiency, decreased production of adenosine triphosphate (ATP), and increased ROS generation [45, 46].
Evidence from studies in humans and in mouse models suggests that TCA cycle activity is increased in NAFLD, with increased cycle flux and replenishment (anaplerosis) of αKG via glutamate. Although the evidence for this is compelling, the technical difficulties of obtaining human tissue samples mean that many of the changes in hepatic TCA cycle flux and anaplerosis which have been described in human studies have been inferred from peripheral blood, and thus the results may be confounded by the release of metabolites from other tissues [23]. Although there are limited data from humans with NASH suggesting that αKG is decreased in diseased hepatic tissue [47], the tissue samples were first embedded in paraffin with downstream metabolomic analysis which may confound some of the results [48], so that further studies would be useful to confirm these findings. Nuclear magnetic resonance (NMR) studies also indicate increased hepatic TCA cycle flux and anaplerosis in human NAFLD [23] and studies in the livers of mice fed a high-fat diet demonstrate increased TCA cycle flux associated with hepatic steatosis [49]. The use of whole tissue to examine the effects of NAFLD on metabolic function can make it difficult to determine which cells within a tissue are exhibiting altered TCA cycle activity. However, a recently developed in vitro model, which uses embryonic stem cell (ESC)-derived hepatocytes to model NAFLD, supports the assertion that intracellular lipid accumulation associates with dysregulation of the TCA cycle and altered abundance of intermediates [50].
The αKG-dependent dioxygenases
In addition to its central role in energy metabolism, components of the TCA cycle can influence the catalytic activity of the αKG-dependent dioxygenase family of enzymes [51]. The αKG-dependent dioxygenases are Fe(II)/O2-dependent enzymes, which catalyse the hydroxylation of a number of substrates, including DNA, histones, and proteins. The enzyme family includes the prolyl-4-hydroxylases (PHD), Jumonji domain-containing histone demethylases (JHDMs), and ten-eleven translocation (TET) dioxygenases [52]. All of the enzymes within this family have two binding domains in their β-sheet core, which are highly specific for αKG, and binding of αKG acts as a positive regulator of dioxygenase activity [53, 54]. Conversely, these two domains can be competitively bound by succinate and fumarate, which inhibit dioxygenase activity [55]
The functions of these enzyme members are numerous but include epigenetic regulation, through DNA methylation, histone methylation, and acetylation, as well as oxygen sensing through the PHDs [56–58]. It is tempting to speculate, therefore, that the αKG-dependent dioxygenases could form a nexus, integrating environmental signals and disseminating them to downstream targets by altering gene transcription to allow cells to maintain homeostasis. Thus, dysfunctional mitochondrial (TCA cycle) metabolism could impact on epigenetic modifications of DNA and chromatin through alterations in JHDM, PHD, and TET enzyme activity, and findings from human breast and colon cancer cell lines open up the possibility that this may also play a role in NAFLD [59–61].
Prolyl-4-hydroxylases, HIF-1α, and NAFLD
PHDs catalyse the most prevalent post-translational modification (PTM) in humans, hydroxylating proline residues to form (2S,4R)-4-hydroxyproline [62]. Whilst PHDs have many substrates, including Argonaut 2 [63], collagen [64], and elastin [65], one of the most well characterised are the hypoxia-inducible factor (HIF) transcription factors, which are targeted by PHD2 [66]. HIF-1 is comprised of two constitutively expressed sub-units: HIF-1α and HIF-1β [62]. Under normal oxygen conditions (normoxia), PHDs hydroxylate two proline residues (Pro402 and Pro564) within the oxygen-dependent degradation domain of HIF-1α [62]. The hydroxylation of these proline residues recruits ubiquitin E3 ligase and leads to the polyubiquitination of HIF-1α, marking it for degradation by the proteasome [67]. Under hypoxic conditions, low oxygen concentrations diminish the ability of PHDs to hydroxylate these proline residues, and thus HIF-1α stabilises, allowing it to translocate to the nucleus where it can dimerise with HIF-1β, bind to hypoxia response elements, and facilitate gene transcription [68] (Fig. 2).
Fig. 2.

TCA cycle metabolites in the cytoplasm can interact with prolyl-4-hydroxylases, to either promote or repress activity. Repression prevents ubiquitination of HIF-1α, promoting protein stabilisation, followed by nuclear accumulation and transcription of target genes
Despite their name, it is not only oxygen that regulates the HIFs. Since PHDs are also regulated by succinate, fumarate, and αKG, HIFs are able to coordinate gene networks in response to metabolic rewiring [69, 70]. Indeed, in human breast cancer cell lines, hypoxia is associated with lipid homeostasis, through coordination of genes regulating lipid droplet formation, de novo lipogenesis, and fatty acid uptake [71]. Whilst there is a lack of data to directly link TCA cycle dysfunction in NAFLD with PHD inhibition and HIF stabilisation, there are limited data from rat models demonstrating altered HIF-1α mRNA and protein levels in steatotic whole liver [72]. A caveat to this study is that it was performed in whole tissue, and perivenous regions of the liver express higher levels of HIF mRNA [73] so that disease-associated alterations in hepatic structure and/or variation in the region of the liver sampled could influence the results. However, in support of this link, studies suggest that there is a relationship between lipid accumulation and HIF-1α stabilisation [74]. Both HIF-1α mRNA and protein levels increase in the adipose tissue of obese subjects, and there is strong evidence that obesity provokes a hypoxia-like response in adipose tissue [75, 76]. Tracer studies in mice, using a 13C-glucose stable isotope, demonstrated an increase in the abundance of αKG, succinate, and fumarate in the adipose tissue of obese animals [24]. Furthermore, this study found increased levels of succinate in the liver of obese mice, suggesting that obesity is associated with alterations in metabolites that have the potential to impact on the activity of PHDs [24].
There are conflicting reports as to whether HIF-1α plays a protective or harmful role in NAFLD [72, 77]. Using a rat model of liver steatosis, Carabelli et al. describe increased expression of HIF-1α mRNA in the liver and suggest that this is responsible for an observed increase in mitochondrial biogenesis [72]. They also speculate that this may form a protective mechanism, whereby hepatocytes are able to increase rates of β-oxidation to process excess intracellular lipids [72]. In contrast, Mesarwi et al. observed that HIF-1α mediates the development of hepatic fibrosis in a mouse model of NAFLD, suggesting a pathogenic role [77]. The discrepancies between these studies could be explained by the use of different animal models. It may also be a result of NAFLD comprising a spectrum of pathology, whereby some molecular mechanisms may be protective during the early stage of disease, but eventually become chronic and harmful. However, there is a lack of data to suggest whether HIF-1α is important for the development of NAFLD in humans and for any impact of TCA cycle dysfunction on PHD catalytic activity. Nevertheless, PHD2 contains a histone-binding domain [78], can translocate to the nucleus, and is found within the chromatin fraction [62], suggesting that it may directly impact on gene transcription.
JHDMs, histone modifications, and NAFLD
Histone proteins form cores around which DNA can condense, allowing it to be packaged efficiently into nucleosomes, to form chromatin [79]. These nucleosomes can be highly compacted, preventing the transcription of DNA to mRNA [80]. Histones are also subject to many PTMs, including acetylation, methylation, phosphorylation, sumoylation, and ubiquitylation [81]. Depending upon the deposition site of PTMs, they can result in chromatin condensation and transcriptional silencing [82] or associate with relaxation of chromatin and active gene transcription [82].
The JHDMs are a large family of αKG-dependent dioxygenases that can catalyse the removal of mono-, di-, and trimethylation marks from specific lysine (K) residues of histone 3 (H3) or histone 4 (H4) [83, 84]. Each JHDM targets a specific histone lysine residue, which impacts on chromatin structure in multiple ways, depending on the resulting methylation pattern. Broadly, methylation of H3K9 and H3K27 is associated with chromatin compaction and transcriptional silencing [85]. In contrast, H3K4 and H3K36 methylation are typically associated with chromatin relaxation and transcriptional activity [85]. However, enrichments of H3K9 di- and trimethylation have been observed in transcriptionally active euchromatic regions of genes [86], demonstrating that the impact of histone methylation on gene transcription is complex and nuanced.
The activity of the JHDMs can be influenced by TCA cycle metabolites, for example, the experimental silencing of succinate dehydrogenase in a human HCC cell line leads to the accumulation of succinate, with resultant inhibition of the JHDMs, altered histone methylation patterns, and changes in transcriptional activity [87]. Although there is no direct evidence to link TCA cycle dysfunction with alterations in JHDM activity in NAFLD, in a mouse model, hepatic steatosis occurred in association with altered JHDM expression and altered H3K4 and H3K9 trimethylation at the peroxisome proliferator-activated receptor alpha (PPARα) gene and in a number of other genes important in lipid catabolism [88].
DNA methylation and NAFLD
DNA methylation is a highly conserved epigenetic modification involving the addition of a methyl group at the C5 position of cytosine, predominantly in cytosine-phosphate-guanine (CpG) dinucleotides, to form 5-methylcytosine (5mC) [89]. The reactions are catalysed by the DNA methyltransferases (DNMTs). Approximately 10% of CpG sites are located in clusters termed CpG islands (CGIs) and are most commonly found in the promoters of genes with important developmental functions [90]. In general, these CGIs are unmethylated, but where methylation does occur, it can be associated with transcriptional repression and heterochromatic regions, as 5mC is, in general, thought to prevent transcription factor binding [91]. During development, it is estimated that fewer than 30% of CpGs are methylated, in contrast to 85% in terminally differentiated cells, illustrating the tight control that is exerted to retain cell homeostasis and phenotype in differentiated cells [92]. The TET enzymes are a subset of the αKG-dependent dioxygenase family, comprised of TET1, TET2, and TET3. These enzymes catalyse the iterative oxidation and demethylation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), before base excision repair (BER) is triggered, resulting in unmodified cytosine [56] (Fig. 3). To achieve this, the double-stranded β-helix at the N-terminus of the TET enzymes brings together Fe(II), αKG, and 5mC to allow oxidation of the methyl group to 5hmC, 5fC, and 5caC [56]. The contiguous cysteine-rich domain then wraps around the double-stranded β-helix to stabilise the interaction between the TET enzyme and the DNA, facilitating the oxidation reaction [56]. Whilst initially thought to be transient, 5hmC is now accepted as a stable cytosine modification [93], and 5hmC enrichment within gene bodies and enhancers broadly correlates with transcriptional activation [94, 95]. In contrast, 5hmC enrichment within gene transcriptional start sites (TSS) is associated with transcriptional repression and the maintenance of CpG hypomethylation [96]. As with other enzymes in the αKG-dependent dioxygenase family, TET catalytic activity is modulated by the TCA cycle metabolites αKG, succinate, and fumarate [55]. This may be important in some human cancers, for example, loss of function mutations in fumarate hydratase and succinate dehydrogenase in human cervical cancer and glioblastoma cell lines result in the accumulation of fumarate and succinate, respectively, with consequent inhibition of TET enzyme activity and altered 5hmC levels [55].
Fig. 3.
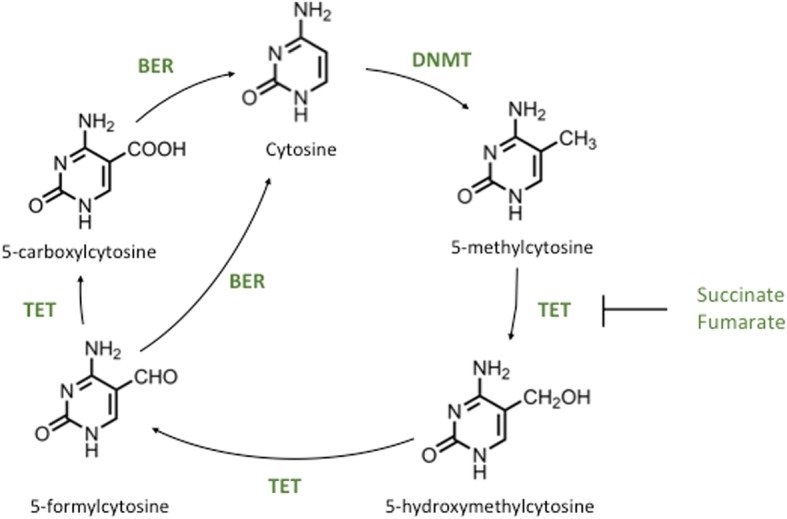
TET1 oxidation of 5mC through to unmodified cytosine. After the deposition of 5-methylcytosine by the DNMT enzymes it can be oxidised by the TETs to 5-hydroxymethylcytosine, 5-formylcytosine, and then 5-carboxylcytosine, before base excision repair resulting in unmodified cytosine. These iterative oxidation steps can be inhibited by the TCA cycle metabolites succinate and fumarate
The development of hepatic steatosis is associated with changes in hepatic DNA methylation in a methionine-deficient diet (MDD) model of NAFLD in mice [97]. In this study, it is not known where in the genome these changes occur, making it difficult to infer any impact on transcriptional regulation. Furthermore, there are concerns as to how well the MDD model recapitulates NAFLD, as shown by suppressed expression of very low-density lipoprotein carboxylesterase mRNA [98]. In contrast, Pirola et al. observed that humans with NASH have increased DNMT1 mRNA and a concurrent increase in DNA methylation of the mitochondrial NADH dehydrogenase 6 gene in the whole liver [99]. The importance of mitochondrial DNA methylation is debated [100, 101], and it is therefore currently uncertain what impact it may have on the pathogenesis of NAFLD.
Evidence of altered nuclear DNA methylation in the liver in NAFLD is supported by a number of studies in humans. Differential methylation has been reported in liver biopsy specimens from patients with NAFLD [102, 103], with some studies showing an inverse correlation between gene expression and DNA methylation [104]. Specific differentially methylated regions have been identified which may associate with NAFLD progression [105]. Notably, some of the changes in DNA methylation may be reversible with weight loss, for example following bariatric surgery [104, 105]. There is also evidence from bariatric surgery patients suggesting that there is a correlation between altered DNA methylation and changes in hepatic insulin signalling [106]. A recent study focused on understanding the link between NAFLD-associated liver fibrosis and cirrhosis and DNA methylation performed bisulfite sequencing in liver biopsies from female patients undergoing bariatric surgery, who had no other liver-related pathologies [107]. The study identified differential CpG island methylation at a number of different genes and gene ontology analysis showed enrichment for a number of pathways related to NAFLD, including reactive oxygen species production and T2D signalling [107]. Whilst compelling, the findings in each of these studies are correlative and do not describe any mechanism(s) by which altered DNA methylation might occur. In addition, the studies used bisulfite treatment to analyse DNA methylation, making it impossible to determine if they were observing changes in 5mC or its oxidised counterpart 5hmC.
There is evidence to suggest that 5hmC levels are altered in NAFLD. Patient studies describe a positive association between global hepatic 5hmC levels and the expression of peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PPARGC1A), a gene at which DNA methylation has previously been associated with NAFLD and IR [99, 108]. The lack of any change in global 5hmC could reflect the use of whole tissue, which may dilute the 5hmC signal through analysis of multiple cell types or alternatively may indicate that alterations in 5hmC in response to NAFLD occur only at specific genes. Indeed, using an in vitro model of NAFLD, we have recently shown that changes in 5hmC levels occur specifically at induced genes associated with lipid synthesis and transport [50]. Further, we found altered expression of several genes encoding TCA cycle enzymes, including succinate dehydrogenase, isocitrate dehydrogenase, and an accumulation of the TCA cycle metabolite oxalosuccinate, suggesting altered cycle activity. The importance of the TET enzymes and 5hmC in liver disease is further supported by studies showing that aberrant TET1 activity in hepatocellular carcinoma is associated with loss of 5hmC and a gain of 5mC, with subsequent transcriptional repression, suggesting that the TETs may normally prevent DNMT binding in these regions in the healthy liver [109].
TET1 and histone methylation
In human ES cells, TET1 may orchestrate the activity of components of the epigenetic machinery by targeting genes for binding by the polycomb recessive complex (PRC). Though they do not appear to physically interact, TET1 appears to recruit PRC2 to particular genes [110], possibly by maintaining a hypomethylated state, as DNA methylation prevents binding by PRC2 [111]. Upon binding to hypomethylated DNA, PRC2 can di- or trimethylate H3K27, an epigenetic mark associated with transcriptional repression [112, 113]. This may be relevant to the pathogenesis of NAFLD, since genome-wide association studies have shown enrichment of PRC2 pathways in humans with NASH [114]. In support of this, data from rat liver and human liver cell lines have demonstrated that downregulation of core components of PRC2 is associated with the de-repression of inflammatory genes and intracellular lipid accumulation [115]. Thus, whilst TET1 may not directly control the DNA-binding activity of PRC2 in the pathogenesis of NAFLD, these data suggest that it may facilitate it.
Post-translational regulation of TET activity
In addition to regulation by TCA cycle metabolites, Fe(II), and oxygen, the TET enzymes are also regulated by PTMs, including phosphorylation and O-GlcNAcylation. The O-GlcNAcylation PTM is deposited by O-linked N-acetylglucosamine transferase (OGT), which attaches an N-acetyl glucosamine to serine and threonine residues within target proteins [116]. Disruption of the OGT binding site in TET1 reduces genomic 5hmC levels, with a concurrent reduction in gene expression [117]. The post-translational addition of O-GlcNAcylation to proteins is promoted by the hexosamine biosynthetic pathway (HBP), which synthesises N-GlcNAc from glucose, linking elevated intracellular glucose to increased O-GlcNAcylation [118]. This generates a positive feedback loop, whereby the HBP diverts glucose away from glycolysis, and lowers αKG levels. This, in turn, promotes stabilisation of HIF-1α via PHD inhibition and promotes increased transcription of the glucose transporter 1 (GLUT1) gene [119], allowing more glucose to enter the cell. The reduction of αKG in response to elevated O-GlcNAcylation could indicate that, whilst TCA cycle intermediates play a direct role in TET enzyme function, they may also play an indirect role through the modulation of PTMs. Indeed, the impact of increased HBP activity, subsequent stabilisation of HIF-1α, and ensuing increase in intracellular glucose may be more important for directing the activity of the TETs. This is relevant, since NAFLD is strongly associated with T2D, hepatic IR, and poor glucose clearance [120]. In addition, studies in mouse models and human tissue show that OGT also promotes the transition from hepatic steatosis to HCC [121]. Whilst this effect is thought to be mediated by palmitic acid production and endoplasmic reticulum stress, other avenues have not yet been explored. In mouse ES cells, OGT can form a complex with TET1 or TET2 in the nucleus, anchoring them to histones and DNA [122], and since genomic hypomethylation is a hallmark of HCCs [123], it is possible that this complex may be involved in its development. This adds to the wealth of evidence suggesting that the TET enzymes and 5hmC may play a role at all stages of NAFLD pathogenesis, and its progression to HCC.
Future perspectives
The study of epigenetic dysregulation in disease is fraught with difficulties and limitations, not least whether changes in epigenetic marks are a cause or consequence of the disease process itself [124]. In addition to this, many studies in the NAFLD field have reported changes in genome methylation, but have arrived at their conclusions based on bisulfite sequencing data [99, 104]. Since bisulfite sequencing cannot distinguish between 5mC and 5hmC [125], it is impossible to determine precisely which changes are occurring during the pathogenesis of NAFLD in much of the current literature. Further to these difficulties are the inherent limitations of the current models of NAFLD, which make it difficult to study the interplay between the TCA cycle and epigenetic modifications. It is technically difficult, and highly invasive, to obtain liver samples from humans, and controls are often not taken from healthy donors but individuals being investigated for pathologies other than NAFLD [104]. The use of whole tissue samples from humans or animals makes it challenging to attribute phenotypes to a particular cell type. This could, typically, be overcome through the use of fluorescence activated cell sorting, but recent studies have shown that this alters the redox status of cells [126], potentially confounding analyses of changes in the TCA cycle. The use of HCC cell lines is also challenging, since malignant transformation often results in metabolic derangement [127]. The development and refinement of 2-D and 3-D in vitro models of ‘NAFLD in a dish’ using a single cell type may be useful for future mechanistic and therapeutic studies, including the careful dissection of the interplay between TCA cycle dysfunction and epigenetic regulation of gene expression in the pathogenesis of the disease [50, 128]
Conclusions
As the obesity pandemic continues, the number of people living with NAFLD will continue to grow, increasing the economic burden on healthcare systems globally [129], making it critical that new strategies for the treatment of the disease are developed. Studies of the interplay between TCA cycle dysfunction and regulation of the αKG-dependent dioxygenases in new models may provide a more comprehensive picture of the processes underlying NAFLD and lead to the development of predictive tests or new diagnostics, alongside novel therapeutics, which are able to reverse the disease.
Acknowledgements
Nil
Abbreviations
- 5caC
5-carboxylcytosine
- 5fC
5-formylcytosine
- 5hmC
5-hydroxymethylcytosine
- ATP
Adenosine triphosphate
- BER
Base excision repair
- CGI
CpG island
- CpG
Cytosine-phosphate-guanine
- DNMT
DNA methyltransferase
- ESC
Embryonic stem cell
- FFA
Free fatty acids
- FOXO1
Fork-head box protein
- GLUT1
Glucose transporter 1
- GLUT4
Glucose transporter 4
- H3
Histone 3
- H4
Histone 4
- HBP
Hexosamine biosynthetic pathway
- HCC
Hepatocellular carcinoma
- HIF
Hypoxia-inducible factor
- IDH
Isocitrate dehydrogenase
- IDH3
Isocitrate dehydrogenase 3
- IR
Insulin resistance
- JHDM
Jumonji domain-containing histone demethylase
- MDD
Methionine-deficient diet
- MDH
Malate dehydrogenase
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- NMR
Nuclear magnetic resonance
- OAA
Oxaloacetate
- OGT
O-linked N-acetylglucosamine transferase
- PC
Pyruvate carboxylase
- PDH
Pyruvate dehydrogenase
- PHD
Prolyl-4-hydroxylase
- PPARGC1A
PPARγ coactivator 1α
- PPARα
Peroxisome proliferator-activated receptor alpha
- PPARγ
peroxisome proliferator-activated receptor γ
- PRC
Polycomb recessive complex
- PTM
Post-translational modification
- ROS
Reactive oxygen species
- T2D
Type 2 diabetes
- TCA
Tricarboxylic acid
- TET
Ten-eleven translocation dioxygenases
- UCP2
Uncoupling protein 2
- αKG
Alpha-ketoglutarate
Authors’ contributions
All authors were involved in the writing of the article and approved the final draft.
Funding
MCS is funded by a British Heart Foundation PhD studentship FS/16/54/32730. DCH was funded by the Chief Scientist Office (TCS/16/37).
Availability of data and materials
No new data/materials are contained in the manuscript.
Ethics approval and consent to participate
This is a review article requiring no ethics approval or consent.
Consent for publication
All authors agree to publication.
Competing interests
David Hay is a co-founder and shareholder in Stemnovate Limited and Higher Steaks Limited. The other authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Abd El-Kader SM, El-Den Ashmawy EMS. Non-alcoholic fatty liver disease: the diagnosis and management. World J Hepatol. 2015;7:846–858. doi: 10.4254/wjh.v7.i6.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepatol. 2015;62:S65–S75. doi: 10.1016/j.jhep.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 3.Valenti L, Bugianesi E, Pajvani U, Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int. 2016;36:1563–1579. doi: 10.1111/liv.13185. [DOI] [PubMed] [Google Scholar]
- 4.George J, Liddle C, Samarasinghe D, Lin R, Karim R, Hui JM, et al. NASH and insulin resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 5.Romero-Gómez M, Zelber-Sagi S, Trenell M. Treatment of NAFLD with diet, physical activity and exercise. J Hepatol. 2017;67:829–846. doi: 10.1016/j.jhep.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 6.Hollebecque A, Pattou F, Pigeyre M, Leteurtre E, Dharancy S, Louvet A, et al. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology. 2009;137:532–540. doi: 10.1053/j.gastro.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 7.Lotia S, Bellamy MC. Anaesthesia and morbid obesity. Contin Educ Anaesthesia Crit Care Pain. 2008;8:151–156. doi: 10.1093/bjaceaccp/mkn030. [DOI] [Google Scholar]
- 8.Lee Y, Doumouras AG, Yu J, Brar K, Banfield L, Gmora S, et al. Complete resolution of nonalcoholic fatty liver disease after bariatric surgery: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2018;17:1040–1060.e11. doi: 10.1016/j.cgh.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD) Metabolism. 2016;65:1038–1048. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Day CP, James OFW. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 11.Fang Yan-Lan, Chen Hong, Wang Chun-Lin, Liang Li. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World Journal of Gastroenterology. 2018;24(27):2974–2983. doi: 10.3748/wjg.v24.i27.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen S, Guo ZK, Johnson CM, Hensrud DD, Jensen MD. Splanchnic lipolysis in human obesity. J Clin Invest. 2004;113:1582–1588. doi: 10.1172/JCI21047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamagate A, Kim DH, Zhang T, Slusher S, Gramignoli R, Strom SC, et al. FoxO1 links hepatic insulin action to endoplasmic reticulum stress. Endocrinology. 2010;151:3521–3535. doi: 10.1210/en.2009-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svedberg J, Björntorp P, Smith U, Lönnroth P. Free-fatty acid inhibition of insulin binding, degradation, and action in isolated rat hepatocytes. Diabetes. 1990;39:570–574. doi: 10.2337/diab.39.5.570. [DOI] [PubMed] [Google Scholar]
- 15.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:5286–5296. doi: 10.3748/wjg.v16.i42.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI200523621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esler WP, Bence KK. Metabolic targets in nonalcoholic fatty liver disease. Cell Mol Gastroenterol Hepatol. 2019. 10.1016/j.jcmgh.2019.04.007. [DOI] [PMC free article] [PubMed]
- 19.Newton AC. Protein kinase C: Structure, function, and regulation. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 20.Hribal ML, D’Alfonso R, Giovannone B, Lauro D, Liu YY, Borboni P, et al. The sulfonylurea glimepiride regulates intracellular routing of the insulin-receptor complexes through their interaction with specific protein kinase C isoforms. Mol Pharmacol. 2018;59:322–330. doi: 10.1124/mol.59.2.322. [DOI] [PubMed] [Google Scholar]
- 21.Tang Y, Chen A. Curcumin prevents leptin raising glucose levels in hepatic stellate cells by blocking translocation of glucose transporter-4 and increasing glucokinase. Br J Pharmacol. 2010;161:1137–1149. doi: 10.1111/j.1476-5381.2010.00956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagao H, Nishizawa H, Bamba T, Nakayama Y, Isozumi N, Nagamori S, et al. Increased dynamics of tricarboxylic acid cycle and glutamate synthesis in obese adipose tissue: in vivo metabolic turnover analysis. J Biol Chem. 2017;292:4469–4483. doi: 10.1074/jbc.M116.770172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys. 2014;68:475–478. doi: 10.1007/s12013-013-9750-1. [DOI] [PubMed] [Google Scholar]
- 26.McCommis KS, Finck BN. Mitochondrial pyruvate transport: a historical perspective and future research directions. Biochem J. 2015;466:443–454. doi: 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277:30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 28.Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med. 2011;17:641–649. doi: 10.1016/j.molmed.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj R, Sharpley MS, Chi F, Braas D, Zhou Y, Kim R, et al. Nuclear localization of mitochondrial TCA cycle enzymes as a critical step in mammalian zygotic genome activation. Cell. 2017;168:210–223.e11. doi: 10.1016/j.cell.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C, Chen H, Zhang J, Hong Y, Ding X, Ying W. Malate-aspartate shuttle mediates the intracellular ATP levels, antioxidation capacity and survival of differentiated PC12 cells. Int J Physiol Pathophysiol Pharmacol. 2014;6:109–114. [PMC free article] [PubMed] [Google Scholar]
- 32.Purohit V, Gao B, Song BJ. Molecular mechanisms of alcoholic fatty liver. Alcohol Clin Exp Res. 2009;33:191–205. doi: 10.1111/j.1530-0277.2008.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldberg M, Singer E, Yogev O, Shaulian E, Fox TD, Pines O, et al. Fumarase: a mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response. PLoS Biol. 2010;8:e1000328. doi: 10.1371/journal.pbio.1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brekke E, Walls AB, Nørfeldt L, Schousboe A, Waagepetersen HS, Sonnewald U. Direct measurement of backflux between oxaloacetate and fumarate following pyruvate carboxylation. Glia. 2012;60:147–158. doi: 10.1002/glia.21265. [DOI] [PubMed] [Google Scholar]
- 35.Sutherland G, Tyson R, Auer R. Truncation of the Krebs cycle during hypoglycemic coma. Med Chem (Los Angeles) 2008;4:379–385. doi: 10.2174/157340608784872235. [DOI] [PubMed] [Google Scholar]
- 36.Tyrakis PA, Yurkovich ME, Sciacovelli M, Papachristou EK, Bridges HR, Gaude E, et al. Fumarate hydratase loss causes combined respiratory chain defects. Cell Rep. 2017;21:1036–1047. doi: 10.1016/j.celrep.2017.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2015;15:8713–8742. doi: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–431. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem. 1980;49:395–420. doi: 10.1146/annurev.bi.49.070180.002143. [DOI] [PubMed] [Google Scholar]
- 40.Handa P, Maliken BD, Nelson JE, Morgan-Stevenson V, Messner DJ, Dhillon BK, et al. Reduced adiponectin signaling due to weight gain results in nonalcoholic steatohepatitis through impaired mitochondrial biogenesis. Hepatology. 2014;60:133–145. doi: 10.1002/hep.26946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satapati S, Kucejova B, Duarte JAG, Fletcher JA, Reynolds L, Sunny NE, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest. 2015;125:4447–4462. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Mailloux RJ, Harper ME. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, et al. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut. 2008;57:957–965. doi: 10.1136/gut.2007.147496. [DOI] [PubMed] [Google Scholar]
- 45.Miwa S, Brand MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans. 2003;31:1300–1301. doi: 10.1042/bst0311300. [DOI] [PubMed] [Google Scholar]
- 46.Kaneko S, Misu H, Nabemoto S, Ando H, Kurita S, Miyamoto K-I, et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–14818. doi: 10.1074/jbc.m901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke JD, Novak P, Lake AD, Shipkova P, Aranibar N, Robertson D, et al. Characterization of hepatocellular carcinoma related genes and metabolites in human nonalcoholic fatty liver disease. Dig Dis Sci. 2014;59:365–374. doi: 10.1007/s10620-013-2873-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huan T, Troyer DA, Li L. Metabolite analysis and histology on the exact same tissue: Comprehensive metabolomic profiling and metabolic classification of prostate cancer. Sci Rep. 2016;6:32272. doi: 10.1038/srep32272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patterson Rainey E., Kalavalapalli Srilaxmi, Williams Caroline M., Nautiyal Manisha, Mathew Justin T., Martinez Janie, Reinhard Mary K., McDougall Danielle J., Rocca James R., Yost Richard A., Cusi Kenneth, Garrett Timothy J., Sunny Nishanth E. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. American Journal of Physiology-Endocrinology and Metabolism. 2016;310(7):E484–E494. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyall MJ, Cartier J, Thomson JP, Cameron K, Meseguer-Ripolles J, O’Duibhir E, et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos Trans R Soc B Biol Sci. 2018;373:20170362. doi: 10.1098/rstb.2017.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–672. doi: 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 52.Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG, et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J Biol Chem. 2016;291:4256–4265. doi: 10.1074/jbc.M115.688762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roach PL, Clifton IJ, Fülöp V, Harlos K, Barton GJ, Hajdu J, et al. Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature. 1995;375:700–704. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 54.Hausinger RP. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 55.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (80- ) 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, et al. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem J. 2008;416:387–394. doi: 10.1016/S0041-1345(99)00166-9. [DOI] [PubMed] [Google Scholar]
- 58.Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels: evidence for an autoregulatory oxygen-sensing system. J Biol Chem. 2006;281:23482–23491. doi: 10.1074/jbc.M601719200. [DOI] [PubMed] [Google Scholar]
- 59.Thyfault JP, Rector RS, Uptergrove GM, Borengasser SJ, Morris EM, Wei Y, et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J Physiol. 2009;587:1805–1816. doi: 10.1113/jphysiol.2009.169060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–736. doi: 10.1016/J.JHEP.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cuyàs E, Fernández-Arroyo S, Verdura S, García RÁ-F, Stursa J, Werner L, et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene. 2018;37:963–970. doi: 10.1038/onc.2017.367. [DOI] [PubMed] [Google Scholar]
- 62.Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qi HH, Ongusaha PP, Myllyharju J, Cheng D, Pakkanen O, Shi Y, et al. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature. 2008;455:421–424. doi: 10.1038/nature07186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hutton JJ, Kaplan A, Udenfriend S. Conversion of the amino acid sequence Gly-Pro-Pro in protein to Gly-Pro-Hyp by collagen proline hydroxylase. Arch Biochem Biophys. 1967;121:384–391. doi: 10.1016/0003-9861(67)90091-4. [DOI] [PubMed] [Google Scholar]
- 65.Bhatnagar RS, Rapaka RS, Urry DW. Interaction of polypeptide models of elastin with prolyl hydroxylase. FEBS Lett. 1978;95:61–64. doi: 10.1016/0014-5793(78)80052-0. [DOI] [PubMed] [Google Scholar]
- 66.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 67.Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1, leading to its oxygen-independent degradation. Mol Cell Biol. 2008;28:7081–7095. doi: 10.1128/MCB.00773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, et al. Stabilization of hypoxia-inducible factor-1α protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–31284. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 70.Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 71.Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68:1003–1011. doi: 10.1158/0008-5472.CAN-07-2489. [DOI] [PubMed] [Google Scholar]
- 72.Carabelli J, Burgueño AL, Rosselli MS, Gianotti TF, Lago NR, Pirola CJ, et al. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J Cell Mol Med. 2011;15:1329–1338. doi: 10.1111/j.1582-4934.2010.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kietzmann T, Cornesse Y, Brechtel K, Modaressi S, Jungermann K. Perivenous expression of the mRNA of the three hypoxia-inducible factor alpha-subunits, HIF1alpha, HIF2alpha and HIF3alpha, in rat liver. Biochem J. 2001;354:531–537. doi: 10.1042/bj3540531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, et al. Dietary obesity-associated hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26:259–270. doi: 10.1101/gad.180406.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 76.Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T-cell infiltration. Int J Obes. 2008;32:451–463. doi: 10.1038/sj.ijo.0803744. [DOI] [PubMed] [Google Scholar]
- 77.Mesarwi OA, Shin M-K, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY. Hepatocyte hypoxia inducible factor-1 mediates the development of liver fibrosis in a mouse model of nonalcoholic fatty liver disease. PLoS One. 2016;11:e0168572. doi: 10.1371/journal.pone.0168572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Musselman CA, Mansfield RE, Garske AL, Davrazou F, Kwan AH, Oliver SS, et al. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem J. 2009;423:179–187. doi: 10.1042/bj20090870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev Proteomics. 2005;2:719–729. doi: 10.1586/14789450.2.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- 81.Nathan D, Sterner DE, Berger SL. Histone modifications: now summoning sumoylation. Proc Natl Acad Sci. 2003;100:13118–13120. doi: 10.1073/pnas.2436173100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tamaru H. Confining euchromatin/heterochromatin territory: Jumonji crosses the line. Genes Dev. 2010;24:1465–1478. doi: 10.1101/gad.1941010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J Biol Chem. 2007;282:35425–35429. doi: 10.1074/jbc.R700027200. [DOI] [PubMed] [Google Scholar]
- 84.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 85.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 86.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 87.Cervera AM, Bayley JP, Devilee P, McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer. 2009;8:89. doi: 10.1186/1476-4598-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jun HJ, Kim J, Hoang MH, Lee SJ. Hepatic lipid accumulation alters global histone H3 lysine 9 and 4 trimethylation in the peroxisome proliferator-activated receptor alpha network. PLoS One. 2012;7:e44345. doi: 10.1371/journal.pone.0044345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 90.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 91.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 92.Leitch HG, Mcewen KR, Turp A, Encheva V, Carroll T, Grabole N, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6:1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin IH, Chen YF, Hsu MT. Correlated 5-hydroxymethylcytosine (5hmC) and gene expression profiles underpin gene and organ-specific epigenetic regulation in adult mouse brain and liver. PLoS One. 2017;12:e0170779. doi: 10.1371/journal.pone.0170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Putiri EL, Tiedemann RL, Thompson JJ, Liu C, Ho T, Choi JH, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol. 2014;15:R81. doi: 10.1186/gb-2014-15-6-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, Ross SA, et al. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J Hepatol. 2009;51:176–186. doi: 10.1016/j.jhep.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lyall MJ, Cartier J, Richards JA, Cobice D, Thomson JP, Meehan RR, et al. Methyl donor deficient diets cause distinct alterations in lipid metabolism but are poorly representative of human NAFLD. Wellcome Open Res. 2017;2:67. doi: 10.12688/wellcomeopenres.12199.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pirola CJ, Scian R, Gianotti TF, Dopazo H, Rohr C, Martino JS, et al. Epigenetic modifications in the biology of nonalcoholic fatty liver disease: the role of DNA hydroxymethylation and TET proteins. Med (United States) 2015;94:e1480. doi: 10.1097/MD.0000000000001480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gao D, Zhu B, Sun H, Wang X. Mitochondrial DNA methylation and related disease. Adv Exp Med Biol. 2017;1038:117–132. doi: 10.1007/978-981-10-6674-0_9. [DOI] [PubMed] [Google Scholar]
- 101.Mechta M, Ingerslev LR, Fabre O, Picard M, Barrès R. Evidence suggesting absence of mitochondrial DNA methylation. Front Genet. 2017;8:166. doi: 10.3389/fgene.2017.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hotta K, Kitamoto T, Kitamoto A, Ogawa Y, Honda Y, Kessoku T, et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol Res. 2018;48:E320–E334. doi: 10.1111/hepr.12992. [DOI] [PubMed] [Google Scholar]
- 103.Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:1076–1087. doi: 10.1053/J.GASTRO.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ahrens M, Ammerpohl O, Von Schönfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18:296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 105.Hotta K, Kitamoto A, Kitamoto T, Ogawa Y, Honda Y, Kessoku T, et al. Identification of differentially methylated region (DMR) networks associated with progression of nonalcoholic fatty liver disease. Sci Rep. 2018;8:13567. doi: 10.1038/s41598-018-31886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Mello VD, Matte A, Perfilyev A, Männistö V, Rönn T, Nilsson E, et al. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics. 2017;12:287–295. doi: 10.1080/15592294.2017.1294305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gerhard GS, Malenica I, Llaci L, Chu X, Petrick AT, Still CD, et al. Differentially methylated loci in NAFLD cirrhosis are associated with key signaling pathways. Clin Epigenetics. 2018;10:93. doi: 10.1186/s13148-018-0525-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology. 2010;52:1992–2000. doi: 10.1002/hep.23927. [DOI] [PubMed] [Google Scholar]
- 109.Thomson JP, Ottaviano R, Unterberger EB, Lempiainen H, Muller A, Terranova R, et al. Loss of tet1-associated5-hydroxymethylcytosine is concomitant with aberrant promoter hypermethylation in liver cancer. Cancer Res. 2016;76:3097–3108. doi: 10.1158/0008-5472.CAN-15-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ge W, Xie W, Coskun V, Tao J, Li E, Yoshikawa K, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science (80- ) 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science (80- ) 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 113.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen QR, Braun R, Hu Y, Yan C, Brunt EM, Meerzaman D, et al. Multi-SNP analysis of GWAS data identifies pathways associated with nonalcoholic fatty liver disease. PLoS One. 2013;8:e65982. doi: 10.1371/journal.pone.0065982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vella S, Gnani D, Crudele A, Ceccarelli S, De Stefanis C, Gaspari S, et al. EZH2 down-regulation exacerbates lipid accumulation and inflammation in in vitro and in vivo NAFLD. Int J Mol Sci. 2013;14:24154–24168. doi: 10.3390/ijms141224154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins: purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase. J Biol Chem. 1992;267:9005–9013. doi: 10.1006/bbrc.1997.6110. [DOI] [PubMed] [Google Scholar]
- 117.Hrit J, Goodrich L, Li C, Wang BA, Nie J, Cui X, et al. OGT binds a conserved C-terminal domain of TET1 to regulate TET1 activity and function in development. Elife. 2018;7. 10.7554/eLife.34870. [DOI] [PMC free article] [PubMed]
- 118.Kim YH, Nakayama T, Nayak J. Glycolysis and the hexosamine biosynthetic pathway as novel targets for upper and lower airway inflammation. Allergy Asthma Immunol Res. 2018;10:6–11. doi: 10.4168/aair.2018.10.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54:820–831. doi: 10.1016/j.molcel.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bedogni G, Gastaldelli A, Tiribelli C, Agosti F, De Col A, Fessehatsion R, et al. Relationship between glucose metabolism and non-alcoholic fatty liver disease severity in morbidly obese women. J Endocrinol Invest. 2014;37:739–744. doi: 10.1007/s40618-014-0101-x. [DOI] [PubMed] [Google Scholar]
- 121.Xu W, Zhang X, Wu JL, Fu L, Liu K, Liu D, et al. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J Hepatol. 2017;67:310–320. doi: 10.1016/j.jhep.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 122.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase OGT to chromatin in embryonic stem cells. Mol Cell. 2013;49:645–656. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 123.Zhang YJ, Wu HC, Yazici H, Yu MW, Lee PH, Santella RM. Global hypomethylation in hepatocellular carcinoma and its relationship to aflatoxin B1 exposure. World J Hepatol. 2012;4:169–175. doi: 10.4254/wjh.v4.i5.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greally JM, Drake AJ. The current state of epigenetic research in humans. JAMA Pediatr. 2016;171:103. doi: 10.1001/jamapediatrics.2016.3508. [DOI] [PubMed] [Google Scholar]
- 125.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Llufrio EM, Wang L, Naser FJ, Patti GJ. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018;16:381–387. doi: 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huang Q, Tan Y, Yin P, Ye G, Gao P, Lu X, et al. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013;73:4992–5002. doi: 10.1158/0008-5472.CAN-13-0308. [DOI] [PubMed] [Google Scholar]
- 128.Rashidi H, Luu NT, Alwahsh SM, Ginai M, Alhaque S, Dong H, et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch Toxicol. 2018;92:3117–3129. doi: 10.1007/s00204-018-2280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data/materials are contained in the manuscript.