

Abstract
A new synthetic approach and full spectral (NMR, IR, MS) and ion chromatographic characterization (IC) of nitrogen‐based ionic liquids bearing allyl‐ or ethyl‐ substituent and triflate, tosylate, methyl sulfate or methanesulfonate anion has been presented. On a sample of 16 new ionic liquids, the versatility of the anion exchange method has been proven. In the metathesis reactions that have been carried out, the halide anion was exchanged in ionic liquid with an alkyl sulfonate based anion using alkylating agents. The results obtained using ion chromatographic analysis on the newly synthesized compounds have been discussed. Also, the utilization of a gaseous methyl halide by‐product, obtained in the metathesis reaction and otherwise difficult to synthesize, has been presented. This approach ensured high atom economy of the overall process, which makes the proposed methodology sustainable and eco‐friendly.
Keywords: ionic liquids, ion chromatography, metathesis reactions, NMR spectroscopy
1. Introduction
The second principle of green chemistry states “synthetic methods should be designed to maximize incorporation of all materials used in the process into the final product.”1 This rule introduces us to the equation for atom economy, which takes into account every used material in the reaction and every obtained product, which then allows for better representation of a chemical process. The main goal is to design the process in a way which maximum amount of all starting materials is incorporated in the final product, with the minimal amount of waste by‐products produced. However, in chemistry in general, when the yield of the reaction is presented, it only shows the efficiency of the process with regards to the desired product, not taking into account the waste produced during the reaction. Thus, the percentage of atom economy is an important information to be provided with conventionally used yield, because it clearly shows how much of the reactants underwent the conversion to the desired product, including all by‐products created in the process.
Over the last few decades research orients itself towards green chemistry. That is why ionic liquids (ILs) have seen an upward trend in the research field. They are generally considered as salts that consist entirely of ions, which melt below 100 °C and remain liquid over a wide temperature range.2 They are known for their low vapor pressure, non‐flammability, low volatility, high chemical inertness, high ionic conductivity and thermal stability.3 As a result of their ionic structure, there are literally millions of ionic liquids with varying properties. This fact gives them the ability to be designer solvents, prepared for a special purpose, in comparison to the organic solvents which can only be changed within their molecular structure, thus their variations are very limited.4 Ionic liquids are also immiscible with many of standard organic solvents used in synthesis, which often allows for easy separation. Moreover, they are highly polar and, most importantly, can be recycled.5 Due to their special properties they can be used in a variety of applications, such as: catalysis,6, 7, 8, 9 electrochemical sensors,10 plant systemic acquired resistance inducers,11 batteries,12, 13 solar cells,14 supercapacitors,15 lubricants,16 gas separation,17 robotics,18 drug delivery,19 biomass conversion,20 coatings21 and many more.
Ionic liquids are most commonly synthesized from amines, phosphines or sulfonates.2 The synthesis can be divided into two paths. First one being quaternization reaction, where e. g. amine is alkylated, using alkyl halide, which leads to formation of desired cation. If the furnished ammonium halide is not the desired ionic liquid, the anion exchange is the second required step, where either metathesis reaction or reaction with Lewis acid takes place (Scheme 1).
Scheme 1.

Routes of synthesis for amine‐based ionic liquids.
After quaternization the next synthetic step is usually metathesis reaction, in which halide anions are exchanged to different ones e. g. [PF6]− or [NTf2]−. In this method molar equivalent amount of halide salt is produced and the purity of such ionic liquids strongly depends on their miscibility with water.22 However, it is almost impossible to obtain completely pure, halide‐free ionic liquid using this method. To surpass this problem, scientists use methyl‐ or ethyl‐ sulfonates, such as methyl triflate, in direct alkylation reaction of organic base.23
Such reaction as below is most often preferred using MeOTf, MeOTs or Me2SO4. However, even though this reaction furnishes ionic liquids with zero halide content, it has some limitations. Main one being methyl‐ or ethyl‐ group always present in ionic liquid, which shortens the possibilities of different substituents on nitrogen atom (Scheme 2).
Scheme 2.

Synthesis of amine‐based sulfonates.
Ionic liquids with methyl‐ or ethyl‐ sulfonate anions can also be obtained via metathesis reaction using alkali metal salt of the corresponding acid.24 However, the disadvantage of this method is the resulted alkali metal halide by‐product, which has to be removed from the mixture by additional purification, and even then the concentration of halide ions in the resulting ionic liquid is not negligible and affects physical properties of mentioned ionic liquids, such as density, viscosity or electrochemical window (EW).25
But what would scientists do if completely halide free 1‐butyl‐3‐ethylimidazolium methyl sulfate or 1‐butyl‐3‐ethylimidazolium triflate was needed? To obtain such ionic liquids, one must go through several synthetic steps and purifications. First, imidazole 1 is used as a starting material in order to obtain 1‐ethylimidazole 3. It can be purchased, but it is worth mentioning that its price is four times higher than 1‐methylimidazole. As for synthesis, the easiest way would be to deprotonate imidazole, using either sodium hydroxide or sodium hydride, and then reaction with ethyl bromide. This synthesis is straightforward, and with precise washing, all of the inorganic salt can be washed off, but atom economy of this reaction is just 48 %. Second reaction involves quaternization of the other nitrogen atom in the imidazole structure, using butyl bromide at elevated temperature. After a few days the reaction is completed and 1‐butyl‐3‐ethylimidazolium bromide 4 is obtained with 100 % atom economy (Scheme 3).
Scheme 3.

Synthesis of 1‐butyl‐3‐ethylimidazolium bromide 4.26
The resulting bromide salt can then be reacted with sulfuric acid (98 %) in dry DCM, in order to exchange halide anion to hydrogen sulfate (Scheme 4). This reaction needs to proceed under reduced pressure to remove resulting HBr. This anion exchange is resulting in 1‐butyl‐3‐ethylimidazolium hydrogen sulfate 5 and has very high yields and atom economy at 75 %. Last step, arguably the hardest one, involves methylation of the hydrogen sulfate anion in ionic liquid (Scheme 4). In order to obtain desired ionic liquid, hydrogen sulfate IL needs to be heated up in methanol for a very long time, while water has to be entirely removed from reaction mixture. This particular step has many disadvantages, such as hard to remove water as by‐product and long period of time needed to obtain small amounts of product. Even though the atom economy is at 93 %, it is very hard to obtain 1‐butyl‐3‐ethylimidazolium methyl sulfate 6 with quantitative yield. Simply, this process is not efficient.
Scheme 4.

Synthesis of 1‐butyl‐3‐ethylimidazolium hydrogen sulfate 5 and 1‐butyl‐3‐ethylimidazolium methyl sulfate 6.27
The synthesis of 1‐butyl‐3‐ethylimidazolium triflate 7 goes through similar path. 1‐Butyl‐3‐ethylimidazolium bromide 4, obtained as stated earlier (Scheme 3), reacts with triflic acid at elevated temperature to give corresponding 1‐butyl‐3‐ethylimidazolium triflate 7, in relatively high yield and atom economy of 79 %, but with HBr being formed as by‐product (Scheme 5). Even though these reactions furnish the desired products, they involve working with dangerous acids, and need to be strictly controlled. To recap, it is possible to obtain 1‐butyl‐3‐ethylimidazolium methyl sulfate 6 or triflate 7, but one requires working with triflic acid, and the other one is consisted of a two‐step process, resulting in the formation of desired product but in unsatisfactory yield and neither reaction has atom economy close to 100 %. There are reports in the literature that present synthesis of ionic liquids with atom economy at 100 % [28–31] that present the method of obtaining methylcarbonate ionic liquids. However, even though those reactions furnish desired products with maximum atom economy, they generate waste such as solvents that need to be evaporated, thus usage of heat is required, hence those type of processes cannot be considered fully green. In 2009 Ignat'ev and co‐workers presented new method of synthesis of ionic liquids with alkyl‐sulfonate and alkyl‐carboxylate anions.32 The invention was related to obtaining tosylates, methanesulfonates and triflates ILs by reaction of any halide‐based organic salts with alkyl‐ or arylsulfonic acid.
Scheme 5.

Synthesis of 1‐butyl‐3‐ethylimidazolium triflate 7.33
This reaction can be carried out at room temperature without use of any solvent. Commercially available substrates are esters of sulfonic acids: trifluoromethanesulfonic acid, p‐toluenesulfonic acid and methanesulfonic acid. All acid listed above are very strong organic acids, e. g. triflic acid (pKa=−14.7) being 100 times stronger than sulfuric acid (pKa=−3). Thanks to their high acidity they have the potential to push chloride anion and bind it with methyl‐ leaving group. All three methylating agents produce by‐product in form of chloro‐ or bromomethane from reactive leaving ‐CH3 group, which compounds can be easily removed from the reaction mixture, due to their low boiling points (they are gaseous at reaction conditions). Emerging gas can be in itself used as a methylating agent, to obtain ionic liquids normally very difficult to synthesize. In 2012 the same group published an article, in which they presented synthesis of triflate anion‐based ionic liquids (Scheme 6).34
Scheme 6.

Preparation of triflate ionic liquids from corresponding halides.
Besides this article and patent mentioned before, there is no literature, according to authors knowledge, mentioning this method for obtaining ionic liquids with sulfonate‐based anions. The great advantage of the use of this approach is that it allows for obtaining ILs with every substituent possible, without the necessity of introduction methyl group into cation structure. Not only that, but the purity levels of such ionic liquids are very high, which gives them great advantage over ionic liquids synthesized using standard procedures. Moreover, this anion exchange protocol allows for obtaining ionic liquids normally unavailable. All of that combined presents great opportunities for preparing new ionic liquids, with low halide content, which would make them great candidates for electrochemical studies or catalysis.
Herein we present 16 ionic liquids obtained using anion exchange method, which assumes the reaction of halide containing ionic liquids with four methyl‐ sulfonates: methyl trifluoromethanesulfonate, methyl p‐toluenesulfonate, methyl methanesulfonate and dimethyl sulfate. As a result of those reactions gaseous halomethane is released and consequently used in the quaternization reaction of an amine. Those two reactions combined provide 100 % atom economy, with no waste produced.
2. Results and Discussion
Our goal was to obtain in pure form and to characterize a series of nitrogen‐based ionic liquids bearing allyl or ethyl group.
Thus, mentioned earlier 1‐butyl‐3‐ethylimidazolium methyl sulfate 6 and 1‐butyl‐3‐ethylimidazolium triflate 7 could be obtained by the implementation of the anion exchange method presented above (Scheme 7). For example, 1‐butyl‐3‐ethylimidazolium bromide 4 could react readily with dimethyl sulfate and methyl triflate, yielding desired products of very high purity. As this reaction produces gaseous by‐product, using it in reaction with amine, 1‐methylimidazole can also allow for synthesis of often difficult to prepare 1,3‐dimethylimidazolium halide 8 with 100 % atom economy. This method, in comparison to those mentioned before, is much more effective, faster and with no waste generated.
Scheme 7.

Synthesis of 1‐butyl‐3‐ethylimidazolium triflate 7 and 1,3‐dimethylimidazolium bromide 8 and 1‐butyl‐3‐ethylimidazolium methyl sulfate 6 and 1,3‐dimethylimidazolium bromide 8 both with 100 % atom economy; in the second stage gaseous CH3X is used.
First, we synthesized three chlorides and one bromide, conducting quaternization reactions of selected aliphatic and aromatic amines 10–12 with allyl chloride 9 or ethyl bromide 16 in MeCN as a reaction medium at elevated temperature (Scheme 8). After the reaction was complete the solvent was evaporated and obtained ionic liquids were placed under vacuum to dry.
Scheme 8.

Syntheses of halide salts 13–15, 17.
As a next step, every chloride and bromide salt was reacted with four methylating agents: dimethyl sulfate, methyl trifluoromethanesulfonate, methyl p‐toluenesulfonate and methyl methanesulfonate in MeCN as a reaction medium at room temperature (Scheme 9). Reactions were finished immediately after the complete addition of methylating agent, nevertheless, the reactions were stirred at room temperature for 30 minutes to ensure total anion exchange. After that time the solvent was evaporated, and the resulting ionic liquids were placed under vacuum to be dried out.
Scheme 9.

General route of synthesis of sulfonate‐based ionic liquids.
Reactions gave desired products in quantitative yields. The absence of chloride or bromide ions was determined using ion chromatography. Figure 1 presents anionic chromatogram of salts based on 1‐allylpirydinium cation paired with different anions: chloride, mesylate, methyl sulfate, tosylate and triflate proving the quantitative yield of reaction (as calculated for Cl− presence in product). Many factors contribute to the retention time of every anion, including their molecular weight, charge with regards to the conductometric detection and polarizability. Furthermore, column type and length have also essential impact on the retention time of the anion and peak shape. Packing material used in our experiments in the separation column was polystyrene‐divinylbenzene copolymer with quaternary ammonium groups. Eluent also has effect on the retention time of the anions, both chemical composition and flow rate, thus in our experiments mixture of acetonitrile (HPLC grade) and aqueous solution containing 3.2 mM of sodium bicarbonate and 1.0 mM of sodium carbonate was used in the ratio of 30 : 70 (v/v), and the flow rate of the eluent was 0.9 mL/min. As it can be observed on the chromatogram (Figure 1) the lowest affinity to the packing material demonstrates methanesulfonate anion. Consequently, both chloride and methyl sulfonate anion are separated next with very similar retention time. In regard to this, to accurately confirm anion exchange from chloride to methyl sulfate, 1H NMR analysis was used to confirm the presence of a methyl group from the methyl sulfate anion and its integration in relation to signals related to protons in cation structure. Accordingly, the evaluation of the chromatographic data suggests that the p‐methylbenzyl ring in the structure of tosylate anion, impacts on lower than predicted affinity to the filling of the column, because of the inert nature and bulky structure of the ring. For those reasons tosylate anion has significantly lower conductivity. Therefore, triflate anion that consists of three fluoride atoms in its structure, with representative high electronegativity, has the strongest affinity to the chemical groups in the packing material of the column. Hence, trifluoromethylsulfonate anion can be characterized with the longest retention time and the lowest conductivity of all analyzed anions with characteristically stretched shape of the chromatographic peak.
Figure 1.

Chromatogram of the 1‐allylpirydinium chloride, 1‐allylpirydinium methyl sulfate, 1‐allylpirydinium triflate, 1‐allylpirydinium tosylate and 1‐allylpirydinium methanesulfonate.
Table 1 contains retention time for every anion for every ionic liquid obtained during anion exchange reaction and their purities.
Table 1.
Retention times and purity of anions of ionic liquids 13–15, 17–33.
| IL number | Retention time [min] | Purity [%] | IL number | Retention time [min] | Purity [%] |
|---|---|---|---|---|---|
| 13 14 15 17 18 19 20 21 22 23 | 3.54 3.48 3.51 7.97 12.11 12.62 12.02 12.42 3.33 3.31 | 99.9 99.9 99.9 99.9 99.827 99.693 99.638 99.840 99.9 99.9 | 24 25 26 27 28 29 30 31 32 33 | 3.31 3.30 1.76 1.76 1.76 1.75 4.88 4.76 4.86 4.83 | 99.9 99.9 99.553 99.838 99.897 99.653 99.414 99.320 99.698 99.739 |
Figure 2 presents all 16 ionic liquids obtained in the anion exchange reaction with four different halide salts and four different methylating agents.
Figure 2.

Ionic liquids obtained in this paper 18–33.
1H and 13C NMR techniques were used for the characterization of all ionic liquids containing −CH3 group, to confirm that the anion exchange took place, together with the confirmation of the presence of the methyl group form the methylating agent used in the reaction. Figure 3 presents comparison of two 1H NMR spectra of 1‐allyltriethylammonium chloride 15 and 1‐allyltriethylammonium methanesulfonate 28. The bottom spectrum represents halide salt and indicates that there are no protic signals from anion of the ionic liquid, whereas the top spectrum presents the exact same cation structure with singlet of −CH3 group (marked with black frame), coming from methanesulfonate anion, confirming that the anion exchange took place. When halide salts were exchanged to triflate anion using methyl triflate, 1H NMR was not useful, that is why 19F NMR experiment was performed, to confirm the presence of −CF3 group.
Figure 3.
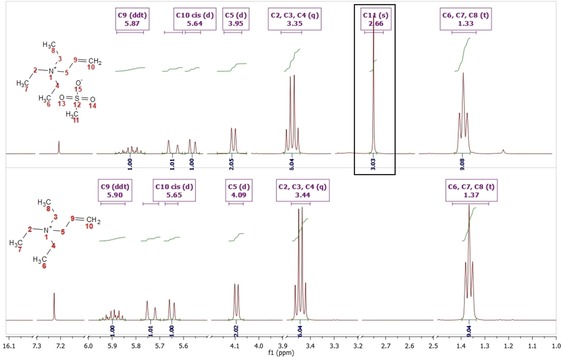
1H NMR spectra of 1‐allyltriethylammonium chloride 15 (bottom) and 1‐allyltriethylammonium methanesulfonate 28 (top).
Ionic liquids obtained from anion exchange method presented in this paper have very high purity levels, which in turn means that they can be of great interest in many scientific fields, such as electrochemistry or catalysis, to name a few.
Halide salt impurities can have influence on the oxidation and reduction potentials of ionic liquids, because halides are easily oxidized in comparison to perfluorinated anions.35 This decreases the anodic potential of an ionic liquid, which in turn decreases its electrochemical window value. The higher the value, the bigger the possibilities of such IL being used as an electrolyte. Another advantage would be liquid nature of products used in electrochemistry. Most of the obtained here ionic liquids are solids at room temperature, but a few are liquid, which fact, combined with their high purity levels, predestines them to be of potential use in many chemical fields, not only electrochemistry.
While synthesizing all ionic liquids with sulfonate‐based anions, very reactive gas is formed (either chloromethane or bromomethane). This gas, as a side product, can be used in another reaction, converting it into methylating agent for another base (amine, phosphine, etc.). This ensures not only 100 % atom economy, but also provides great opportunity for synthesis of ILs that are usually difficult to obtain.
Thus, with one closed system, two different ionic liquids can be obtained, both being not available in regard to conventional synthetic routes. In our example, after the straightforward quaternization reaction of triethylamine 11 with ethyl bromide 16, the resulting ionic liquid was used as a substrate in the reaction with methyl triflate to yield tetraethylammonium bromide 17, and generating the readily reacting gaseous bromomethane 35, that was used as a substrate in reaction with triethylamine 11, yielding triethylmethylamine bromide 36 (Scheme 10).
Scheme 10.

Synthesis of tetraethylammonium triflate 21 and triethhylmethylammonium bromide 36.
Tetraethylammonium bromide 17 was dissolved in MeCN and placed in the two‐necked flask and cooled down in liquid N2/acetone bath. The solution of triethylamine 11 in MeCN was also placed in the two‐necked flask, with two condensers placed at one another. The flasks were connected by silicone tube, for gaseous bromomethane 35 to travel from one flask to the other. After the solution of bromide ionic liquid was cooled down, and the triethylamine solution heated up to 80 °C, the slow addition of methyl triflate 34 to the tetraethylammonium bromide 17 began. Figure 4 represents scheme of the reaction set, which was used in this particular reaction.
Figure 4.

Scheme of reaction set for simultaneous synthesis of two different ionic liquids with utilization of gaseous by‐product (IL 1 – tetraethylammonium triflate 21, IL 2 – triethylmethylammonium bromide 36).
The idea was to introduce gaseous bromomethane to the amine solution as slowly as possible to ensure as high conversion of triethylamine to the product as possible. After complete addition of methyl triflate to the tetraethylammonium bromide the temperature of this cold mixture was slowly brought up to room temperature. This allowed for the chemicals in the solution to slowly react, which in turn meant slow production of gaseous by‐product. Bromomethane, which has a very low boiling point of 3.56 °C, was introduced to the very hot triethylammonium solution, so that reaction could take place immediately after contacting with amine solution.
Two condensers up top of the flask provided long path for the gas to travel, also ensuring the condensation of bromomethane in case it did not react straight away with amine in the solution. The reaction was carried out at these conditions for 5 hours, and after that time the acetonitrile was evaporated yielding a white crystal solid – triethylmethylammonium bromide 36 with yield of 60 %. The structure of the product was confirmed by ion chromatography and 1H and 13C NMR spectroscopy. Figure 5 presents two chromatograms of triethylmethylammonium bromide 36. The top ionic chromatogram presents cation analysis, where the product has retention time of 6.70 min, and purity level at 96.5 %. Unreacted triethylamine is present, as a smaller peak with retention time of 3.99 min. The bottom chromatogram shows anion separation, and it can be observed that the bromide anion has retention time of 7.53 min with 100 % purity.
Figure 5.

Chromatograms of triethylmethylammonium bromide 36, cation (top), anion (bottom).
With regards to the entire two‐step process (metathesis reaction and quaternization of amine using gaseous by‐product), if it was considered as one single synthesis, the atom economy could be then calculated to 100 %. However, if considering both reactions as separate processes, where the by‐product of one reaction is to be stored in order to be used later as substrate for the second reaction, then the atom economy of the processes should be considered separately for each reaction carried out. In such case the atom economy of the first reaction is 75 % and the atom economy of the second reaction is 100 %. Nevertheless, the intention of the authors was to present the entire procedure of synthesis of two difficult to be prepared salts as single synthetic process.
3. Conclusions
A series of sulfonate anion based ionic liquid has been synthesized and their spectral properties (NMR, IR, MS) have been fully characterized. Synthetic method presented in this paper is based on the anion exchange method, employing sulfonates as substrates, but not as it is commonly known for the quaternization reaction, e. g. delivering methyl group to the cation structure and remaining alkyl sulfonate as an anion, but rather using it is very reactive methyl leaving group as a binding medium for halide anion. This method of synthesis of ionic liquids containing anions such as triflate, tosylate, methyl sulfate, methyl methanesulfonate has only been presented in one patent29, and has not been fully examined and introduced in the scientific field. Ionic liquids obtained in this manner have been characterized with use of ion chromatography technique, which sustained the claim provided in the article,30 that the anion exchange takes place exceptionally readily and quantitatively. Thus, purity of those ionic liquids has been established, based on the peak areas in the chromatograms of each anion. All of the ionic liquids studied in frames of this work had the anion purity levels above 99.3 %. In the described metathesis reactions, gaseous haloalkane by‐product is formed and released from the reaction medium. Thus, we developed the method to utilize this by‐product as a substrate in consecutive reaction, leading to another ionic liquid. Haloalkane can be used in the reaction with amines to yield ionic liquids normally very difficult and expensive to obtain. Thus, reaction of anion exchange and quaternization of amine, by utilization of by‐product, ensures almost 100 % atom economy and high level of the process sustainability.
Experimental Section
General Information
All used reagents and solvents of high purity were commercially available, purchased from Sigma Aldrich and Iolitec and used as received.
NMR spectra were recorded using Bruker Ascend 400 and 500 MHz spectrometers in commercially available deuterated solvents. IR measurements were conducted with utilization of diamond single crystal single reflection ATR system.
MS spectra and HRMS measurements were recorded using QTOF type mass spectrometer – Impact HD, Bruker.
The chromatographic experiments were carried out using a Metrohm Eco IC system (Metrohm, Herisau, Switzerland) equipped with an 863 Compact IC Autosampler, a 10.0 μl injection loop and a conductometric detector (maintained at room temperature). A self‐regenerating Suppressor Module (MSM) (Metrohm, Herisau, Switzerland), regenerated with distilled water and sulfuric acid, were used to separate and determine the anions in ionic liquids. All data were recorded by Metrohm software. Anion ion chromatograph apparatus was equipped with a Metrosep A Supp 10 ion exchange column (150×4.0 mm) coupled with Metrosep A Supp Guard. The packing material was polystyrene‐divinylbenzene copolymer with quaternary ammonium groups. A flow rate of 0.9 ml/min was used. Anion separation was performed with an eluent composed of 30 : 70, v/v proportions of acetonitrile (HPLC purity grade) and an aqueous solution containing 3.2 mM of sodium bicarbonate and 1.0 mM of sodium carbonate (Merck). using eluent composed of 30 : 70 ratio of acetonitrile (HPLC purity grade, Merck) and an aqueous solution containing 0.7 mM of dipicolinic acid and 1.7 mM of nitric acid (Merck). All aqueous solutions were prepared carefully using distilled water (conductivity=0.05 μS/cm). During cation separation, average pressure in analytical system was maintained at the level of 3.5 MPa, whereas during anion analysis system worked under pressure of 13.5 MPa. Samples for anion analysis were prepared by dissolving about 15 mg of ionic liquid in eluent solution (3 ml), which was then filtered through syringe filter. After that, 200 μl of filtrate was taken for analysis into plastic vial and refilled with 10 ml of eluent. Sample for cation analysis was prepared by uptake of 200 μl of filtered substance to a vial and then fulfilled with 2.0 ml of acetone (HPLC grade, Merck), 3.0 ml of nitric acid (2.0 mM) and 5.0 ml of proper eluent. Before filtration, cation samples were dissolved in acetone (3 mL) and protonated with nitric acid (2.0 mM). Purity of prepared ionic liquids was determined by taking into the account both cation and anion chromatographic analysis for each compound. Methodology used for calculation of purity of ionic liquids is presented in ESI, and the values are included in the section below.
Synthesis of Halogen Anion Based Ionic Liquids
General Procedure A
A solution of amine (1 eq) in MeCN was placed in two‐neck round‐bottomed flask equipped with a reflux condenser in one neck and septum in the other and heated to 60 °C with simultaneous stirring on magnetic stirrer. Then allyl chloride or ethyl bromide (1 eq) was added dropwise (addition rate 5 mL/h). A resulting mixture was stirred at 60 °C for time required to complete the reaction. Next, the solvent was evaporated using rotary evaporator, and obtained product was washed with ethyl acetate and dried under vacuum to furnish pure product.
1‐Allyl‐2,3‐Dimethylimidazolium Chloride (13)
Compound was obtained according to general procedure A with use of 1,2‐dimethylimidazole (13.92 g, 0.145 mol), allyl chloride (11.10 g, 0.145 mol) and MeCN (150 mL). Reaction time was equal to 96 h and the reaction gave pure product as a white solid (24.25 g, 97 %). Purity: 98.66 %.
1H NMR (400 MHz, DMSO‐d6): δ=7.84 (AB, d, 3 J=2.0 Hz, 1H), 7.77 (AB, d, 3 J=2.0 Hz, 1H), 5.95 (ddt, 3 J=17.1 Hz, 3 J=10.7 Hz, 3 J=5.2 Hz, 1H), 5.25 (dd, 3 J=10.4 Hz, 2 J=1.0 Hz, 1H), 5.14 (dd, 3 J=17.2 Hz, 2 J=1.1 Hz, 1H), 4.91 (d, 3 J=5.5 Hz, 2H), 3.81 (s, 3H), 2.59 (s, 3H); 13C{1H} NMR (100 MHz, DMSO‐d6): δ=144.8, 132.0, 122.7, 121.3, 119.0, 49.8, 35.1, 9.7; IC− (min): tR=3.54.
Allyl‐Triethylammonium Chloride (14)
Compound was obtained according to general procedure A with use of triethylamine (6.00 g, 0.060 mol), allyl chloride (4.60 g, 0.060 mol) and MeCN (40 mL). Reaction time was equal to 72 h and the reaction gave pure product as a yellow solid (10.50 g, 99 %). Purity: 97.72 %.
1H NMR (600 MHz, CDCl3): δ=5.55 (ddt, 3 J=17.2 Hz, 3 J=10.0 Hz, 3 J=7.3 Hz, 1H), 5.37 (dd, 3 J=16.8 Hz, 2 J=0.6 Hz, 1H), 5.27 (d, 3 J=10.1 Hz, 1H), 3.69 (d, 3 J=7.3 Hz, 2H), 3.06 (q, 3 J=7.3 Hz, 6H), 0,98 (t, 3 J=7.3 Hz, 9H); 13C{1H} NMR (125 MHz, CDCl3): δ=128.6, 123.8, 59.5, 52.9, 7.7; IC− (min): tR=3.48.
1‐Allylpyrydinium Chloride (15)
Compound was obtained according to general procedure A with use of pyridine (5.08 g, 0.064 mol), allyl chloride (4.89 g, 0.064 mol) and MeCN (50 mL). Reaction time was equal to 72 h and the reaction gave pure product as a brown solid (9.69 g, 97 %). Purity: 98.00 %.
1H NMR (400 MHz, DMSO‐d6): δ=9.40 (AA′BB′, dd, 3 J=6.6 Hz, 4 J=1.2 Hz, 2H), 8.69 (tt, 3 J=7.9 Hz, 4 J=1.3 Hz, 1H), 8.24 (AA′BB′, dd, 3 J=7.7 Hz, 3 J=6.7 Hz, 2H), 6.16 (ddt, 3 J=16.5 Hz, 3 J=10.2 Hz, 3 J=6.3 Hz, 1H), 5.53 (d, 3 J=6.3 Hz, 2H), 5.43 (dq, 3 J=17.0 Hz, 2 J=1.3 Hz, 1H), 5.36 (dd, 3 J=10.2 Hz, 2 J=1.2 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO‐d6): δ=146.2, 145.2, 132.3, 128.5, 122.1, 62.0; IC− (min): tR=3.51.
Tetraethylammonium Bromide (17)
Compound was obtained according to general procedure A with use of triethylamine (10.00 g, 0.099 mol), ethyl bromide (10.79 g, 0.099 mol) and MeCN (50 mL). Reaction time was equal to 72 h and the reaction gave pure product as a white solid (20.60 g, 98 %). Purity: 99.00 %.
1H NMR (500 MHz, CDCl3): δ=3.43 (q, 3 J=7.3 Hz, 8H), 1.32 (t, 3 J=7.3 Hz, 12H); 13C{1H} NMR (100 MHz, CDCl3): δ=52.52, 7.69; IC− (min): tR=7.97.
Synthesis of Sulfonate Anion Based Ionic Liquids
Methyl Sulfates, General Procedure B
A solution of corresponding halide salt (1 eq.) in MeCN was placed in round‐bottomed flask and stirred at room temperature on magnetic stirrer and dimethyl sulfate (1 eq.) was added dropwise over 5 minutes. After 30 minutes the solvent was evaporated using rotatory evaporator and obtained product was dried under vacuum to furnish pure product.
Triflates, General Procedure C
A solution of corresponding halide salt (1 eq.) in MeCN was placed in round‐bottomed flask and stirred at room temperature on magnetic stirrer and methyl triflate (1 eq.) was added dropwise over 5 minutes. After 30 minutes the solvent was evaporated using rotatory evaporator and obtained product was dried under vacuum to furnish pure product.
Tosylates, General Procedure D
A solution of corresponding halide salt (1 eq.) in MeCN was placed in round‐bottomed flask and stirred at room temperature on magnetic stirrer and the solution of methyl tosylate (1 eq.) in MeCN was added dropwise over 5 minutes. After 30 minutes the solvent was evaporated using rotatory evaporator and obtained product was dried under vacuum to furnish pure product.
Methanesulfonates, General Procedure E
A solution of corresponding halide salt (1 eq.) in MeCN was placed in round‐bottomed flask and stirred at room temperature on magnetic stirrer and methyl methanesulfonate (1 eq.) was added dropwise over 5 minutes. After 30 minutes the solvent was evaporated using rotatory evaporator and obtained product was dried under vacuum to furnish pure product.
1‐Allyl‐2,3‐Dimethylimidazolium Methyl Sulfate (22)
Compound was obtained according to general procedure B with use of 1‐allyl‐2,3‐dimethylimidazolium chloride (1.00 g, 6.31 mmol), dimethyl sulfate (0.79 g, 6.31 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow oil (1.40 g, a quantitative yield). Purity: 99.06 %.
1H NMR (400 MHz, CDCl3): δ=7.51 (d, 3 J=2.1 Hz, 1H), 7.38 (d, 3 J=2.1 Hz, 1H), 5.95 (ddt, 3 J=16.1 Hz, 3 J=10.3 Hz, 3 J=5.8 Hz, 1H), 5.36 (d, 3 J=10.3 Hz, 1H), 5.25 (d, 3 J=17.1 Hz, 1H), 4.81 (dt, 3 J=5.8 Hz, 2 J=1.4 Hz, 2H), 3.89 (s, 3H), 3.63 (s, 3H), 2.66 (s, 3H); 13C {1H} NMR (100 MHz, DMSO‐d6): δ=145.06, 132.04, 122.93, 121.49, 119.27, 53.32, 50.05, 35.18, 9.61; IR (ATR, νmax, cm−1): 3135 (w), 2947 (w), 1647 (w), 1589 (w), 1538 (w), 1452 (w), 1419 (w), 1218 (vs), 1058 (m), 1003 (s), 943 (w), 733 (s), 676 (w), 608 (w); MS (ESI)+: m/z (%)=137 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H13N2 + 137.1079; Found 137.1078, ▵ 0.7294; MS (ESI)−: m/z (%)=111([M+], 100), 96 ([M−CH3]+, 30); HRMS (ESI)− m/z: Calcd for CH3O4S− 110.9752; Found 110.9758, Δ −5.4066; IC− (min): tR=3.33.
1‐Allyl‐2,3‐Dimethylimidazolium Triflate (18)
Compound was obtained according to general procedure C with use of 1‐allyl‐2,3‐dimethylimidazolium chloride (1.00 g, 6.31 mmol), methyl triflate (1.04 g, 6.31 mmol) and MeCN (10 mL). The reaction gave pure product as an orange solid (1.60 g, a quantitative yield). Purity: 99.07 %.
1H NMR (500 MHz, CDCl3): δ=7.27 (d, 3 J=2.1 Hz, 1H), 7.19 (d, 3 J=2.2 Hz, 1H), 5.86 (dd, 3 J=17.1 Hz, 3J=10.3 Hz, 1H), 5.34 (dt, 3 J=10.4 Hz, 2 J=1.5 Hz, 1H), 5.20 (dt, 3 J=17.1 Hz, 2 J=1.7, Hz, 1H), 4.67 (dt, 3 J=5.8 Hz, 2 J=1.5 Hz, 2H), 3.78 (s, 3H), 2.56 (s, 3H); 13C {1H} NMR (100 MHz, DMSO‐d6): δ=145.03, 131.94, 122.92, 121.48, 121.17 (q, 1 J CF=322.8 Hz), 119.27, 50.07, 35.19, 9.59; 19F NMR (376 MHz, CDCl3): δ=‐78.50; IR (ATR, νmax, cm−1): 3142 (w), 3119 (w), 1650 (w), 1592 (w), 1544 (w), 1460 (w), 1415 (w), 1254 (vs), 1224 (s), 1150 (s), 1027 (s), 992 (w), 917 (m), 775 (m), 677 (w); MS (ESI)+: m/z (%)=137 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H13N2 + 137.1079; Found 137.1078, ▵ 0.7294; MS (ESI)−: m/z (%)=149 ([M+], 100); HRMS (ESI)− m/z: Calcd for CF3O3S− 148.9520; Found 148.9520, Δ 0; IC− (min): tR=12.11.
1‐Allyl‐2,3‐Dimethylimidazolium Tosylate (30)
Compound was obtained according to general procedure D with use of 1‐allyl‐2,3‐dimethylimidazolium chloride (1.00 g, 6.31 mmol), methyl tosylate (1.18 g, 6.31 mmol) and MeCN (15 mL). The reaction gave pure product as a beige solid (1.75 g, a quantitative yield). Purity: 98.18 %.
1H NMR (500 MHz, CDCl3): δ=7.60 (d, 3 J=8.1 Hz, 2H), 7.45 (d, 3 J=2.1 Hz, 1H), 7.28 (d, 3 J=2.1 Hz, 1H), 7.03 (d, 3 J=7.9 Hz, 2H), 5.79 (ddt, 3 J=16.1 Hz, 3 J=10.3 Hz, 3 J=5.8 Hz, 1H), 5.23 (d, 3 J=10.3 Hz, 1H), 5.10 (d, 3 J=17.1 Hz, 1H), 4.68 (dt, 3 J=5.7 Hz, 2 J=1.4 Hz, 2H), 3.76 (s, 3H), 2.53 (s, 3H), 2.24 (s, 3H); 13C {1H} NMR (100 MHz, DMSO‐d6): δ=146.19, 145.01, 138.1, 132.03, 128.52, 125.92, 122.93, 121.49, 119.27, 50.05, 35.19, 21.25, 9.62; IR (ATR, νmax, cm−1): 3130 (s), 3100 (s), 3086 (s), 3016 (w), 1645 (w), 1587 (m), 1539 (m), 1493 (w), 1457 (m), 1417 (m), 1366 (w), 1282 (m), 1243 (m), 1206 (s), 1186 (vs), 1118( s), 1030 (m), 1009 (m), 995 (s), 938 (m), 816 (s), 785 (s), 763 (m), 679 (s), 640 (m); MS (ESI)+: m/z (%)=137 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H13N2 + 137.1079; Found 137.1078, ▵ 0.7294; MS (ESI)−: m/z (%)=171 ([M+], 100); HRMS (ESI)− m/z: Calcd for C7H7O3S− 171.0116; Found 171.0118, Δ −1.1695; IC− (min): tR=4.88.
1‐Allyl‐2,3‐Dimethylimidazolium Methanesulfonate (26)
Compound was obtained according to general procedure E with use of 1‐allyl‐2,3‐dimethylimidazolium chloride (1.00 g, 6.31 mmol), methyl methanesulfonate (0.70 g, 6.31 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow oil (1.75 g, a quantitative yield). Purity: 98.30 %.
1H NMR (500 MHz, CDCl3): δ=7.55 (d, 3 J=2.1 Hz, 1H), 7.40 (d, 3 J=2.1 Hz, 1H), 5.89 (ddd, 3 J=16.2 Hz, 3 J=10.9 Hz, 3 J=5.8 Hz, 1H), 5.31 (d, 3 J=10.3 Hz, 1H), 5.19 (d, 3 J=17.1 Hz, 1H), 4.80 (d, 3 J=5.7 Hz, 2H), 3.87 (s, 3H), 2.64 (s, 3H), 2.57 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3): δ=144.2, 130.19, 122.88, 121.34, 120.25, 50.61, 39.43, 35.38, 9.83; IR (ATR, νmax, cm−1): 3444 (w), 3133 (m), 3010 (w), 2933 (w), 1647 (w), 1588 (m), 1538 (m), 1459 (w), 1419 (m), 1332 (w), 1177 (vs), 1037 (vs), 946 (m), 766 (s), 676 (w); MS (ESI)+: m/z (%)=137 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H13N2 + 137.1079; Found 137.1078, ▵ 0.7294; MS (ESI)−: m/z (%)=95 ([M+], 100); HRMS (ESI)− m/z: Calcd for CH3O3S− 94.9803; Found 94.9807, Δ −4.2114; IC− (min): tR=1.76.
Allyltriethylammonium Methyl Sulfate (23)
Compound was obtained according to general procedure B with use of allyltriethylammonium chloride (1.00 g, 4.51 mmol), dimethyl sulfate (0.57 g, 4.51 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow oil (1.40 g, a quantitative yield). Purity: 98.18 %.
1H NMR (400 MHz, CDCl3): δ=5.94 (ddt, 3 J=17.1 Hz, 3 J=10.0 Hz, 3 J=7.2 Hz, 1H), 5.76 (dd, 3 J=16.8 Hz, 2 J=0.9, Hz, 1H), 5.69 (d, 3 J=10.2 Hz, 1H), 3.95 (d, 3 J=7.2 Hz, 2H), 3.70 (s, 3H), 3.37 (q, 3 J=7.3 Hz, 6H), 1.38 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=128.53, 124.03, 59.43, 54.58, 52.86, 7.52; IR (ATR, νmax, cm−1): 3182 (s), 3141 (vs), 3090 (m), 3057 (s), 3020 (s), 2985 (vs), 2947 (s), 2164 (m), 1591 (m), 1559 (m), 1520 (m), 1477 (s), 1429 (m), 1397 (s), 1379 (s); MS (ESI)+: m/z (%)=142 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C9H20N+ 142.1596; Found 142.1597, ▵ −0.7034; MS (ESI)−: m/z (%)=111([M+], 100), 96 ([M−CH3]+, 27); HRMS (ESI)− m/z: Calcd for CH3O4S− 110.9752; Found 110.9756, Δ −3.6044; IC− (min): tR=3.31.
Allyltriethylammonium Triflate (19)
Compound was obtained according to general procedure C with use of allyltriethylammonium chloride (2.00 g, 11.30 mmol), methyl triflate (1.85 g, 11.30 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow solid (3.30 g, a quantitative yield). Purity: 96.40 %.
1H NMR (600 MHz, CDCl3): δ=5.91 (ddt, 3 J=17.3 Hz, 3 J=10.0 Hz, 3 J=7.3 Hz, 1H), 5.74 (dd, 3 J=21.7 Hz, 3 J=5.4 Hz, 2H), 3.87 (d, 3 J=7.3 Hz, 2H), 3.32 (q, 3 J=7.3 Hz, 6H), 1.36 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=128.97, 123.59, 120.75 (q, 1JCF=322.10 Hz), 59.58, 52.95, 7.49; 19F NMR (376 MHz, CDCl3): δ=‐78.44; IR (ATR, νmax, cm−1): 2999 (w), 1459 (w), 1258 (vs), 1224 (s), 1150 (s), 1029 (s), 950 (w), 800 (w), 636 (m), 572 (m), 516 (s); MS (ESI)+: m/z (%)=142 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C9H20N+ 142.1596; Found 142.1597, ▵ −0.7034; MS (ESI)−: m/z (%)=149 ([M+], 100); HRMS (ESI)− m/z: Calcd for CF3O3S− 148.9520; Found 148.9521, Δ −0.6714; IC− (min): tR=12.62.
Allyltriethylammonium Tosylate (31)
Compound was obtained according to general procedure D with use of allyltriethylammonium chloride (2.00 g, 11.30 mmol), methyl tosylate (2.10 g, 11.30 mmol) and MeCN (15 mL). The reaction gave pure product as a yellow solid (3.50 g, a quantitative yield). Purity: 98.16 %.
1H NMR (600 MHz, CDCl3): δ=7.68 (d, 3 J=8.1 Hz, 2H), 7.05 (d, 3 J=8.0 Hz, 2H), 5.77 (ddt, 3 J=17.2 Hz, 3 J=10.0 Hz, 3 J=7.2 Hz, 1H), 5.60 (dd, 3 J=16.8 Hz, 2 J=0.5 Hz, 1H), 5.53 (d, 3 J=10.2 Hz, 1H), 3.81 (d, 3 J=7.2 Hz, 2H), 3.21 (q, 3 J=7.3 Hz, 6H), 2.25 (s, 3H), 1.20 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=144.52, 138.86, 128.43, 128.30, 125.89, 124.06, 59.33, 52.75, 21.20, 7.54; IR (ATR, νmax, cm−1): 2985 (m), 2946 (w), 1642 (w), 1598 (w), 1478 (m), 1458 (m), 1397 (w), 1300 (m), 1192 (vs), 1119 (s), 1030 (s), 1010 (s), 817 (m), 797 (m); MS (ESI)+: m/z (%)=142 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C9H20N+ 142.1596; Found 142.1597, ▵ −0.7034; MS (ESI)−: m/z (%)=171 ([M+], 100); HRMS (ESI)− m/z: Calcd for C7H7O3S− 171.0116; Found 171.0107, Δ 5.2628; IC− (min): tR=4.76.
Allyltriethylammonium Methanesulfonate (27)
Compound was obtained according to general procedure E with use of allyltriethylammonium chloride (2.00 g, 11.30 mmol), methyl methanesulfonate (1.25 g, 11.30 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow solid (2.65 g, a quantitative yield). Purity: 96.13 %.
1H NMR (500 MHz, CDCl3): δ=5.87 (ddt, 3 J=17.2 Hz, 3 J=10.0 Hz, 3 J=7.2 Hz, 1H), 5.71 (d, 3 J=16.8 Hz, 1H), 5.64 (d, 3 J=10.1 Hz, 1H), 3.95 (d, 3 J=7.2 Hz, 2H), 3.35 (q, 3 J=7.3 Hz, 6H), 2.66 (s, 3H), 1.33 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=128.58, 124.03, 59.45, 52.86, 39.45, 7.61; IR (ATR, νmax, cm−1): 3004 (w), 1943 (w), 1477 (m), 1459 (m), 1403 (m), 1324 (m), 1308 (m), 1194 (vs), 1164 (s), 1038 (s), 950 (m), 807 (m), 764 (m); MS (ESI)+: m/z (%)=142 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C9H20N+ 142.1596; Found 142.1597, ▵ −0.7034; MS (ESI)−: m/z (%)=95 ([M+], 100); HRMS (ESI)− m/z: Calcd for CH3O3S− 94.9803; Found 94.9804, Δ −1.0528; IC− (min): tR=1.76.
Tetraethylammonium Methyl Sulfate (25)
Compound was obtained according to general procedure B with use of tetraethylammonium bromide (2.00 g, 9.50 mmol), dimethyl sulfate (1.20 g, 9.50 mmol) and MeCN (10 mL). The reaction gave pure product as a beige solid (2.30 g, a quantitative yield). Purity: 97.40 %.
1H NMR (500 MHz, CDCl3): δ=3.71 (s, 3H), 3.38 (q, 3 J=7.3 Hz, 6H), 1.36 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=54.28, 52.33, 7.45; IR (ATR, νmax, cm−1): 2992 (w), 2949 (w), 1462 (m), 1407 (w), 1243 (s), 1226 (vs), 1182 (s), 1059 (m), 1009 (s); MS (ESI)+: m/z (%)=130 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H20N+ 130.1596; Found 130.1594, ▵ 1.5366; MS (ESI)−: m/z (%)=111([M+], 100), 96 ([M−CH3]+, 72); HRMS (ESI)− m/z: Calcd for CH3O4S− 110.9752; Found 110.9758, Δ −5.4066; IC− (min): tR=3.30.
Tetraethylammonium Triflate (21)
Compound was obtained according to general procedure C with use of tetraethylammonium bromide (1.00 g, 4.80 mmol), methyl triflate (0.79 g, 4.80 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow solid (1.33 g, a quantitative yield). Purity: 97.85 %.
1H NMR (400 MHz, CDCl3): δ=3.24 (q, 3 J=7.3 Hz, 6H), 1.27 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=120.58 (q, 1 J CF=319.9 Hz), 52.5, 7.36; 19F NMR (376 MHz, CDCl3): δ=−78.50; IR (ATR, νmax, cm−1): 3000 (w), 2955 (w), 1703 (w), 1477 (m), 1461 (m), 1404 (m), 1261 (vs), 1224 (s), 1151 (vs), 1028 (vs), 791 (m), 754 (w); MS (ESI)+: m/z (%)=130 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H20N+ 130.1596; Found 130.1594, ▵ 1.5366; MS (ESI)−: m/z (%)=149 ([M+], 100); HRMS (ESI)− m/z: Calcd for CF3O3S− 148.9520; Found 148.9522, Δ −1.3424; IC− (min): tR=12.42.
Tetraethylammonium Tosylate (33)
Compound was obtained according to general procedure D with use of tetraethylammonium bromide (1.00 g, 4.80 mmol), methyl tosylate (0.89 g, 4.80 mmol) and MeCN (15 mL). The reaction gave pure product as a white solid (1.43 g, a quantitative yield). Purity: 97.46 %.
1H NMR (500 MHz, CDCl3): δ=7.71 (d, 3 J=8.1 Hz, 2H), 7.06 (d, 3 J=7.9 Hz, 2H), 3.28 (q, 3 J=7.3 Hz, 6H), 2.26 (s, 3H), 1.24 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=144.54, 138.88, 128.44, 125.90, 52.28, 21.22, 7.48; IR (ATR, νmax, cm−1): 3021 (w), 2997 (w), 2982 (w), 2947 (w), 2923 (w), 1595 (w), 1488 (m), 1441 (w), 1396 (m), 1365 (w), 1213 (s), 1190 (vs), 1176 (s), 1116 (s), 1101 (m), 1056 (w), 1030 (s), 1007 (s), 1001 (s), 855 (w), 821 (m), 795 (m), 713 (w); MS (ESI)+: m/z (%)=130 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H20N+ 130.1596; Found 130.1594, ▵ 1.5366; MS (ESI)−: m/z (%)=171 ([M+], 100); HRMS (ESI)− m/z: Calcd for C7H7O3S− 171.0116; Found 171.0117, Δ −0.5848; IC− (min): tR=4.83.
Tetraethylammonium Methanesulfonate (29)
Compound was obtained according to general procedure E with use of tetraethylammonium bromide (1.00 g, 4.80 mmol), methyl methanesulfonate (0.53 g, 4.80 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow solid (1.00 g, a quantitative yield). Purity: 98.33 %.
1H NMR (500 MHz, CDCl3): δ=3.38 (q, 3 J=7.3 Hz, 6H), 2.69 (s, 3H), 1.34 (t, 3 J=7.3 Hz, 9H); 13C {1H} NMR (100 MHz, CDCl3): δ=52.19, 39.28, 7.36; IR (ATR, νmax, cm−1): 2953 (s), 2925 (vs), 2854 (m), 2163 (m), 1463 (m), 1307 (s), 1234 (s), 1203 (vs), 1043 (vs), 1019 (s); MS (ESI)+: m/z (%)=130 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H20N+ 130.1596; Found 130.1594, ▵ 1.5366; MS (ESI)−: m/z (%)=95 ([M+], 100); HRMS (ESI)− m/z: Calcd for CH3O3S− 94.9803; Found 94.9809, Δ −6.33717; IC− (min): tR=1.75.
1‐Allylpyridinium Methyl Sulfate (24)
Compound was obtained according to general procedure B with use of 1‐allylpyridinium chloride (2.00 g, 12.85 mmol), dimethyl sulfate (1.62 g, 12.85 mmol) and MeCN (10 mL). The reaction gave pure product as a brown oil (2.95 g, a quantitative yield). Purity: 99.52 %
1H NMR (400 MHz, DMSO‐d6): δ=9.06 (d, 3 J=5.4 Hz, 2H), 8.65 (tt, 3 J=7.9 Hz, 2 J=1.2 Hz, 1H), 8.20 (m, 2H), 6.18 (ddt, 3 J=16.7 Hz, 3 J=10.3 Hz, 3 J=6.3 Hz, 1H), 5.44 (m, 2H), 5.30 (d, 3 J=6.3 Hz, 2H), 3.40 (s, 3H); 13C {1H} NMR (100 MHz, DMSO‐d6): δ=145.94, 144.83, 131.69, 128.28, 122.01, 62.30, 52.97; IR (ATR, νmax, cm−1): 3131 (w), 3061 (w), 2978 (w), 2946 (w), 2832 (w), 1632 (m), 1487 (m), 1449 (w), 1423 (w), 1312 (w), 1217 (vs), 1158 (m), 1057 (m), 1002 (vs), 821 (w); MS (ESI)+: m/z (%)=120 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H10N+ 120.0813; Found 120.0813, ▵ 0; MS (ESI)−: m/z (%)=111([M+], 100), 96 ([M−CH3]+, 40); HRMS (ESI)− m/z: Calcd for CH3O4S− 110.9752; Found 110.9755, Δ −2.7033; IC− (min): tR=3.31.
1‐Allylpyridinium Triflate (20)
Compound was obtained according to general procedure C with use of 1‐allylpyridinium chloride (2.00 g, 12.85 mmol), dimethyl sulfate (1.62 g, 12.85 mmol) and MeCN (10 mL). The reaction gave pure product as a brown oil (2.95 g, a quantitative yield). Purity: 98.50 %.
1H NMR (400 MHz, DMSO‐d6): δ=9.06 (d, 3 J=5.4 Hz, 2H), 8.65 (tt, 3 J=7.9 Hz, 2 J=1.2 Hz, 1H), 8.20 (m, 2H), 6.18 (ddt, 3 J=16.7 Hz, 3 J=10.3 Hz, 3 J=6.3 Hz, 1H), 5.44 (m, 2H), 5.30 (d, 3 J=6.3 Hz, 2H), 3.40 (s, 3H); 13C {1H} NMR (100 MHz, DMSO‐d6): δ=146.34, 145.24, 132.05, 128.69, 122.38, 121,37 (q, 1 J CF=321.53 Hz), 62.92; 19F NMR (376 MHz, CDCl3): δ=‐77.78; IR (ATR, νmax, cm−1): 3134 (w), 3089 (w), 3068 (w), 1633 (w), 1488 (m), 1448 (w), 1255 (vs), 1224 (s), 1152 (s), 1027 (s), 951 (w), 821 (w), 771 (w), 686 (m); MS (ESI)+: m/z (%)=120 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H10N+ 120.0813; Found 120.0813, ▵ 0; MS (ESI)−: m/z (%)=149 ([M+], 100); HRMS (ESI)− m/z: Calcd for CF3O3S− 148.9520; Found 148.9516, Δ 2.6854; IC− (min): tR=12.02.
1‐Allylpyridinium Tosylate (32)
Compound was obtained according to general procedure D with use of 1‐allylpyridinium chloride (2.00 g, 12.85 mmol), methyl tosylate (2.40 g, 12.85 mmol) and MeCN (15 mL). The reaction gave pure product as a brown solid (3.75 g, a quantitative yield). Purity: 95.51 %.
1H NMR (500 MHz, CDCl3): δ=9.20 (d, 3 J=5.7 Hz, 2H), 8.39 (t, 3 J=7.8 Hz, 1H), 7.99 (t, 3 J=7.1 Hz, 2H), 7.74 (d, 3 J=8.1 Hz, 2H), 7.14 (d, 3 J=8.0 Hz, 2H), 6.06 (ddt, 3 J=16.8 Hz, 3 J=10.0 Hz, 3 J=6.7 Hz, 1H), 5.53 (d, 3 J=17.2 Hz, 1H), 5.43 (m, 3H), 2.34 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3): δ=145.32, 145.19, 143.94, 139.36, 130.32, 128.71, 128.36, 125.77, 123.77, 63.27, 21.25; IR (ATR, νmax, cm−1): 3028 9w), 3080 (m), 3056 (m), 3032 (m), 2966 (w), 2942 (w), 2861 (w), 1646 (w), 1631 (m), 1598 (w), 1486 (s), 1444 (w), 1330 (w), 1216 (s), 1184 (vs), 1148 (m), 1120 (s), 1031 (s), 1008 (s), 941 (m), 874 (w); MS (ESI)+: m/z (%)=120 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H10N+ 120.0813; Found 120.0813, ▵ 0; MS (ESI)−: m/z (%)=171 ([M+], 100); HRMS (ESI)− m/z: Calcd for C7H7O3S− 171.0116; Found 171.0122, Δ −3.5085; IC− (min): tR=4.86.
1‐Allylpyridinium Methanesulfonate (28)
Compound was obtained according to general procedure E with use of 1‐allylpyridinium chloride (2.00 g, 12.85 mmol), methyl methanesulfonate (1.42 g, 12.85 mmol) and MeCN (10 mL). The reaction gave pure product as a yellow solid (2.70 g, a quantitative yield). Purity: 98.79 %.
1H NMR (500 MHz, CDCl3): δ=9.27 (d, 3 J=5.7 Hz, 2H), 8.50 (t, 3 J=7.9 Hz, 1H), 8.10 (t, 3 J=7.0 Hz, 2H), 6.15 (ddd, 3 J=23.5 Hz, 3 J=13.3 Hz, 3 J=6.7 Hz, 1H), 5.62 (d, 3 J=17.2 Hz, 1H), 5.51 (m, 3H), 2.75 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3): δ=145.53, 145.31, 130.40, 128.52, 123.88, 63.38, 39.65; IR (ATR, νmax, cm−1): 3127 (w), 3078 (m), 3055 (m), 3031 (m), 2933 (w), 1885 (w), 1644 (w), 1630 (m), 1487 (s), 1411 (m), 1329 (m), 1221 (m), 1186 (vs), 1146 (vs), 1037 (s), 1000 (s), 939 (s), 819 (w), 768 (m), 682 (m), 657 (w), 551 (m), 522 (m); MS (ESI)+: m/z (%)=120 ([M]+, 100); HRMS (ESI)+ m/z: Calcd for C8H10N+ 120.0813; Found 120.0813, ▵ 0; MS (ESI)−: m/z (%)=95 ([M+], 100); HRMS (ESI)− m/z: Calcd for CH3O3S− 94.9803; Found 94.9808, Δ −5.2642; IC− (min): tR=1.76.
Triethylmethylammonium Bromide (36)
Previously obtained tetraethylammonium bromide (2.00 g, 9.50 mmol) was dissolved in MeCN (10 mL), placed in a two‐necked flask and cooled down in water bath. Triethylamine (0,96 g, 9.50 mmol) was dissolved in MeCN (3 mL) and placed in a two‐necked flask with two condensers on top of each other and heated up to 80 °C. Flasks were connected by a silicon tube, which one end was immersed in triethylmine solution to allow the gas bubbling. Methyl triflate (1.56 g, 9.50 mmol) was added slowly to the solution of tetraethylammonium bromide. The reaction was carried out at these conditions for 5 h, after which MeCN was evaporated from both flasks, yielding in the first one tetraethylammonium triflate and in the second one triethylmethylammonium bromide as a white crystalline solid (1.12 g, 60 %). Purity: 94.49 %.
1H NMR (400 MHz, CDCl3): δ=3.60 (q, 3 J=7.3 Hz, 8H), 3.25 (s, 3H), 1.39 (t, 3 J=7.3 Hz, 12H); 13C {1H} NMR (100 MHz, CDCl3): δ=56.05, 47.29, 8.26; IC+ (min): tR=6.70; IC− (min): tR=7.53.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge financial support from The National Science Centre (Poland), project OPUS 8 (No. 2014/15/B/ST5/04257) entitled “Rhodium and platinum complexes with ionic liquids as ligands – new catalysts for hydrosilylation processes”.
The authors acknowledge financial support from The National Science Centre (Poland), project OPUS 10 (No. 2015/19/B/ST8/02761) entitled “Fabrication and optimization of photonic components by electron and ion beam induced polymerization of ionic liquids monomers”.
A. Szpecht, A. Zajac, D. Zielinski, H. Maciejewski, M. Smiglak, ChemistryOpen 2019, 8, 972.
Contributor Information
Prof. Hieronim Maciejewski, Email: maciejm@amu.edu.pl.
Prof. Marcin Smiglak, Email: marcin.smiglak@gmail.com.
References
- 1. Ivankovic A., Dronjic A., Martinovic Bevanda A., Talic S., Int. J. Sustain. Energ. 2017, 6, 39–48. [Google Scholar]
- 2. Wasserscheid P., Welton T., Ionic Liquids in Synthesis, Wiley-VCH, Stuttgart, 2002, pp. 1–19. [Google Scholar]
- 3. Wilkes J. S., J. Mol. Catal. A 2004, 214, 11–17. [Google Scholar]
- 4. Earle M. J., Seddon K. R., Pure Appl. Chem. 2000, 72, 1391–1398. [Google Scholar]
- 5. Seddon K. R., Kinet. Catal. 1996, 37, 693–697. [Google Scholar]
- 6. Hardacre C., Parvulescu V., Catalysis in Ionic Liquids: From Catalyst Synthesis to Application, Royal Society of Chemistry, Cambridge: 2014, pp. 44–308. [Google Scholar]
- 7. Kukawka R., Pawlowska-Zygarowicz A., Dzialkowska J., Pietrowski M., Maciejewski H., Bica K., Smiglak M., ACS Sustainable Chem. Eng. 2019, 7, 4699–4706. [Google Scholar]
- 8. Kukawka R., Januszewski R., Kownacki I., Smiglak M., Maciejewski H., Catal. Commun. 2018, 108, 59–63. [Google Scholar]
- 9. Kukawka R., Pawlowska-Zygarowicz A., Dutkiewicz M., Maciejewski H., Smiglak M., RSC Adv. 2016, 6, 61860–61868. [Google Scholar]
- 10. Gebicki J., Kloskowski A., Chrzanowski W., Stepnowski P., Namiesnik J., Crit. Rev. Anal. Chem. 2015, 46, 122–138. [DOI] [PubMed] [Google Scholar]
- 11. Zajac A., Kukawka R., Pawlowska-Zygarowicz A., Stolarska O., Smiglak M., Green Chem. 2018, 20, 4764–4789. [Google Scholar]
- 12. Yang Q., Zhang Z., Sun X. G., Hu Y. S., Xing H., Dai S., Chem. Soc. Rev. 2018, 47, 2020–2064. [DOI] [PubMed] [Google Scholar]
- 13. Basile A., Bhatt A. I., O'Mullane A. P., Nat. Commun. 2016, 7, ncomms11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hashmi S. G., Ozkan M., Halme J., Misic K. D., Zakeeruddin S. M., Paltakari J., Gratzel M., Lund P. D., Nano Energy 2015, 17, 206–215. [Google Scholar]
- 15. Watanabe M., Thomas M. L., Zhang S., Ueno K., Yasuda T., Dokko K., Chem. Rev. 2017, 117, 7190–7239. [DOI] [PubMed] [Google Scholar]
- 16. Zhou Y., Qu J., ACS Appl. Mater. Interfaces 2017, 9, 3209–3222. [DOI] [PubMed] [Google Scholar]
- 17. Dai Z., Noble R. D., Gin D. L., Zhang X., Deng L., J. Membr. Sci. 2019, 497, 1–20. [Google Scholar]
- 18. Choi D. Y., Kim M. H., Oh Y. S., Jung S.-H., Sung H. J., Lee H. W., Lee H. M., ACS Appl. Mater. Interfaces 2017, 9, 1770–1780. [DOI] [PubMed] [Google Scholar]
- 19. Williams H. D., Sahbaz Y., Ford L., Nguyen T. H., Scammells P. J., Porter C. J. H., Chem. Commun. 2014, 50, 1688–1690. [DOI] [PubMed] [Google Scholar]
- 20. George A., Brandt A., Tran K., Zahari S. M. S. Nizan S., Klein-Marcuschamer D., Sun N., Sathitsuksanoch N., Shi J., Stavila V., Parthasarathi R., Singh S., Holmes B. M., Welton T., Simmons B. A., Hallett J. P., Green Chem. 2015, 17, 1728–1734. [Google Scholar]
- 21. Endres F., Abbott A., MacFarlane D., Electrodeposition from Ionic Liquids, Wiley-VCH, Stuttgart: 2017, pp. 17–53. [Google Scholar]
- 22. Srour H., Rouault H., Santini C. C., Chauvin Y., Green Chem. 2013, 15, 1341–1347. [Google Scholar]
- 23. Bonhôte P., Dias A. P., Papageorgiou N., Kalyanasundaram K., Grätzel M., Inorg. Chem. 1996, 35, 1168. [DOI] [PubMed] [Google Scholar]
- 24. Huddleston J. G., Willauer H. D., Swatlowski R. P., Visser A. E., Rogers R. D., Chem. Commun. 1998, 16, 1765. [Google Scholar]
- 25. Seddon K. R., Stark A., Torres M. J., Pure Appl. Chem. 2000, 72, 2275–2287. [Google Scholar]
- 26. Dimitrijevic A., Trtic-Petrovic T., Vranes M., Papovic S., Tot A., Dozic S., Gadzuric S., J. Chem. Eng. Data 2016, 61, 549–555. [Google Scholar]
- 27. Wolfenden R., Yuan Y., P. Natl.Acad. Sci. USA 2007, 104, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R. S. Kalb, W. Wesner, R. Hermann, M. Kotschan, M. Schelch, W. Staber, WO2005021484, 2005, Proionic Production of Ionic Substances & Cokg GmbH, Grambach, Austria.
- 29.R. S. Kalb, WO2008052860, 2008, Proionic Production of Ionic Substances & Cokg GmbH, Grambach, Austria.
- 30.R. S. Kalb, WO2008052863, 2008, Proionic Production of Ionic Substances & Cokg GmbH, Grambach, Austria.
- 31.R. S. Kalb, WO 2008052861, 2008, Proionic Production of Ionic Substances & Cokg GmbH, Grambach, Austria.
- 32.N. Ignat'ev, U. Welz-Biermann, A. I. Kucheryna, H. Willner, WO 2006/063656 A1, 2006, Merck Patent GmbH, Darmstadt, Germany.
- 33. Howells R. D., Cown J. D. Mc, Chem. Rev. 1975, 77, 69–92. [Google Scholar]
- 34. Ignat'ev N., Barthen P., Kucheryna A., Willner H., Sartori P., Molecules 2012, 17, 5319–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rogers E. I., Ijukic B. S., Hardacre C., Compton R. G., J. Chem. Eng. Data 2009, 54, 2049–2053. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary