

Abstract
Colchicine binding site inhibitors (CBSIs) hold great potential in developing new generations of antimitotic drugs. Unlike existing tubulin inhibitors such as paclitaxel, they are generally much less susceptible to resistance caused by the overexpression of drug efflux pumps. The 3,4,5-trimethoxyphenyl (TMP) moiety is a critical component present in many CBSIs, playing an important role in maintaining suitable molecular conformations of CBSIs and contributing to their high binding affinities to tubulin. Previously reported modifications to the TMP moiety in a variety of scaffolds of CBSIs have usually resulted in reduced antiproliferative potency. We previously reported a potent CBSI, VERU-111, that also contains the TMP moiety. Herein, we report the discovery of a VERU-111 analogue 13f that is significantly more potent than VERU-111. The X-ray crystal structure of 13f in complex with tubulin confirms its direct binding to the colchicine site. In addition, 13f exhibited a strong inhibitory effect on tumor growth in vivo.
Graphical Abstract
INTRODUCTION
Microtubule-targeting agents (MTAs) such as paclitaxel interrupt the cell cycle, result in mitotic arrest at the metaphase/anaphase transition, and eventually induce apoptosis of cancer cells.1,2 Although MTAs have achieved great success in cancer treatment, their clinical efficacy is often limited by the development of drug resistance.3 Therefore, MTAs that can overcome multidrug resistance (MDR) are highly desirable for the treatment of resistant phenotypes.
Currently, all FDA approved tubulin inhibitors for cancer treatment bind to either the paclitaxel site or the vinca alkaloids site. Colchicine binding site inhibitors (CBSIs) destabilize microtubules and have shown significant ability to overcome clinically relevant MDR mechanisms.4–6 A number of CBSIs also effectively circumvent drug resistance resulting from the overexpression of the β-III tubulin isoform in cancer cells.7 Therefore, CBSIs hold great potential as a new generation of tubulin inhibitors.
3,4,5-Trimethoxyphenyl (TMP) is a common moiety shared by many CBSIs, as shown in Figure 1. This TMP moiety is crucial for maintaining suitable molecular conformations that are needed for optimal interactions with tubulin8 and for producing the maximum antiproliferative activities. Attempts to modify this TMP moiety usually lead to significantly reduced anticancer potency.4,9–11 For example, substituting the methoxy with a bulky group or demethylating the methoxy on the TMP moiety of colchicine were reported to reduce the potency.12,13 In addition, removing or adding methoxy to TMP significantly impaired the antimitotic activity (>10-fold).14 However, some isosteric modifications of the TMP in colchicine or Combretastatin A4 (CA-4) have been shown to maintain the potency to that of parent natural products colchicine or CA-4. For instance, Semenov et al. have shown that isosteric replacement of the TMP of CA-4 with [1.3] dioxole or [1,4]dioxane did not significantly reduce antimitotic activity when compared to CA-4.14 Cornigerine, a [1,3] dioxole analogue of colchicine, displayed equipotent antimitotic activity to that of the parent compound.15
Figure 1.

Examples of CBSIs with TMP moieties shown in blue.
We have previously reported the discovery of a 2-aryl-4-benzoyl-imidazole (ABI) scaffold as a new class of CBSI.16 Subsequent structural optimization of the benzoyl moiety, aryl moiety, and imidazole fragment in this scaffold resulted in analogues with improved potency.17–20 VERU-111 (Figure 1) is the best analogue from that series of structural optimizations. It has an average IC50 value of 5.2 nM against a panel of cancer cell lines, is not a substrate of P-glycoprotein (P-gp), and effectively inhibits tumor growth in paclitaxel-resistant models.18 However, consistent with the structure–activity relationships for many CBSIs, our previous attempts to modify the TMP moiety in the ABI scaffold have all led to significantly reduced activity.
On the basis of our recently reported high-resolution crystal structure of DJ101 (Figure 1),6 a close analogue of VERU-111, we discovered that (1) only one methoxy of the TMP moiety in DJ101 was involved in the formation of a hydrogen bond interaction with the β-Cys241 residue of tubulin, and (2) there is very limited space around the TMP moiety to accommodate any larger moieties.6 We hypothesized that two of the three methoxys of the TMP moiety could be optimized to improve antiproliferative activity without damaging the interactions to tubulin. To test this hypothesis, we carried out a focused SAR investigation of VERU-111 by modifying the TMP moiety by specifically linking two adjacent methoxy moieties into a conformationally restricted ring system. Among the eight VERU-111 analogues synthesized, 13f contains a unique 3- methoxybenzo[4,5]-dioxene moiety and exhibits the greatest improvement of antiproliferative activity against a panel of melanoma cell lines with IC50 values ranging from 1.1 to 3.3 nM compared with those of VERU-111 (5.6–8.1 nM). We solved the crystal structure of 13f in complex with tubulin to confirm its direct binding to the colchicine site and to understand the structural basis of its potency.
CHEMISTRY
Scheme 1 shows the synthetic method we followed to access the commercially expensive or unavailable benzoyl chlorides. In brief, commercially available methyl 3-methoxy-4,5-dihydroxybenzoate was treated with dibromomethane or 1,3-dibromopropane to form 1a and 1b in the presence of potassium carbonate. The cyclized benzoates were then hydrolyzed under basic conditions to provide carboxylic acids 2a and 2b; 2a and 2b were subsequently refluxed with thionyl chloride in DCM to generate benzoyl chlorides 3a and 3b, which were used directly for next step without purification.
Scheme 1. Synthesis of Benzoyl Chlorides 3a and 3ba.

aReagents and conditions: (a) dibromomethane or 1,3-dibromopropane, K2CO3, acetonitrile, reflux; (b) LiOH, dioxane-H2O (2:1), 50 °C; (c) SOCl2, DCM, reflux.
Another commercially unavailable benzoyl chloride 8 was prepared following Scheme 2. Methyl 3-methoxy-4,5-dihydroxybenzoate was refluxed together with allyl bromide in the presence of potassium carbonate to generate alkylated methyl benzoate 4. An isomerization/ring closing metathesis strategy as reported in the literature21 was followed to furnish the dioxene moiety in 6. In brief, 4 was treated with a catalytic equivalent of (Ph3P)3Ru(CO)(Cl)H in toluene to provide 5, which was subjected to Grubbs reaction to afford 6. Hydrolysis of the methyl ester under basic conditions provided 7, which was converted to benzoyl chloride 8 by refluxing with thionyl chloride.
Scheme 2. Synthesis of the Benzoyl Chloride 8a.

aReagents and conditions: (a) allyl bromide, K2CO3, acetonitrile, reflux; (b) (Ph3P)3Ru(CO)(Cl)H, toluene, reflux; (c) Grubbs catalyst second generation, toluene, reflux; (d) LiOH, dioxane-H2O (2:1), 50 °C; (e) SOCl2, DCM, reflux.
ABI structure was constructed through a Suzuki coupling/Grignard reaction strategy as depicted in Scheme 3. In short, 2-(trimethylsilyl)ethoxymethyl (SEM)-protected compound 9 was obtained from the treatment of 2,4,5-tribromoimidazole with SEMCl in the presence of sodium hydride. Compound 9 coupled with 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester in the presence of Pd2(dba)3 and 2-dicyclohexylphosphi- no-2′,4′,6′-triisopropylbiphenyl to provide 10 in 34% yield. It is worth mentioning that the Suzuki coupling reaction of this tribromo substrate under current conditions was not regioselective. Coupling reactions happening on the 4- or 5-bromo were also observed. Compound 10 was treated with i-PrMgCl(LiCl) and benzoyl chlorides to furnish compounds 11a-f in 31–48% yields. Bromo on the imidazole moiety and benzenesulfonyl were simultaneously removed in the presence of Pd(OAc)2, PPh3, and K2CO3 in n-BuOH under reflux conditions, giving 12a-f in 76–89% yields. Deprotection of SEM using TFA in dichloromethane finally provided VERU- 111 analogues 13a-f in 79–92% yields.
Scheme 3. Synthesis of the VERU-111 Analogues 13a-fa.

aReagents and conditions: (a) SEMCl, NaH, THF, 0 °C to rt; (b) Pd2(dba)3, 2-dicydohexylphosphmo-2′,4′,6′-triisopropylbiphenyl, Na2CO3, toluene-MeOH-H2O (20:4:1), reflux; (c) benzoyl chlorides, i-PrMgCl(LiCl), THF, rt to reflux; (d) Pd(OAc)2, K2CO3, PPh3, n-BuOH, reflux; (e) TFA, DCM, rt.
In a parallel SAR investigation focusing on modification of the indole moiety of VERU-111 (manuscript in preparation), we demonstrate that replacing the 3-indolyl with either a 4-indolyl or 4-methyl-3-indolyl moiety greatly improved the antiproliferative activity. Thus, we herein also produced analogues 19 and 25 to determine the combinational effect of the unique 3-methoxybenzo[4,5]-dioxene moiety with the 4-indolyl or 4-methyl-3-indolyl moiety. Analogue 19 was synthesized according to Scheme 4. To access 19, dibromo species 16 was generated beforehand. In brief, commercially available 4-bromoindole was treated with benzenesulfonyl chloride in the presence of sodium hydride to provide 14 in 93% yield; 14 was then subjected to Miyaura borylation, which was catalyzed by Pd(pddf)2Cl2-CH2Cl2 to generate 15 in 86% yield. Boronic ester 15 was subsequently subjected to Suzuki coupling in the presence of Pd2(dba)3 and 2-dicyclohex- ylphosphino-2′,4′,6′-triisopropylbiphenyl to produce dibromo intermediate 16 in 31% yield. With 16 in hand, it was then treated with 8 in the presence of i-PrMgCl(LiCl) to form 17, which was subjected to deprotection to eventually yield analogue 19 in 67% yield over two steps. To synthesize analogue 25, we followed the synthetic method as shown in Scheme 5. Imidazole intermediate 21 was prepared by following a reported procedure22 that involved imidazole formation (in the presence of ammonium hydroxide and glyoxal); 21 was subjected to bromination (in the presence of N-bromosuccinimide) and subsequently treated with SEMCl and sodium hydride to afford intermediate 22 in 70% yield over two steps. Compound 22 was then subjected to Grignard reaction in the presence of i-PrMgCl(LiCl) and 8 to provide compound 23 in 37% yield. Treatment of 23 with Pd(OAc)2, K2CO3, and PPh3 under reflux conditions followed by TFA successfully afforded analogue 25 in 69% over two steps.
Scheme 4. Synthesis of 19a.

aReagents and conditions: (a) PhSO2Cl, NaH, THF, 0 °C to rt; (b) bis(pinacolato)diboron, Pd(dppf)2.CH2Cl2, KOAc, dioxane, 80 °C; (c) Pd2(dba)3, 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl, Na2CO3, toluene-MeOH-H2O (20:4:1), reflux; (d) 8, i-PrMgCl(LiCl), THF, rt to reflux; (e) Pd(OAc)2, K2CO3, PPh3, n-BuOH, reflux; (f) TFA, DCM, rt.
Scheme 5. Synthesis of 25a.

aReagents and conditions: (a) PhSO2Cl, NaH, THF, 0 °C to rt; (b) NH4OH, glyoxal, ethanol, reflux; (c) i) NBS, THF, 0 °C to rt; ii) SEMCl, NaH, THF; (d) 8, i-PrMgCl(LiCl), THF, rt to reflux; (e) Pd(OAc)2, K2CO3, PPh3, n-BuOH, reflux; (f) TFA, DCM, rt.
RESULTS AND DISCUSSION
VERU-111 is a clinical candidate initiated by VERU Healthcare to develop as third line hormonal therapy, but it also exhibits potent antiproliferative activities against melanoma cell lines and is currently being actively evaluated in many other cancer types both in vitro and in vivo, such as for prostate, pancreatic, and breast cancer (unpublished data). To compare the activity of VERU-111 that was reported previously22 to those of our new analogues, their antiproliferative effects were evaluated in three human melanoma cell lines (A375, M14, and RPMI7951). Colchicine was used as a positive control (n = 4). The in vitro cell viability following 72 h exposure to the analogues is shown in Table 1.
Table 1.
In Vitro Growth Inhibitory Effects (nM) of VERU-111 Analogues Modifying the TMP Moiety
| ||||
|---|---|---|---|---|
| compound | R | A375 | M14 | RPMI7951 |
| 13a |
|
158.7 ± 16.4 | 118.8 ± 14.3 | 213.7 ± 17.1 |
| 13b |
|
190.6 ± 16.8 | 154.6 ± 10.4 | 235.6 ± 19.3 |
| 13c |
|
21.0 ± 1.6 | 11.3 ± 0.8 | 29.2 ± 1.7 |
| 13d |
|
3.5 ± 0.4 | 5.6 ± 0.6 | 5.6 ± 0.5 |
| 13e |
|
32.2 ± 3.4 | 38.2 ± 3.5 | 47.7 ± 3.9 |
| 13f |
|
1.1 ± 0.1 | 1.2 ± 0.2 | 3.3 ± 0.3 |
| 19 |
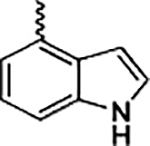
|
17.1 ± 1.1 | 13.8 ± 0.9 | 34.8 ± 2.2 |
| 25 |
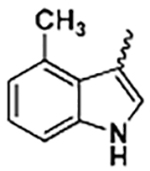
|
6.1 ± 0.2 | 6.1 ± 0.2 | 8.8 ± 0.5 |
| VERU-111 | 8.1 ± 1.6 | 5.6 ± 0.9 | 7.2 ± 0.9 | |
Benzo[4,5]-dioxane analogue 13a lacks the 3-methoxy present in the TMP of VERU-111. It exhibited remarkable loss of cytotoxicity (IC50 ranges from 119 to 218 nM). Ring contraction of the six-member benzo[4,5]-dioxane ring to the five-member benzo[4,5]-dioxole ring resulted in analogue 13b, which showed comparable antiproliferative activity to that of 13a. In contrast to 13a, 3-methoxybenzo[4,5]-dioxane analogue 13c increased the antiproliferative potency and had IC50 values ranging from 11.3 to 29.2 nM. This is consistent with our previous SAR, where we discovered that removing one or more of the methoxy groups in the TMP moiety negatively affects the antiproliferative potency for this scaffold. Similarly, introducing the 3-methoxy to benzo-[4,5]-dioxole analogue 13b led to the formation of 13d, which restored the potency (IC50 ranges from 3.5 to 5.6 nM, 30-fold more active than 13b). Interestingly, this five-membered ring analogue was ~4-fold more potent than the six-membered ring analogue 13c. Thus, although the 3-methoxy was crucial for the activities of VERU-111 analogues, the size of the cyclic rings on the phenyl moiety also played an important role (13c vs 13d). Consistent with this finding, further increasing the ring size to a seven-membered ring resulted in 3-methoxybenzo[4,5]dioxepin analogue 13e, which had significantly decreased potency (IC50 ranging from 32.2 to 47.7 nM). By comparing 13c, 13d, and 13e, it was revealed that the activity for the size of this saturated ring is 5 > 6 > 7 (Table 1).
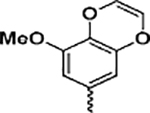
We next introduced an unsaturation to this ring by synthesizing 13f, a unique 3-methoxybenzo[4,5]-dioxene analogue. Interestingly, 13f (IC50 values ranging from 1.1 to 3.3 nM) exhibited the most potent antiproliferative activity among the analogues solely modifying the TMP moiety (13a–f, Table 1). To determine the combination modifications to both the TMP and indole moieties on 13f, we further produced analogues 19 and 25. Although the 4-indolyl analogue 19 exhibited ~ 11-fold reduced antiproliferative activity when compared to that of 13f, the 4-methyl-3-indolyl analogue 25 (IC50 values ranging from 6.1 to 8.8 nM) was only slightly less potent than 13f. We also expanded our assessment of 13f into prostate cancer cell lines and taxane-resistant prostate cancer cell lines to determine the efficacy in another cell type, and the result is shown in Figure S1. Other colchicine binding site inhibitors are reportedly less susceptible to taxane-related drug resistance, and we therefore are keen to determine if 13f would still be efficacious in taxane-resistant models.23–25 Compound 13f was tested against paclitaxel in PC-3, paclitaxel-resistant PC-3/TxR, DU145, and docetaxel-resistant DU145/TxR prostate cancer cell lines. Paclitaxel was more potent in the parental PC-3 cell lines with an IC50 of 1.1 ± 0.2 nM compared to 15.2 ± 1.3 nM for 13f, which is not surprising given paclitaxel’s potency and clinical success. However, in resistant PC-3/TxR cell lines, paclitaxel had a resistance index of 103.5 and an IC50 of 113.9 ± 4.3 nM, whereas 13f was more potent and demonstrated an IC50 of 7.6 ± 0.5 nM and resistance index of 0.5. Although paclitaxel achieved significant potency in the sensitive DU145 cell line (1.5 ± 0.2 nM), no effect was observed at all in the DU145/TxR cells at concentrations up to 1 μM. In contrast, 13f demonstrated IC50s of 42.2 ± 3.6 and 81.5 ± 11.6 nM against DU145 and DU145TxR cells, respectively, and had a resistance index of only 1.9. These data support the development of this scaffold as an alternative treatment for cancers resistant to taxane treatment. Overall, our SAR results show that the TMP moiety in VERU-111 can be modified without negatively impacting antiproliferative activities.
To confirm that 13f maintains its mechanism of action as a tubulin polymerization inhibitor, we performed a tubulin polymerization assay (Figure 2). The vehicle control caused an extensive increase in tubulin polymerization, giving a Vmax = 27. Colchicine, which was used as a positive control, and 13f both inhibited tubulin polymerization with calculated Vmax values of 0.5 and 0, respectively. These results confirm that 13f maintains its mode of action by inhibiting tubulin assembly and interfering with tubulin dynamics.
Figure 2.

Inhibition of tubulin polymerization with polymerization of purified tubulin in a cell-free assay. Tubulin (3.33 mg/mL) was exposed to vehicle control (n = 2), 10 μM of 13f, or 10 μM of colchicine. Absorbance at 340 nm was monitored at 37 °C every minute for 60 min.
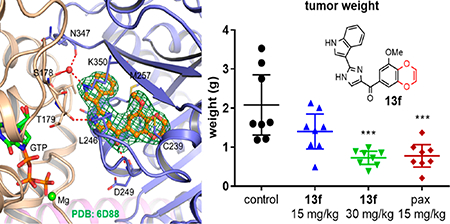
To understand the molecular basis of the strong interaction of 13f with tubulin, we determined the crystal structure of the T2R-TTL (consisting of α/β-tubulin, the stathmin-like domain of RB3, and tubulin tyrosine ligase)26,27 in complex with 13f at a resolution of 2.85 Å (PDB ID: 6D88). Details of the X-ray diffraction data collection and structure refinement statistics are provided in Table 2. As expected, 13f occupies the colchicine binding site at the interface of the α- and β-tubulin, mostly confined in a deep pocket in β-tubulin opposite the GTP molecule that is bound in a pocket in the α-tubulin (Figure 3A and Figure S2). There are two α/β-tubulin heterodimers in this complex, and both interfaces are occupied by the small molecule. In both heterodimers (chain A/B and C/D), 13f forms virtually identical interactions. First, it forms three hydrogen bonds with the surrounding amino acids: an imidazole NH to carbonyl of Thr179 (α-tubulin), an indole NH to Ser178 (α-tubulin) and Asn347 (β-tubulin) via a water molecule, and a carbonyl linker to Asp249 (β-tubulin) (Figure 3B). Second, the 3-methoxybenzo[4,5]-dioxene moiety is stacked between Cys239 and Leu253 from β-tubulin, the latter making a pi-H interaction with the ring. Finally, the imidazole ring is surrounded by Leu246 and Asn256 from β-tubulin while the indole ring is surrounded by both Lys350 and Met257 from β-tubulin and Val181 from α-tubulin. Colchicine targets the β subunit and keeps the tubulin from adopting a straight conformation, thus inhibiting microtubule assembly; 13f also blocks the curved-to-straight conformational change of tubulin by steric clashes with surrounding secondary structure elements (Figure 3C) and therefore shares the same mechanism of action as that of colchicine.
Table 2.
X-Ray Data Collection and Refinement Statistics for Tubulin-RB3_SLD-TTL (T2R-TTL) Complex Bound with 13f (PDB ID: 6D88)a
| data collection | |
|---|---|
| space group | P212121 |
| Cell Dimensions | |
| a, b, c (Å) | 105.54, 157.89, 182.03 |
| α, β, γ (deg) | 90, 90, 90 |
| resolution (Å) | 50–2.85 (2.92–2.85) |
| Rmeas, | 0.246 (0.90) |
| I/σ(I) | 7.9 (2.0) |
| completeness (%) | 100.0 (100.0) |
| redundancy | 6.8 (7.1) |
| Refinement | |
| resolution (Å) | 50–2.85 (2.92–2.85) |
| no. reflections | 69437 (5895) |
| Rwork/Rfree | 0.1919/0.2461 |
| no. atoms | 17530 |
| protein | 17202 |
| ligand/ion | 236 |
| water | 92 |
| B Factors | |
| protein | 59.26 |
| ligand/ion | 57.44 |
| water | 43.29 |
| R.M.S. Deviations | |
| bond lengths (Å) | 0.007 |
| bond angles (deg) | 1.19 |
| Ramachandran Plot | |
| favored (%) | 96.98 |
| allowed (%) | 2.88 |
| outliers (%) | 0.14 |
Values in parentheses are for the highest-resolution shell.
Figure 3.

T2R-TTL in complex with 13f. (A) Surface representation of the complex. Various components are marked with arrows. (B) Close-up view of first α/β -tubulin heterodimer (chain A and B) interface occupied with 13f. The compound is shown in ball and stick model with orange carbons. Water and Mg are shown in ball model with GTP in thick sticks and amino acids in thin sticks. 2Fo-Fc map of the compound drawn at 1.0 σ is shown as green mesh. (C) Interference of 13f with the tubulin straight conformation. The close-up view of superimposition of the tubulin—13f complex (blue ribbons—orange carbon balls, PDB ID: 6D88) and tubulin as found in straight protofilaments (gray ribbons, PDB ID: IJFF) shows that 13f binding is not compatible with the straight conformation. Conformational changes of the secondary elements in both subunits upon binding of 13f are labeled.
Prior to the in vivo study, the in vitro metabolic stabilities of analogues 13a–f were examined by measuring their half-lives upon incubation with mouse, rat, and human liver microsomes in the presence of an NADPH regeneration system. The results are summarized in Table 3. Overall, 13f showed the highest stability in rat liver microsomes and also exhibited satisfying stabilities against mouse and human liver microsomes. Because it is also the most potent analogue, we selected 13f for in vivo efficacy evaluation in an A375 melanoma xenograft model in nude mice. After tumors were established, mice were randomized and treated every other day for 15 days by i.p. injection with 15 mg/kg of 13f, 30 mg/kg of 13f, 15 mg/kg of paclitaxel, or a vehicle control solution (Figure 4A). All drug-treated groups caused a significant decrease in tumor growth based on both tumor volume and tumor weight (Figure 4B and C). The average tumor growth inhibitions (TGIs) for the groups treated with 15 and 30 mg/kg of 13f were 54.8 and 73.9%, respectively. These results were similar to the paclitaxel-treated group, which was used as a positive control and had an average TGI of 70.7%. The inhibitory action of 13f was also demonstrated by the reduction in tumor weight, where the tumors of the 30 mg/kg group weighed 65.0% less than the control group and the paclitaxel group averaged 62.3% less (Figure 4C). Mouse weights and activities were monitored to assess for apparent acute toxicities, and significant deviations of animal weight were not observed (Figure 4D). Therefore, we conclude that 13f is effective at inhibiting tumor growth in this xenograft model without causing apparent toxicity at doses up to 30 mg/kg.
Table 3.
In Vitro Microsomal Stabilities of Compounds 13a–f
| metabolic stability (mouse) |
metabolic stability (rat) |
metabolic stability (human) |
||||
|---|---|---|---|---|---|---|
| compound | t1/2 (h) | clint (ml min−1 kg−1) | t1/2 (h) | clint (ml min−1 kg−1) | t1/2 (h) | clint (ml min−1 kg−1) |
| Verapamil | 0.94 ± 0.06 | 60.66 | 1.29 ± 0.09 | 36.18 | 1.68 ± 0.16 | 12.41 |
| VERU-111 | 3.76 ± 0.24 | 15.20 | 6.74 ± 0.59 | 6.94 | 5.13 ± 0.26 | 4.05 |
| 13a | 0.37 ± 0.01 | 154.12 | 0.85 ± 0.01 | 54.94 | 3.50 ± 0.10 | 5.95 |
| 13b | 2.28 ± 0.19 | 25.10 | 2.55 ± 0.18 | 18.3 | 3.77 ± 0.34 | 5.50 |
| 13c | 2.03 ± 0.05 | 28.19 | 0.90 ± 0.02 | 51.92 | 2.13 ± 0.06 | 9.76 |
| 13d | 0.82 ± 0.03 | 69.56 | 0.18 ± 0.01 | 253.35 | 1.28 ± 0.03 | 16.26 |
| 13e | 2.48 ± 0.22 | 23.00 | 2.95 ± 0.15 | 15.90 | 0.71 ± 0.02 | 29.20 |
| 13f | 1.42 ± 0.14 | 40.30 | 3.57 ± 0.43 | 13.10 | 2.69 ± 0.26 | 7.70 |
Figure 4.

Compound 13f inhibits tumor growth in vivo. (A) A375 xenograft model in nude mice. The graph represents mean tumor volume ± SEM (n = 8). (B) Individual tumor final volumes ± SEM. (C) Tumor weights ± 95% CI. Statistical significance for tumor volume and weight was determined by one-way ANOVA analysis followed by Dunnett’s multiple comparison test for the treatment group compared with the corresponding results of the control group. (D) Mouse body weights are represented as a percent of weight change compared to initial weight ± SEM (***p < 0.001, ****p < 0.0001).
CONCLUSION
Modifying the TMP moiety of CBSIs is usually unsuccessful for improving antiproliferative activities. On the basis of our recently discovered tubulin inhibitor VERU-111 and the crystal structure of its close analogue DJ101, we described the synthesis of eight new VERU-111 analogues and their SAR evaluation by focusing on the TMP modification. Our results showed that isosteric (conformationally restricted) replacement of the TMP is feasible to significantly increase the antiproliferative activities in this scaffold. We identified the analogue 13f, which contains a unique 3-methoxybenzo[4,5]-dioxene moiety, that demonstrated more potent antiproliferative activity than VERU-111 and achieved IC50 values ranging from 1.1 to 3.3 nM in melanoma cell lines. Although this improvement in antiproliferative activity is not dramatic, these results clearly demonstrate the feasibility of modifying this TMP moiety. In addition, compared to paclitaxel, 13f had a significantly improved resistance index in parental and taxane-resistance prostate cancer cell lines, suggesting its potential against drug-resistant phenotypes. Crystallographic analysis revealed that the interactions between 13f and tubulin is centered on a hydrogen-bonding network, which provides potential avenues for future modifications to improve potency. Compound 13f maintained the same mechanism of action as its prototype VERU-111 as an inhibitor of tubulin polymerization. Moreover, 13f significantly inhibited tumor growth in vivo without observable toxicity in a mouse melanoma xenograft model. Overall, our study provides a successful example of modifying the TMP moiety of CBSIs in this scaffold to enhance antiproliferative activity while not affecting the mechanistic and safety profile.
EXPERIMENTAL SECTION
General Chemistry
Tetrahydrofuran was distilled from sodium benzophenone. All other solvents and chemical reagents were obtained from commercial sources and used directly without further purification. Glassware was oven-dried before use. All reactions were performed under an argon atmosphere. TLC was performed on silica gel 60 GF254 and monitored under UV light or visualized using phosphomolybdic acid reagent. Flash chromatography was performed on 230–400 mesh silica gel (Fisher Scientific). Melting points were recorded on an MPA100 Automated Melting Point Apparatus. NMR spectra were obtained on a Bruker Ascend 400 (Billerica, MA) spectrometer or a Varian Inova-500 spectrometer (Agilent Technologies, Santa Clara, CA). HR-MS were obtained on Waters Acquity UPLC linked to a Waters Acquity Photodiode Array Detector and Waters qTof mass detector. All compounds reported herein with biological data had purities ≥95% as determined by HPLC. The purity of associated compounds was verified by the HPLC study performed on BEH C18 (2.1 × 50 mm, 1.7 μm) column using a mixture of solvent acetonitrile/water (with 0.1% formic acid) at a flow rate of 0.3 mL/min and monitoring by UV absorption at the appropriate wavelength. Chemical shifts are given in ppm with tetramethylsilane (TMS) as an internal reference. All coupling constants (J) are given in hertz (Hz).
General Procedure A for the Synthesis of Alkylated Methyl Benzoate 1a, 1b, and 4
To a solution of methyl 3-methoxy-4,5-dihydroxybenzoate in acetonitrile were added potassium carbonate and the appropriate bromide. The mixture was refluxed overnight and then cooled to room temperature; the precipitate was filtered off and washed with dichloromethane. The combined filtration was evaporated under vacuum to give the oily crude product, which was further purified with flash chromatography on silica. Elution with hexane/ethyl acetate (5:1–2:1) gave 1a, 1b, and 4.
General Procedure B for the Synthesis of Benzoic Acids 2a, 2b, and 7
To a solution of the methyl benzoate 1a, 1b, or 6 (1.0 mmol) in dioxane (5 mL) was added a solution of lithium hydroxide (1.5 mmol) in water (3 mL). The mixture was stirred at 50 °C until TLC indicated completion of the reaction. The solvents were removed under reduced pressure; the resulting residue was partitioned between water (10 mL) and DCM (10 mL), and the pH value was adjusted to 5 using 1 M HCl solution. The organic solvents were then combined, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:1) gave 2a, 2b, and 7.
General Procedure C for the Synthesis of Benzoyl Chloride 3a, 3b, and 8
To a solution of benzoic acid 2a, 2b, or 7 (0.8 mmol) in DCM (5 mL) was added thionyl chloride (1.5 mL). The mixture was stirred at 50 °C for 3 h. The solvents were then removed under reduced pressure, and the corresponding crude benzoyl chloride was used directly for the next step.
General Procedure D for the Synthesis of 11a–f, 17, and 23
To a stirred solution of compound 10, 16, or 21 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h, and benzoyl chloride (1.3 mmol) was added in anhydrous THF (1 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11a–11f, 17, and 22.
General Procedure E for the Synthesis of 12a–12f, 18, and 24
To a suspension of 11a–f, 17, or 23 (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12a–12f, 18, and 24.
General Procedure F for the Synthesis of 13a–f, 19, and 25
To a solution of 12a–12f, 18, or 24 in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13a-13f, 19, and 25.
Methyl 7-Methoxybenzo[d][1,3]dioxole-5-carboxylate (1a)
Following general procedure A, to a solution of methyl 3-methoxy-4,5-dihydroxybenzoate (2.5 mmol) in acetonitrile (10 mL) was added potassium carbonate (6.0 mmol) and dibromomethane (2.75 mmol). The mixture was refluxed overnight and then cooled to room temperature; the precipitate was filtered off and washed with dichloromethane. The combined filtration was evaporated under vacuum to give the oily crude, which was further purified with flash chromatography on silica. Elution with hexane/ethyl acetate (5:1–2:1) gave 1a as colorless oil in 42% yield. 1H NMR (400 MHz, chloroform-d) δ 7.32 (d, J = 1.4 Hz, 1H), 7.19 (d, J = 1.4 Hz, 1H), 6.04 (s, 2H), 3.93 (s, 3H), 3.87 (s, 3H). HRMS: calcd for C10H11O5 [M + H]+ 211.0606, found 211.0607.
Methyl 9-Methoxy-3,4-dihydro-2H-benzo[b][1,4]dioxepine-7-carboxylate (1b)
Following general procedure B, to a solution of methyl 3-methoxy-4,5-dihydroxybenzoate (2.5 mmol) in acetonitrile (10 mL) were added potassium carbonate (6.0 mmol) and 1,3-dibromopropane (2.75 mmol). The mixture was refluxed overnight and then cooled to room temperature; the precipitate was filtered off and washed with dichloromethane. The combined filtration was evaporated under vacuum to give the oily crude product, which was further purified with flash chromatography on silica. Elution with hexane/ethyl acetate (5:1–2:1) gave 1b as colorless oil in 36% yield. 1H NMR (400 MHz, chloroform-d) δ 7.31 (d, J = 2.0 Hz, 1H), 7.25 (d, J = 2.0 Hz, 1H), 4.39–4.22 (m, 6H), 3.88 (s, 3H), 2.22 (q, J = 5.8 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). HRMS: calcd for C12H15O5 [M + H]+ 239.0919, found 239.0917.
7-Methoxybenzo[d][1,3]dioxole-5-carboxylic Acid (2a)
Following general procedure B, to a solution of methyl benzoate 1a (1.0 mmol) in dioxane (5 mL) was added a solution of lithium hydroxide (1.5 mmol) in water (3 mL). The mixture was stirred at 50 °C until the TLC indicated completion of the reaction. The solvents were removed under reduced pressure; the resulting residue was partitioned between water (10 mL) and DCM (10 mL), and the pH value was adjusted to 5 using 1 M HCl solution. The organic solvents were then combined, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:1) gave 2a as colorless oil in 88% yield. 1H NMR (400 MHz, acetone-d6) δ 11.11 (s, 1H), 7.34 (d, J = 1.5 Hz, 1H), 7.15 (d, J = 1.5 Hz, 1H), 6.10 (s, 2H), 3.93 (s, 3H). HRMS: calcd for C9H9O5 [M + H]+ 197.0450, found 197.0451.
9-Methoxy-3,4-dihydro-2H-benzo[b][1,4]dioxepine-7-carboxylic Acid (2b)
Following general procedure B, to a solution of the methyl benzoate 1b (1.0 mmol) in dioxane (5 mL) was added a solution of lithium hydroxide (1.5 mmol) in water (3 mL). The mixture was stirred at 50 °C until TLC indicated completion of the reaction. The solvents were removed under reduced pressure; the resulting residue was partitioned between water (10 mL) and DCM (10 mL), and the pH value was adjusted to 5 using 1 M HCl solution. The organic solvents were then combined, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:1) gave 2b as a colorless oil in 91% yield. 1H NMR (400 MHz, chloroform-d) δ 10.87 (s, 1H), 7.39 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 2.0 Hz, 1H), 4.41 (t, J = 5.8 Hz, 2H), 4.29 (t, J = 5.9 Hz, 2H), 3.90 (s, 3H), 2.25 (p, J = 5.8 Hz, 2H). HRMS: calcd for C11H13O5 [M + H]+ 225.0763, found 225.0765.
Methyl 3,4-Bis(allyloxy)-5-methoxybenzoate (4)
Following general procedure A, to a solution of methyl 3-methoxy-4,5-dihydroxybenzoate (2.5 mmol) in acetonitrile (10 mL) were added potassium carbonate (6.0 mmol) and allyl bromide (6.0 mmol). The mixture was refluxed overnight and then cooled to room temperature; the precipitate was filtered off and washed with dichloromethane. The combined filtration was evaporated under a vacuum to give the oily crude product, which was further purified with flash chromatography on silica. Elution with hexane/ethyl acetate (5:1−2:1) gave 4 as a colorless oil in 93% yield. 1H NMR (400 MHz, chloroform-d) δ 7.27 (s, 2H), 6.12–5.99 (m, 2H), 5.42 (dq, J = 17.3, 1.6 Hz, 1H), 5.345.24 (m, 2H), 5.17 (ddt, J = 10.3, 1.9, 1.1 Hz, 1H), 4.59 (ddt, J = 6.1, 5.0, 1.5 Hz, 4H), 3.87 (d, J = 0.7 Hz, 6H). HRMS: calcd for C15H19O5 [M + H]+ 279.1232, found 279.1236.
Methyl Methyl 3-Methoxy-4,5-bis(prop-1-en-1-yloxy)benzoate (5)
To a solution of 4 (279 mg, 1.0 mmol) in toluene (5 mL) was added carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (95 mg, 0.1 mmol). The mixture was refluxed for 36 h. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous Na2SO4. The combined extracts were evaporated under vacuum to give a crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (8:1–3:1) gave 5 as a colorless oil (218 mg, 78%). 1H NMR (400 MHz, chloroform-d) δ 7.35 (s, 2H), 6.47–6.11 (m, 2H), 5.44–4.60 (m, 2H), 3.90 (d, J = 2.0 Hz, 6H), 1.80–1.55 (m, 6H). HRMS: calcd for C15H19O5 [M + H]+ 279.1232, found 279.1234.
Methyl 8-Methoxybenzo[b][1,4]dioxine-6-carboxylate (6)
To a solution of 5 (200 mg, 0.72 mmol) in toluene (5 mL) was added Grubbs catalyst second generation (95 mg, 0.072 mmol). The mixture was refluxed for 24 h. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous Na2SO4. The combined extracts were evaporated under a vacuum to give a crude product that was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (8:1–3:1) gave 6 as a colorless oil (131 mg, 82%). 1H NMR (400 MHz, chloroform-d) δ 7.14 (d, J =1.9 Hz, 1H), 6.89 (d, J = 1.9 Hz, 1H), 5.91 (d, J = 3.6 Hz, 1H), 5.83 (d, J = 3.6 Hz, 1H), 3.81 (d, J = 0.9 Hz, 6H). HRMS: calcd for C11H11O5 [M + H]+ 223.0606, found 223.0608.
8-Methoxybenzo[b][1,4]dioxine-6-carboxylic Acid (7)
Following general procedure B, to a solution of the methyl benzoate 4 (1.0 mmol) in dioxane (5 mL) was added a solution of lithium hydroxide (1.5 mmol) in water (3 mL). The mixture was stirred at 50 °C until the TLC indicated completion of the reaction. The solvents were removed under reduced pressure; the resulting residue was partitioned between water (10 mL) and DCM (10 mL), and the pH value was adjusted to 5 using 1 M HCl solution. The organic solvents were then combined, dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:1) gave 7 as a colorless oil in 84% yield. 1H NMR (400 MHz, acetone-d6) δ 7.25 (d, J = 1.8 Hz, 1H), 6.91 (d, J = 1.8 Hz, 1H), 6.15 (d, J = 3.6 Hz, 1H), 6.12 (d, J = 3.6 Hz, 1H), 3.86 (s, 3H). HRMS: calcd for C10H9O5 [M + H]+ 209.0450, found 209.0447.
2,4,5-Tribromo-1-((2-(trimethylsi:lyl)ethoxy)methyl)-1H-imidazole (9)
To a stirred solution of 2,4,5-tribromoimidazole (9.5 g, 31.1 mmol) in anhydrous THF (100 mL) at ice temperature was added sodium hydride (1.5 g, 40.6 mmol) in portions under argon. The mixture was stirred for 1 h at this temperature and to it was added 2-(trimethylsilyl)ethoxymethyl chloride (6.7 mL, 35.8 mmol) dropwise. The reaction was then warmed to room temperature and stirred for another 1.5 h. Water was then added at ice temperature carefully, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the oily residue, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:0–10:1) gave 9 as a slightly yellowish solid (12.6 g, 93%). 1H NMR (400 MHz, chloroform-d) δ 5.30 (s, 2H), 3.65–3.49 (m, 2H), 0.94–0.87 (m, 2H), −0.03 (s, 9H). HRMS: calcd for C9H16Br3OSi [M + H]+ 432.8582, found 432.8588.
3-(4,5-Dibromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)-1-(phenylsulfonyl)-1H-indole (10)
To a mixture of 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester (5.0 g, 13.1 mmol), 9 (6.8 g, 15.7 mmol), and sodium carbonate (2.8 g, 26.1 mmol) in toluene (20 mL) and methanol (4 and 1 mL) were added 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (935 mg, 2.0 mmol) and tris(dibenzylideneacetone)dipalladium (600 mg, 0.66 mmol) under argon. The mixture was refluxed overnight. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous Na2SO4. The combined extracts were evaporated under a vacuum to give a crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (15:1–4:1) gave 10 as a pale-yellow solid in 34% yield. 1H NMR (400 MHz, chloroform-d) δ 8.20 (s, 1H), 8.17 (dt, J = 7.7, 1.1 Hz, 1H), 8.05–8.00 (m, 1H), 7.92–7.85 (m, 2H), 7.58–7.51 (m, 1H), 7.47–7.36 (m, 3H), 7.32 (td, J = 7.7, 1.1 Hz, 1H), 5.35 (s, 2H), 3.80–3.68 (m, 2H), 1.09–0.98 (m, 2H), 0.05 (s, 9h). HRMS: calcd for C23H26Br2N3O3SSi [M + H]+ 609.9831, found 609.9812.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(2,3-dihydrobenzo-[b][1,4]dioxin-6-yl)methanone (11a)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl chloride (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4O solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under a vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11a as a pale-yellow solid in 37% yield. 1H NMR (400 MHz, chloroform-d) δ 8.30 (s, 1H), 8.19 (ddd, J = 7.7, 1.3, 0.7 Hz, 1H), 8.05 (dt, J = 8.4, 1.0 Hz, 1h), 7.97–7.87 (m, 2H), 7.59–7.54 (m, 1H), 7.49–7.32 (m, 6H), 5.62 (s, 2H), 4.42–4.33 (m, 2H), 4.30 (dt, J = 5.7, 1.6 Hz, 2H), 3.64–3.53 (m, 2H), 0.98–0.90 (m, 2H), −0.07 (s, 9H). HRMS: calcd for C32H33BrN3O6SSi [M + H]+ 694.1043, found 694.1063.
Benzo[d][1,3]dioxol-5-yl(4-bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)-methanone (11b)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added benzo[d][1,3]dioxole-5-carbonyl chloride (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under a vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11b as a pale-yellow solid in 31% yield. 1H NMR (400 MHz, chloroform-d) δ 8.29 (s, 1H), 8.19 (ddd, J = 7.9, 1.4, 0.7 Hz, 1H), 8.05 (dt, J = 8.3, 1.0 Hz, 1H), 7.95–7.87 (m, 2H), 7.59–7.50 (m, 2H), 7.49–7.32 (m, 5H), 6.90 (dd, J = 8.2, 1.3 Hz, 1H), 6.10 (s, 2H), 5.62 (s, 2H), 3.66–3.50 (m, 2h), 0.99–0.90 (m, 2H), −0.06 (s, 9h). HRMS: calcd for C31H31BrN3O6SSi [M + H]+ 680.0886, found 680.0871.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(8-m ethoxy-2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methanone (11c)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 8-methoxy-2,3-dihydrobenzo[b][1,4]dioxine-6-carbonyl chloride (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under a vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11c as pale-yellow solid in 34% yield. 1H NMR (400 MHz, chloroform-d) δ 8.34 (s, 1H), 8.15 (d, J = 7.8 Hz, 1H), 8.04 (dd, J = 8.4, 1.0 Hz, 1H), 7.97–7.89 (m, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.49–7.39 (m, 3H), 7.35 (td, J = 7.6, 1.1 Hz, 1H), 7.20–7.11 (m, 3H), 5.62 (s, 2H), 4.41 (dd, J = 3.8, 1.8 Hz, 2H), 4.30 (dd, J = 3.8, 1.8 Hz, 2H), 3.95 (s, 3H), 3.62–3.53 (m, 2H), 0.96–0.89 (m, 2H), −0.08 (s, 9H). HRMS: calcd for C33H35BrN3O7SSi [M + H]+ 724.1148, found 724.1124.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl) (7-methoxybenzo-[d][1,3]dioxol-5-yl)methanone (11d)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 3a (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11d as a pale-yellow solid in 39% yield. 1H NMR (400 MHz, chloroform-d) δ 8.30 (s, 1H), 8.20 (ddd, J = 7.9, 1.4, 0.7 Hz, 1H), 8.05 (dt, J = 8.2, 1.0 Hz, 1H), 7.94–7.90 (m, 2H), 7.60–7.53 (m, 1H), 7.48–7.33 (m, 4H), 7.22 (d, J = 1.5 Hz, 1H), 7.12 (d, J = 1.5 Hz, 1H), 6.11 (s, 2H), 5.63 (s, 2H), 3.95 (s, 3H), 3.65–3.57 (m, 2H), 1.00–0.91 (m, 2H), −0.06 (s, 9H). HRMS: calcd for C32H33BrN3O7SSi [M + H]+ 710.0992, found 710.0979.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)eth oxy)methyl)-1H-imidazol-5-yl)(9-methoxy-3,4-di-hydro-2H-benzo[b][1,4]dioxepin-7-yl)methanone (11e)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 3b (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 11e as a pale-yellow solid in 43% yield. 1H NMR (400 MHz, chloroform-d) δ 8.30 (s, 1H), 8.23–8.17 (m, 1H), 8.05 (dt, J = 8.4, 1.0 Hz, 1H), 7.94–7.89 (m, 2H), 7.60–7.53 (m, 1H), 7.49–7.32 (m, 4H), 7.22 (d, J = 2.0 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1h), 5.63 (s, 2H), 4.46 (t, J = 5.7 Hz, 2H), 4.30 (t, J = 5.9 Hz, 2H), 3.93 (s, 3H), 3.64–3.56 (m, 2H), 2.28 (p, J = 5.8 Hz, 2H), 0.99–0.90 (m, 2H), −0.06 (s, 9H). HRMS: calcd for C34H37BrN3O7SSi [M + H]+ 738.1305, found 738.1283.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(8-methoxybenzo-[b][1,4]dioxin-6-yl)methanone (11f)
Following general procedure D, to a stirred solution of compound 10 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 8 (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ ethyl acetate (10:1–3:1) gave pure 11f as a yellowish solid in 48% yield. 1H NMR (400 MHz, acetone-d6) δ 8.54 (s, 1H), 8.35 (ddd, J = 8.0, 1.3, 0.7 Hz, 1H), 8.18–8.09 (m, 3H), 7.79–7.73 (m, 1H), 7.72–7.63 (m, 2H), 7.52 (ddd, J = 8.4, 7.3, 1.4 Hz, 1H), 7.44 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.31 (d, J = 1.9 Hz, 1H), 6.92 (d, J = 1.9 Hz, 1H), 6.24 (d, J = 3.6 Hz, 1H), 6.20 (d, J = 3.6 Hz, 1H), 5.81 (s, 2H), 3.93 (s, 3H), 3.76–3.67 (m, 2H), 1.06–0.95 (m, 2H), 0.00 (s, 9H). HRMS: calcd for C33H33BrN3O7SSi [M + H]+ 722.0992, found 722.0979.
(2-(1H-lndol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(benzo[d][1,3]dioxol-5-yl)methanone (12b)
Following general procedure E, to a suspension of 11b (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12b as a pale-yellow solid in 88% yield. 1H NMR (400 MHz, chloroform-d) δ 9.35 (s, 1H), 8.30–8.17 (m, 1H), 7.94 (d, J = 2.8 Hz, 1H), 7.74 (s, 1H), 7.51 (dd, J = 8.1, 1.7 Hz, 1H), 7.41 (dd, J = 7.2, 1.7 Hz, 2H), 7.29–7.18 (m, 3H), 6.91 (d, J = 8.1 Hz, 1H), 6.08 (s, 2H), 5.88 (s, 2H), 3.78–3.64 (m, 2H), 1.00–0.86 (m, 2H), −0.06 (s, 9H). HRMS: calcd for C25H28N3O4Si [M + H]+ 462.1849, found 462.1842.
(2-(1H-Indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(8-methoxy-2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-methanone (12c)
Following general procedure E, to a suspension of 11c (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12c as a pale-yellow solid in 76% yield. 1H NMR (400 MHz, chloroform-d) δ 9.24 (s, 1H), 8.19 (d, J = 7.2 Hz, 1H), 7.95 (s, 1H), 7.81 (s, 1H), 7.50–7.42 (m, 1H), 7.30–7.20 (m, 2H), 7.18 (d, J = 2.0 Hz, 1h), 7.11 (d, J = 2.0 Hz, 1H), 5.88 (s, 2H), 4.42 (dd, J = 3.9, 1.7 Hz, 2H), 4.33 (dd, J = 3.7, 1.7 Hz, 2H), 3.96 (s, 3H), 3.77–3.64 (m, 2h), 1.00–0.87 (m, 2H), −0.05 (s, 9H). HRMS: calcd for C27H32N3O5Si [M + H]+ 506.2111, found 506.2123.
(2-(1H-Indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(7-methoxybenzo[d][1,3]dioxol-5-yl)methanone (12d)
Following general procedure E, to a suspension of 11d (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12d as a pale-yellow solid in 89% yield. 1H NMR (400 MHz, chloroform-d) δ 8.79 (s, 1H), 8.35–8.25 (m, 1H), 8.03 (d, J = 2.8 Hz, 1H), 7.77 (s, 1H), 7.46–7.42 (m, 1H), 7.30–7.27 (m, 1H), 7.20 (d, J = 1.5 Hz, 1H), 7.13 (d, J = 1.5 Hz, 1H), 6.10 (s, 2H), 5.91 (s, 2H), 3.96 (s, 3H), 3.80–3.70 (m, 2H), 0.94 (d, J = 8.3 Hz, 2H), −0.05 (s, 9H). HRMS: calcd for C26H30N3O5Si [M + H]+ 492.1955, found 492.1950.
(2-(1H-Indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(9-methoxy-3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)-methanone (12e)
Following general procedure E, to a suspension of 11e (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12e as pale-yellow solid in 85% yield. 1H NMR (400 MHz, chloroform-d) δ 8.66 (s, 1H), 8.36–8.23 (m, 1H), 8.05 (d, J = 2.8 Hz, 1H), 7.79 (s, 1H), 7.51–7.40 (m, 1H), 7.33–7.24 (m, 4H), 7.22 (d, J = 2.1 Hz, 1H), 7.16 (d, J = 2.0 Hz, 1H), 5.92 (s, 2H), 4.44 (t, J = 5.7 Hz, 2H), 4.32 (t, J = 5.9 Hz, 2H), 3.92 (s, 3H), 3.81–3.71 (m, 2H), 2.34–2.20 (m, 2H), 1.00–0.91 (m, 2H), −0.04 (s, 9H). HRMS: calcd for C28H34N3O5Si [M + H]+ 520.2268, found 520.2262.
(2-(1H-Indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(8-methoxybenzo[b][1,4]dioxin-6-yl)methanone (12f)
Following general procedure E, to a suspension of 11f (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 12f as a yellowish solid in 79% yield. 1H NMR (400 MHz, chloroform-d) δ 10.02 (d, J = 2.8 Hz, 1H), 8.36–8.27 (m, 1H), 7.94 (d, J = 2.8 Hz, 1H), 7.87 (s, 1H), 7.38–7.31 (m, 1H), 7.30–7.22 (m, 2H), 7.16 (d, J = 1.9 Hz, 1H), 6.95 (d, J = 1.9 Hz, 1H), 6.04 (d, J = 3.6 Hz, 1H), 5.98–5.88 (m, 3H), 3.92 (s, 3H), 3.83–3.67 (m, 2H), 1.03–0.89 (m, 2H), 0.00 (s, 9h). HRMS: calcd for C27H30N3O5Si [M + H]+ 504.1955, found 504.1932.
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(2,3-dihydrobenzo[b][1,4]-dioxin-6-yl)methanone (13a)
Following general procedure F, to a solution of 12a (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13a as a yellowish solid in 88% yield. 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 11.62 (s, 1H), 8.47–8.36 (m, 1H), 8.30–8.17 (m, 1H), 7.87 (s, 1H), 7.68 (s, 2H), 7.48 (dt, J = 8.2, 0.9 Hz, 1H), 7.25–7.12 (m, 2H), 7.04 (d, J = 8.8 Hz, 1H), 4.52–4.27 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 182.34, 147.19, 143.08, 136.31, 131.36, 125.85, 124.94, 122.99, 122.13, 121.06, 120.17, 118.17, 116.93, 111.84, 105.51, 64.50, 64.01. HRMS: calcd for C20H16N3O3 [M + H]+ 346.1192, found 346.1205. Purity: 100.0% by HPLC.
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(benzo[d][1,3]dioxol-5-yl)-methanone (13b)
Following general procedure F, to a solution of 12b (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13b as a yellowish solid in 92% yield. 1H NMR (400 MHz, DMSO-d6) δ 13.36 (s, 1H), 11.67 (s, 1H), 8.42–8.32 (m, 1H), 8.23 (d, J = 2.7 Hz, 1H), 7.92 (s, 1H), 7.81 (d, J = 7.1 Hz, 1H), 7.71–7.56 (m, 1H), 7.49 (dt, J = 8.2, 0.9 Hz, 1H), 7.19 (dtd, J = 17.2, 7.1, 1.3 Hz, 2H), 7.11 (d, J = 8.1 Hz, 1H), 6.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 182.19, 150.85, 147.53, 136.31, 132.20, 126.18, 125.20, 124.82, 122.20, 120.93, 120.25, 111.90, 108.79, 108.03, 105.05, 101.88. HRMS: calcd for C19H14N3O3 [M + H]+ 332.1035, found 332.1050. Purity: 97.5% by HPLC.
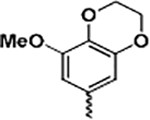
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(8-methoxy-2,3-dihydrobenzo[b][1,4]dioxin-6-yl)methanone (13c)
Following general procedure F, to a solution of 12c (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13c as a yellowish solid in 79% yield. 1H NMR (400 MHz, chloroform-d) δ 8.02 (s, 1H), 7.92 (s, 1H), 7.81 (s, 1H), 7.40 (dd, J = 10.9, 7.7 Hz, 1H), 7.16 (dd, J = 7.0, 3.0 Hz, 3H), 7.07 (s, 1H), 4.41–4.32 (m, 2H), 4.32–4.22 (m, 2H), 3.91 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 182.90, 149.30, 147.56, 143.77, 137.96, 136.52, 130.62, 129.30, 128.47, 124.17, 123.16, 121.50, 119.33, 112.37, 112.24, 104.74, 102.80, 64.99, 64.18, 56.44. HRMS: calcd for C21H18N3O4 [M + H]+ 376.1297, found 376.1309. Purity: 98.2% by HPLC.
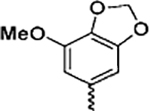
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(7-methoxybenzo[d][1,3]-dioxol-5-yl)methanone (13d)
Following general procedure F, to a solution of 12d (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13d as a yellowish solid in 85% yield. 1H NMR (400 MHz, chloroform-d) δ 8.16–8.07 (m, 1H), 7.90 (s, 1H), 7.74 (s, 1H), 7.46–7.37 (m, 1H), 7.25–7.20 (m, 3H), 7.14 (d, J = 1.5 Hz, 1H), 6.06 (s, 2H), 3.93 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 182.72, 148.95, 148.33, 143.59, 139.51, 136.49, 132.27, 130.88, 126.65, 124.37, 122.93, 121.26, 119.79, 111.98, 109.78, 105.09, 103.46, 102.39, 56.68. HRMS: calcd for C20H16N3O4 [M + H]+ 362.1141, found 362.1146. Purity: 99.7% by HPLC.
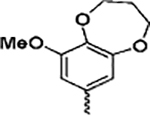
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(9-methoxy-3,4-dihydro-2H-benzo[b][1,4]dioxepin-7-yl)methanone (13e)
Following general procedure F, to a solution of 12e (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13e as a yellowish solid in 80% yield. 1H NMR (400 MHz, chloroform-d) δ 8.16 (dd, J = 6.5, 2.9 Hz, 1H), 7.87 (d, J = 3.0 Hz, 1H), 7.74 (d, J = 3.4 Hz, 1H), 7.62–7.48 (m, 1H), 7.42 (dt, J = 6.0, 3.0 Hz, 1H), 7.25–7.20 (m, 2H), 7.20–7.13 (m, 1H), 4.38 (t, J = 5.8 Hz, 2H), 4.34–4.23 (m, 2H), 3.89 (s, 3H), 2.24 (dd, J = 7.0, 4.5 Hz, 2H). 13C NMR (101 MHz, chloroform-d) δ 183.11, 151.77, 151.18, 144.62, 136.51, 132.03, 131.93, 131.78, 128.67, 128.55, 126.17, 124.48, 122.84, 121.17, 119.92, 115.51, 111.88, 106.65, 70.82, 70.50, 56.37, 30.97. HRMS: calcd for C22H20N3O4 [M + H]+ 390.1454, found 390.1472. Purity: 100.0% by HPLC.
(2-(1H-Indol-3-yl)-1H-imidazol-5-yl)(8-methoxybenzo[b][1,4]-diOXin-6-yl)methanone (13f)
Following general procedure F, to a solution of 12f (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 13f as a yellowish solid in 84% yield. 1H NMR (400 MHz, DMSO-d6) δ 13.02 (s, 1H), 11.55 (s, 1H), 8.40 (d, J = 7.8 Hz, 1H), 7.94 (s, 2h), 7.47 (d, J = 7.9 Hz, 1H), 7.16 (dtd, J = 16.1, 8.1, 7.6, 6.4 Hz, 2H), 6.30 (d, J = 9.2 Hz, 2H), 3.88 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 183.83, 153.01, 148.97, 141.70, 136.51, 133.30, 126.13, 124.53, 122.77, 121.05, 120.00, 111.88, 106.31, 105.74, 56.18. HRMS: calcd for C21H16N3O4 [M + H]+ 374.1141, found 374.1157. Purity: 99.4% by HPLC.
4-Bromo-1-(phenylsulfonyl)-1H-indole (14)
To a solution of 4- bromoindole (1.0 g, 5.1 mmol) in THF (10 mL) was added sodium hydride (314 mg, 7.6 mmol) in portions under ice temperature. After 1 h, benzenesulfonyl chloride (0.81 mL, 6.1 mmol) was added dropwise. The reaction was stirred at room temperature for 2 h. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous Na2SO4. The combined extracts were evaporated under a vacuum to give a crude product that was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (20:1–6:1) gave 14 as a colorless solid (1.6 g, 93%). 1H NMR (400 MHz, chloroform-d) δ 7.96 (dd, J = 8.4, 0.8 Hz, 1H), 7.91–7.84 (m, 2H), 7.64 (d, J = 3.7 Hz, 1H), 7.57–7.50 (m, 1H), 7.44 (dd, J =8.5, 7.1 Hz, 2H), 7.39 (d, J = 7.8 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 6.74 (d, J = 3.7 Hz, 1H). HRMS: calcd for C14H11BrNO2S [M + H]+ 335.9694, found 335.9691.
1-(Phenylsulfonyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (15)
To a solution of 14 (0.86 g, 2.5 mmol) in dioxane (10 mL) was added bis(pinacolato)diboron (1.9 g, 7.5 mmol), potassium acetate (0.75 g, 7.5 mmol), and [1,1′-bis-(diphenylphosphino)ferrocene]dichloropalladium complex with di- chloromethane (220 mg, 0.125 mmol). The mixture was heated to reflux and stirred overnight. Dioxane was removed under reduced pressure, and the resulting mixture was partitioned between water and EtOAc. The combined organic layer was evaporated under a vacuum to give a crude product that was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (20:1) gave 15 as a colorless solid (2.47 g, 86%). 1H NMR (400 MHz, chloroform-d) δ (dt, J = 8.4, 0.9 Hz, 1H), 7.88–7.82 (m, 2H), 7.71 (dd, J = 7.2, 1.0 Hz, 1H), 7.60 (d, J = 3.7 Hz, 1H), 7.53–7.47 (m, 1H), 7.40 (dd, J = 8.5, 7.0 Hz, 2H), 7.32 (dd, J = 8.3, 7.3 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H), 1.35 (s, 12H). HRMS: calcd for C20H23BNO4S [M + H]+ 384.1441, found 384.1438.
4-(4,5-Dibromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-im idazol-2-yl)-1-(phenylsulfonyl)-1H-indole (16)
To a mixture of 15 (5.0 g, 13.1 mmol), 9 (6.8 g, 15.7 mmol), and sodium carbonate (2.8 g, 26.1 mmol) in toluene (20 mL) and methanol (4 and 1 mL) were added 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (935 mg, 2.0 mmol) and tris(dibenzylideneacetone)dipalladium (600 mg, 0.66 mmol) under argon. The mixture was refluxed overnight. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with anhydrous Na2SO4. The combined extracts were evaporated under a vacuum to give a crude product that was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (15:1–4:1) gave 16 as a pale-yellow solid in 31% yield. 1H NMR (400 MHz, chloroform-d) S 8.11 (dd, J = 8.4, 1.0 Hz, 1H), 7.86 (dd, J = 8.0, 1.4 Hz, 2H), 7.64 (d, J = 3.7 Hz, 1H), 7.57–7.50 (m, 2H), 7.41 (dt, J = 20.0, 7.8 Hz, 3h), 6.97 (d, J = 3.7 Hz, 1H), 5.25 (s, 2H), 3.49 (dd, J = 8.8, 7.6 Hz, 2H), 0.85 (dd, J = 8.8, 7.6 Hz, 2h), −0.06 (s, 9H). HRMS: calcd for C23H26Br2N3O3SSi [M + H]+ 609.9831, found 609.9844.
(4-Bromo-2-(1-(phenylsulfonyl)-1H-indol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)(8-methoxybenzo-[b][1,4]diOXin-6-yl)methanone (17)
Following general procedure D, to a stirred solution of compound 16 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 8 (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ ethyl acetate (10:1–3:1) gave pure 17 as a yellowish solid in 41% yield. 1H NMR (400 MHz, chloroform-d) δ 8.16 (d, J = 8.3 Hz, 1H), 7.88 (dd, J = 8.4, 1.3 Hz, 2H), 7.68 (d, J = 3.7 Hz, 1H), 7.59–7.53 (m, 2H), 7.48–7.39 (m, 3H), 7.13 (d, J = 1.9 Hz, 1H), 6.96 (d, J = Hz, 1H), 6.88 (d, J = 1.8 Hz, 1H), 6.00 (d, J = 3.6 Hz, 1h), 5.91 (d, J = 3.5 Hz, 1H), 5.50 (s, 2H), 3.90 (s, 3H), 3.34–3.27 (m, 2H), 0.76–0.67 (m, 2H), −0.15 (s, 9H). HRMS: calcd for C33H33BrN3O7SSi [M + H]+ 722.0992, found 722.0986.
(2-(1H-lndol-4-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1 H-imida- zol-5-yl)(8-methoxybenzo[b][1,4]dioxin-6-yl)methanone (18)
Following general procedure E, to a suspension of 17 (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenylphosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 18 as a yellowish solid in 77% yield. 1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 7.76 (s, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.34–7.29 (m, 2H), 7.15 (d, J = 1.8 Hz, 1H), 6.93 (d, J = Hz, 1H), 6.73 (s, 1H), 6.01 (d, J = 3.6 Hz, 1H), 5.92 (d, J = 3.7 Hz, 1H), 5.78 (s, 2H), 3.91 (s, 3H), 3.43–3.33 (m, 2H), 0.80–0.73 (m, 2H), −0.13 (s, 9H). HRMS: calcd for C27H30N3O5Si [M + H]+ 504.1955, found 504.1962.
(2-(1H-Indol-4-yl)-1H-imidazol-5-yl)(8-methoxybenzo[b][ 1,4]-diOXin-6-yl)methanone (19)
Following general procedure F, to a solution of 18 (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 19 as a yellowish solid in 87% yield. 1H NMR (400 MHz, chloroform-d) δ 10.59 (s, 1H), 8.51 (s, 1H), 7.90 (s, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.38 (t, J = 2.9 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 7.19 (s, 1H), 7.01 (s, 1H), 6.01 (d, J = 3.6 Hz, 1H), 5.93 (d, J = 3.6 Hz, 1H), 3.90 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 182.88, 147.69, 142.96, 138.98, 136.74, 136.27, 132.77, 127.10, 126.76, 126.16, 125.32, 121.66, 121.46, 120.51, 119.05, 113.47, 110.49, 108.86, 102.13, 101.84, 56.52. HRMS: calcd for C21H16N3O4 [M + H]+ 374.1141, found 374.1151. Purity: 95.1% by HPLC.
3-(1H-lmidazoI-2-yl)-4-methyl-1-(phenylsulfonyl)-1H-indole (21)
Compound 21 was synthesized following our previously reported procedure.22
3-(4,5-Dibromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-2-yl)-4-methyl-1-(phenylsulfonyl)-1H-indole (22)
To a solution of 21 (1.0 g, 3.3 mmol) in THF (10 mL) was added N-bromosuccinimide (1.07 g, 6.0 mmol) in portions. The reaction was stirred for 1.5 h, quenched with saturated Na2S2O3 solution, and extracted with EtOAc. The combined extracts were evaporated under reduced pressure and dried under vacuum to give a crude product. The crude product was dissolved in anhydrous THF (10 mL) at ice temperature, and sodium hydride (122 mg, 3.3 mmol) was added in portions under argon. The mixture was stirred for another 1 h at this temperature and to it was added 2-(trimethylsilyl)ethoxymethyl chloride (0.62 mL, 3.3 mmol) dropwise. The reaction was then warmed to room temperature and stirred for 1.5 h. Water was then added, and the reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under a vacuum to give an oily residue that was purified with flash chromatography on silica. Elution with hexane/ ethyl acetate (10:0–10:1) gave 22 as a pale-yellow solid (1.4 g, 70%). 1H NMR (400 MHz, chloroform-d) δ 7.97–7.86 (m, 4H), 7.63–7.54 (m, 1H), 7.53–7.43 (m, 2H), 7.29 (dd, J = 8.4, 7.3 Hz, 1H), 7.04 (dt, J = 7.4, 1.0 Hz, 1H), 5.19 (s, 2H), 3.56–3.47 (m, 2h), 2.18 (s, 3H), 0.96–0.84 (m, 2H), 0.00 (s, 9H). HRMS: calcd for C24H28Br2N3O3SSi [M + H]+ 623.9987, found 623.9976.
(4-Bromo-2-(4-methyl-1-(phenylsulfonyl)-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl) (8-methoxybenzo-[b][1,4]dioxin-6-yl)methanone (23)
Following general procedure D, to a stirred solution of compound 22 (1.0 mmol) in anhydrous THF (3.0 mL) under argon was added isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF, 0.92 mL, 1.2 mmol) at room temperature. The mixture was stirred for 1 h and to it was added 8 (1.3 mmol) in anhydrous THF (1.0 mL). The reaction was kept stirring at room temperature for 1 h and then refluxed for 30 min. A saturated NH4Cl solution was then added to quench the reaction. The reaction mixture was extracted with ethyl acetate, washed with brine, and dried with Na2SO4. The combined extracts were evaporated under a vacuum to give the crude product, which was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 23 as a yellowish solid in 37% yield. 1H NMR (400 MHz, chloroform-d) δ 7.92–7.83 (m, 4H), 7.54 (dd, J = 8.3, 6.5 Hz, 1H), 7.47–7.39 (m, 2H), 7.25–7.20 (m, 1H), 7.10 (d, J = 1.9 Hz, 1H), 7.01 (d, J = 7.5 Hz, 1H), 6.84 (s, 1H), 6.00 5.91 (m, 1H), 5.88 (dd, J = 3.3, 1.8 Hz, 1H), 5.36 (d, J = 1.8 Hz, 2H), 3.87 (d, J = 1.7 Hz, 3H), 3.39–3.29 (m, 2h), 2.18 (s, 3H), 0.79–0.66 (m, 2H), −0.14 (d, J = 1.5 Hz, 9H). HRMS: calcd for C34H35BrN3O7SSi [M + H]+ 736.1148, found 736.1125.
(8-Methoxybenzo[b][1,4]dioxin-6-yl) (2-(4-methyl-1H-indol-3-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)methanone (24)
Following general procedure E, to a suspension of 23 (0.5 mmol), potassium carbonate (276 mg, 2.0 mmol), and triphenyl- phosphine (26 mg, 0.1 mmol) in n-BuOH (3 mL) was added palladium acetate (5.6 mg, 0.025 mmol). The mixture was heated to reflux for 4 h. n-BuOH was removed under reduced pressure. The residue was partitioned between water (10 mL) and EtOAc (10 mL). The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (10:1–3:1) gave pure 24 as a yellowish solid in 82% yield. 1H NMR (400 MHz, chloroform-d) δ 9.17 (s, 1H), 7.36 (s, 1H), 7.21 (d, J = 8.2 Hz, 1H), 7.15 (s, 1H), 7.11 (d, J = 7.4 Hz, 1H), 6.94 (d, J = 7.2 Hz, 1H), 6.90 (q, J = 1.6 Hz, 1H), 6.01 (t, J = 2.4 Hz, 1H), 5.92 (t, J = 2.8 Hz, 1H), 5.46 (s, 2H), 3.90 (s, 3H), 3.31 (t, J = 8.1 Hz, 2H), 2.34 (s, 3H), 0.70 (t, J = 8.1 Hz, 2H), −0.14 (d, J = 1.5 Hz, 9h). HRMS: calcd for C28H32N3O5Si [M + H]+ 518.2111, found 518.2118.
(8-Methoxybenzo[b][1,4]dioxin-6-yl) (2-(4-methyl-1H-indol-3-yl)-1H-imidazol-5-yl)methanone (25)
Following general procedure F, to a solution of 24 (0.3 mmol) in DCM (1 mL) was added trifluoroacetic acid (1 mL). The reaction was stirred for 2 h, and the solvent was evaporated under reduced pressure. The residue was partitioned between saturated NaHCO3 solution and EtOAc. The combined organic solvents were then evaporated under reduced pressure. The crude product was purified with flash chromatography on silica. Elution with hexane/ethyl acetate (4:1–1:2) gave pure 25 as a yellowish solid in 84% yield. 1H NMR (400 MHz, methylene chloride-d2) δ 9.11 (s, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.30 (s, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.19 (s, 1H), 7.10 (t, J = 7.8 Hz, 1h), 6.98 (s, 1H), 6.90 (d, J = 7.1 Hz, 1H), 6.00 (d, J = 2.7 Hz, 1H), 5.95 (t, J = 2.8 Hz, 1H), 3.83 (s, 3H), 2.47 (s, 3H). 13C NMR (101 MHz, chloroform-d) δ 182.91, 148.66, 147.80, 142.95, 136.54, 136.27, 132.74, 132.02, 130.45, 127.12, 126.81, 124.54, 122.81, 122.36, 110.47, 109.71, 108.75, 105.70, 56.56, 20.76. HRMS: calcd for C22H18N3O4 [M + H]+ 388.1297, found 388.1297. Purity: 96.8% by HPLC.
Cell Culture and Reagents
Human melanoma cell lines A375, M14, and RPMI7951 (American Type Culture Collection or ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Corning, Manasas, VA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and 1% antibiotic/antimycotic mixture (Sigma-Aldrich, St. Louis, MO). Parental prostate cancer PC-3, its paclitaxel-resistant daughter line PC-3/TxR, parental prostate cancer DU-145, and its docetaxel-resistant daughter line DU-145/TxR are gifts from Dr. Evan Keller at the University of Michigan Medical School. PC-3 and DU-145 cell lines were cultured in RPMI 1640 medium (Gibco by Life Technologies, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and 1% antibiotic/antimycotic mixture (Sigma-Aldrich, St. Louis, MO). Taxane-resistant PC-3/TxR and DU-145/TxR cell lines were cultured in the same media and additionally supplemented with 10 nM paclitaxel or docetaxel, respectively. Drugs were not included in the media for PC-3/TxR or DU-145/TxR for at least 1 week prior to in vitro testing. All cell lines were authenticated by ATCC by short tandem repeat profiling. Cultures were maintained to 80–90% confluency at 37 °C in a humidified atmosphere containing 5% CO2. Compounds were dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO) to make a stock solution of 20 mM. Compound solutions were freshly prepared by diluting stocks with cell culture medium before use.
Cytotoxicity Assay
Logarithmic growth phase cells were seeded in 96-well plates at a concentration of 1,800–3,500 cells per well depending on te growth rate of the cell line. After overnight incubation, the media was replaced, and cells were treated with the test compounds at 10 concentrations ranging from 0.03 nM to 1 μM plus a vehicle (DMSO) control for 72 h in four replicates. Following treatment, the MTS reagent (Promega, Madison, WI) was added to the cells and incubated in the dark at 37 °C for at least 1 h. Absorbance at 490 nm was measured using a plate reader (BioTek Instraments Inc., Winooski, VT). IC50 values were calculated by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA).
Protein Expression and Purification
Porcine brain tubulin (Catalog # T-238P) was obtained from Cytoskeleton, Inc. The stathmin-like domain of RB3(RB3-SLD) was transformed into and overexpressed in E. coli. The protein was purified by anion-exchange chromatography and gel filtration chromatography. The peak fractions from gel filtration column were concentrated to 10 mg/mL and stored at −80 °C.28–30 TTL protein was expressed and purified from the E. coli expression system as described in the previous reference.26 Briefly, the protein was expressed in E. coli using LB and purified through Ni-NTA affinity chromatography and gel filtration chromatography (buffer: bis-tris propane pH 6.5, 200 mM NaCl, 2.5 mM MgCl2, 5 mM β-Me, 1% glycerol). The peak fractions of the target protein were collected and concentrated to 20 mg/mL and saved at −80 °C. Porcine brain tubulin was supplied at 10 mg/mL in G-PEM (general tubulin buffer: 80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP) as a frozen liquid and saved at −80 °C until use.
Crystallization and Crystal Soaking
The process to getting crystals of T2R-TTL followed the previously reported procedure.2,31 Briefly, the complex containing tubulin (10 mg/mL), TTL (20 mg/mL), and RB3 (10 mg/mL) at the molar ratio of 2:1.3:1.2 (tubulin:RB3:TTL) was incubated on ice with an additional 1 mM AMPPCP, 5 mM tyrosinol, and 10 mM DTT, and then the final sample was concentrated to 20 mg/mL at 4 °C. Crystallization of the T2R-TTL complex was carried out at 20 °C using the sitting drop vapor diffusion method by mixing equal volumes of the protein complex and crystallization buffer containing 6% PEG, 5% glycerol, 0.1 M MES, 30 mM CaCl2, 30 mM MgCl2 pH 6.7. Seeding was used to optimizing the crystal conditions. Initial crystals were observed after 2 days, and crystals reached their final size of 200–300 μm within 3–5 days.
For crystal soaking, 0.1 μL of the ligand solution (l3f dissolved in 100% DMSO) was added to the 2 μL crystal-containing drop for 12 h at 20 °C. The best crystals were selected and frozen in liquid nitrogen in the presence of cryoprotectant (crystallization buffer containing 20% glycerol).
X-ray Data Collection and Structure Determination
Crystals of the T2R-TTL-13f complexes were mounted in nylon loops and flash-cooled in a nitrogen stream at 100 K. The diffraction data were collected on beamlines BL19U1 at Shanghai Synchrotron Radiation Facility (SSRF) in Shanghai, China. Data were indexed, integrated, and scaled using the HKL2000 program package.32 The structure of T2R-TTL-13f complex was solved by molecular replacement using the previously published T2R-TTL structure (PDB ID: 4I55) as the search model. The rotation and translation function searches were performed by the program PHASER. The model was manually built with Coot33 and then refined using the phenix refine module of the Phenix program.34 The model quality was checked with the PROCHECK program and showed good stereochemistry according to the Ramachandran plot.
Tubulin Polymerization Assay
Bovine brain tubulin (0.4 mg, >97% pure) (Cytoskeleton, Denver, CO) was mixed with 10 μM of the test compounds and incubated in 100 μL of general tubulin buffer (80 mM PIPES, 2.0 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP) at pH 6.9. The absorbance of the wavelength at 340 nm was monitored every 1 min for 20 min by the SYNERGY 4 Microplate Reader (BioTek Instruments, Winooski, VT). BioTek Gen5 data analysis software was used to calculate the Vmax values. The spectrophotometer was set at 37 °C for tubulin polymerization.
In Vivo Xenograft Model
All animal experiments were performed in accordance with the NIH animal use guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee Health Science Center (UTHSC, Memphis, TN). Nude mice, 6–8 weeks old, were purchased from Evigo.
Logarithmic growth phase A375 cells (5 × 107 cells per ml) were prepared in phenol red-free, FBS-free media and mixed with Matrigel immediately before injecting into mice. Tumors were established by injecting 100 μL of this mixture subcutaneously in the dorsal flank of each mouse (2.5 × 106 cells). After tumor volumes reached ~100 mm3, mice were randomized into control or treatment groups (n = 8). Compound 13f or paclitaxel was dissolved in a 1:1 ratio of PEG300:PBS solution to produce the desired concentrations. The vehicle control solution was formulated with equal parts PEG300 and PBS only. THen, 100 μL of the drug treatment or vehicle control was administered via i.p. injection every other day for 2 weeks.
Tumor volume was measured three times a week with a caliper and calculated using the formula a × b2 × 0.5, where a and b represent the larger and smaller diameters, respectively. Tumor growth inhibition (TGI) at the conclusion of the experiments was calculated as 100 – 100((T – T0)/(C – C0)), where T, T0, C, and C0 are the mean tumor volume for the specific group on the last day of treatment, mean tumor volume of the same group on the first day of treatment, mean tumor volume for the vehicle control group on the last day of treatment, and mean tumor volume for the vehicle control group on the first day of treatment, respectively. Animal activity and body weights were monitored during the entire experiment period to assess potential acute toxicity. At the end of the experiment, mice were sacrificed, and the tumors were weighed.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by NIH/NCI grant R01CA148706 to W.L. and D.D.M and NIH grants 1S10OD010678–01 and 1S10RR026377–01 and the Guangzhou key medical discipline construction project to W.L. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional support was from the University of Tennessee College of Pharmacy Drug Discovery Center. We thank Dr. Lei Yang at St. Jude Children’s Research Hospital for the metabolic stability evaluation for new VERU-111 analogues. We also thank Dr. Benoit Gigant (Institute for Integrative Biology of the Cell (I2BC), CEA, CNRS, Univ. Paris-Sud, Université Paris-Saclay, France) and Dr. Michel O. Steinmetz (Paul Scherrer Institute, Switzerland) for kindly providing the plasmids of RB3-SLD and TTL. The X-ray work was supported by National Natural Science Foundation of China (No. 81703553), China Postdoctoral Science Foundation (No. 2017M610607), and Postdoctoral Science Foundation of Sichuan University (No. 2017SCU12045). G.K. and S.W.W. acknowledge the support of American Lebanese Syrian Associated Charities (ALSAC).
ABBREVIATIONS
- CBSIs
colchicine binding site inhibitors
- TMP
3,4,5-trimethoxyphenyl
- MTAs
microtubule-targeting agents
- TGI
tumor growth inhibition
- CA-4
Combretastatin A4
- SEM
2-(trimethylsilyl)ethoxymethyl
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.8b00827.
Full synthetic procedure and characterization data for all intermediates and VERU-111 analogues 13a–f, 19, and 25; close-up view of the conformational changes at the colchicine-binding sites between colchicine and 13f; and spectral data for synthetic intermediates and VERU-111 analogues (PDF)
Molecular formula strings and biological data (CSV)
REFERENCES
- (1).Jordan MA; Wilson L Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4 (4), 253–265. [DOI] [PubMed] [Google Scholar]
- (2).Etienne-Manneville S From signaling pathways to microtubule dynamics: the key players. Curr. Opin. Cell Biol 2010, 22 (1), 104–111. [DOI] [PubMed] [Google Scholar]
- (3).Kavallaris M; Verrills NM; Hill BT Anticancer therapy with novel tubulin-interacting drugs. Drug Resist. Updates 2001, 4 (6), 392–401. [DOI] [PubMed] [Google Scholar]
- (4).Wang Z; Chen J; Wang J; Ahn S; Li CM; Lu Y; Loveless VS; Dalton JT; Miller DD; Li W Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm. Res 2012, 29 (11), 3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kanthou C; Tozer GM Microtubule depolymerizing vascular disrupting agents: novel therapeutic agents for oncology and other pathologies. Int. J. Exp. Pathol 2009, 90 (3), 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Arnst KE; Wang Y; Hwang DJ; Xue Y; Costello T; Hamilton D; Chen Q; Yang J; Park F; Dalton JT; Miller DD; Li W A Potent, Metabolically stable tubulin inhibitor targets the colchicine binding site and overcomes taxane resistance. Cancer Res. 2018, 78 (1), 265–277. [DOI] [PubMed] [Google Scholar]
- (7).Stengel C; Newman SP; Leese MP; Potter BVL; Reed MJ; Purohit A Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2010, 102 (2), 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen J; Liu T; Dong XW; Hu YZ Recent development and SAR analysis of colchicine binding site inhibitors. Mini-Rev. Med. Chem 2009, 9 (10), 1174–1190. [DOI] [PubMed] [Google Scholar]
- (9).Yue QX; Liu XA; Guo DA Microtubule-binding natural products for cancer therapy. Planta Med. 2010, 76 (11), 1037–1043. [DOI] [PubMed] [Google Scholar]
- (10).Wu XX; Wang QH; Li W Recent advances in heterocyclic tubulin inhibitors targeting the colchicine binding site. Anti-Cancer Agents Med. Chem 2016, 16 (10), 1325–1338. [DOI] [PubMed] [Google Scholar]
- (11).Dong M; Liu F; Zhou H; Zhai S; Yan B Novel natural product- and privileged scaffold-based tubulin inhibitors targeting the colchicine binding site. Molecules 2016, 21 (10), 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rosner M; Capraro HG; Jacobson AE; Atwell L; Brossi A; Iorio MA; Williams TH; Sik RH; Chignell CF Biological effects of modified colchicines - improved preparation of 2-demethylcolchicine, 3-demethylcolchicine, and (+)-colchicine and reassignment of the position of the double-bond in dehydro-7-deacetamidocolchicines. J. Med. Chem 1981, 24 (3), 257–261. [DOI] [PubMed] [Google Scholar]
- (13).Ringel I; Jaffe D; Alerhand S; Boye O; Muzaffar A; Brossi A Fluorinated colchicinoids - antitubulin and cytotoxic properties. J. Med. Chem 1991, 34 (11), 3334–3338. [DOI] [PubMed] [Google Scholar]
- (14).Semenov VV; Kiselyov AS; Titov IY; Sagamanova IK; Ikizalp NN; Chernysheva NB; Tsyganov DV; Konyushkin LD; Firgang SI; Semenov RV; Karmanova IB; Raihstat MM; Semenova MN Synthesis of antimitotic polyalkoxyphenyl derivatives of combretastatin using plant allylpolyalkoxybenzenes. J. Nat. Prod 2010, 73 (11), 1796–1802. [DOI] [PubMed] [Google Scholar]
- (15).Hamel E; Ho HH; Kang GJ; Lin CM Cornigerine, a potent antimitotic colchicum alkaloid of unusual structure - interactions with tubulin. Biochem. Pharmacol 1988, 37 (12), 2445–2449. [DOI] [PubMed] [Google Scholar]
- (16).Chen JJ; Wang Z; Li CM; Lu Y; Vaddady PK; Meibohm B; Dalton JT; Miller DD; Li W Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. J. Med. Chem 2010, 53 (20), 7414–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen JJ; Li CM; Wang J; Ahn S; Wang Z; Lu Y; Dalton JT; Miller DD; Li W Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization. Bioorg. Med. Chem 2011, 19 (16), 4782–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen J; Ahn S; Wang J; Lu Y; Dalton JT; Miller DD; Li W Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem 2012, 55 (16), 7285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Xiao M; Ahn SJ; Wang J; Chen JJ; Miller DD; Dalton JT; Li W Discovery of 4-aryl-2-benzoyl-imidazoles as tubulin polymerization inhibitor with potent antiproliferative properties. J. Med. Chem 2013, 56 (8), 3318–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hwang DJ; Wang J; Li W; Miller DD Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med. Chem. Lett 2015, 6 (9), 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).van Otterlo WAL; Ngidi EL; de Koning CB Sequential isomerization and ring-closing metathesis: masked styryl and vinyloxyaryl groups for the synthesis of benzo-fused heterocycles. Tetrahedron Lett. 2003, 44 (34), 6483–6486. [Google Scholar]
- (22).Chen JJ; Ahn S; Wang J; Lu Y; Dalton JT; Miller DD; Li W Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem 2012, 55 (16), 7285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Banerjee S; Arnst KE; Wang Y; Kumar G; Deng S; Yang L; Li G.-b.; Yang J; White SW; Li W; Miller DD Heterocyclicfused pyrimidines as novel tubulin polymerization inhibitors targeting the colchicine binding site: structural basis and antitumor efficacy. J. Med. Chem 2018, 61 (4), 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lu Y; Chen J; Xiao M; Li W; Miller DD An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res 2012, 29 (11), 2943–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Safa AR Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem.: Anti-Cancer Agents 2004, 4 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Prota AE; Bargsten K; Zurwerra D; Field JJ; Diaz JF; Altmann KH; Steinmetz MO Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339 (6119), 587–590. [DOI] [PubMed] [Google Scholar]
- (27).Prota AE; Magiera MM; Kuijpers M; Bargsten K; Frey D; Wieser M; Jaussi R; Hoogenraad CC; Kammerer RA; Janke C; Steinmetz MO Structural basis of tubulin tyrosination by tubulin tyrosine ligase. J. Cell Biol 2013, 200 (3), 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Charbaut E; Curmi PA; Ozon S; Lachkar S; Redeker V; Sobel A Stathmin family proteins display specific molecular and tubulin binding properties. J. Biol. Chem 2001, 276 (19), 16146–16154. [DOI] [PubMed] [Google Scholar]
- (29).Dorleans A; Gigant B; Ravelli RB; Mailliet P; Mikol V; Knossow M Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (33), 13775–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang Y; Zhang H; Gigant B; Yu Y; Wu Y; Chen X; Lai Q; Yang Z; Chen Q; Yang J Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016, 283 (1), 102–111. [DOI] [PubMed] [Google Scholar]
- (31).Wang Y; Yu Y; Li GB; Li SA; Wu C; Gigant B; Qin W; Chen H; Wu Y; Chen Q; Yang J Mechanism of microtubule stabilization by taccalonolide AJ. Nat. Commun 2017, 8, 15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (33).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (34).Adams PD; Grosse-Kunstleve RW; Hung LW; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.