

Abstract
Introduction:
Inflammatory myofibroblastic tumor (IMT), a locally aggressive neoplasm capable of metastasis, may show an IgG4-rich lymphoplasmacytic infiltrate. Prior reports suggest that storiform-fibrosis and obliterative phlebitis aid in the distinction of IMT from IgG4-related diseases. Herein, we highlight the morphologic overlap between the two diseases, and emphasize the importance of a multiplex fusion assay in the distinction of IgG4-RD from IMT.
Methods:
We identified 7 IMTs with morphologic and immunohistochemical features of IgG4-RD; 3 patients were originally diagnosed with IgG4-RD. Demographic, clinical and morphologic data was recorded. We also re-evaluated 56 patients with IgG4-RD. We performed immunohistochemistry for IgG4, IgG, ALK and ROS1. In situ hybridization for IgG4 and IgG was performed in selected cases. A multiplex next-generation sequencing (NGS) based RNA assay for gene fusions was performed to detect all known IMT-related gene fusions.
Results:
All 7 IMTs showed a dense lymphoplasmacytic infiltrate and storiform-type fibrosis, with obliterative phlebitis noted in 3 cases. The neoplastic stromal cells constituted <5% of overall cellularity and stromal atypia was either absent or focal and mild. Elevated numbers of IgG4 positive cells and increased IgG4 to IgG ratio was identified in all cases. Four cases showed ALK related abnormalities; while two patients showed ROS1 and NTRK3 fusions. One tumor was negative for known IMT-related gene fusions. All 56 IgG4-RD cases were negative for ALK and ROS1 on immunohistochemistry; 6 cases were negative on the fusion assay.
Conclusion:
Highly-inflamed IMTs are indistinguishable from IgG4-RD both histologically and on immunohistochemistry for IgG4. We advocate scrutinizing patients with presumptive single organ IgG4-RD for IMT and the diagnostic algorithm should include ALK and ROS1 immunohistochemistry and, in selected cases, a NGS-based fusion assay that covers known IMT-associated gene fusions.
Keywords: Inflammatory myofibroblastic tumor, IgG4-related disease, ALK, ROS1
Introduction:
Inflammatory myofibroblastic tumor (IMT), a mesenchymal neoplasm of intermediate biological potential characterized by a prominent inflammatory infiltrate, is increasingly defined by recurring genomic alterations.(1–4) Among these, fusions involving ALK and ROS1 have emerged as common genetic abnormalities, although recent efforts have uncovered a range of other alterations.(1–4) The morphologic heterogeneity is widely recognized in IMTs, ranging from low-grade spindle cell neoplasms to high-grade sarcomas and tumors with an epithelioid phenotype (5–7); the wide morphologic spectrum increases reliance on immunohistochemical and/or genetic testing. The success of kinase inhibitors such as Crizotinib have greatly increased the importance of a precise diagnosis of IMT.(8)
IgG4-related disease (IgG4-RD), a multi-system IgG4-rich fibroinflammatory disease, predominantly affects elderly patients and responds, generally swiftly, to immunosuppressive therapy. IMT, a close mimic of IgG4-RD (9, 10), may also show elevated numbers of IgG4 positive cells (11–14). However, storiform-type fibrosis and obliterative phlebitis, believed to represent characteristic histologic features of IgG4-RD, are generally not observed in IMT.(11–14)
The study was prompted by the identification of IMT-associated genetic abnormalities in two patients initially diagnosed as IgG4-RD. Herein, we illustrate a series of IMTs with histologic features indistinguishable from IgG4-RD and evaluate the value of next-generation sequencing (NGS) in this scenario. The study also surveys a cohort of IgG4-RD patients evaluated at this institution to identify undiagnosed IMTs.
Materials and Methods
We identified 7 IMTs histologically and immunohistochemically resembling IgG4-RD; 3 received in consultation by one of the authors (VD) and the other 4 from our institutional files. Three cases were initially diagnosed as IgG4-RD. To uncover other IMTs that may have been misinterpreted as IgG4-RD, we assembled a cohort of 56 patients diagnosed and treated as IgG4-RD at this institution. We also evaluated 4 additional pulmonary IMTs and 5 additional patients with IgG4-related pulmonary disease. The hematoxylin and eosin stained slides were reviewed for histological features of IgG4-RD; specifically, storiform type fibrosis and obliterative phlebitis. Elastic stains were performed on selected cases.
Immunohistochemistry
Immunohistochemistry for IgG4 and IgG was performed using antibodies to IgG4 (1:200 dilution, Zymed) and IgG (1;3000 dilution, Dako).(15) Antigen retrieval was conducted after protease digestion, and antigen detection was achieved using UltraView diaminobenzidine chromogen (Ventana Medical Systems; Tucson, AZ). Three high power fields (HPF) with the highest number of IgG4-positive cells were identified and the mean counts in these fields were recorded. The number of IgG-positive plasma cells within these 3 fields was also recorded, enabling derivation of IgG4 to IgG ratio. Immunohistochemistry for ALK (clone 5A4, 1:50 dilution, Novacastra) and ROS1 (clone D4D6, 1:200, Cell Signaling Technology) was performed.
An in-situ hybridization approach was used in selected cases in which either the IgG4 or IgG preparation showed marked non-specific background reactivity. Briefly, RNA in-situ hybridization probes (Affymetrix, Santa Clara, CA) were designed against the IgG4 and IgG transcripts as identified in the NCBI nucleotide database. The IgG4 probe is isotype-specific, while the IgG probe targets RNA sequences to all subclasses of IgG. Additional details regarding this in-situ hybridization platform are available in a prior publication.(16)
Fluorescence in-situ hybridization (FISH)
ALK FISH was conducted on formalin-fixed and paraffin-embedded tissue using a dual-color break-apart probe specific to the ALK locus (Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe; Abbott Molecular). To detect ROS1 rearrangement we used a break-apart FISH approach using BAC clones corresponding to the 5′ (RP11-835I21) and 3′(RP11-1036C2) sequences flanking the ROS1 gene labeled by nick translation in green and red. Samples were considered positive if more than 15% of lesional cells showed split signals.
Fusion assay
We used a clinically validated laboratory-developed assay based on Anchored Multiplex PCR (AMP) for targeted fusion transcript detection involving 51 genes using NGS.(17) cDNA was obtained off total nucleic acid extracted from formalin fixed paraffin embedded tumor tissue. ArcherDx FusionPlex Solid Tumor Kit primers were used in 2 hemi-nested PCR reactions and the library so prepared was sequenced on an Illumina NextSeq (2 × 150 base paired-end sequencing). A laboratory-developed algorithm was used for fusion transcript detection and annotation (version 2.1.0). The assay is designed to detect fusions involving the following genes: ALK (19-22, intron 19), BRAF (7-12, 15), EGFR (2-7 exon skipping/vIII variant, 7-9, 16, 20, 24, 25), EWSR1 (4-14), FGFR2 (2, 8-10, 17), MAML2 (2,3 ), MET (exon 14 skipping), NRG1 (1-3, 6), NUTM1 (3), RET (8-13), ROS1 (31-37), AKT3 (1-3), ARHGAP26 (2, 10-12), AXL (19,20), BRAF (7-12, 15), BRD3 (9-12), BRD4 (10, 11), ERG (2-11), ESR1 (3-6), ETV1 (3-13), ETV4 (2, 4-10), ETV5 (2, 3, 7-9), ETV6 (1-7), FGFR1 (2, 8-10, 17), FGFR3 (8-10, 17, intron 17), FGR (2), INSR (12-22), JAZF1 (2-4), MAML2 (2,3 ), MAST1 (7-9, 18-21), MAST2 (2, 3, 5, 6), MET (13, 15), MSMB (2-4), MUSK (7-9, 11-14), MYB (7-9, 11-16), NOTCH1 (2, 4, 26-31, internal exon 3-27 deletion), NOTCH2 (5-7, 26-28), NRG1 (1-3, 6), NTRK1 (8,10-13), NTRK2 (11-17), NTRK3 (13-16), NUMBL (3), PDGFRA (7, exon 8 deletion, 10-14), PDGFRB (8-14), PIK3CA (2), PKN1 (10-13), PPARG (1-3), PRKCA (4-6), PRKCB (3), RAF1 (4-7, 9-12), RELA (3, 4), RSPO2 (1, 2), RSPO3 (2), TERT (2), TFE3 (2-8), TFEB (1,2), THADA (28), and TMPRSS2 (1-6).
Results:
IMTs mimicking IgG4-RD (n=7)
This cohort showed a male: female ratio of 1:1.33 with a mean age of 38 years (range: 10-75 years) (table 1). There were 5 pulmonary tumors, 2 of which predominantly involved the trachea. One tumor each involved the pharynx and stomach. Three tumors were initially diagnosed as IgG4-RD (cases #1, 2 and 6). Multicentric involvement was not noted, and serum IgG4 was normal (n=3).
Table 1:
Clinical, histologic and molecular feature of inflammatory myofibroblastic sarcoma mimicking IgG4 related disease
| case | Age | Gender | Tumor size | Location | Histology | IgG4 counts | IgG4/IgG (%) | IHC for ALK/ROS1 | FISH/Translocation assay | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 49 | F | 1.5 cm | Lung | Storiform fibrosis, obliterative phlebitis, dense LP infiltrate | 52 | 42% | Negative | ETV6-NTRK fusion; FISH for ALK/ROS1 negative | ANED 26 months |
| 2 | 27 | F | 9.3 | Lung | Storiform fibrosis, obliterative phlebitis, dense LP infiltrate | 63 | 79% | Negative | TFG-ROS1 fusion ROS1 FISH positive | Recurrent tumor * |
| 3 | 33 | M | 2.9 | Trachea | Subtle storiform fibrosis, dense LP infiltrate | 77 | 55% | Negative | FISH for ALK negative; Fusion assay negative | Tumor recurred, LTFU after resection of recurrence |
| 4 | 44 | M | 2.0 | Trachea | Subtle storiform fibrosis, dense LP infiltrate | 82 | 61% | ALK positive, ROS1 negative | TFG-ALK fusion | ANED - LTFU after 12 months |
| 5 | 10 | M | 2.8 | Pharynx | Marked fibrosis with subtle storiform fibrosis, dense LP infiltrate | 344 | 85% | ALK positive, ROS1 negative | THBS1-ALK fusion | ANED 38 months |
| 6 | 75 | F | 2.5 cm | Stomach | Storiform fibrosis, obliterative phlebitis, dense LP infiltrate | 99 | 45% | ALK positive, ROS1 negative | FISH for ALK negative; fusion assay negative | ANED 15 months |
| 7. | 27 | F | 3.6 cm | Lung | Dense LP, storiform fibrosis | 86 | 45% | ALK positive, ROS1 negative | DCTN1-ALK fusion | ANED 24 months |
LP= lymphoplasmacytic
ANED = alive no evidence of disease
LTFU = lost to follow-up
following resection and discontinuing Crizotinib the tumor recurred. The recurrent tumor responded to Crizotinib. Currently the patient has no evidence of tumor.
Morphological and immunophenotypic overlap:
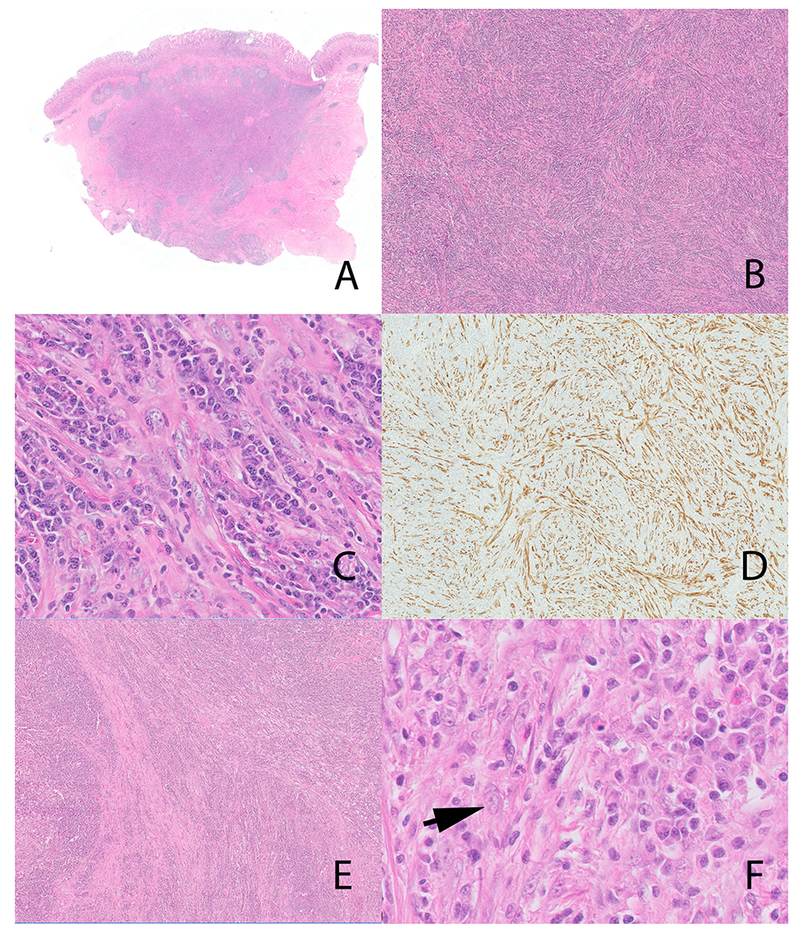
All 7 tumors showed a dense lymphoplasmacytic infiltrate with inconspicuous stromal cells. All tumors showed storiform fibrosis (Figures 1C; 2A; 3B,C), while obliterative phlebitis (Figure 1E, F) was noted in 3 cases. The atypia in stromal cells varied from absent (n=2) (Figure 1B) to mild (n=5) (Figure 2B, 3C). Even with cases showing mild atypia, the overwhelming majority of stromal cells lacked atypia. The stromal cells were virtually indistinguishable from similar cells seen in IgG4-RD. Notably, lymphocytes, plasma cells and occasional eosinophils constituted the most prevalent cells, outnumbering stromal cells, often by a ratio of >20:1 (Figure 1B, 2A, 3C).
Figure 1:
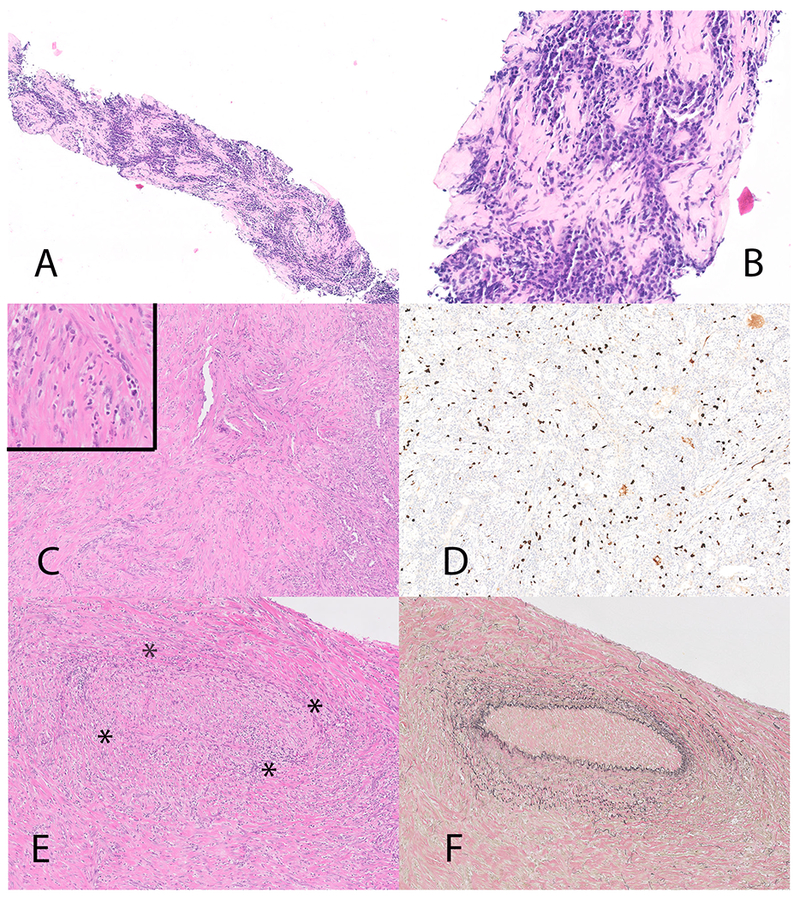
Inflammatory myofibroblastic tumor with TFG-ROS1 fusion (case 2). Biopsy on Panel A and B, thoracoscopic biopsy on Panel C, D, E and F. The needle biopsy (Panel A and B) showed a dense plasma cell infiltrate and elevated numbers of IgG4 positive cells (not illustrated). The thoracoscopic biopsy (panel C and inset) although less inflamed, shows prominent stromal cells with atypia. Diffuse increase in IgG4 positive plasma cells (immunohistochemical stain for IgG4) (Panel D). Panel E inset shows a focus of obliterative phlebitis (* highlights the outlines of the obliterated vein). The obliterated vein is highlighted on an elastic stain (Panel F).
Figure 2.
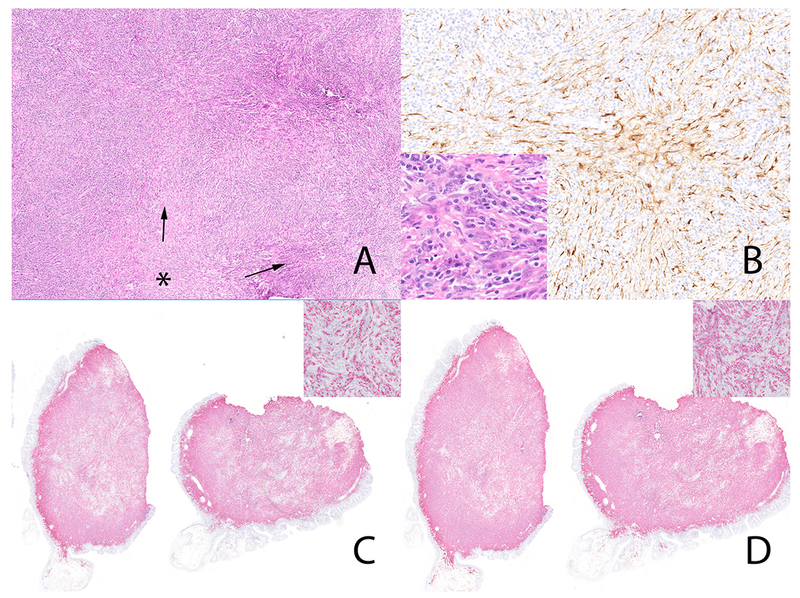
Inflammatory myofibroblastic tumor (case 5) with THBS1-ALK fusion. Storiform-type fibrosis (Panel A). * marks the center with fascicles of fibroinflammatory cells (arrow) emanating from this region. The neoplastic cells are diffusely positive for ALK (Panel B) Note the occasional stromal cells with vesicular nuclei (Panel B, inset). The plasma cells are diffusely positive for IgG4 in-situ hybridization (Panel C and inset) and IgG in-situ hybridization (Panel D and inset). The IgG4 to IgG ratio was > 90%.
Figure 3.
Inflammatory myofibroblastic tumor with ETV6-NTRK3 fusion (case 1). The lung lesion is well circumscribed (Panel A). Note the storiform-type fibrosis (Panel B). On high-power the inflammatory cells dominate with only occasional stromal cells with vesicular nuclei (Panel C); storiform-type fibrosis is also seen. Immunohistochemical stain for IgG4 showing an IgG4-rich infiltrate (Panel D).
All cases showed increased numbers of IgG4 positive plasma cells as well as an IgG4 to IgG ratio that exceeded 40% (table 1) (Figure 1D; 2C, D; 3D). Four tumors (cases #4-7) were positive for ALK (Figure 2B). All tumors were negative for ROS1.
Genetic alterations:
The genetic alterations included three ALK related fusions, and one patient each with ROS1 and NTRK3 fusions (Figure 5). Although showing morphologic overlap with IgG4-RD, the stromal cells in three cases with ALK alterations showed strong ALK reactivity on immunohistochemistry, distinguishing this disease from IgG4-RD. NGS-based assay identified 3 unique fusion transcripts: TFG-ALK, THBS1-ALK and DCTN1-ALK (Figure 5B). One notable tumor (case 6), although positive for ALK immunohistochemistry, lacked genetic alteration on FISH as well as NGS, raising the possibility of an alternative mechanism of ALK activation (see discussion). Finally, case #3, classified as an IMT, lacked a detectable genetic alteration, but showed stromal atypia beyond that typically seen in IgG4-RD (Figure 4F).
Figure 5:
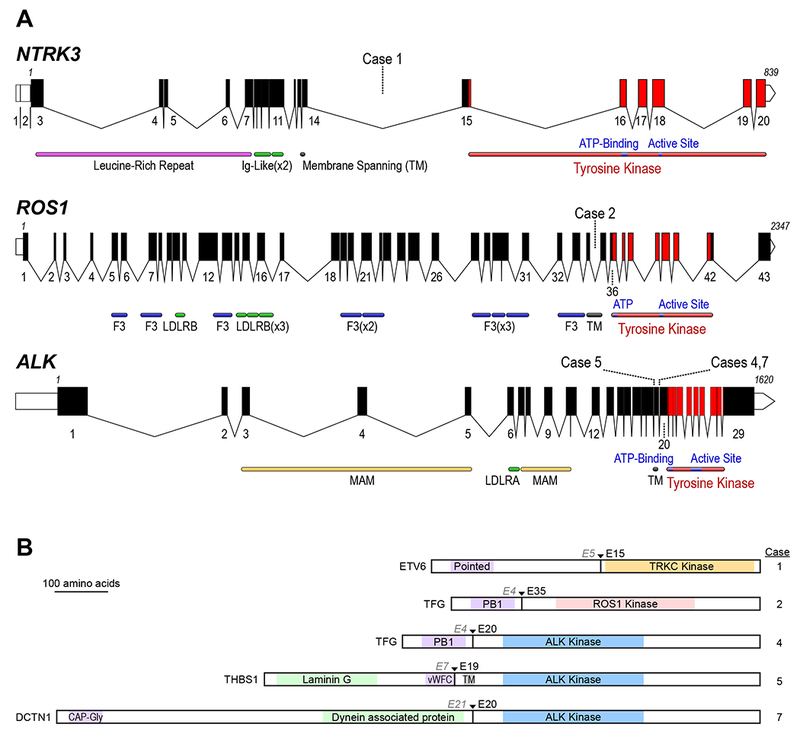
A Schematic of the human ALK, ROS1, and NTRK3 loci. Exon numbers are shown below their respective boxes for RefSeq ALK transcript variant 1 (NM_004304.4), ROS1 (NM_006180.4), and NTRK3 transcript variant 1 (NM_001012338.2). Fusion breakpoints are shown as dotted lines for the indicated cases; all breakpoints are intronic. Note that the three schematics are not at the same scale (amino acid residue numbers are listed above first and last exons), exons are drawn at a larger scale than introns, and introns are not drawn to the same scale for each gene (ALK locus is ~739 kB, ROS1 is ~137 kB, and NTRK3 is ~384 kB). Domains LDLR: LDL Receptor (A/B); F3: Fibronectin type III; MAM: meprin, A-5, mu-receptor; TM: Transmembrane Helix). B: Schematic of predicted fusion protein products (see also Table 1). Triangles and Ex notation indicate the fusion breakpoints, preceding partner gene exon, and subsequent kinase exon. Purple shaded domains are those predicted or shown to induce dimerization or trimerization in the fusion partner (Pointed: sterile alpha motif (SAM) / helix loop helix (HLH) oligomerization domain; PB1: Phox and Bem1p interaction domain; vWFC von Willebrand Factor C domain; CAP-Gly: cytoskeleton-associated protein glycine-rich domain); Green shaded domains are other annotated sequence features. Gray shaded TM: transmembrane domain. Proteins are drawn to scale (DCTN1-ALK fusion = 1383 AA).
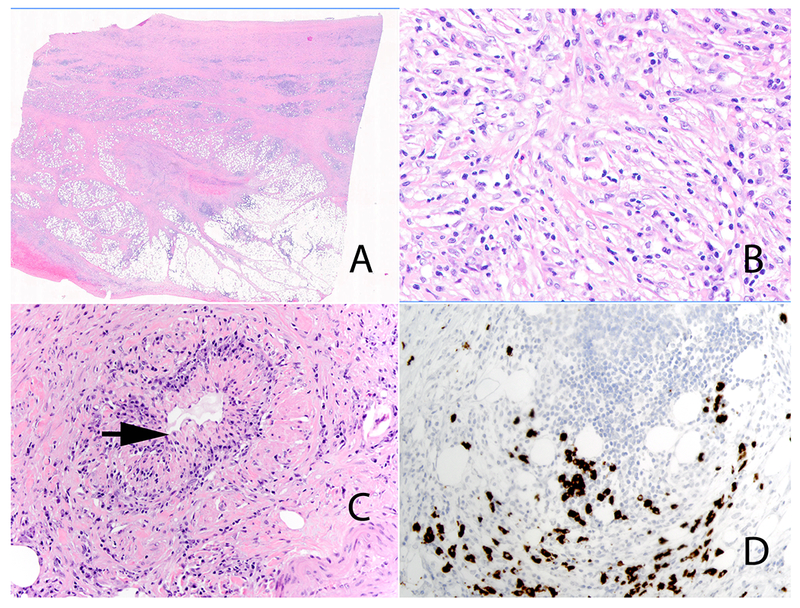
Figure 4:
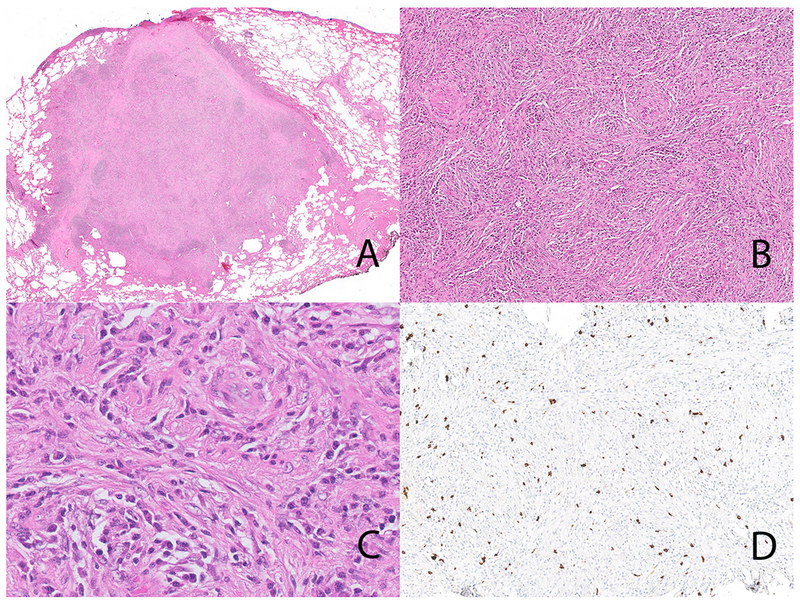
Gastric inflammatory myofibroblastic tumor (case #6), Panel A-D. The tumor involved the submucosa and muscularis propria (Panel A) and showed a prominent storiform-type pattern (Panel B). Occasional larger cells with vesicular nuclei are present (Panel C). These cells are diffusely positive for ALK (Panel D, immunohistochemistry). Tracheal tumor, likely representing an inflammatory myofibroblastic tumor (case #3) (Panel E and F). The tumor shows a prominent storiform-type pattern (Panel E). Note the stromal cells, similar to those depicted in figures 1–3 (Panel F) (arrow). However, the fusion panel was negative for IMT-related genetic alterations.
Both patients with non-ALK rearrangements were originally diagnosed as IgG4-RD. Case 1 showed histological (dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis) and immunohistochemical (elevated numbers of IgG4 plasma cells and elevated IgG4 to IgG ratio) features of IgG4-RD. The diagnoses was revised following the identification of ETV6-NTRK3 and TFG-ROS1 fusion.
Case #2, a 27-year old female, presented with difficulty in swallowing. A PET-CT showed a lobulated mass in the left lower lung measuring 9.3×5.7 cm. A needle biopsy revealed a dense lymphoplasmacytic infiltrate with storiform-type fibrosis and increased IgG4 positive cells, prompting a diagnosis of IgG4-RD (Figure 1A, B). She received 60 mg per day of prednisone. The lack of radiographic response prompted weekly rituximab for 8 weeks. Post-rituximab CT showed no improvement. Her serum IgG4 was not elevated [70.6 mg/dL (normal=3.9-86.4mg/dL)]. Areas indistinguishable from IgG4-RD were identified on thoracoscopic biopsy (dense lymphoplasmacytic infiltrate, increased IgG4 positive plasma cells and elevated IgG4 to IgG ratio). Foci with prominent stromal cells were also detected, a minority showed mild cellular atypia (Figure 1C,D,F). Next-generation sequencing identified a TFG-ROS1 transcript, a finding supported by FISH for ROS1 (Figure 5B). She was treated with oral Crizotinib for 3 months with marked symptomatic improvement and underwent esophagus sparing resection. Histologically, the mass, replaced by large zones of fibrosis and calcification, lacked viable tumor cells.
IgG4-RD cohort (n=56)
We also re-evaluated surgical resections from a cohort of 56 patients with established IgG4-RD. The mean age was 56 years and the male: female ratio was 2.2:1. The organs examined included the pancreas (22), lung, airways and pleura (10), salivary gland (7), lymph node (6), gallbladder (3), liver (2), retroperitoneum (2), bile duct (1), ampulla (1), appendix (1) and breast (1). Forty-seven (84%) cases showed storiform fibrosis, while obliterative phlebitis was identified in 41 (73%) (table 2). As with IMTs, inflammatory cells dominated, outnumbering stromal cells by a ratio of >20:1. The mean IgG4 count was 143 per HPF (range 22-550); the mean IgG4 to IgG ratio was 60.7% (range 21-99%). The patients responded favorably to steroids and/or Rituximab. The cases were also critically evaluated for atypia in the stromal compartment; atypia ranged from absent to mild (Figure 6B). Immunohistochemical stains for ALK and ROS1 were negative in all cases. NGS-based RNA assay for gene fusions was performed on a subset of cases (n=6) with mild nuclear atypia, similar to that observed in the IMT cohort; the 6 cases were negative for fusions involving ALK, ROS1, NTRK3, RET and PDGFRB.
Table 2:
Histology and immunohistochemistry data on patients with an established diagnosis of IgG4 related disease. All cases were negative for ALK and ROS1 immunohistochemistry
| No. | Site | Storiform fibrosis | Obliterative phlebitis | IgG4 | IgG | IgG4/IgG |
|---|---|---|---|---|---|---|
| 1 | Breast | Yes | Yes | 47 | 187 | 25 |
| 2 | Lung | Yes | Yes | 121 | 458 | 27 |
| 3 | Pancreas | Yes | Yes | 121 | 140 | 87 |
| 4 | Pancreas | Yes | Yes | 93 | 210 | 44 |
| 5 | Pancreas | Yes | Yes | 37 | 50 | 75 |
| 6 | Pancreas | Yes | Yes | 187 | 294 | 63 |
| 7 | Ampulla | Yes | Yes | 160 | 336 | 48 |
| 8 | Appendix | Yes | Yes | 61 | 173 | 35 |
| 9 | Lymph node | No | No | 383 | 850 | 45 |
| 10 | Gallbladder | Yes | No | 51 | 60 | 86 |
| 11 | Arytenoid | Yes | No | 84 | 336 | 25 |
| 12 | Pancreas | Yes | Yes | 154 | 262 | 59 |
| 13 | Lymph node | No | No | 138 | 190 | 73 |
| 14 | Gallbladder | Yes | Yes | 32 | 55 | 58 |
| 15 | Pancreas | Yes | Yes | 187 | 196 | 95 |
| 16 | Pancreas | Yes | Yes | 126 | 175 | 72 |
| 17 | Trachea | Yes | Yes | 47 | 112 | 42 |
| 18 | Lung | Yes | Yes | 200 | 220 | 91 |
| 19 | Salivary gland | Yes | Yes | 112 | 178 | 63 |
| 20 | Pancreas | Yes | Yes | 290 | 304 | 95 |
| 21 | Pancreas | Yes | Yes | 257 | 336 | 76 |
| 22 | Lymph node | No | No | 147 | 200 | 74 |
| 23 | Pancreas | Yes | Yes | 136 | 290 | 47 |
| 24 | Pancreas | Yes | Yes | 220 | 458 | 48 |
| 25 | Pancreas | Yes | Yes | 154 | 187 | 83 |
| 26 | Retroperitoneum | Yes | Yes | 112 | 234 | 48 |
| 27 | Salivary gland | Yes | Yes | 70 | 173 | 41 |
| 28 | Pancreas | Yes | Yes | 364 | 369 | 99 |
| 29 | Pancreas | Yes | Yes | 28 | 37 | 75 |
| 30 | Pancreas | Yes | No | 35 | 74 | 47 |
| 31 | Pancreas | Yes | No | 178 | 238 | 75 |
| 32 | Lymph node | No | No | 176 | 224 | 79 |
| 33 | Pancreas | Yes | Yes | 138 | 148 | 93 |
| 34 | Salivary gland | Yes | Yes | 93 | 215 | 43 |
| 35 | Salivary gland | Yes | Yes | 290 | 322 | 90 |
| 36 | Lymph node | No | No | 134 | 353 | 38 |
| 37 | Pancreas | Yes | Yes | 65 | 313 | 21 |
| 38 | Pancreas | Yes | Yes | 260 | 270 | 96 |
| 39 | Pancreas | Yes | Yes | 134 | 320 | 42 |
| 40 | Bile duct | Yes | No | 94 | 157 | 60 |
| 41 | Gallbladder | Yes | Yes | 41 | 75 | 55 |
| 42 | Liver | Yes | No | 94 | 114 | 82 |
| 43 | Trachea | Yes | Yes | 145 | 282 | 51 |
| 44 | Salivary gland | Yes | Yes | 160 | 190 | 84 |
| 45 | Lung | Yes | Yes | 164 | 210 | 78 |
| 46 | Pancreas | Yes | Yes | 41 | 67 | 61 |
| 47 | Pancreas | Yes | Yes | 110 | 240 | 46 |
| 48 | Retroperitoneum | Yes | Yes | 69 | 200 | 35 |
| 49 | Pleura | No | No | 105 | 200 | 53 |
| 50 | Orophaynx | No | No | 75 | 210 | 36 |
| 51 | Lung | Yes | Yes | 200 | 350 | 57 |
| 52 | Salivary gland | Yes | Yes | 126 | 177 | 71 |
| 53 | Liver | No | No | 82 | 150 | 55 |
| 54 | Salivary gland | Yes | Yes | 110 | 250 | 44 |
| 55 | Lung | Yes | Yes | 230 | 500 | 46 |
| 56 | Lymph node | No | No | 550 | 850 | 65 |
Figure 6:
IgG4-related retroperitoneal fibrosis. Low power view (Panel A). High-power view highlights the prominent population of stromal cells with mild nuclear atypia in the background of lymphocytes and plasma cells (Panel B). Obliterative phlebitis (Panel C) (arrow). Immunohistochemical stain for IgG4 showing increased numbers of IgG4 positive plasma cells (Panel D).
Comparison of Pulmonary IMT cases with Pulmonary IgG4-RD
We compared the 6 pulmonary IMTs (2 from this series and 4 non-IgG4-RD mimics) with seven patients with IgG4-related pulmonary disease (2 from this series and 5 independent cases). The most distinctive histologic difference between IgG4-related pulmonary disease and IMT lay in the characteristic pattern of involvement of the adjacent lung: patients with IgG4-RD showed extension of the cellular fibroinflammatory infiltrate (5 of 7 cases) along the airways, interlobular septa and accompanying pulmonary veins, a finding not observed in IMTs. It should be noted that perilesional acellular fibrous septa were seen in both IMTs and IgG4-RD.
Discussion
IgG4-RD and IMT can involve virtually every organ, with both diseases characterized by a rich inflammatory infiltrate. Prior studies have alluded to the immunohistochemical overlap between IgG4-RD and IMT; a subset of IMTs may show an IgG4-rich inflammatory infiltrate. (11, 12, 14) Nevertheless, there has been a tacit understanding that the two diseases could be distinguished on routine histologic evaluation: 1) IgG4-RD is characterized by storiform-type fibrosis, obliterative phlebitis and an increase in IgG4 positive cells, 2) in contrast, IMTs lack these features and instead show a prominent population of spindle/epithelioid cells with nuclear atypia.(11, 12, 13), The current study underscores the inadequacy of some of these assumptions: highly-inflamed IMTs may be morphologically indistinguishable from IgG4-RD, particularly on needle biopsies. Both diseases may show storiform-type fibrosis, obliterative phlebitis, IgG4-rich plasma cell infiltrates and mild atypia in the stromal compartment. In these circumstances, the distinction is reliant on additional ancillary testing, including immunohistochemistry and/or FISH for ALK and ROS1. Our current generation NGS-based RNA assay for gene fusions detects all known IMT-related fusions: ALK, ROS1, NTRK3, RET, and PDGFRA. Of note, the assay is agnostic to the fusion partner, for example, the test can identify the more than a dozen fusions involving ALK.
In the largest study to date, Antonescu and co-workers evaluated 62 IMTs including 25 children and 37 adults. (1) Among these, 35 patients (56%) showed ALK gene rearrangement, 6 (10%) showed ROS1 gene rearrangement, with a single case of RET rearrangement. Other reported IMT-related kinase fusions include NTRK3 and PDGFRB.(3, 4) Thirty-two percent of IMTs lacked detectable gene fusions with adults accounting for 90% of fusion-negative IMTs.(1) Notably, two cases in the current series lacked detectable genetic alterations. Patient #6, showed strong unequivocal reactivity with ALK on immunohistochemistry but lacked a genetic correlate. However, given the morphologic appearance, the specificity of the ALK antibody, and alternative means of activating ALK (including amplification, point mutation, alternate transcription initiation), all of which would be difficult to diagnose given the very low cellularity of the neoplastic cells(18), the lesion is best characterized as an IMT. Patient #3 highlights the current challenge associated with distinguishing IgG4-RD from fusion negative IMT. Although negative for known fusions, the morphologic appearance, particularly stromal atypia, and the knowledge that 49% of adults lacked known genetic alterations, would support the diagnosis of an IMT.(1)
Immunohistochemistry for ALK is a robust surrogate marker for the corresponding fusion.(2,19) The sensitivity and specificity of ROS1 immunohistochemistry is >90%, although only a limited number of IMTs have been examined.(20, 21) In one study all 3 ROS1-rearranged IMTs showed immunohistochemical reactivity for ROS1.(2) However, another study reported 1 ROS1 immunohistochemistry negative IMT among 4 ROS-rearranged IMTs while a second case showed weak reactivity.(1) Notably, case#2 in our series, a ROS1-rearranged IMT, was negative on immunohistochemistry and the patient was only identified after she failed to respond to treatment for IgG4-RD. The antibody clone used in this study was similar to that used in prior publications.(1, 2) Collectively, while ALK serves as a robust marker of ALK-rearranged IMTs, ROS1 immunohistochemistry may be a less reliable surrogate marker in this context. Pan-TRK immunohistochemistry has been reported as a sensitive method to detect NTRK fusions, (22) although we have not evaluated it in the context of a highly-inflamed IMT. There are no reliable immunohistochemical markers for other IMT-associated kinase fusions: PDGFRB and RET.
In addition to IMTs, NTRK fusions have been identified in high-grade uterine sarcomas (NTRK1 and NTRK3 fusion)(23), lipofibromatosis-like neural tumors (NTRK1 fusion)(24), myopericytic neoplasms (NTRK1 fusion)(25) and infantile fibrosarcoma (NTRK3 fusion). Given that recurrent NTRK fusions are identified in a variety of epithelial and mesenchymal neoplasms, the ultimate classification would necessarily rely on a careful morphologic evaluation. (26) Based on the diffuse lymphoplasmacytic infiltrate, the bland appearance of neoplastic cells and overlap with other highly-inflamed IMTs, case #1 is best classified as an IMT. Ultimately, given the remarkable success of tropomyosin receptor kinase (TRK) inhibitor that is agnostic to tumor type, the precise classification may prove a secondary consideration, with therapy driven by the fusion.(26)
Immunohistochemistry for ALK and ROS1 would nevertheless be the first line assay in biopsies showing a dense lymphoplasmacytic infiltrate with fibrosis/storiform fibrosis. NGS-based RNA assay for gene fusions, a powerful means of evaluating multiple genes, some with no immunohistochemical surrogates, may overcome false negative results of ROS1 immunohistochemistry. Although a second line test, NGS is nevertheless an essential component of the diagnostic algorithm. Finally, it must be emphasized that almost half of all adult IMTs currently lack a distinctive genetic signature. Morphology remains a critical element and any degree of stromal atypia should be viewed with suspicion, and a thorough work-up to exclude a diagnosis of IMT and identify other evidence of IgG4-RD would be warranted. An alternative approach would be to perform FISH for ALK and ROS1. A significant shortcoming of FISH testing is the low percentage of neoplastic cells in highly-inflamed IMTs; the neoplastic cells generally constitute <10% of total cellularity. In contrast, the sensitivity of the NGS-based RNA fusion is 5% or less. The evidence thus favors multiplex NGS-based assay over a FISH-based approach.
The prototypic patient with IgG4-RD is an elderly male with multiorgan involvement.(9, 27) However, a significant proportion of patients are younger than 50; although, pediatric disease remains uncommon. (9, 27) Thus, there is significant overlap in the demographic features of the two diseases. Additionally, one-third of patients show disease isolated to a single organ.(28) Furthermore, serum IgG4 is not elevated in about 50% of patients with IgG4-related disease; elevated serum IgG4 is more likely in patients with multi-organ disease.(28) In a study by Ebbo and co-workers, only 10% of patients with elevated serum IgG4 levels were diagnosed with IgG4-RD, highlighting the lack of specificity of this assay.(29) Given the significant clinical and histologic overlap, and the relatively large proportion of IMTs lacking detectable genetic alterations, the current study raises questions regarding our current diagnostic approach to IgG4-RD. While we did not identify additional misdiagnosed patients in our IgG4-RD cohort, it is conceivable that IMTs may have been mischaracterized as IgG4-RD. Our patients with IgG4-RD had long term follow-up and/or responded favorably to immunosuppression. We would therefore advocate that the possibility of an IMT should be strongly considered in young/middle aged patients and those with single organ disease, regardless of the histologic appearance. This distinction is important because IMT is a neoplasm with intermediate biological behavior with a potential for metastasis and surgical resection and/or targeted therapy is the mainstay of treatment. Conversely, IgG4-RD respond swiftly to steroids and surgical resection is unwarranted. Ironically, in the pre-IgG4-RD era, this disease was occasionally misdiagnosed as IMT.
The majority of our cases involved the respiratory system. The characteristic pattern of pulmonary involvement in IgG4-RD, extension of a cellular and inflamed component along the airway and interlobular septa would support the diagnosis of this fibroinflammatory disease. (30) Conversely, IMTs do not spread along lymphatic planes. However, these features are appreciable only on a resection specimen.
In conclusion, highly-inflamed IMTs are histologically and immunohistochemically indistinguishable from IgG4-RD. In a young patient or a patient with single organ disease, the pathologic diagnostic algorithm for IgG4-RD necessitates the use of immunohistochemistry for ALK and ROS1. NGS-based RNA fusion assay, a powerful means of distinguishing IMT from its inflammatory mimics, overcomes many of the shortcomings of the immunohistochemical platform and FISH based assays, and could represent a first-line ancillary test, albeit in selected cases. These guidelines are particularly applicable to biopsies, given that the stromal atypia may not be captured on a needle biopsy. The availability of highly effective targeted therapy for actionable kinases underscores the importance of distinguishing IgG4-RD from IMT, as illustrated by the dramatic response to Crizotinib in case 2. Conversely, IgG4-RD responds swiftly to immunosuppressive agents. Patients diagnosed with IgG4-RD but failing to respond to treatment should also prompt diagnostic reconsideration.
References:
- 1.Antonescu CR, Suurmeijer AJ, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hornick JL, Sholl LM, Dal Cin P, et al. Expression of ROS1 predicts ROS1 gene rearrangement in inflammatory myofibroblastic tumors. Mod Pathol. 2015;28:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016;69:72–83. [DOI] [PubMed] [Google Scholar]
- 5.Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61:428–437. [DOI] [PubMed] [Google Scholar]
- 6.Cessna MH, Zhou H, Sanger WG, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Mod Pathol. 2002;15:931–938. [DOI] [PubMed] [Google Scholar]
- 7.Marino-Enriquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35:135–144. [DOI] [PubMed] [Google Scholar]
- 8.Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363:1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–551. [DOI] [PubMed] [Google Scholar]
- 10.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–1192. [DOI] [PubMed] [Google Scholar]
- 11.Bhagat P, Bal A, Das A, et al. Pulmonary inflammatory myofibroblastic tumor and IgG4-related inflammatory pseudotumor: a diagnostic dilemma. Virchows Arch. 2013;463:743–747. [DOI] [PubMed] [Google Scholar]
- 12.Chougule A, Bal A, Das A, et al. A Comparative Study of Inflammatory Myofibroblastic Tumors and Tumefactive IgG4-related Inflammatory Lesions: the Relevance of IgG4 Plasma Cells. Appl Immunohistochem Mol Morphol. 2016;24:721–728. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto H, Yamaguchi H, Aishima S, et al. Inflammatory myofibroblastic tumor versus IgG4-related sclerosing disease and inflammatory pseudotumor: a comparative clinicopathologic study. Am J Surg Pathol. 2009;33:1330–1340. [DOI] [PubMed] [Google Scholar]
- 14.Saab ST, Hornick JL, Fletcher CD, et al. IgG4 plasma cells in inflammatory myofibroblastic tumor: inflammatory marker or pathogenic link? Mod Pathol. 2011;24:606–612. [DOI] [PubMed] [Google Scholar]
- 15.Ferry JA, Klepeis V, Sohani AR, et al. IgG4-related Orbital Disease and Its Mimics in a Western Population. Am J Surg Pathol. 2015;39:1688–1700. [DOI] [PubMed] [Google Scholar]
- 16.Shahid M, Mubeen A, Tse J, et al. Branched chain in situ hybridization for albumin as a marker of hepatocellular differentiation: evaluation of manual and automated in situ hybridization platforms. Am J Surg Pathol. 2015;39:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]
- 18.Wiesner T, Lee W, Obenauf AC, et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature. 2015;526:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha YJ, Lee JS, Kim HR, et al. Screening of ROS1 rearrangements in lung adenocarcinoma by immunohistochemistry and comparison with ALK rearrangements. PLoS One. 2014;9:e103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sholl LM, Sun H, Butaney M, et al. ROS1 immunohistochemistry for detection of ROS1-rearranged lung adenocarcinomas. Am J Surg Pathol. 2013;37:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Y, Goncalves T, Dias-Santagata D, et al. Immunohistochemical Detection of ROS1 Fusion. Am J Clin Pathol. 2017;147:77–82. [DOI] [PubMed] [Google Scholar]
- 22.Hechtman JF, Benayed R, Hyman DM, et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am J Surg Pathol. 2017;41:1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang S, Cotzia P, Hyman DM, et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype With Features of Fibrosarcoma. Am J Surg Pathol. 2018;42:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am J Surg Pathol. 2016;40:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haller F, Knopf J, Ackermann A, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol. 2016;238:700–710. [DOI] [PubMed] [Google Scholar]
- 26.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med. 2018;378:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahajan VS, Mattoo H, Deshpande V, et al. IgG4-related disease. Annu Rev Pathol. 2014;9:315–347. [DOI] [PubMed] [Google Scholar]
- 28.Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-Related Disease: Clinical and Laboratory Features in One Hundred Twenty-Five Patients. Arthritis Rheumatol. 2015;67:2466–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebbo M, Grados A, Bernit E, et al. Pathologies Associated with Serum IgG4 Elevation. Int J Rheumatol. 2012;2012:602809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui S, Yamamoto H, Minamoto S, et al. Proposed diagnostic criteria for IgG4-related respiratory disease. Respir Investig. 2016;54:130–132. [DOI] [PubMed] [Google Scholar]