

Abstract
α,β-dehydroamino acids (dhAAs) are noncanonical amino acids that are found in a wide array of natural products and can be easily installed into peptides and proteins. dhAAs exhibit remarkable synthetic flexibility, readily undergoing a number of reactions, such as polar and single-electron additions, transition metal catalyzed cross-couplings, and cycloadditions. Because of the relatively mild conditions required for many of these reactions, dhAAs are increasingly being used as orthogonal chemical handles for late-stage modification of biomolecules. Still, only a fraction of the chemical reactivity of dhAAs has been exploited in such biorthogonal applications. Herein, we provide an overview of the broad spectrum of chemical reactivity of dhAAs, with special emphasis on recent efforts to adapt such transformations for biomolecules such as natural products, peptides, and proteins. We also discuss examples of enzymes from natural product biosynthetic pathways that have been found to catalyze many similar reactions; these enzymes provide mild, regio- and stereoselective, biocatalytic alternatives for future development. We anticipate that the continued investigation of the innate reactivity of dhAAs will furnish a diverse portfolio dhAA-based chemistries for use in chemical biology and drug discovery.
Introduction.
α,β-dehydroamino acids (dhAAs, figure 1A) are not counted among the 20 proteinogenic amino acids yet they are abundant in nature (figure 1C–D).1 This seemingly simple functional group has unique physical and chemical properties that can significantly impact the structure and function of biomolecules that contain them. Structurally, dhAAs adopt a roughly planar conformation, with distinctive trans orientations about the phi and psi torsion.1–4 Overall, this makes dhAAs strong turn inducers and they promote peptide conformations that are not permitted with typical, saturated residues (figure 1B).5 In the context of natural products/peptides this translates to three-dimensional structures with increased structural rigidity and tighter target complementarity. Antibiotics such as the thiopeptides, thiocillin or cyclothiazomycin (figure 1D, 3) rely on the rigidifying effects of the.dhAAs, dehydroalanine (Dha) and dehydrobutyrine (Dhb), for their tight target engagement and potent activity.6,7 dhAAs can also increase proteolytic stability of peptides8 and can play direct chemical roles in the activity of natural products. For example, the dhAA in the cyanobacterial toxin Microcystin-LR, (figure 1D, 1) helps the macrocycle adopt a high affinity conformation for its targets, the protein phosphatase-1 and 2-Α, but also allows electrophilic trapping of a nucleophilic cysteine residue in the target.9,10 This is only some of the biological chemistry accessible to dhAAs.
Figure 1.
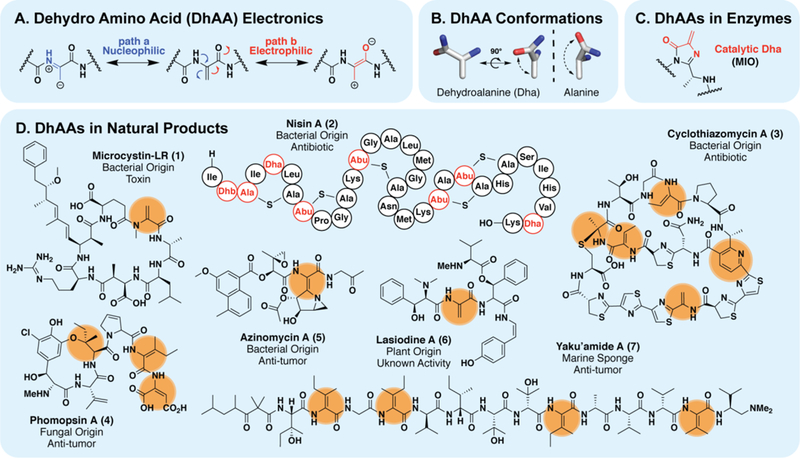
(A) Push-pull electronic structure of dhAAs makes them act as electrophiles or nucleophiles under given conditions. (B) dhAAs exhibit unusual conformational proclivity and restricted rotation relative to natural amino acids. (C) Some enzymes are armed with modified dhAAs that act as catalytic electrophiles to perform chemistry. (D) dhAAs are abundant in natural products from bacteria, fungi and plants. Additional chemistry can be performed on dhAAs to install more complex functionality, such as the pyridine ring in thiopeptides like cyclothiazomycin (3). The thioether and phenolic ether bonds, from cyclothiazomycin A (3) and phomopsin A (4) respectively, may also originate from dhAA modifications.
Figure 3.
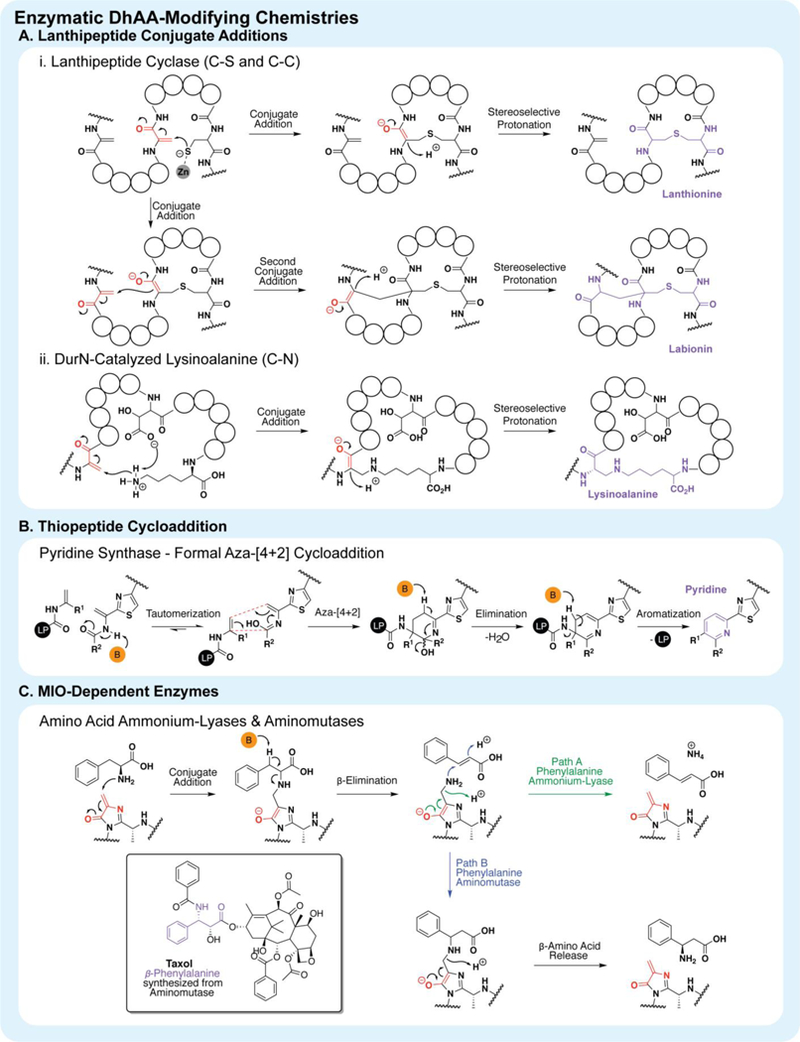
dhAAs also undergo enzymatic modification. (A) Enzymes involved in lantibiotic biosynthesis catalyze conjugate additions using S, C, and N nucleophiles. (B) Pyridine synthases from thiopeptide biosynthesis install pyridine rings via a formal aza-[4+2] cycloaddition between two Dhas. LP = leader peptide, B = base. (C) Amino acid ammonium-lyases and aminomutases use a modified dhAA to breakdown amino acids and generate β-amino acids, as in the cancer drug, Taxol.
dhAAs contain uniquely polar double bonds that are at once electron-rich due to conjugation with the amide nitrogen lone pair of electrons and electron poor due to the electron-withdrawing carbonyl. This distinctive electronic architecture makes dhAAs competent partners for a variety of chemical transformations. Notably, dhAAs have been implicated in nucleophilic additions, radical additions, transition metal catalyzed cross-coupling reactions, and cycloadditions, among others. The synthetic potential of dhAAs is now being realized in biological contexts due to the identification of milder and more selective activating reagents that work at ambient temperature and in aqueous media. Radical additions to dhAAs have been particularly important in this regard because such reactions are highly specific for dhAAs and result in new, unnatural amino acid derivatives. Certain enzymes are also capable of much of the same dhAA chemistry and key mechanistic insights have added new biocatalysts, in particular enzymes from natural product biosynthesis, to the toolbox dhAA-modifying chemistries. Thus, dhAAs are proving versatile chemical handles to incorporate designer modifications into peptides, natural products, and proteins.11–14 The chemical diversity that can be accessed through dhAA-modification has also spurred use in peptide display technologies and new applications in chemical biology and drug discovery.
In light of the expanding application of dhAAs in chemical biology, we have chosen here to review the fundamental chemistry of dhAAs with an emphasis on new, biorthogonal conditions. There are a handful of very excellent and useful reviews that provide comprehensive examples of dhAA chemistry in synthetic methodologies and total synthesis, but as yet, few that analyze such reactivity in the context of potential applications and challenges in chemical biology. Additionally, we cover examples of enzyme catalyzed dhAA modification from natural product biosynthesis and primary metabolism, as biocatalysis and synthetic biology are playing an increasingly important role in chemical biology. A detailed descriptions of methods for site-selective introduction of dhAAs into proteins and peptides is beyond the scope of this review. Many robust strategies have been reported, including oxidation of phenylselenocysteine incorporated by genetic codon expansion,15,16 chemical modification of protein cysteine side-chains,17 and enzymatic dehydration of serine and threonine residues,18 among others; interested readers are directed to a number of other useful reviews in this area.19–22 Ultimately, we hope this review will help to identify new opportunities for the unique reactivity of dhAAs to be further leveraged in the lab for elucidating new biology and augmenting drug discovery efforts.
I. Polar additions.
The combined electronic contributions of nitrogen and carbonyl substituents results in some complex reactivity between dhAAs and nucleophiles. Under sparingly acidic conditions with pi-aromatic nucleophiles and haloacids, the electron-rich character can dominate, leading preferentially to nucleophilic substitutions at the α-position of the dhAA (figure 2A).23 This dhAA reaction manifold has been used sparingly in select total syntheses and methodologies.23–25 Conjugate addition of diverse nucleophiles, which results from the electron poor character of dhAAs and proceeds under mildly basic conditions, leads to substitution at the β-carbon and allow access to unnatural amino acid derivatives. Thus, conjugate additions to dhAAs are versatile and valuable transformations that have been used widely in the literature and are finding new applications in late stage modifications of proteins, peptides, and dhAA-containing natural products.19,21,26–36
Figure 2.
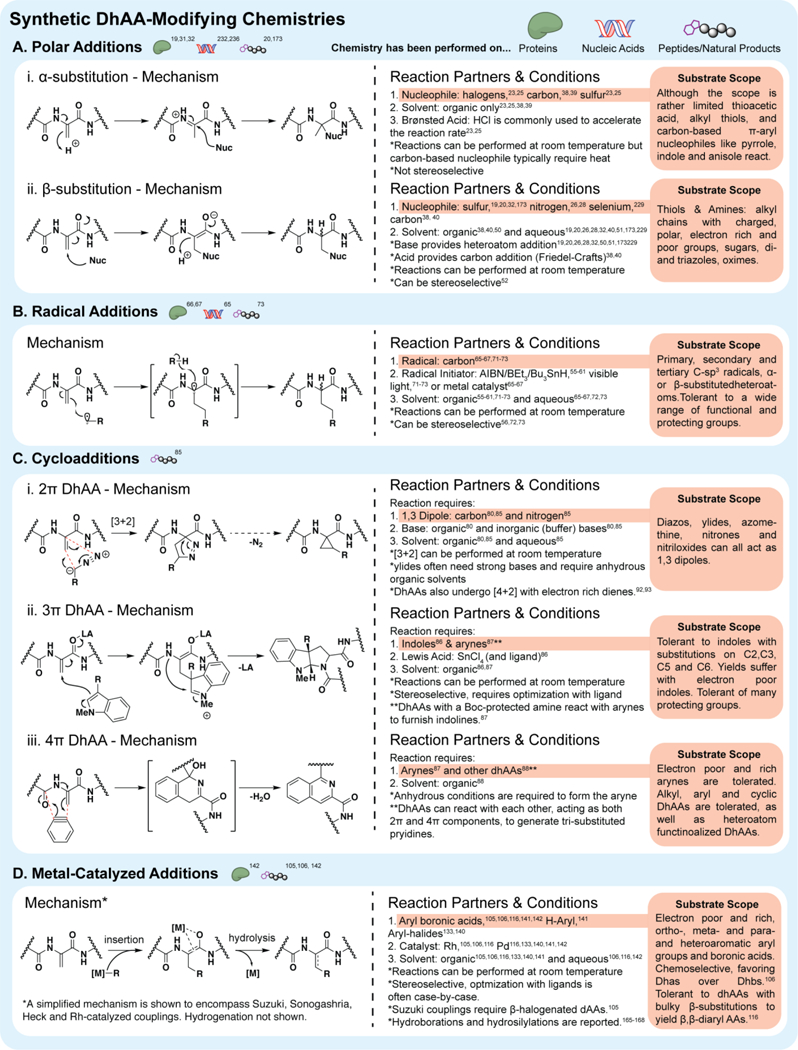
Common dhAA modifying chemistries include (A) nucleophilic additions, (B) radical additions, (C) cycloadditions and (D) metal-catalyzed cross-couplings. Representative or simplified mechanisms, general reaction conditions, substrate scope and additional notes are presented for each.
dhAAs readily undergo Friedel-Crafts type arylation reactions to give predominantly α-substituted products. Early reports showed that under dilute acidic conditions with haloacids, such as HBr and HCl, β-protonation can precede α-halogenation with dhAAs to give an α-halo amino acid.23 Alternatively, under Friedel-Crafts conditions with heat, in the presence of π-aryl nucelophiles, such as indole or anisole, direct α-arylation can be achieved.37–39 This chemistry has been exploited in several cases to prepare diverse α-heteroaromatic amino acids as anticonvulsants or selectively substituted diketopiperazines, en route to epipolythiodioxopiperazines.23–25 Although a detailed mechanistic analysis of this chemistry has not yet been reported, it seems apparent that the α-alkylation may in fact be a reaction manifold under thermodynamic control, as separate reports, under less forcing Friedel-Crafts conditions yield access to the β-arylated product, which has been used to make clavicipitic acid and Fischer indole fragments.38,40–45 Despite the limited uses of this chemistry, the value in new α,α-disubstituted amino acids bares potential for future applications.
As opposed to the aromatic substitution chemistry, conjugate additions to dhAAs with carbon, nitrogen, and sulfur nucelophiles have been very well studied (figure 2A). As early as 1965, Zahn and colleagues reported the conjugate addition of lysine to N-acetyldehydroalanine ethyl ester in presence of sodium hydroxide.46,47 Since then, the reaction has been used extensively to make a variety of substituted alanine derivatives. The most extensive work on substrate scope was done by Ferreira and coworkers, who compared conversions for reaction of N,N-diacyl dhAAs with thiols, dicarbonyl carbon nucleophiles, amines, and heterocycles under mild basic conditions.48–51 As expected, the reaction efficiency correlates with the strength of the nucleophile, although steric effects also play an important role, with primary amines (3–15 hours) for example reacting faster than secondary amines (up to 192 hours). Similarly, thiols react much faster than amines in the presence of organic bases such as triethylamine. Broadly, these findings have been further supported by recent computational work.26 In a particularly practical study, Ueda and coworkers showed that the conjugate addition of amines and thiols to dhAAs is greatly accelerated in water.51 In this case, the reaction between a model dhAA and benzylamine failed in DMF or THF after 48 hours, while the same reaction in methanol gives 77% yield after 120 hours and 91% after only 15 hours in water. The authors provide several explanations for the dramatic rate increase including reinforced hydrophobic interactions between reactants and/or stabilization of an activated complex through solvation or hydrogen bonding. They developed the scope of these reaction conditions with select amines and thiols, demonstrating how conjugate addition to dhAAs is could be particularly well-suited for protein modification.
One remaining challenge for dhAA conjugate addition chemistry is the lack of stereoselectivity and several groups have been working to address this shortcoming. Some of the most promising results in this regard have come from chiral thiourea catalysts, which were used to direct stereoselective conjugate additions between dhAAs and azalactones to access α,γ-diamino diacids.52 The thiourea catalyst is proposed to coordinate and activate each component by (1) acting as a base to promote enolization of the azalactone and (2) acting as a Brønsted acid to activate the dhAA β-carbon, while (3) only allowing attack from one face of the dhAA. While control over stereochemistry is impressive, it is highly sensitive to substitutions on the amine.
Given the robustness of conjugate additions to dhAAs in aqueous conditions, this chemistry has found wide application in peptide and protein modification.19,21,26–35 Optimized conditions for dhAAs in proteins have now been developed for thiols, as well as a range of nitrogen-nucleophiles such as amines, heterocycles, hydroxylamines and hydrazines.26,28 Overall, this chemistry allows site selective installation of unnatural amino acids and mimics of protein post translational modification (PTMs). Conjugate addition to dhAAs is increasingly preferred over other protein modifying techniques such as isothiocyanates and click chemistries because it results in less “unnatural scarring” and the chemical diversity that can be achieved far surpasses that of other modification techniques.11 These features have given rise to numerous applications.
One important application of thiol conjugate addition to dhAAs has been to explore biophysical impacts of protein PTMs. In a pair of proof-of-concept publications, groups lead separately by Davis19 and Schultz32 demonstrated site-specific incorporation of common PTMs via dehydroalanine handles. The former described the functionalization of a model protein, subtilisin (a serine protease), via a conjugate addition with a focused library of thiol nucleophiles after mild and site-selective installation of the dhAA.19 Thiols were specifically chosen to mimic the most common PTMs and included sodium thiophosphate, glycocysteine derivatives, glutathione, mono- di- and tri-methylamine ethane thiols and farnesyl thiol; all thiols readily reacted with the dhAA-bearing protein, exhibiting >95% conversion in 90 minutes in dilute and slightly basic phosphate buffer at 4°C. Similarly, the Schultz group introduced an acetylated lysine mimic along with mono-, di- and trimethyl-lysine analogs into histone H3 under comparable reaction conditions.32 To provide more evidence that the acetylated lysine PTM mimic is a good functional representation of the natural modification the authors showed that histone deacetylase (HDAC) 3 is capable of removing the acetyl group. The Davis group later went one step further with modified H3 proteins, incorporating several K9 PTM mimics (methylations and acetylation) and a phosphoserine 10 mimic.19 All modifications could be recognized and confirmed by primary antibodies to the natural PTMs and the phosphoserine mimic could be recognized by a chromatin “reader” protein. Together these data verify this technology could be used to identify and assess interaction of both “reader” and “eraser” chromatin-modifiers despite giving diastereomeric mixtures.
The facile incorporation of thiol nucleophiles into dhAAs can also be used to study enzyme function and design. For instance, ubiquitin-based dhAA probes have been used extensively to profile the families of enzymes involved in the modification of protein ubiquitinylation states. Brik and coworkers exploited the reactivity of dhAAs by developing it into an electrophilic trap to capture deubiquitinases by their catalytic cysteines.30 In this way, di-ubiquitin (di-Ub) probes bearing reactive dhAAs near different cleavage sites were prepared and could be used to pull-down and profile the specificity of deubiquitinating enzymes. Similarly, introduction of a reactive dhAA “warhead” on the c-terminus of a ubiquitin probe could be used to sequentially trap E1, E2 and E3 enzymes.29 Alternatively, dhAA conjugate additions allow new mechanistic insights by providing novel isosteres of active site residues. Thus, aza-michael additions with simple azoles granted access to isosteres of key histidines 44 and 47 in the active site of pantothenate synthetase (PanC).28 Subsequent kinetic analyses of these iso-histidine mutants emphasized the importance of these residues in activation of ATP through key binding interactions. Since histidine is a common catalytic residue, this approach could have profound impact across many important enzyme families. Lastly, Pearson and coworkers have used thiol-dhAA conjugate addition to alter substrate specificity and produce novel activity in the aldolase, N-acetylneuraminic acid lyase (NAL).27 By individually introducing a dhAA into 12 positions in the active site of NAL and individually modifying them with 13 thiol nucleophiles, the authors produced a library of 156 mutant NALs. Subsequent kinetic assays revealed an NAL mutant bearing a 2,3-dihydroxypropyl cysteine that exhibited a 10-fold increase in kcat/Km relative to wild type for processing an otherwise poorly accepted substrate (erythrose). Crystallographic data of the more active mutant allowed the authors to rationalize the activity based on transition state stabilization.
Cumulatively, these examples of protein modification demonstrate key advantages over other protein modification techniques. Libraries of mutant proteins can be efficiently produced with a single enzyme preparation, whereas a technique like amber codon suppression requires the expression and purification of each mutant individually, often times with a significant sacrifice in yield. Additionally, new mutants require involved artificial evolution experiments. Chemical mutagenesis is also capable of a wide range of protein mutants which allows the researcher to propose more focused hypotheses. Conjugate additions into dhAAs have proven to be a powerful strategy to synthesize new unnatural amino acids and incorporate unnatural functionality into proteins as cargo, probes, and catalytic residues.
II. Radical additions.
Given the olefinic character of dhAAs, it is not surprising that they can undergo facile addition of alkyl radicals.53,54 Importantly, these radical additions give access to new C-C bonds, which are not readily available by polar conjugate additions (figure 2B). Addition occurs almost exclusively at the less substituted, exo-methylene carbon and proceeds through an intermediate α-radical. The latter is stabilized by a captodative effect between the α-nitrogen and carbonyl and can readily be quenched to give β-substituted α-amino acids. Thus, dhAAs are exceptional SOMO-philes and react with alkyl radicals generated under a variety of conditions, including common radical initiators such as AIBN, BEt3, and iodosobenzene, frequently with Bu3SnH as a propagator.55–61 Because many of these initiator/propagator pairs require heating in toluene and can be strongly reactive, radical additions have limited application to complex biomolecules. However, Luche and co-workers uncovered water-compatible conditions for radical addition to dhAAs by generating the alkyl radicals from the parent alkyl halides by reduction with zinc and copper; an adaptation of the Giese reaction, suggesting its potentially broad applicability.62–64
Recently, the now seminal Luche conditions have been used to breathe new life into radical additions to dhAAs with applications in protein and peptide modification as well as DNA-encoded libraries (DELs).65–67 Independent reports by Davis and Park showed that alkyl radicals prepared by zinc reduction of alkyl halides under optimized conditions could chemoselectively add into dhAAs in complex proteins and peptides.66,67 These groups were able to achieve >90% yields in as little as 30 minutes, using a range of primary, secondary and tertiary alkyl halides. Importantly, the addition of NaBH4 effectively suppressed oxidative cleavage and di-substitution side reactions resulting from the long-lived radical species.66 These reaction conditions displayed a remarkable level of functional group tolerance and were used to introduce an impressive array of natural and unnatural amino acid side chains into proteins. In particular, the authors demonstrated chemical modification at multiple sites of eight different proteins (tallying >50 synthetic variants), which exhibit diverse secondary structures and protein types. Although these radical additions still lacked stereoselectivity, the approach allowed the authors to glean fundamental insights into the functional roles of PTMs on histones and kinases. In a related method, the Baran and Blackmond labs, in conjunction with researchers at Pfizer described a radical addition into dhAAs via zinc-catalyzed radical decomposition of an activated ester that could be performed in the presence of nucleic acids (with the intent for use with DELs). 65 Although dhAAs were not used as the primary radical acceptor, 17 of the over 70 analogs made used dhAAs, revealing an exceptionally high functional group tolerance. The zero-order rate behavior of the substrates allowed the reaction to be performed in dilute aqueous conditions making it imminently translatable to applications on DELs and perhaps proteins as well.
Visible light can provide an alternative and potentially milder entrance into alkyl and acyl radicals.68–70 Importantly, the redox potentials of many photoredox catalysts (Ir, Ru, Cu, etc.) can be tuned with metal ligands to provide greater functional group selectivity. This translates to reactions with typically high functional group tolerance that can generally be done in aqueous solution at ambient temperatures, with benchtop-stable reagents. The Dixon and Jui labs have both leveraged visible light photoredox chemistries to initiate nucleophilic radical attack on dhAAs in complex peptide substrates.71–73 In both cases, the reactions were performed at room temperature, employed blue light and iridium based catalysts. Preliminary results suggest that these reactions could also be performed in aqueous media.72,73 Dixon described photoredox conditions for preparing nucleophilic α-amino radicals from normally electrophilic imines generated in situ, and coupling them with dhAAs to create 1,3-diamines.71 Interestingly, when the imine was substituted for the parent aldehyde, the resultant ketyl radicals also underwent addition to the dhAA to produce 1,3-aminoalcohols in excellent yields. Additionally, a derivative bearing a boronic acid pinacol ester derivative proved to be a capable substrate allowing the possibility for further downstream derivatization. Similarly, the Jui lab used the Ir[dF(CF3)ppy]2dtbbpy·PF6 catalyst to generate α-amino and pyridyl radicals from alkyl amines and 3-bromo-pyridines respectively.72,73 These radicals can be reacted with dhAAs to form unnatural (e.g. β-heteroaryl) amino acids and conjugate complex bioactive payloads such as dextromethorphan and strychnine onto peptides. Reactions using the pyridyl radicals proved particularly robust, proceeding in 93% yield when open to air and performed in bourbon as solvent.72 Notably, this radical conjugate addition was tolerant of amines substituted with indole, phenol, sodium propionate, and imidazole, as useful isosteres for tryptophan, tyrosine, carboxylates, and histidine, respectively. The reaction’s robust nature and broad substrate scope make it a promising strategy for translation into more complex settings (e.g. natural products, proteins, nucleic acids)
Clearly, based on these new reports, the scope of many of these radical additions to dhAAs can be quite broad with exceptional functional group tolerance. The main barrier to developing these chemistries was the identification of biomolecule-compatible radical initiators. Likely, there will be many new uses for this chemistry. One remaining hurdle, as is with polar conjugate additions, is stereoselectivity, although there are promising preliminary reports. Jui could obtain >95% diastereomeric excess (de) with a chiral auxiliary in their work and early work by Sibi showed good stereoinduction when using chiral bisoxazoline ligands with a magnesium catalyst.56,73 Still, new advances will be needed to extend these results to more complex substrates. Furthermore, heteroatom centered radicals may complement the C-centered ones used in these reports, to provide access to new and unique functional derivatives.
III. Cycloadditions
The unique, push-pull electronic structures of dhAAs also allows them to participate in a variety of cycloaddition reactions.53,54,74–78 Together, the competing electronic forces of the pi-donating nitrogen substituent and electron withdrawing carbonyl polarize the dhAA exo-methylene and compress the HOMO-LUMO gap, allowing it to serve as a 2π partner to a variety of electron rich 3π and 4π elements, as well as nucleophilic carbenes (figure 2C).79–84 As opposed to conjugate addition chemistry, these cycloadditions provide access to α,α-disubstituted amino acid products, which have unique conformational properties. Alternatively, extended conjugation with the amide nitrogen and/or carbonyl also allows dhAAs to act as pseudo-1,3-dipoles for [3+2] cycloadditions or aza-dienes in [4+2] cycloadditions.85–88 Importantly, N-acyl substituents can have a pronounced effect on which of these roles (2π, 3π, or 4π) dhAAs play in a reaction and their product profiles.87 Recent work has focused on all three modes of reactivity, providing new routes to complex fused ring structures and leading to more mild and selective reaction conditions that could be relevant to peptide, protein, and natural product contexts.
dhAAs have perhaps been best investigated as dienophiles or 2π synthons. Here, dhAAs typically give regioselectivity consistent with an electrophilic α-carbon and nucleophilic β-carbon, although select conditions provide mixed regioisomers. For example, the simple dhAA methyl 2-acetamidoacrylate reacts readily with Danishefsky’s diene under mild conditions to yield the α-alkoxy cyclohexene products.89–91 In related cycloadditions, electron-withdrawing groups on the N-acyl substituent accentuate the activity of the β-carbon, allowing reactions to proceed more smoothly at lower temperatures and shorter reaction times.92,93 2π reactivity can also be enhanced by coordination with a metal catalyst. In a novel example, Hirano and co-workers used a rhodium/bisphosphine complex to react dhAAs with 1,6-diynes via the bidentate coordinate with an in situ generated metallacyclopentadiene.79 The catalyst provided excellent stereoselectivity in this net [2+2+2] cycloaddition reaction. This type of 2π reactivity can also extend to nucleophilic carbenes and ylides to yield 1-amino-2-cyclopropane carboxylic acids (ACCAs), although yields and conditions seem to vary widely.84,94,95 Thus, stereoselective cyclopropanations of dhAAs with sulfur and phosphorus-based ylides have been described, as have reactions with rhodium and iron carbenes.81–84,96–98 Neither electron withdrawing nor electron donating groups had significantly influenced on yields, however electron-deficient ylides displayed poor reactivity and required more forceful conditions.80 Similarly, dhAAs can undergo [3+2] cycloadditions with azomethine ylides and other 1,3-dipoles, such as nitrones and nitrileoxides.96,97,99,100 These reactions proceed smoothly at room temperature however, reaction conditions are often dictated by those necessary to generate the 1,3-dipole. Of note, the Raines group could achieve clean and quick 1,3 polar cycloadditions between dhAAs and electron-rich diazoacetamides in water/acetonitrile solvent systems.85 Under more forcing conditions, certain versions of this reaction undergo a loss of nitrogen gas to give the cyclopropane product, but here the reaction can be stopped at the intermediate pyrrazolines. This reaction successfully labels dhAAs in the lantibiotic nisin in a crude mixture of denatured proteins, suggesting a broad scope of application in natural product derivatization.85
With some help, dhAAs can participate in cycloadditions as 3π components. Isolated reports from Reisman and Stoltz disclosed chemistry that makes indolines from dhAAs via proposed [3+2] cycloadditions.86,87 In the former, a SnCl4/BINOL Lewis acid catalyst was employed to activate dhAAs toward nucleophilic addition by C3 of the indole. Subsequent collapse of the amide nitrogen onto the C2 imine and enantioselective protonation gave pyrroloindolines - common components of many natural products.86 In contrast, the Stoltz work exploited the potent reactivity of in situ generated arynes with Boc-protected dhAAs to forge mono-, di-, and tri-substituted indolines.87 These examples highlight the opportunity to use the reactivity of the β-carbon of dhAAs as a means to access the amide nitrogen through proximity. Further exploration of this chemistry may yield alternative ways to derivatize dhAAs in peptide contexts.
In select cases dhAAs can also act as 4π components in formal [4+2] cycloadditions. For instance, Stoltz showed that dhAAs with acetyl rather than carbamoyl protecting groups react with arynes to yield isoquinolines via a proposed [4+2] reaction.87 The change in reactivity is presumably due to the reduced nucleophilicity of the amide nitrogen and increased electrophilicity of the carbonyl. Although yields were fairly modest the substrate scope proved to be quite broad. Additionally, the reagents needed to generate arynes somewhat limits applications in late stage derivatizations. In related chemistry, Moody and coworkers use dhAAs as both the 2π and 4π components in a unique formal [4+2] cycloaddition.88 In this case, the dhAA amide carbonyl was converted to the imidate ester via O-alkylation to provide the 4π component and intense heat or microwave irradiation were required to push the reaction. Still, a similar mechanism to the Stoltz chemistry can be invoked, suggesting that this reaction manifold may be broadly accessible to dhAAs.
Overall, dhAAs are flexible cycloaddition partners, enabling clean formation of a variety of novel heterocycles and amino acid derivatives from a single functional group. Given that cycloadditions often occur with relatively fast kinetics and that most dipoles and dienes are unreactive with peptide sidechains and other biomolecules, there may be significant opportunity yet for dhAAs as cycloaddition handles in biomolecules. Still, few of these conditions have been adapted for reaction in aqueous environments. This could bring about new orthogonal reactions to diversify peptides, proteins and nucleic acid-based display technologies with complex fused ring systems, common in natural products and drugs alike.
IV. Transition metal catalyzed reactions.
Modern transition metal catalysts with tightly coordinated transition states tuned by designer ligands offer unique reactivity and a high degree of stereoselectivity that are often hard to achieve with the naked radical and polar additions described above.101–104 dhAAs have proven competent substrates for a number of transition metal catalyzed modifications and increasingly, these reactions are being adapted for stereoselective modification of biomolecules and natural products (figure 2D).53,54,105,106 Indeed, dhAAs were some of the first substrates for the Nobel prize-winning catalysts developed by Knowles and Noyori.107–112 Since then, dhAAs have been widely exploited as olefins for metal catalyzed transfer hydrogenations113–117 and C-C bond forming reactions, such as Pd-catalyzed Suzuki,116,118–124 Sonogashira,125–127 and Heck-type couplings124,128–142 and perhaps most effectively, Rh-catalyzed conjugate additions.103,105,106,143–147 These reactions provide significant stereocontrol and can often be conducted in aqueous buffer, leading to new routes for late stage diversification of dhAAs.148,149
dhAAs are readily reduced by rhodium catalysts armed with a variety of chiral, electron-donating phosphine ligands. For example, in one of the very first applications, William Knowles, while working at Monsanto, employed a [Rh((R,R)- DIPAMP)] complex in the enantioselective hydrogenation of a β-aryl-substituted α-(acylamino)acrylate derivative, en route to L-3,4-dihydroxyphenylalanine (L-DOPA).110 Numerous advancements have been made on these initial conditions and catalysts and new chiral rhodium complexes perform exceptionally well in terms of efficiency (i.e. turnover number or frequency), enantioselectivity (i.e. enantiomeric excess [ee]) and solvent profiles, being used extensively in early stages of natural product total synthesis and industrial process development.117,150 The stereoselective rhodium-catalyzed reduction of β-aryl dhAAs has become an industry standard for entrance into chiral amino acid synthons in large scale production of drugs like Levetiracetam (Keppra),151 Indiniavir (Crixivan),152 and Sitagliptin (Januvia).117,153,154 Given the already extensive scope and application of this chemistry, it makes sense that it might eventually be applied to late stage modification of dhAAs in larger natural product scaffolds and polypeptides. In a pioneering example of the potential for this chemistry, Dong and co-workers recently applied transfer hydrogenation to the stereoselective reduction of a suite of four dehydropheylalanines in a cyclopentapeptide in protic solvents.155 Combined experimental and theoretical work suggests that the catalyst binds and sequentially reduces successive residues in a C-N direction. The initial 1,3-bis(diphenylphosphino)propane (dppp) ligand used provided alternating D- and L- stereochemistry at the four centers, but appropriate choice of chiral ligand could override this stereocontrol to provide the all-D stereoisomer. The Dong work suggests that similar levels of stereocontrol may be obtained with these robust catalysts and larger and still more complex peptide substrates.
In addition to hydrogenations, Rh-catalyzed conjugate additions have been developed into reliable and functional group tolerant methods for preparing C-C bonds between arylboronic acids and dhAAs with excellent stereocontrol.105,106,143–145,147 Based on the seminal work of Hayashi, the catalytic cycle is generally accepted to occur in three major steps: (1) the organoboron reagent undergoes transmetalation with the ligand-complexed hydroxyrhodium to generate an aryl-rhodium species; (2) the electron-deficient alkene then inserts into the aryl-Rh bond to give an oxa-π-allylrhodium intermediate; (3) hydrolysis of this unstable intermediate protonates the enolate, releasing and regenerating the hydroxyrhodium catalyst.103,156 A number of groups have investigated stereoselective Rh-catalyzed 1,4 conjugate additions to dhAAs.105,106,143–145,147 Importantly, the dhAAs typically react under similar conditions to the more typical enones, despite the different electronic and steric substitution pattern.103 For example, early work demonstrated that Rh-catalyzed conjugate additions to dhAAs could be carried out in dioxanes:water mixtures at elevated temperatures and that stereoselectivity could be achieved with Karady-Beckwith chiral auxiliaries similar to radical additions.157 More recent work by Genet and Darses has provided additional mechanistic insight and heightened stereoselectivity.158,159 This group showed that potassium trifluoroorganoborates react smoothly with simple dhAAs in the presence of [Rh(cod)2][PF6] and chiral ligands to give unnatural isosteres of aromatic amino acids in toluene at 110°C for 20 hours. They further revealed that a cryptic β-hydride transfer from the amide-nitrogen to the α-carbon via the metal center is both rate limiting and stereodetermining. Based on this insight, the use of more electron-deficient phosphine ligands increased yields and stereoselectivity substantially (>91% ee). This example emphasizes how the unique and innate structure and reactivity of dhAAs can be exploited for greater regio- and stereocontrol.
Still more contemporary work has moved this rhodium chemistry into more complex peptide and natural products territory. Separately, Willis and Frost used Rh(acac)(C2H4)2 or [RhCl(C2H4)2]2 catalysts and BINAP-based ligands to modify dhAAs in di- and tripeptides.105,144,145,147 A variety of boronic acids were tolerated and reactions could be performed on peptides with unprotected tryptophans, tyrosines and thioethers in good yields and selectivity.105,145 Miller demonstrated the extreme versatility of this chemistry by developing conditions that allow the site- and stereoselective functionalization of the complex natural product thiostrepton.106 Although standard conditions involving prolonged heating were incompatible with the sensitive functional groups on thiostrepton, the group found that reactions could be run at room temperature by increasing catalyst loading to 50 mol%, without degrading the natural product. Of the four dhAAs present in thiostrepton, these conditions proved highly selective for Dha 16. This is distinct from the chemoselectivity of uncatalyzed conjugate additions with thiol nucleophiles which typically favor terminal Dha 17.160 Further development of this chemistry may allow site selective modification of other dhAAs within complex natural products that are difficult to modify otherwise. Interestingly, while the substrate scope was similar to prior work, the stereoselectivity proved highly dependent on Na and K salts, although the basis of this selectivity is unknown.
dhAAs also undergo a number of common Pd-catalyzed olefin chemistries, including Suzuki, Sonogashira, and Heck-type coupling reactions. Only a handful of examples of Suzuki or Sonogoshira couplings have been reported and these are of limited scope due to the need for the β-halogenated dhAA coupling partner to the boronic acids or alkynes, respectively.116,118–127 However, the Heck reaction has been used rather extensively to modify small molecule dhAAs.124,128–142 In contrast to the rhodium-catalyzed conjugate additions described above, the Heck products still contain the double bond, but with new substituents at the β-carbon, making Heck-type cross-couplings viable strategies to synthesize new alkyl and aryl dhAAs. A number of examples have been reported in the literature, often providing, selectively, the Z-product with the new substituent trans to the dhAA carbonyl group.53 The Heck chemistry can be performed in tandem with heteroannulation to allow access to isoquinolines,129,130,161 pyrroles,162,163 and oxazoles.164 Due to the solubility needs of the catalyst and elevated reaction temperatures, these reactions have mostly been limited to small molecule dhAAs being modified in typically organic solvents. Recently, Roelfes and co-workers reported on the development of an EDTA (ethylenediaminetetraacetic acid)-based complex to allow palladium-mediated cross coupling of arylboronic acids with dhAAs in peptides and proteins.142 Although the catalyst gives a mixture of the Heck-type olefin product and a conjugate addition product, the authors demonstrated good tolerance of aqueous buffer and examples of robust modification of the natural product nisin, as well as a dhAA-bearing version of the small protein SUMO (small ubiquitin-like modifier). In this case, concerns about stereoselectivity of the conjugate addition product are secondary to the overall regioselectivity of the reaction, but Pd-based catalysts are well known for allowing excellent stereocontrol with ligand optimization.
Cumulatively, it is clear that with careful choice of metal catalyst, chiral ligands, and other conditions, robust, controlled conjugate additions into complex molecule dhAAs can be achieved. Although significant progress has been made in making reaction conditions milder, the high temperatures, organic solvents, prolonged reaction times and limitation to aryl conjugates (by virtue of the otherwise labile boron compounds) are less than ideal for use in a protein context. However, the interesting stereoselective proton transfer dhAAs may offer opportunities to expand its orthogonality.146 Perhaps a ligand/Rh complex that promotes internal hydride transfer significantly faster than water may (1) allow it to be performed in water and (2) increase the reaction rate enough that high temperatures are not needed, making it useful in a wider range of chemical biology-oriented applications. Copper-catalyzed hydroboration and hydrosilylation of dhAAs have also been recently described.165–168 These modifications are particularly useful in a pharmaceutical context because they can be derivatized further to access valuable noncanonical amino acids and often afford greater bioavailability.166,168
V. Enzymatic modification.
Many of the dhAA chemistries described above have also been found in enzymatic contexts (figure 3). Some of the most well studied and recent examples of enzymatic dhAA modification are from metabolism and peptide natural product biosynthetic pathways. In particular, enzymes use dhAAs as catalytic residues to perform chemistry in amino acid metabolism.169 Additionally, dhAAs are used heavily as reactive handles for enzymatic diversification in ribosomally synthesized and post-translationally modified peptide (RiPPs) natural products.170 RiPP enzymes perform chemistry on precursor peptide substrates by recognizing an N-terminal leader peptide and modifying a diverse array of typically C-terminal core peptides.171 RiPP enzymes have been found to perform numerous reactions on dhAAs, including stereoselective conjugate addition of cysteine thiols, lysine amines, select examples of α-enolate carbanions, conjugate reductions, novel intramolecular cycloadditions, and even non-natural carbene chemistries.
In the biosynthesis of lanthipeptides, dhAAs can undergo enzyme-catalyzed intramolecular 1,4-conjugate additions using sulfur, carbon and nitrogen nucleophiles (figure 3A).8 There are four major types of enzymes that catalyze the formation of these dhAA-based crosslinks. Type-I, II and IV are zinc-dependent lanthipeptide cyclases that use zinc to activate cysteine thiols and promote nucleophilic attack on the dhAA.172 Distinct from the polar conjugate additions discussed above, enzymes catalyze such chemistry with exquisite chemo- and stereoselective control. NisC, a type-I lanthipeptide cyclase, installs five thioether bridges (one lanthionine, derived from dha, and four methyllanthionines from dhbs) in the antibiotic Nisin (figure 1c) with specific ring topology and stereochemistry - making a single lanhtipeptide natural product out of nearly a million possibilities.172,173 Despite exhibiting such exceptional chemo- and stereoselectivity, lanthipeptide cyclases also display impressive substrate promiscuity/plasticity. Type-II cyclase ProcM, from the prochlorosins biosynthetic pathway, has 29 natural substrates and installs distinct yet specific ring topology and stereochemistry on each substrate, yielding 29 discrete natural products.174 Similar to types-I and II, type-III cyclases install thioether crosslinks however, they are distinguished by “catching” the enolate in a second conjugate addition with another dhAA (figure 3).175,176 Stereoselective protonation of the second enolate furnishes the final labionin crosslink. Nitrogen-based conjugate addition of dhAAs has also been observed in Nature. A recent report showed the enzyme DurN, from duramycin biosynthesis, is responsible for the installation of an unusual lysinoalanine bridge.177 Mutational and structural data supports a substrate-assisted mechanism of catalysis, whereby another unnatural residue within duramycin itself, β-hydroxyl aspartic acid, activates the amine by deprotonation and coordinates stereoselective enol protonation via sterics.
dhAAs can be stereoselectively reduced to form D-amino acids. Two enzyme families found in lanthipeptide biosynthetic pathways, have been discovered to display this activity; zinc and NADPH-dependent hydrogenases (LtnJ, PenN, NpnJ and SacJ) and flavin-dependent oxidoreductases (LasJ, BsjJ and CrnJ).8,178,179 The mechanism is assumed to occur by conjugate addition of a hydride, transferred by either the NADPH or flavin cofactor. Stereoselective protonation yields the D-amino acid. While no crystal structures of these enzymes have been solved, extensive mutagenesis on LtnJ (from lacticin 3147 biosynthesis) has begun to reveal important aspects of its stereoselectivity.180 In particular, LtnJ K359A mutant loses stereoselectivity. This led the authors to believe that K359 acts either to stabilize the negative charge on the α-carbon, or as a proton source. Only CrnJ and BsjJ, from the biosynthetic pathways of carnolysin and bicereucin, respectively, have been shown to reduce Dhbs in addition to Dhas.181,182 All enzymes seem to act in a leader peptide independent manner and often reduce Dhas (or Dhbs) that are positioned distant from charged/polar groups and steric congestion (thioethers or other bridges).8
dhAAs can also undergo enzyme-catalyzed cycloadditions in thiopeptide biosynthesis.183–186 They serve as both 2π and 4π components in an intramolecular aza-[4+2] cycloaddition to furnish a tri-substituted pyridine ring (figure 3B).187 The mechanism is thought to involve the tautomerization of a C-terminal amide bond, adjacent to a Dha, to an imidic acid, forming the 4π component. This is followed by an aza-[4+2]-cycloaddition with an N-terminal Dha. Subsequent elimination of water and aromatization yields the final pyridine core (figure 3).188–190 This reaction is entirely-enzyme dependent, and through this process the substrate is simultaneously macrocyclized and cleaved from its leader peptide, releasing a mature thiopeptide.187 To date, three pyridine synthases from thiopeptide biosynthetic pathways, TclM (thiocillin), TbtD (thiomuracin) and PbtD (GE2270), have been shown to independently catalyze this reaction.187,191,192 Pyridine synthases undoubtedly exploit the polarizable nature of dhAAs to manipulate each Dha differently; forcing one to exhibit a more electrophilic character, and the other, a more nucleophilic character. The structures of TbtD and PbtD have revealed a leader peptide binding site distant from the expected active site however, no structural data describing how pyridine synthases manipulate either Dha has emerged.193 Pyridine synthases exhibit broad substrate promiscuity and are capable of cyclizing substrates with a wide variety of different macrocycle sizes and core sequences.6,7,187,194–199 Still, other classes of thiopeptides harbor distinct nitrogen-containing cores that originate from dhAAs and the enzymes responsible for installing these modifications may prove to be a rich source of novel dhAA chemistry.183–186
Analogous to the synthetic cyclopropanations of dhAAs, enzymes have been used to achieve stereoselective cyclopropanation of dhAAs. Although there are few natural examples of enzyme-catalyzed carbene insertions, engineered heme-dependent cytochromes P450 have been shown to cyclopropanate aryl olefins with iron carbenes generated from electron-deficient diazo compounds (such as ethyldiazoacetate).200–203 Acting as aryl olefin equivalents, dhAAs provide natural handles for this kind of unnatural chemistry. Two P450s, TbtJ1 and J2 (from the biosynthesis of the thiopeptide thiomuracin), were engineered to carry out the cyclopropanation of dhAAs embedded in thiopeptide scaffolds to yield the ACCA derivatives.204 Interestingly, because these enzymes already carry out site- and stereoselective oxidations of thiopeptide substrates, cyclopropanations with the engineered enzymes were also regio- and stereoselective with moderately modified versions of their native substrates. Similar Rh-catalyzed reactions on thiopeptide scaffolds only yielded trace amounts cycloproponated product. Redirecting natural oxidation chemistries towards orthogonal handles such as dhAAs could prove a broad strategy for late-stage natural product modification.
Nature has employed dhAAs in enzymes to perform chemistry. A modified and particularly electrophilic dhAA, 4-methylidene-imidazole-5one (MIO), acts as a catalytic residue during amino acid metabolism and natural product biosynthesis (figure 3C).169,205,206 Two main mechanisms have been proposed for the MIO-catalyzed breakdown of amino acids by ammonium lyase enzymes.169,205,207 In the first, MIO undergoes a nucleophilic attack by the amine of the amino acid. β-elimination of the amine cleaves the amino acid and collapse of the enolate releases ammonia and reforms the dhAA (figure 3). Alternatively, amino acids like tyrosine (or phenylalanine) may proceed by a Friedel-Crafts type mechanism, whereby the δ-carbon performs the attack on the MIO. Abstraction of the β-proton followed by collapse of the enolate, eliminates ammonia and releases p-coumaric acid and MIO. Nearly identical chemistry is performed by the closely-related enzyme family of MIO-dependent aminomutases, however instead of releasing ammonia, the MIO-amine adduct performs a conjugate addition on the newly formed alkene followed by stereoselective protonation. Collapse of the MIO-enolate releases a β-amino acid.208,209 Aminomutases are important enzymes involved in the biosynthesis of several natural products including C-1027,210 taxol211 and the polyketide andrimid.212 Additionally, these enzymes have been used to stereoselectively generate unnatural α- and β-amino acids.213–215
The above examples clearly demonstrate there are enzymatic counterparts to many of synthetic dhAA chemistries. The same polar conjugate addition chemistry that has proven to be so useful in chemical biology is catalyzed by a large family of lanthipeptide cyclyases, however enzymes have the added benefit of chemo- and stereoselectivity. The Stoltz [4+2] chemistry is analogous to the chemistry performed by pyridine synthases. The generation of the arynes however, requires strictly anhydrous conditions and uses fairly harsh fluoride sources, limiting its applicability. Pyridine synthases catalyze an equivalent [4+2] in buffer at room temperature with no additional cofactors. These mild conditions make dhAA-modifying enzymes suitable biocatalysts for broad application in chemical biology and drug discovery. The biosynthesis of RiPPs natural products is rich with dhAA modifying enzymes, however these types of enzymes may be more widespread. For example, the fungal natural product phomopsin A contains several unique dhAAs (figure 1), yet none of the enzymes in its biosynthetic gene cluster resemble known dhAA-installing enzymes.216,217 Moreover, the presence of dehydroisoleucine makes it conceivable that the aryl-ether may result from conjugate addition of the m-oxygen into another dehydroisoleucine. Continued exploration of the natural products made by other biosynthetic pathways, such as non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) may lead to new dhAA modifying enzymes. For instance, since dhAAs are known radical acceptors and radical chemistry is so prevalent in Nature, enzymes capable of modifying dhAAs through radical mechanisms may also exist.218,219 These enzymes may provide next generation biocatalysts to mildly install exotic functionality through the flexible dhAA handles.
Conclusion.
In summary, dhAAs are versatile amino acids. The unique push-pull electronic architecture of dhAAs can be leveraged in many chemical transformations, including conjugate additions, cycloadditions, transfer hydrogenations, radical reactions and metal-mediated cross-couplings, allowing access to unnatural amino acid derivatives and complex ring systems. Continued exploration of this innate reactivity is turning dhAAs into robust chemical handles for peptide, protein, and natural product modification.11–14 For example, early work demonstrated that dhAAs were susceptible to regioselective radical additions, but only under harsh conditions with strong activating reagents, such as AIBN/Bu3SnH. As discussed in this review, the use of new, more functional group tolerant activators by Davis, Park and others has expanded the applicability of radical-dhAA chemistry which can now be performed on complex natural products and proteins and in the presence of nucleic acid tags. Similarly, dhAA-cross couplings were, until recently, considered an “unanswered challenge” in peptide and protein modification, but are succumbing to new, more water-soluble catalysts and other reactions are beginning to follow suit.220 Although there are still significant hurdles in chemical modification of dhAAs, it clearly provides a powerful toolbox for late stage chemical diversification.
In retrospect, the chemical potential of dhAAs has already been on display for a long time in the structures and biosynthetic pathways of numerous complex natural products. dhAA-modifying enzymes from natural product biosynthesis present significant opportunities for biorthogonal tailoring. Such enzymes already function under mild conditions, in aqueous media, and display exceptional chemo-, regio- and stereoselectivity on complex substrates. Moreover, enzymes can be engineered to install non-native functionality while retaining their natural substrate specificity, as shown with TbtJ1 and J2. However, this activity comes at a price and many of these enzymes can be slow or exhibit low turnover numbers, necessitating some degree of further optimization for large-scale use.221–223 Still, with the rapidly increasing number of sequenced genomes, new dhAA-modifying enzymes are close at hand.224 The structures of natural products like phomopsin A,216,217 cyclothiazomycin A225,226 and theonellamide F227 may already hint at new dhAA-modifying chemistries. For example, biosynthetic evidence suggests that a rare tertiary thioether bridge in cyclothiazomycin A might arise from a novel enzyme-catalyzed rearrangement following attack at a dhAA. Similarly, the phenolic ether in phomopsins may derive from a radical addition to a dhAA, a chemical transformation not previously seen in natural product biosynthesis. These and other new biocatalysts may be waiting in the genomes of many newly sequenced organisms.
Importantly, the versatility of dhAA-modifications provides remarkable new tools for research both in basic biology and drug discovery efforts. 11–13,36,228,229 For instance, the chemical modification of dhAAs provides reliable routes for the selective introduction of mimics of protein post-translational modification (PTMs), such as methylation, acetylation, and phosphorylation in order to interrogate the chemical cross-talk mediated by these modifications.19,21,26–35 New reactions will allow the detailed investigation of structure-activity relationships related to these PTMs. Also, these chemistries can and have been used to enhance phage,230,231 yeast,231 mRNA display,232–236 and DNA-encoded libraries.65 dhAA-modifying enzymes from RiPP biosynthetic pathways seem to be especially useful in this regard because their innate substrate plasticity allows them to make vast libraries of complex natural product-like compounds. In a recent example, authors were able to pull out nanomolar integrin binders from large libraries of peptides displayed on yeast or phage and modified by enzyme catalyzed conjugate additions of cysteine thiols to dhAAs.231 Cumulatively, dhAA modification chemistries appear poised to address major biological questions and uncover new therapeutic molecules.
References
- 1.Siodłak D, Amino Acids, 2014, 47, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siodlak D, Rzeszotarska B, Broda MA, Koziol AE and Kolodziejczyk E, Acta Biochim. Pol, 2004, 51, 145–152. [PubMed] [Google Scholar]
- 3.Siodłak D, Broda MA and Rzeszotarska B, J. Mol. Struct.: THEOCHEM, 2004, 668, 75–85. [Google Scholar]
- 4.Palmer DE, Pattaroni C, Nunami K, Chadha RK, Goodman M, Wakamiya T, Fukase K, Horimoto S and Kitazawa M, J. Am. Chem. Soc, 1992, 114, 5634–5642. [Google Scholar]
- 5.Ajò D, Granozzi G, Tondello E and Del Prà A, Biopolymers, 2004, 19, 469–475. [Google Scholar]
- 6.Tran HL, Lexa KW, Julien O, Young TS, Walsh CT, Jacobson MP and Wells JA, J. Am. Chem. Soc, 2017, 139, jacs.6b10792–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowers AA, Acker MG, Koglin A and Walsh CT, J. Am. Chem. Soc, 2010, 132, 7519–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Repka LM, Chekan JR, Nair SK and van der Donk WA, Chem. Rev, 2017, 117, 5457–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacKintosh RW, Dalby KN, Campbell DG, Cohen PT, Cohen P and MacKintosh C, FEBS Letters, 1995, 371, 236–240. [DOI] [PubMed] [Google Scholar]
- 10.McLellan NL and Manderville RA, Toxicol. Res, 2017, 6, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spicer CD and Davis BG, Nature Communications, 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- 12.Chalker JM, Bernardes GJL and Davis BG, Acc. Chem. Res, 2011, 44, 730–741. [DOI] [PubMed] [Google Scholar]
- 13.Dadová J, Galan SR and Davis BG, Current Opinion in Chemical Biology, 2018, 46, 71–81. [DOI] [PubMed] [Google Scholar]
- 14.Nadal S, Raj R, Mohammed S and Davis BG, Current Opinion in Chemical Biology, 2018, 45, 35–47. [DOI] [PubMed] [Google Scholar]
- 15.Levengood MR and van der Donk WA, Nat Protoc, 2006, 1, 3001–3010. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Schiller SM and Schultz PG, Angew. Chem. Int. Ed, 2007, 46, 6849–6851. [DOI] [PubMed] [Google Scholar]
- 17.Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernández-González M, Bernardes GJL, Griffin L, Hailu H, Schofield CJ and Davis BG, Chem. Sci, 2011, 2, 1666. [Google Scholar]
- 18.Repka LM, Chekan JR, Nair SK and van der Donk WA, Chem. Rev, 2017, acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed]
- 19.Bernardes GJL, Chalker JM, Errey JC and Davis BG, J. Am. Chem. Soc, 2008, 130, 5052–5053. [DOI] [PubMed] [Google Scholar]
- 20.Okeley NM, Zhu Y and van der Donk WA, Org. Lett, 2000, 2, 3603–3606. [DOI] [PubMed] [Google Scholar]
- 21.Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernández-González M, Bernardes GJL, Griffin L, Hailu H, Schofield CJ and Davis BG, Chem. Sci, 2011, 2, 1666–12. [Google Scholar]
- 22.Morrison PM, Foley PJ, Warriner SL and Webb ME, Chemical Communications, 2015, 51, 13470–13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machin PJ and Sammes PG, J. Chem. Soc., Perkin Trans. 1, 1974, 698–5.
- 24.Kohn H, Sawhney KN, Bardel P, Robertson DW and Leander JD, J. Med. Chem, 1993, 36, 3350–3360. [DOI] [PubMed] [Google Scholar]
- 25.Jin S and Liebscher J, Synlett, 1999, 1999, 459–461. [Google Scholar]
- 26.Freedy AM, Matos MJ, Boutureira O, Corzana F, Guerreiro A, Akkapeddi P, Somovilla VJ, Rodrigues T, Nicholls K, Xie B, Jiménez-Osés G, Brindle KM, Neves AA and Bernardes GJL, J. Am. Chem. Soc, 2017, 139, 18365–18375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windle CL, Simmons KJ, Ault JR, Trinh CH, Nelson A, Pearson AR and Berry A, Proceedings of the National Academy of Sciences, 2017, 114, 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dadová J, Wu K-J, Isenegger PG, Errey JC, Bernardes GJL, Chalker JM, Raich L, Rovira C and Davis BG, ACS Cent. Sci, 2017, 3, 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulder MPC, Witting K, Berlin I, Pruneda JN, Wu K-P, Chang J-G, Merkx R, Bialas J, Groettrup M, Vertegaal ACO, Schulman BA, Komander D, Neefjes J, El Oualid F and Ovaa H, Nat Chem Biol, 2016, 12, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haj-Yahya N, Hemantha HP, Meledin R, Bondalapati S, Seenaiah M and Brik A, Org. Lett, 2013, 16, 540–543. [DOI] [PubMed] [Google Scholar]
- 31.Chalker JM, Lercher L, Rose NR, Schofield CJ and Davis BG, Angew. Chem. Int. Ed, 2012, 51, 1835–1839. [DOI] [PubMed] [Google Scholar]
- 32.Guo J, Wang J, Lee JS and Schultz PG, Angew. Chem. Int. Ed, 2008, 47, 6399–6401. [DOI] [PubMed] [Google Scholar]
- 33.Wang ZU, Wang Y-S, Pai P-J, Russell WK, Russell DH and Liu WR, Biochemistry, 2012, 51, 5232–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber A, Elliott PR, Pinto-Fernandez A, Bonham S, Kessler BM, Komander D, El Oualid F and Krappmann D, Cell Chemical Biology, 2017, 24, 1299–1313.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Yang R, Huang J, Liang Q, Guo Y, Bian W, Luo L and Li H, FEBS Letters, 2015, 589, 3648–3653. [DOI] [PubMed] [Google Scholar]
- 36.Yu Y, Hu C, Xia L and Wang J, ACS Catal, 2018, 8, 1851–1863. [Google Scholar]
- 37.de la Hoz A, Díaz-Ortiz A, Victoria Gómez M, Antonio Mayoral J, Moreno A, Sánchez-Migallón AM and Vázquez E, Tetrahedron, 2001, 57, 5421–5428. [Google Scholar]
- 38.Angelini E, Balsamini C, Bartoccini F, Lucarini S and Piersanti G, J. Org. Chem, 2008, 73, 5654–5657. [DOI] [PubMed] [Google Scholar]
- 39.Righi M, Bartoccini F, Lucarini S and Piersanti G, Tetrahedron, 2011, 67, 7923–7928. [Google Scholar]
- 40.Bartoccini F, Bartolucci S, Mari M and Piersanti G, Org. Biomol. Chem, 2016, 14, 10095–10100. [DOI] [PubMed] [Google Scholar]
- 41.Bartoccini F, Casoli M, Mari M and Piersanti G, J. Org. Chem, 2014, 79, 3255–3259. [DOI] [PubMed] [Google Scholar]
- 42.Mari M, Lucarini S, Bartoccini F, Piersanti G and Spadoni G, Beilstein Journal of Organic Chemistry, 2014, 10, 1991–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mari M, Bartoccini F and Piersanti G, J. Org. Chem, 2013, 78, 7727–7734. [DOI] [PubMed] [Google Scholar]
- 44.Lucarini S, Bartoccini F, Battistoni F, Diamantini G, Piersanti G, Righi M and Spadoni G, Org. Lett, 2010, 12, 3844–3847. [DOI] [PubMed] [Google Scholar]
- 45.Bartolucci S, Bartoccini F, Righi M and Piersanti G, Org. Lett, 2011, 14, 600–603. [DOI] [PubMed] [Google Scholar]
- 46.Ferreira PMT, Maia HLS, Monteiro LS, Sacramento J and Sebastião J, Perkin 1, 2000, 3317–3324.
- 47.Okuda T and Zahn H, Chem. Ber, 1965, 98, 1164–7. [Google Scholar]
- 48.Ferreira PMT, Maia HLS and Monteiro LS, Tetrahedron Letters, 1999, 40, 4099–4102. [Google Scholar]
- 49.Ferreira PMT, Maia HLS, Monteiro LS, Sacramento J and Sebastião J, J. Chem. Soc., Perkin Trans. 1, 2000, 3317–3324.
- 50.Ferreira PMT, Maia HLS, Monteiro LS and Sacramento J, J. Chem. Soc., Perkin Trans. 1, 2001, 3167–3173.
- 51.Naidu BN, Sorenson ME, Connolly TP and Ueda Y, J. Org. Chem, 2003, 68, 10098–10102. [DOI] [PubMed] [Google Scholar]
- 52.Yang J, Sun W, He Z, Yu C, Bao G, Li Y, Liu Y, Hong L and Wang R, Org. Lett, 2018, 20, 7080–7084. [DOI] [PubMed] [Google Scholar]
- 53.Kotha S, Bandarugattu VB and Krishna NG, Tetrahedron, 2014, 70, 5361–5384. [Google Scholar]
- 54.Bonauer C, Walenzyk T and König B, Synthesis, 2006, 2006, 1–20. [Google Scholar]
- 55.Sutherland A and Vederas JC, Chemical Communications, 2002, 224–225. [DOI] [PubMed]
- 56.Sibi MP, Asano Y and Sausker JB, Angew. Chem. Int. Ed. Engl, 2001, 40, 1293–1296. [DOI] [PubMed] [Google Scholar]
- 57.Beckwith ALJ and Chai CLL, J. Chem. Soc., Chem. Commun, 1990, 1087–8.
- 58.Axon JR and Beckwith ALJ, J. Chem. Soc., Chem. Commun, 1995, 24, 549–550. [Google Scholar]
- 59.Jones RCF, Berthelot DJC and Iley JN, Chemical Communications, 2000, 2131–2132.
- 60.Jones RCF, Berthelot DJC and Iley JN, Tetrahedron, 2001, 57, 6539–6555. [Google Scholar]
- 61.Kabat MM, Tetrahedron Letters, 2001, 42, 7521–7524. [Google Scholar]
- 62.Dupuy C, Petrier C, Sarandeses LA and Luche JL, Synthetic Communications, 1991, 21, 643–651. [Google Scholar]
- 63.Luche JL and Allavena C, Tetrahedron Letters, 1988, 29, 5369–5372. [Google Scholar]
- 64.Petrier C, Dupuy C and Luche JL, Tetrahedron Letters, 1986, 27, 3149–3152. [Google Scholar]
- 65.Wang J, Lundberg H, Asai S, Martín-Acosta P, Chen JS, Brown S, Farrell W, Dushin RG, O’Donnell CJ, Ratnayake AS, Richardson P, Liu Z, Qin T, Blackmond DG and Baran PS, Proceedings of the National Academy of Sciences, 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright TH, Bower BJ, Chalker JM, Bernardes GJL, Wiewiora R, Ng WL, Raj R, Faulkner S, Vallee MRJ, Phanumartwiwath A, Coleman OD, Thezenas ML, Khan M, Galan SRG, Lercher L, Schombs MW, Gerstberger S, Palm-Espling ME, Baldwin AJ, Kessler BM, Claridge TDW, Mohammed S and Davis BG, Science, 2016, 354, aag1465–aag1465. [DOI] [PubMed] [Google Scholar]
- 67.Yang A, Ha S, Ahn J, Kim R, Kim S, Lee Y, Kim J, Soll D, Lee HY and Park HS, Science, 2016, 354, 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schultz DM and Yoon TP, Science, 2014, 343, 1239176–1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Staveness D, Bosque I and Stephenson CRJ, Acc. Chem. Res, 2016, 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koike T and Akita M, Inorg. Chem. Front, 2014, 1, 562–576. [Google Scholar]
- 71.Rossolini T, Leitch JA, Grainger R and Dixon DJ, Org. Lett, 2018, 20, 6794–6798. [DOI] [PubMed] [Google Scholar]
- 72.Aycock RA, Vogt DB and Jui NT, Chemical Science, 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aycock RA, Pratt CJ and Jui NT, ACS Catal, 2018, 8, 9115–9119. [Google Scholar]
- 74.Mehta G and Kotha S, Tetrahedron, 2001, 57, 625–659. [Google Scholar]
- 75.Chai CLL, Johnson RC and Koh J, Tetrahedron, 2002, 58, 975–982. [Google Scholar]
- 76.Souchet M, Guilhem J and Le Goffic F, Tetrahedron Letters, 1987, 28, 2371–2374. [Google Scholar]
- 77.Burkett BA and Chai CLL, Tetrahedron Letters, 2000, 41, 6661–6664. [Google Scholar]
- 78.Avenoza A, Busto JH, Canal N, Corzana F, Peregrina JM, Perez-Fernandez M and Rodriguez F, J. Org. Chem, 2010, 75, 545–552. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka K, Takahashi M, Imase H, Osaka T, Noguchi K and Hirano M, Tetrahedron, 2008, 64, 6289–6293. [Google Scholar]
- 80.Contreras-Cruz DA, Sánchez-Carmona MA, Vengoechea-Gómez FA, Peña-Ortíz D and Miranda LD, Tetrahedron, 2017, 73, 6146–6156. [Google Scholar]
- 81.Ung AT, Schafer K, Lindsay KB, Pyne SG, Amornraksa K, Wouters R, Van der Linden I, Biesmans I, Lesage ASJ, Skelton BW and White AH, J. Org. Chem, 2002, 67, 227–233. [DOI] [PubMed] [Google Scholar]
- 82.Pyne SG, Schafer K, Skelton BW and White AH, Aust. J. Chem, 1998, 51, 127–135. [Google Scholar]
- 83.Bunuel E, Bull SD, Davies SG, Garner AC, Savory ED, Smith AD, Vickers RJ and Watkin DJ, Org. Biomol. Chem, 2003, 1, 2531–2542. [DOI] [PubMed] [Google Scholar]
- 84.Adams LA, Aggarwal VK, Bonnert RV, Bressel B, Cox RJ, Shepherd J, De Vicente J, Walter M, Whittingham WG and Winn CL, J. Org. Chem, 2003, 68, 9433–9440. [DOI] [PubMed] [Google Scholar]
- 85.Aronoff MR, Gold B and Raines RT, Org. Lett, 2016, 18, 1538–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Repka LM, Ni J and Reisman SE, J. Am. Chem. Soc, 2010, 132, 14418–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gilmore CD, Allan KM and Stoltz BM, J. Am. Chem. Soc, 2008, 130, 1558–1559. [DOI] [PubMed] [Google Scholar]
- 88.Moody CJ, Hughes RA, Thompson SP and Alcaraz L, Chem. Commun, 2002, 225, 1760–1761. [DOI] [PubMed] [Google Scholar]
- 89.Avenoza A, Cativiela C, Busto JH, Fernandez-Recio MA, Peregrina JM and Rodríguez F, Tetrahedron, 2001, 57, 545–548. [Google Scholar]
- 90.Avenoza A, Barriobero JI, Cativiela C, Fernandez-Recio MA, Peregrina JM and Rodríguez F, Tetrahedron, 2001, 57, 2745–2755. [Google Scholar]
- 91.Avenoza A, Cativiela C, Fernandez-Reico MA and Peregrina JM, J. Chem. Soc., Perkin Trans. 1, 1999, 3375–3379.
- 92.Crossley MJ and Stamford AW, Aust. J. Chem, 1993, 46, 1443–6. [Google Scholar]
- 93.Crossley MJ and Stamford AW, Aust. J. Chem, 1994, 47, 1695–711. [Google Scholar]
- 94.Adams LA, Charmant JPH, Cox RJ, Walter M and Whittingham WG, Org. Biomol. Chem, 2004, 2, 542–553. [DOI] [PubMed] [Google Scholar]
- 95.Aggarwal VK, Alonso E, Fang G, Ferrara M, Hynd G and Porcelloni M, Angew. Chem. Int. Ed. Engl, 2001, 40, 1433–1436. [DOI] [PubMed] [Google Scholar]
- 96.Pyne SG, Safaei-G J and F. Koller in part, Tetrahedron Letters, 1995, 36, 2511–2514. [Google Scholar]
- 97.Pyne SG, Safaei-G J, Schafer K, Javidan A, Skelton BW and White AH, Aust. J. Chem, 1998, 51, 137–158. [Google Scholar]
- 98.Pyne SG, Schafer K, Skelton BW and White AH, Chemical Communications, 1997, 2267–2268.
- 99.Chai CLL, Edwards AJ, Wilkes BA and Woodgate RCJ, Tetrahedron, 2003, 59, 8731–8739. [Google Scholar]
- 100.Pyne SG, Safaei-G J, Skelton BW and White AH, Aust. J. Chem, 1995, 48, 1–23. [Google Scholar]
- 101.Nicolaou KC, Bulger PG and Sarlah D, Angew. Chem. Int. Ed. Engl, 2005, 44, 4442–4489. [DOI] [PubMed] [Google Scholar]
- 102.Beletskaya IP and Cheprakov AV, Chem. Rev, 2000, 100, 3009–3066. [DOI] [PubMed] [Google Scholar]
- 103.Hayashi T and Yamasaki K, Chem. Rev, 2003, 103, 2829–2844. [DOI] [PubMed] [Google Scholar]
- 104.Edwards HJ, Hargrave JD, Penrose SD and Frost CG, Chem. Soc. Rev, 2010, 39, 2093–13. [DOI] [PubMed] [Google Scholar]
- 105.Chapman CJ, Matsuno A, Frost CG and Willis MC, Chem. Commun, 2007, 43, 3903–4. [DOI] [PubMed] [Google Scholar]
- 106.Key HM and Miller SJ, J. Am. Chem. Soc, 2017, 139, 15460–15466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Noyori R, Angew. Chem. Int. Ed, 2002, 41, 2008–2022. [PubMed] [Google Scholar]
- 108.Noyori R and Takaya H, Acc. Chem. Res, 2002, 23, 345–350. [Google Scholar]
- 109.Noyori R, Chem. Soc. Rev, 1989, 18, 187–22. [Google Scholar]
- 110.Knowles WS, J. Chem. Educ, 1986, 63, 222–4. [Google Scholar]
- 111.Knowles WS, Acc. Chem. Res, 2002, 16, 106–112. [Google Scholar]
- 112.Knowles WS and Sabacky MJ, Chem. Commun. (London), 1968, 1445–2.
- 113.Peña D, Minnaard AJ, de Vries JG and Feringa BL, J. Am. Chem. Soc, 2002, 124, 14552–14553. [DOI] [PubMed] [Google Scholar]
- 114.Ohashi A and Imamoto T, Org. Lett, 2001, 3, 373–375. [DOI] [PubMed] [Google Scholar]
- 115.Wang X and Ding K, J. Am. Chem. Soc, 2004, 126, 10524–10525. [DOI] [PubMed] [Google Scholar]
- 116.Molinaro C, Scott JP, Shevlin M, Wise C, Ménard A, Gibb A, Junker EM and Lieberman D, J. Am. Chem. Soc, 2015, 137, 999–1006. [DOI] [PubMed] [Google Scholar]
- 117.Etayo P and Vidal-Ferran A, Chem. Soc. Rev, 2013, 42, 728–754. [DOI] [PubMed] [Google Scholar]
- 118.Abreu AS, Ferreira PMT, Queiroz M-JRP, Ferreira ICFR, Calhelha RC and Estevinho LM, Eur. J. Org. Chem, 2005, 2005, 2951–2957. [Google Scholar]
- 119.Queiroz M-JRP, Begouin A, Pereira G and Ferreira PMT, Tetrahedron, 2008, 64, 10714–10720. [Google Scholar]
- 120.Roff GJ, Lloyd RC and Turner NJ, J. Am. Chem. Soc, 2004, 126, 4098–4099. [DOI] [PubMed] [Google Scholar]
- 121.Burk MJ, Allen JG, Kiesman WF and Stoffan KM, Tetrahedron Letters, 1997, 38, 1309–1312. [Google Scholar]
- 122.Hoerrner RS, Askin D, Volante RP and Reider PJ, Tetrahedron Letters, 1998, 39, 3455–3458. [Google Scholar]
- 123.Abreu AS, Silva NO, Ferreira PMT and Queiroz M-JRP, Tetrahedron Letters, 2003, 44, 6007–6009. [Google Scholar]
- 124.Prieto M, Mayor S, Lloyd-Williams P and Giralt E, J. Org. Chem, 2009, 74, 9202–9205. [DOI] [PubMed] [Google Scholar]
- 125.Miossec B, Danion-Bougot R and Danion D, Synthesis, 1994, 1994, 1171–1174. [Google Scholar]
- 126.Abreu AS, Ferreira PMT, Queiroz M-JRP, Gatto E and Venanzi M, Eur. J. Org. Chem, 2004, 2004, 3985–3991. [Google Scholar]
- 127.Singh J, Kronenthal DR, Schwinden M, Godfrey JD, Fox R, Vawter EJ, Zhang B, Kissick TP, Patel B, Mneimne O, Humora M, Papaioannou CG, Szymanski W, Wong MKY, Chen CK, Heikes JE, DiMarco JD, Qiu J, Deshpande RP, Gougoutas JZ and Mueller RH, Org. Lett, 2003, 5, 3155–3158. [DOI] [PubMed] [Google Scholar]
- 128.Crestey F, Collot V, Stiebing S and Rault S, Synthesis, 2006, 2006, 3506–3514. [Google Scholar]
- 129.Jeffery T, J. Chem. Soc., Chem. Commun, 1984, 1287–1289.
- 130.Jeffery T, Tetrahedron Letters, 1985, 26, 2667–2670. [Google Scholar]
- 131.Basu B, Chattopadhyay SK, Ritzen A and Frejd T, Tetrahedron: Asymmetry, 1997, 8, 1841–1846. [Google Scholar]
- 132.de Azambuja F and Correia CRD, Tetrahedron Letters, 2011, 52, 42–45. [Google Scholar]
- 133.Harrington PJ, Hegedus LS and McDaniel KF, J. Am. Chem. Soc, 1987, 109, 4335–4338. [Google Scholar]
- 134.Lange G, Lesuisse D, Deprez P, Schoot B, Loenze P, Benard D, Marquette J-P, Broto P, Sarubbi E and Mandine E, J. Med. Chem, 2003, 46, 5184–5195. [DOI] [PubMed] [Google Scholar]
- 135.Lesuisse D, Lange G, Deprez P, Benard D, Schoot B, Delettre G, Marquette J-P, Broto P, Jean-Baptiste V, Bichet P, Sarubbi E and Mandine E, J. Med. Chem, 2002, 45, 2379–2387. [DOI] [PubMed] [Google Scholar]
- 136.De Lombaert S, Blanchard L, Stamford LB, Tan J, Wallace EM, Satoh Y, Fitt J, Hoyer D, Simonsbergen D, Moliterni J, Marcopoulos N, Savage P, Chou M, Trapani AJ and Jeng AY, J. Med. Chem, 2000, 43, 488–504. [DOI] [PubMed] [Google Scholar]
- 137.Berteina S, Wendeborn S, Brill WKD and De Mesmaeker A, Synlett, 1998, 1998, 676–678. [Google Scholar]
- 138.Bozell JJ, Vogt CE and Gozum J, J. Org. Chem, 1991, 56, 2584–2587. [Google Scholar]
- 139.Costa A, Najera C and Sansano JM, J. Org. Chem, 2002, 67, 5216–5225. [DOI] [PubMed] [Google Scholar]
- 140.Garcia-Gonzalez MC, Hernandez-Vazquez E, Gordillo-Cruz RE and Miranda LD, Chemical Communications, 2015, 51, 11669–11672. [DOI] [PubMed] [Google Scholar]
- 141.Bartoccini F, Cannas DM, Fini F and Piersanti G, Org. Lett, 2016, 18, 2762–2765. [DOI] [PubMed] [Google Scholar]
- 142.de Bruijn AD and Roelfes G, Chemistry – A European Journal, 2018, 24, 12728–12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Phelan JP and Ellman JA, Beilstein Journal of Organic Chemistry, 2016, 12, 1203–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chapman CJ, Wadsworth KJ and Frost CG, Journal of Organometallic Chemistry, 2003, 680, 206–211. [Google Scholar]
- 145.Chapman CJ, Hargrave JD, Bish G and Frost CG, Tetrahedron, 2008, 64, 9528–9539. [Google Scholar]
- 146.Navarre L, Martinez R, Genet J-P and Darses S, J. Am. Chem. Soc, 2008, 130, 6159–6169. [DOI] [PubMed] [Google Scholar]
- 147.Hargrave JD, Bish G, Köhn GK and Frost CG, Org. Biomol. Chem, 2010, 8, 5120–5125. [DOI] [PubMed] [Google Scholar]
- 148.Christoffel F, Catalysis Letters, 2018, 148, 489–511. [Google Scholar]
- 149.Lipshutz BH, Isley NA, Moser R, Ghorai S, Leuser H and Taft BR, Advanced Synthesis & Catalysis, 2012, 354, 3175–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brock S, Hose DRJ, Moseley JD, Parker AJ, Patel I and Williams AJ, Org. Process Res. Dev, 2008, 12, 496–502. [Google Scholar]
- 151.Kenda BM, Matagne AC, Talaga PE, Pasau PM, Differding E, Lallemand BI, Frycia AM, Moureau FG, Klitgaard HV, Gillard MR, Fuks B and Michel P, J. Med. Chem, 2004, 47, 530–549. [DOI] [PubMed] [Google Scholar]
- 152.Kuwano R and Ito Y, J. Org. Chem, 1999, 64, 1232–1237. [Google Scholar]
- 153.Desai AA, Angew. Chem. Int. Ed, 2011, 50, 1974–1976. [DOI] [PubMed] [Google Scholar]
- 154.Hansen KB, Hsiao Y, Xu F, Rivera N, Clausen A, Kubryk M, Krska S, Rosner T, Simmons B, Balsells J, Ikemoto N, Sun Y, Spindler F, Malan C, Grabowski EJJ and Armstrong JD III., J. Am. Chem. Soc, 2009, 131, 8798–8804. [DOI] [PubMed] [Google Scholar]
- 155.Le DN, Hansen E, Khan HA, Kim B, Wiest O and Dong VM, Nature Chemistry, 2018, 1–6. [DOI] [PMC free article] [PubMed]
- 156.Hayashi T, Takahashi M, Takaya Y and Ogasawara M, J. Am. Chem. Soc, 2002, 124, 5052–5058. [DOI] [PubMed] [Google Scholar]
- 157.Sakai M, Hayashi H and Miyaura N, Organometallics, 1997, 16, 4229–4231. [Google Scholar]
- 158.Navarre L, Martinez R, Genet J-P and Darses S, J. Am. Chem. Soc, 2008, 130, 6159–6169. [DOI] [PubMed] [Google Scholar]
- 159.Navarre L, Darses S and Genet J-P, Angew. Chem. Int. Ed. Engl, 2004, 43, 719–723. [DOI] [PubMed] [Google Scholar]
- 160.Myers CL, Hang PC, Ng G, Yuen J and Honek JF, Bioorganic & Medicinal Chemistry, 2010, 18, 4231–4237. [DOI] [PubMed] [Google Scholar]
- 161.Ture A, Rubina K, Rozhkov E and Kauss V, Chem. Heterocycl. Compd. (N. Y., NY, U. S.), 2011, 47, 838–844. [Google Scholar]
- 162.Crawley ML, Goljer I, Jenkins DJ, Mehlmann JF, Nogle L, Dooley R and Mahaney PE, Org. Lett, 2006, 8, 5837–5840. [DOI] [PubMed] [Google Scholar]
- 163.Queiroz M-JRP, Castanheira EMS, Carvalho MSD, Abreu AS, Ferreira PMT, Karadeniz H and Erdem A, Tetrahedron, 2008, 64, 382–391. [Google Scholar]
- 164.Ferreira PMT, Castanheira EMS, Monteiro LS, Pereira G and Vilaca H, Tetrahedron, 2010, 66, 8672–8680. [Google Scholar]
- 165.Chea H, Sim H-S and Yun J, Advanced Synthesis & Catalysis, 2009, 351, 855–858. [Google Scholar]
- 166.Chung JYL, Shevlin M, Klapars A and Journet M, Org. Lett, 2016, 18, 1812–1815. [DOI] [PubMed] [Google Scholar]
- 167.He Z-T, Zhao Y-S, Tian P, Wang C-C, Dong H-Q and Lin G-Q, Org. Lett, 2014, 16, 1426–1429. [DOI] [PubMed] [Google Scholar]
- 168.Bartoccini F, Bartolucci S, Lucarini S and Piersanti G, Eur. J. Org. Chem, 2015, 2015, 3352–3360. [Google Scholar]
- 169.Cooke HA, Christianson CV and Bruner SD, Current Opinion in Chemical Biology, 2009, 13, 460–468. [DOI] [PubMed] [Google Scholar]
- 170.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian K-D, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Müller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJT, Rebuffat S, Ross RP, Sahl H-G, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Süssmuth RD, Tagg JR, Tang G-L, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM and van der Donk WA, Nat. Prod. Rep, 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Oman TJ and van der Donk WA, Nat Meth, 2010, 6, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li B, Yu JPJ, Brunzelle JS, Moll GN, van der Donk WA and Nair SK, Science, 2006, 311, 1464–1467. [DOI] [PubMed] [Google Scholar]
- 173.Zhu Y, Gieselman MD, Zhou H, Averin O and van der Donk WA, Org. Biomol. Chem, 2003, 1, 3304–3315. [DOI] [PubMed] [Google Scholar]
- 174.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW and van der Donk WA, Proceedings of the National Academy of Sciences, 2010, 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang H and van der Donk WA, ACS Chem. Biol, 2012, 7, 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Völler GH, Krawczyk JM, Pesic A, Krawczyk B, Nachtigall J and Süssmuth RD, ChemBioChem, 2012, 13, 1174–1183. [DOI] [PubMed] [Google Scholar]
- 177.An L, Cogan DP, Navo CD, Jiménez-Osés G, Nair SK and Donk WA, Nat Chem Biol, 2018, 1–11. [DOI] [PMC free article] [PubMed]
- 178.Cotter PD, O’Connor PM, Draper LA, Lawton EM, Deegan LH, Hill C and Ross RP, Proceedings of the National Academy of Sciences, 2005, 102, 18584–18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lohans CT, Li JL and Vederas JC, J. Am. Chem. Soc, 2014, 136, 13150–13153. [DOI] [PubMed] [Google Scholar]
- 180.Suda S, Lawton EM, Wistuba D, Cotter PD, Hill C and Ross RP, Journal of Bacteriology, 2012, 194, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lohans CT and Vederas JC, The Journal of Antibiotics, 2013, 67, 23–30. [DOI] [PubMed] [Google Scholar]
- 182.Huo L and van der Donk WA, J. Am. Chem. Soc, 2016, 138, 5254–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xavier Just-Baringo FAMÁ, Marine Drugs, 2014, 12, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Just-Baringo X, Albericio F and Álvarez M, Angew. Chem. Int. Ed, 2014, 53, 6602–6616. [DOI] [PubMed] [Google Scholar]
- 185.Walsh CT, Acker MG and Bowers AA, J. Biol. Chem, 2010, 285, 27525–27531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zheng Q, Fang H and Liu W, Org. Biomol. Chem, 2017, 15, 3376–3390. [DOI] [PubMed] [Google Scholar]
- 187.Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC and Bowers AA, J. Am. Chem. Soc, 2015, 137, 3494–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bycroft BW and Gowland MS, J. Chem. Soc., Chem. Commun, 1978, 256. [Google Scholar]
- 189.Mocek U, Knaggs AR, Tsuchiya R, Nguyen T, Beale JM and Floss HG, J. Am. Chem. Soc, 1993, 115, 7557–7568. [Google Scholar]
- 190.Mocek U, Zeng Z, O’Hagan D, Zhou P, Fan LDG, Beale JM and Floss HG, J. Am. Chem. Soc, 1993, 115, 7992–8001. [Google Scholar]
- 191.Hudson GA, Zhang Z, Tietz JI, Mitchell DA and van der Donk WA, J. Am. Chem. Soc, 2015, 137, 16012–16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA and Mitchell DA, J. Am. Chem. Soc, 2016, 138, 15511–15514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cogan DP, Hudson GA, Zhang Z, Pogorelov TV, van der Donk WA, Mitchell DA and Nair SK, Proceedings of the National Academy of Sciences, 2017, 114, 12928–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wever WJ, Bogart JW and Bowers AA, J. Am. Chem. Soc, 2016, jacs.6b05389. [DOI] [PubMed]
- 195.Bowers AA, Acker MG, Young TS and Walsh CT, J. Am. Chem. Soc, 2012, 134, 10313–10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bowers AA, Walsh CT and Acker MG, J. Am. Chem. Soc, 2010, 132, 12182–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Acker MG, Bowers AA and Walsh CT, J. Am. Chem. Soc, 2009, 131, 17563–17565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Young TS, Dorrestein PC and Walsh CT, Chemistry & Biology, 2012, 19, 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zhang F, Li C and Kelly WL, ACS Chem. Biol, 2016, 11, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Coelho PS, Brustad EM, Kannan A and Arnold FH, Science, 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- 201.Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH and Brustad EM, Nat Chem Biol, 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gober JG and Brustad EM, Current Opinion in Chemical Biology, 2016, 35, 124–132. [DOI] [PubMed] [Google Scholar]
- 203.Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS and Arnold FH, Angew. Chem. Int. Ed, 2014, 53, 6810–6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gober JG, Ghodge SV, Bogart JW, Wever WJ, Watkins RR, Brustad EM and Bowers AA, ACS Chem. Biol, 2017, acschembio.7b00358. [DOI] [PubMed]
- 205.Asano Y, Kato Y, Levy C, Baker P and Rice D, Biocatalysis and Biotransformation, 2009, 22, 133–140. [Google Scholar]
- 206.Kudo F, Miyanaga A and Eguchi T, Nat. Prod. Rep, 2014, 31, 1056–1073. [DOI] [PubMed] [Google Scholar]
- 207.Pinto GP, Ribeiro AJM, Ramos MJ, Fernandes PA, Toscano M and Russo N, Archives of Biochemistry and Biophysics, 2015, 582, 107–115. [DOI] [PubMed] [Google Scholar]
- 208.Wu B, Szymański W, Heberling MM, Feringa Ben L and Janssen DB, Trends in Biotechnology, 2011, 29, 352–362. [DOI] [PubMed] [Google Scholar]
- 209.Wybenga GG, Szymański W, Wu B, Feringa BL, Janssen DB and Dijkstra BW, Biochemistry, 2014, 53, 3187–3198. [DOI] [PubMed] [Google Scholar]
- 210.Christenson SD, Liu W, Toney MD and Shen B, J. Am. Chem. Soc, 2003, 125, 6062–6063. [DOI] [PubMed] [Google Scholar]
- 211.Walker KD, Klettke K, Akiyama T and Croteau R, J. Biol. Chem, 2004, 279, 53947–53954. [DOI] [PubMed] [Google Scholar]
- 212.Jin M, Fischbach MA and Clardy J, J. Am. Chem. Soc, 2006, 128, 10660–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Turner NJ, Current Opinion in Chemical Biology, 2011, 15, 234–240. [DOI] [PubMed] [Google Scholar]
- 214.Parmeggiani F, Weise NJ, Ahmed ST and Turner NJ, Chem. Rev, 2017, 118, 73–118. [DOI] [PubMed] [Google Scholar]
- 215.Cui JD, Qiu JQ, Fan XW, Jia SR and Tan ZL, Critical Reviews in Biotechnology, 2013, 34, 258–268. [DOI] [PubMed] [Google Scholar]
- 216.Ding W, Liu W-Q, Jia Y, Li Y, van der Donk WA and Zhang Q, Proceedings of the National Academy of Sciences, 2016, 113, 3521–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ye Y, Minami A, Igarashi Y, Izumikawa M, Umemura M, Nagano N, Machida M, Kawahara T, Shin-ya K, Gomi K and Oikawa H, Angew. Chem. Int. Ed, 2016, 55, 8072–8075. [DOI] [PubMed] [Google Scholar]
- 218.Grove TL, Himes PM, Hwang S, Yumerefendi H, Bonanno JB, Kuhlman B, Almo SC and Bowers AA, J. Am. Chem. Soc, 2017, 139, 11734–11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Broderick JB, Duffus BR, Duschene KS and Shepard EM, Chem. Rev, 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.deGruyter JN, Malins LR and Baran PS, Biochemistry, 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Thibodeaux CJ, Ha T and van der Donk WA, J. Am. Chem. Soc, 2014, 136, 17513–17529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW and Schmidt EW, J. Am. Chem. Soc, 2010, 132, 15499–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McIntosh JA and Schmidt EW, ChemBioChem, 2010, 11, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Schwalen CJ, Hudson GA, Kille B and Mitchell DA, J. Am. Chem. Soc, 2018, 140, 9494–9501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cox CL, Tietz JI, Sokolowski K, Melby JO, Doroghazi JR and Mitchell DA, ACS Chem. Biol, 2014, 9, 2014–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wang J, Yu Y, Tang K, Liu W, He X, Huang X and Deng Z, Applied and Environmental Microbiology, 2010, 76, 2335–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mori T, Cahn JKB, Wilson MC, Meoded RA, Wiebach V, Martinez AFC, Helfrich EJN, Albersmeier A, Wibberg D, Dätwyler S, Keren R, Lavy A, Rückert C, Ilan M, Kalinowski J, Matsunaga S, Takeyama H and Piel J, Proceedings of the National Academy of Sciences, 2018, 115, 1718–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chalker JM, Lin YA, Boutureira O and Davis BG, Chem. Commun, 2009, 47, 3714–3. [DOI] [PubMed] [Google Scholar]
- 229.Lin YA, Boutureira O, Lercher L, Bhushan B, Paton RS and Davis BG, J. Am. Chem. Soc, 2013, 135, 12156–12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Urban JH, Moosmeier MA, Aumüller T, Thein M, Bosma T, Rink R, Groth K, Zulley M, Siegers K, Tissot K, Moll GN and Prassler J, Nature Communications, 2017, 1–10. [DOI] [PMC free article] [PubMed]
- 231.Hetrick KJ, Walker MC and van der Donk WA, ACS Cent. Sci, 2018, 4, 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Goto Y, Iwasaki K, Torikai K, Murakami H and Suga H, Chem. Commun, 2009, 13, 3419–3. [DOI] [PubMed] [Google Scholar]
- 233.Jongkees SAK, Umemoto S and Suga H, Chemical Science, 2017, 8, 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bashiruddin NK and Suga H, Current Opinion in Chemical Biology, 2015, 24, 131–138. [DOI] [PubMed] [Google Scholar]
- 235.Seebeck FP and Szostak JW, J. Am. Chem. Soc, 2006, 128, 7150–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hofmann FT, Szostak JW and Seebeck FP, J. Am. Chem. Soc, 2012, 134, 8038–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]