

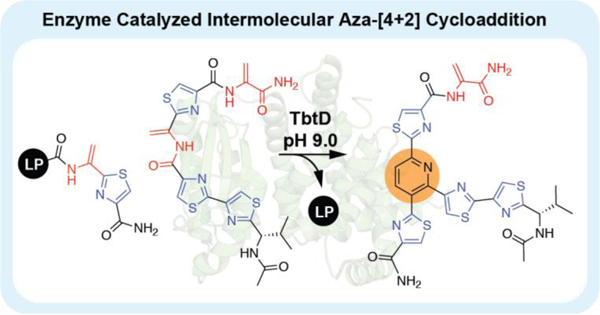
Abstract
Thiopeptide pyridine synthases catalyze a multistep reaction involving a unique and non-spontaneous intramolecular aza-[4+2] cycloaddition between two dehydroalanines to forge a tri-substituted pyridine core. We discovered that the in vitro activity of pyridine synthases from the thiocillin and thiomuracin pathways are significantly enhanced by general base catalysis and that this broadly expands the enzymes substrate tolerance. Remarkably, TbtD is competent to perform an intermolecular cyclization in addition to its cognate intramolecular reaction, underscoring its versatility as a biocatalyst. These data provide evidence that pyridine synthases use a two-site substrate recognition model to engage and process their substrates.
Graphical Abstract
The Diels-Alder (DA) reaction has been a staple of organic synthesis since its discovery by Otto Diels and Kurt Alder in 1928.1 Numerous total syntheses have exploited different versions of this typically concerted cycloaddition reaction and related transformations to efficiently construct carbon frameworks and stitch together complex organic scaffolds.2 In recent years, it has become apparent that similar chemistry is employed widely in the biosynthetic pathways of complex natural products.3–6 Several enzymes have been found to catalyze DA or DA-like reactions, including PyrI4 in pyroindomycin biosynthesis, SpnF in spinosyn biosynthesis, and LovB in lovastatin biosynthesis, among others.7–11 In many of these cases, the enzymes work to activate, accelerate, and/or direct the stereochemical outcome of an otherwise spontaneous cycloaddition reaction. As a result, reactions catalyzed by these Diels-Alderases can be highly substrate dependent and their biocatalytic utility is limited.
Pyridine synthases in thiopeptide biosynthesis provide unique examples of what is thought to be an enzyme catalyzed aza-DA. The biosyntheses of thiopeptides are similar to other ribosomally synthesized and post-translationally modified peptides (RiPPs) in that the C-terminal ‘core’ of a precursor peptide is transformed into the mature natural product by a suite of peptide modifying enzymes that are recruited by an N-terminal ‘leader’ peptide. 12,13 Pyridine synthases are responsible for the late stage formation of the class-defining nitrogenous heterocycles at the core of the thiopeptide pharmacophores.14–16 The key reaction is thought to entail an intramolecular formal [4+2] cycloaddition between two dehydroalanines (Dhas) followed by elimination of water and leader peptide (LP) to yield a tri-substituted pyridine core (Fig. 1a).17,18 In contrast to other DAs, this reaction is non-spontaneous and incurs a significant thermodynamic penalty due to the inactivated nature of the reaction subcomponents. Synthetic versions of this cycloaddition are low yielding and require high temperatures.19
Figure 1.
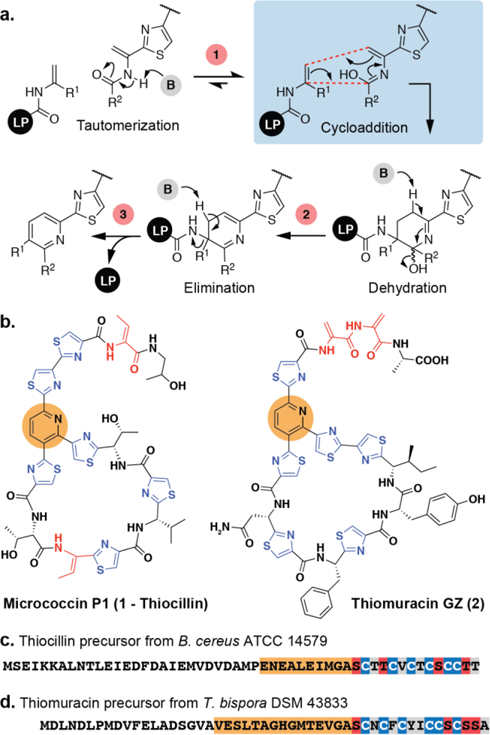
(a) Proposed mechanism of pyridine synthases. Steps potentially influenced by base (1–3) are highlighted. (b) Thiopeptides thiocillin (left) and thiomuracin (right) and their precursor peptide sequences (c, d). Orange regions highlight pyridine synthase recognition sequences used in this work. Core residues in red are converted to Dhas/Dhbs and residues in blue are converted to thiazoles.
The in vivo manipulation of thiopeptide biosynthetic gene clusters has demonstrated that pyridine synthases are very promiscuous enzymes and can be used to generate a variety of thiopeptide derivatives with different sized macrocycles and pyridine substituents.20–25 More specifically, enzymatic macrocyclization is independent of any structural preorganization imposed by the core peptide, as neither its length nor rigidity prevent cyclization.23 Thus we reasoned that pyridine synthases might be able to catalyze an intermolecular reaction between isolated 2π and 4π components. Such a reaction would provide perhaps the most extreme example of substrate promiscuity and support a two-site model of substrate recognition. Therefore, we sought to test whether pyridine synthases TclM or TbtD, from the biosynthetic pathways of thiocillin (1) and thiomuracin (2), respectively, could catalyze an intermolecular reaction (Fig. 1b). Optimization of the enzyme reaction conditions was an important initial step to test this hypothesis because our previous work showed that the enzymes could be slow and several modifications known to be permitted in vivo either refused to be cyclized in vitro and/or generated by-products.26
We first looked to improve the in vitro activity of TclM and TbtD by investigating the influence of pH on the rate of pyridine formation. Several steps of the proposed mechanism could be influenced by pH: (1) tautomerization of the enamide to iminol, (2) elimination of water and (3) aromatization (Fig. 1a). We used our previously described solid-phase strategy to prepare substrates for TclM and TbtD (compounds 3 and 4, respectively, Fig. 2a, b) based on their respective precursor peptides, TclE and TbtA (Fig. 1c).26 Each substrate consisted of a simplified core with a minimal LP fragment that was previously found to be sufficient for complete processing.14,26 The TclM substrate (3), was synthesized with a 10-residue LP, while the TbtD substrate (4), had a 15-residue LP (Fig. 2a, b). Enzymatic reactions were monitored at neutral and basic pH by measuring the distinctive absorbance of the newly forming, tri-substituted pyridine at 350nm over time. Reaction rates were calculated based on standard curve (see SI). A significant increase in rate was observed for each enzyme at pH 9.0 (Fig. 2c). Specifically for TbtD, a nearly 10-fold increase in kcat was observed from pH 7.2 to 9.0 (Fig. 2d, S1). Although kcat at pH 9.0 is still generally considered low, it is comparable to other late stage RIPP-modifying enzymes.27–32
Figure 2.
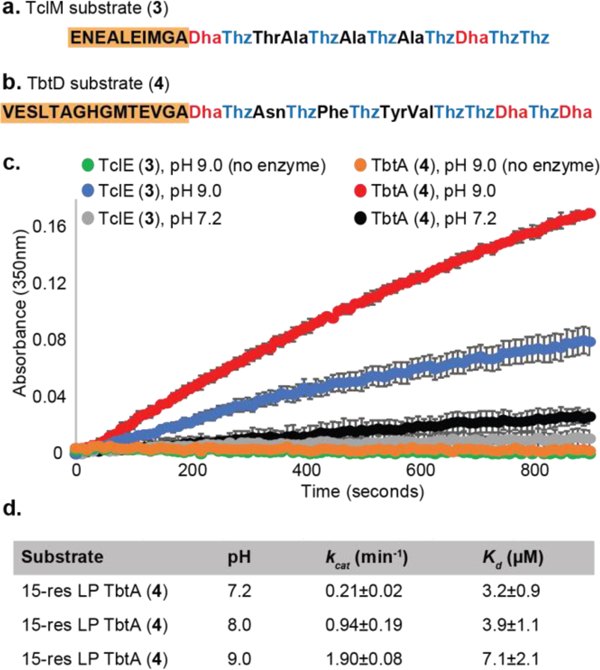
Simplified TclM (a) and TbtD substrates (b). (c) The rate of TclM or TbtD catalyzed pyridine formation is enhanced under basic pH. (b) Impact of pH on kcat and Kd.
Next, we assessed whether pH influenced substrate binding. We measured the dissociation constant, Kd for TbtA bearing the 15-residue LP and MBP-TbtD via fluorescence polarization assay. A non-reactive substrate for TbtD, with alanines instead of Dhas, was synthesized and derivatized with fluorescein isothiocyanate (FITC) at the N-terminus (5). Kd’s were determined at pH 7.2, 8.0 and 9.0 (Fig. 2d, S2). Effects of pH on Kd were minor compared to those on kcat. At pH 7.2, a Kd of 3.2μM was measured whereas values of 3.9 and 7.1μM were observed at pH 8.0 and 9.0 respectively. These data suggest that the pH does not significantly influence substrate binding.
Due to the pronounced binding affinity of TbtA’s N-terminal 16-residue LP for TbtD,33 we sought to determine whether this region of the LP might further enhance the reaction rate by allosteric regulation in trans. Studies have shown that this region contribute much of the full-length leader LP binding affinity to TbtD (Kd = 1.3μM),33 but is not strictly essential for enzymatic processing.16,26 The 16-residue N-terminal fragment (6) was synthesized by SPPS and reaction rate for substrate 4 was measured at pH 7.2 and 9.0 in presence or absence of 6. No appreciable change in rate was observed (Fig. S3). These data suggest that the N-terminal LP fragment does not act as an allosteric modulator. In the context of full length TbtA, this fragment’s higher affinity may allow the LP to bind first, thereby increasing local concentration of the core around the active site to facilitate faster modification.
As an initial test of TbtD’s improved substrate promiscuity under the optimized reaction conditions we assessed its ability to cyclize TbtA LP mutants. Previously, alanine scanning mutagenesis had been used to investigate LP binding to TbtD16,33 but not substrate processing. We reasoned that similar experiments might identify residues within the LP that are important for catalysis in addition to acting as a metric for promiscuity. A suite of 16, largely alanine-substituted, LP variants were prepared in parallel by solid-phase peptide synthesis (SPPS, see SI). The LP variants were separately tested at pH 7.2 and 9.0, and LCMS analysis revealed several differences (Table 1, Fig. S4a–q). Overall, 12 of the 16 substrates were processed at neutral pH, while 15 were cyclized under basic pH. Interestingly, only the H27A LP variant failed to cyclize under either condition. These data indicate that TbtD exhibits broader substrate promiscuity under basic pH.
Table 1.
Cyclization efficiency of TbtA LP mutants.
| % conversion | |||||
|---|---|---|---|---|---|
| TbtA Leader | pH 7.2 | pH 9.0 | TbtA Leader | pH 7.2 | pH 9.0 |
| 16-res WT | 85 | 96 | 16-res G28A | 83 | 86 |
| 16-res E21Q | 74 | 69 | 16-res M29A | 0 | 45 |
| 16-res E21A | 71 | 65 | 16-res T30A | 74 | 81 |
| 16-res S22A | 82 | 74 | 16-res E31Q | 30 | 14 |
| 16-res L23A | 2 | 51 | 16-res E31A | 65 | 18 |
| 16-res T24A | 72 | 77 | 16-res V32A | 49 | 76 |
| 16-res A25G | 80 | 72 | 16-res G33A | 84 | 87 |
| 16-res G26A | 1 | 47 | 16-res A34G | 68 | 81 |
| 16-res H27A | 0 | 0 | |||
With the enhanced activity of pyridine synthases confirmed, we sought to test whether TclM or TbtD could catalyze an intermolecular reaction. 2π components for both TclM and TbtD were prepared by SPPS and consisted of the corresponding minimal leader sequences with a C-terminal Dha-thiazole. The TclM 4π component was synthesized via SPPS following the same synthetic strategy that was used to prepare the intramolecular substrates. The TbtD 4π component was synthesized in solution from thiazole building blocks following standard peptide coupling procedures (see SI). In either case, each 4π component was designed to present the amide bond and Dha proposed to be involved in the cycloaddition plus two-to-three modified residues flanking either side. Incubation of each set of substrates with their respective pyridine synthases at pH 7.2 for 21 hrs yielded only trace amounts of product by LCMS. At pH 9.0, we again observed only trace amounts of product in reactions with TclM (Table S1). However, TbtD cleanly consumes both components and produces a new UV peak with characteristic absorbance at 350 nm and [m/z] consistent with a fully formed pyridine product (Fig. 3, S5a). The intermolecular reaction was scaled up and the identity of the product was confirmed by 1H and 13C NMR. TbtD is capable of recognizing the 2π and 4π reaction partners separately, without need for tethering between them, suggesting that an exceptionally broad array of macrocycles could likely be prepared by this enzyme.
Figure 3.
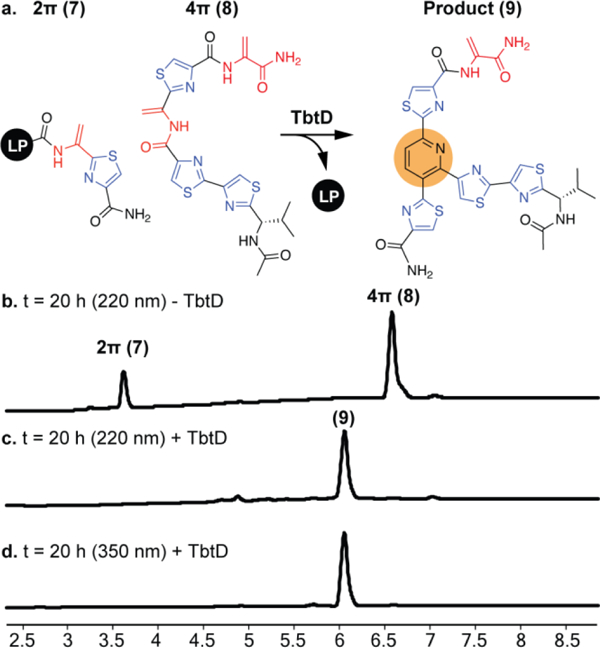
(a) TbtD-catalyzed intermolecular reaction. (b-d) UV traces of 2π and 4π substrates after 20 h in the presence (c and d) and absence (b) of TbtD.
In order to further define the limits of the TbtD-catalyzed intermolecular reaction, we prepared a small library of 2π and 4π substrates. With respect to the 2π component, previous LP truncation experiments and the above LP variants have defined the important residues in the LP so we focused further variation on the C-terminal side on the Dha. The thiazole (Thz) was changed to an oxazole (Oxz, Fig. 4b, entry 2) or removed entirely (Fig. 4b, entry 3). These 4π substrates were again synthesized via solution-phase chemistry and Dhas were installed either through alkylation-elimination chemistry of cysteine thiols or via mesylation-elimination of serine alcohols (see SI). As with the 2π substrates, thiazoles were iteratively changed to oxazoles and residues flanking the necessary Dha were removed to isolate the crucial components of the 4π substrate. For each new reaction, a percent conversion was calculated based on LCMS analysis and the rate of cyclization was measured by change in absorbance at 350 nm over time relative to a standard curve of purified product (Fig. 4b, S5a–h, S6). The results illustrate several key requirements for intermolecular substrates (summarized in Fig. 4a): (1) the C-terminal heterocycle must be present in the 2π component (Fig. 4b, entry 3), but a thiazole can be exchanged for an oxazole without significantly perturbing the rate or conversion (Fig. 4b, entry 2); (2) TbtD is sensitive to changes to both the N- and C-termini of the 4π component (comparing entry 1 to 6, 7 and 8 in Fig. 4b); (3) the bisazole motif is critical for cyclization; truncation to a single thiazole results in no product (Fig. 4b, entry 8); (4) removal of the C-terminal Dha results in no product – this Dha can be replaced with an alanine, albeit at significant cost to overall rate (Fig. 4b, entries 6 and 7); (5) in all 4π substrates, exchanging thiazoles for oxazoles is tolerated but there is a decrease in efficiency in each case (comparing entry 1 to 2, 4 and 5 in Fig. 4b). Ultimately, all permissible reactions could be forced to full conversion with higher enzyme loadings, allowing isolation and structural confirmation of products by 1H and 13C NMR. These data demonstrate TbtD is tolerant of major changes to nearly every residue outside of the two Dhas undergoing pyridine ring formation.
Figure 4.
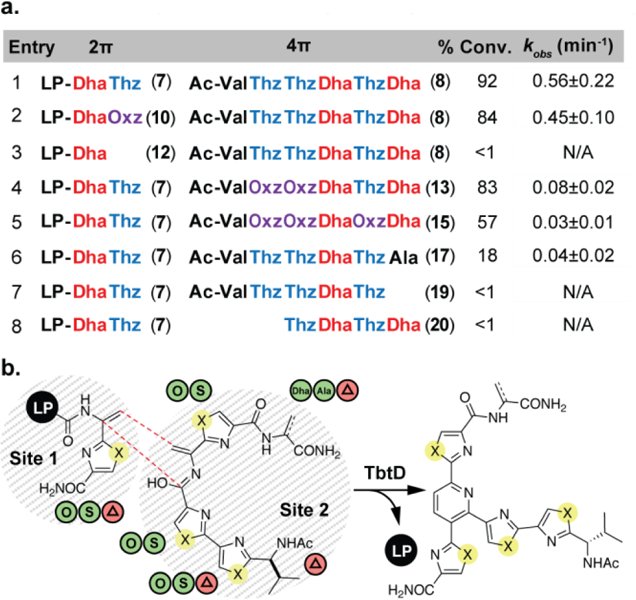
Substrate scope of the intermolecular reaction. (a) Consistent with intramolecular substrate 4, a 15-residue LP was used for all 2π components. Compound numbers for 2π and 4πs components are listed next to each sequence. See SI for calculations regarding percent conversion and kobs. (b) Green circles represent permissible changes (S for thiazoles, O for oxazoles). Removing residues with red triangles prevents cyclization.
In conclusion, we found that the thiomuracin pyridine synthase TbtD is capable of catalyzing an intermolecular pyridine formation in addition to its cognate intramolecular reaction. The intermolecular reaction proved possible only under rate-enhancing alkaline conditions, which allow an approximate 10-fold increase in enzyme turnover (kcat) while minimally affecting substrate binding (Kd). The thiocillin pyridine synthase, TclM, observed similar base-catalyzed rate-enhancement but could not perform an intermolecular reaction, suggesting that different families of pyridine synthases may have distinct substrate requirements. The intermolecular chemistry of TbtD provides evidence that pyridine synthases use a two-site model to engage and process their precursor peptides; this reaction is a strong indicator of this enzyme family’s broad promiscuity, suggesting that their use as biocatalysts could be extended beyond natural thiopeptide scaffolds, to allow larger ring sizes, incorporation of non-natural functionality or even novel linear substrates. The intermolecular reaction may be of use in elucidating the mechanism of these enzymes and may provide a barometer for evaluating new pyridine synthases for biotechnology applications.
Supplementary Material
ACKNOWLEDGMENT
We thank N. J. Kramer, R. Wolfenden, C. Neumann and B. Li for informative discussions. JWB is recipient of an AFPE pre-doctoral fellowship and a GSK pre-doctoral fellowship. This work was supported by NIH Grant GM125005 (AAB). AAB is a Beckman Young Investigator and acknowledges support by Arnold & Mabel Beckman Foundation.
Footnotes
ASSOCIATED CONTENT
Experimental details, synthetic schemes, figures available at http://pubs.acs.org.
Funding Sources
No competing financial interests have been declared.
REFERENCES
- (1).Diels O; Alder K Synthesen in der hydroaromatischen Reihe Justus Liebigs Ann. Chem 1928, 460, 98. [Google Scholar]
- (2).Nicolaou K; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed 2002, 41, 1668. [DOI] [PubMed] [Google Scholar]
- (3).Jamieson CS; Ohashi M; Liu F; Tang Y; Houk K The Expanding World of Biosynthetic Pericyclases: Cooperation of Experiment and Theory for Discovery. Nat. Prod. Rep 2018, 1, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jeon B; Wang S-A; Ruszczycky MW; Liu H Natural [4 + 2]-Cyclases. Chem. Rev 2017, 117, 5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Minami A; Oikawa H Recent Advances of Diels–Alderases Involved in Natural Product Biosynthesis. J. Antibiotics 2016, 69, 500. [DOI] [PubMed] [Google Scholar]
- (6).Klas K; Tsukamoto S; Sherman DH; Williams RM Natural Diels–Alderases: Elusive and Irresistable. J. Org. Chem 2015, 80, 11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zheng Q; Guo Y; Yang L; Zhao Z; Wu Z; Zhang H; Liu J; Cheng X; Wu J; Yang H; et al. Enzyme-Dependent [4 + 2] Cycloaddition Depends on Lid-like Interaction of the N-Terminal Sequence with the Catalytic Core in PyrI4. Cell Chem. Biol 2016, 23, 352. [DOI] [PubMed] [Google Scholar]
- (8).Fage CD; Isiorho EA; Liu Y; Wagner DT; Liu H; Keatinge-Clay AT The Structure of SpnF, a Standalone Enzyme That Catalyzes [4 + 2] Cycloaddition. Nat. Chem. Biol 2015, 11, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim H; Ruszczycky MW; Choi S; Liu Y; Liu H Enzyme-Catalysed [4+2] Cycloaddition Is a Key Step in the Biosynthesis of Spinosyn A. Nature 2011, 473, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ma SM; Li J; Choi JW; Zhou H; Lee MK; Moorthie VA; Xie X; Kealey JT; Silva NA; Vederas JC; et al. Complete Reconstitution of a Highly Reducing Iterative Polyketide Synthase. Science 2009, 326, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Auclair K; Sutherland A; Kennedy J; Witter DJ; Van den Heever JP; Hutchinson CR; Vederas JC Lovastatin Nonaketide Synthase Catalyzes an Intramolecular Diels-Alder Reaction of a Substrate Analogue. J. Am. Chem. Soc 2000, 122, 11519. [Google Scholar]
- (12).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep 2012, 30, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Brown LC; Acker MG; Clardy J; Walsh CT; Fischbach MA Thirteen Posttranslational Modifications Convert a 14-Residue Peptide into the Antibiotic Thiocillin. Proc. Natl. Acad. Sci. USA 2009, 106, 2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hudson GA; Zhang Z; Tietz JI; Mitchell DA; van der Donk WA In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin. J. Am. Chem. Soc 2015, 137, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wever WJ; Bogart JW; Baccile JA; Chan AN; Schroeder FC; Bowers AA Chemoenzymatic Synthesis of Thiazolyl Peptide Natural Products Featuring an Enzyme-Catalyzed Formal [4 + 2] Cycloaddition. J. Am. Chem. Soc 2015, 137, 3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang Z; Hudson GA; Mahanta N; Tietz JI; van der Donk WA; Mitchell DA Biosynthetic Timing and Substrate Specificity for the Thiopeptide Thiomuracin. J. Am. Chem. Soc 2016, 138, 15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mocek U; Knaggs AR; Tsuchiya R; Nguyen T; Beale JM; Floss HG Biosynthesis of the Modified Peptide Antibiotic Nosiheptide in Streptomyces actuosus. J. Am. Chem. Soc 1993, 115 7557. [Google Scholar]
- (18).Bycroft BW; Gowland MS The Structures of the Highly Modified Peptide Antibiotics Micrococcin P1 and P2. J. Chem. Soc., Chem. Commun 1978, 256. [Google Scholar]
- (19).Moody CJ; Hughes RA; Thompson SP; Alcaraz L Biosynthesis Inspired Diels–Alder Route to Pyridines: Synthesis of the 2,3-Dithiazolylpyridine Core of the Thiopeptide Antibiotics. Chem. Commun 2002, 0, 1760. [DOI] [PubMed] [Google Scholar]
- (20).Tran H; Lexa K; Julien O; Young TS; Walsh CT; Jacobson MP; Wells JA SAR and Molecular Mechanics Reveal the Importance of Ring Entropy in the Biosynthesis and Activity of a Natural Product. J. Am. Chem. Soc 2017, 139, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang F; Kelly WL Saturation Mutagenesis of TsrA Ala4 Unveils a Highly Mutable Residue of Thiostrepton A. ACS. Chem. Biol 2015, 10, 998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Young TS; Dorrestein PC; Walsh CT Codon Randomization for Rapid Exploration of Chemical Space in Thiopeptide Antibiotic Variants. Chem. Biol 2012, 19, 1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bowers AA; Acker MG; Young TS; Walsh CT Generation of Thiocillin Ring Size Variants by Prepeptide Gene Replacement and in Vivo Processing by Bacillus Cereus. J. Am. Chem. Soc 2012, 134, 10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bowers AA; Acker MG; Koglin A; Walsh CT Manipulation of Thiocillin Variants by Prepeptide Gene Replacement: Structure, Conformation, and Activity of Heterocycle Substitution Mutants. J. Am. Chem. Soc 2010, 132, 7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bowers AA; Walsh CT; Acker MG Genetic Interception and Structural Characterization of Thiopeptide Cyclization Precursors from Bacillus Cereus. J. Am. Chem. Soc 2010, 132, 12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wever WJ; Bogart JW; Bowers AA Identification of Pyridine Synthase Recognition Sequences Allows a Modular Solid-Phase Route to Thiopeptide Variants. J. Am. Chem. Soc 2016, 138, 13461. [DOI] [PubMed] [Google Scholar]
- (27).Lee J; McIntosh J; Hathaway BJ; Schmidt EW Using Marine Natural Products to Discover a Protease That Catalyzes Peptide Macrocyclization of Diverse Substrates. J. Am. Chem. Soc 2009, 131, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).McIntosh JA; Schmidt EW Marine Molecular Machines: Heterocyclization in Cyanobactin Biosynthesis. ChemBioChem 2010, 11, 1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).McIntosh JA; Robertson CR; Agarwal V; Nair SK; Bulaj GW; Schmidt EW Circular Logic: Nonribosomal Peptide-like Macrocyclization with a Ribosomal Peptide Catalyst. J. Am. Chem. Soc 2010, 132, 15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Agarwal V; Pierce E; McIntosh J; Schmidt EW; Nair SK Structures of Cyanobactin Maturation Enzymes Define a Family of Transamidating Proteases. Chem. Biol 2012, 19, 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Thibodeaux CJ; Ha T; van der Donk WA A Price to Pay for Relaxed Substrate Specificity: A Comparative Kinetic Analysis of the Class II Lantipeptide Synthetases ProcM and HalM2. J. Am. Chem. Soc 2014, 136, 17513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Roy RS; Belshaw PJ; Walsh CT Mutational Analysis of Posttranslational Heterocycle Biosynthesis in the Gyrase Inhibitor Microcin B17: Distance Dependence from Propeptide and Tolerance for Substitution in a GSCG Cyclizable Sequence. Biochemistry 1998, 37, 4125. [DOI] [PubMed] [Google Scholar]
- (33).Cogan DP; Hudson GA; Zhang Z; Pogorelov TV; van der Donk WA; Mitchell DA; Nair SK Structural Insights into Enzymatic [4+2] Aza-Cycloaddition in Thiopeptide Antibiotic Biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.