

Abstract
CRISPR-Cas9 gene-editing strategies have revolutionized our ability to engineer the human genome for robust functional interrogation of complex biological processes. We have recently adapted this technology for use in primary human CD4+ T cells to create a high-throughput platform for analyzing the role of host factors in HIV infection and pathogenesis. Briefly, CRISPR-Cas9 ribonucleoproteins (crRNPs) are synthesized in vitro and delivered to activated CD4+ T cells by nucleofection. These cells are then assayed for editing efficiency and expanded for use in downstream cellular, genetic, or protein-based assays. This platform supports the rapid, arrayed generation of multiple gene manipulations and is widely adaptable across culture conditions, infection protocols, and downstream applications. Here, we present detailed protocols for crRNP synthesis, primary T-cell culture, 96-well nucleofection, molecular validation, and HIV infection, and discuss additional considerations for guide and screen design, as well as crRNP multiplexing. Taken together, this procedure allows high-throughput identification and mechanistic interrogation of HIV host factors in primary CD4+ T cells by gene knockout, validation, and HIV spreading infection in as little as 2–3 weeks.
Introduction
Viral pathogens depend on the specialized microenvironments of their hosts for optimal replication and transmission. As obligate intracellular parasites, viruses rely on host proteins or ‘dependency factors’ to successfully replicate and infect new cells. In turn, the host has evolved molecular defenses known as ‘restriction factors’ to interfere with the replicative cycle of such pathogens1–3. Inhibition of host dependency factors or activation of host restriction factors can severely limit virus replication, and so these avenues represent promising candidates for the development of next-generation ther-apeutics4,5. Thus far, most systematic efforts to identify and characterize such host-pathogen interactions have relied on the manipulation of immortalized human cell lines in vitro with RNAi reagents. Although readily scalable for high-throughput screening, such experimental systems often fail to recapitulate and identify authentic in vivo relationships, owing to limitations in both cell line models and RNAi technology.
Immortalized human cell lines provide readily scalable, genetically tractable, and relatively cost-effective model systems for the study of human disease, but they often fail to recapitulate the normal cellular physiology that pathogens encounter during the course of natural infection6–8. The process of immortalization, selection, and expansion of these lines often markedly changes cellular expression profiles as well as responses to complex stimuli such as infection9,10. Thus, although the use of immortalized human cell lines has yielded an extraordinary wealth of information on host-pathogen relationships, many of the findings resulting from these systems have failed to translate in vivo11–13. These experimental concerns and the limited translational capacity of the resultant findings have driven interest in primary cell models of disease; however, primary cells are often difficult to maintain and manipulate. Only recently have advances in genome engineering made some types of primary cells genetically tractable ex vivo, allowing the development of model systems that are not only malleable, but also more reflective of in vivo cellular physiology than immortalized human cells14,15.
In addition to the limitations of using cell line models, unbiased genetic screening approaches to uncover host-pathogen interactions have also been complicated by limitations in the tools used for genetic perturbation, such as RNAi7,16–18. Although RNAi gene knockdown methodologies have provided an invaluable tool to biologists, they often suffer from low penetrance, transient efficacy, and high incidence of off-target effects6,19. These characteristics are of particular concern when the targeted gene products either have a long half-life or are required at only low abundance (such as is the case with the retroviral integrase interactor, LEDGF20–23). In such cases, even if a set of efficacious RNAi reagents are identified, validated, and stably integrated, knocking down the gene product may not be sufficient to reveal its functional significance6,19. Furthermore, off-target effects can compromise the specificity of the screen, leading to the need for all hits to be extensively validated24,25.
Owing to these and other limitations, most attempts to comprehensively and systematically define host-pathogen interactions have yielded only a limited number of verifiable associations. Meta-analysis of three genome-wide RNAi screens for human-HIV host-pathogen interacting factors found a <7% overlap in candidates between any two studies and an overlap of only three genes among all three studies6. Likewise, meta-analysis of eight genome-wide RNAi screens for human influenza A virus host-pathogen interacting factors found only a 7% overlap in candidates between any two studies26. In both cases, the variation among studies has been ascribed to differences in the RNAi libraries used to screen host candidate genes, the in vitro systems used to model infection, the readout of pathogen infectivity, and the strain of the pathogen itself6,19,26. Although pathway- and complex-level analyses of these same datasets have revealed some novel insights6,26, new genetic tools and more functionally relevant models are required for the systematic identification and improved understanding of human host-pathogen relationships8,27–30.
The recent advent of CRISPR-Cas9 genome editing offers an alternative strategy for gene manipulation that corrects for some key failings of previous approaches. Cas9 is a programmable DNA endonuclease that can be directed to a complementary region of the genome by its associated CRISPR (or guide) RNA31–33. Unlike RNAi-based approaches, which suppress messenger RNA, Cas9 targets the DNA directly for cleavage. Imperfect repair of the resultant double-strand break can result in insertions or deletions (indels) within coding regions, permanently ablating protein expression34,35. Alternatively, concurrent delivery of a specific repair template can result in the precise incorporation of new sequences by homology-directed repair15,31,32. As these edits occur at the DNA level, they are stable, allowing long-term expansion of edited populations without selection and the complete depletion of even long-lived proteins8,14. Furthermore, CRISPR-Cas9 editing has been shown to display fewer off-target effects than RNAi manipulation36–40.
Here, we describe a protocol for high-efficiency genome engineering in primary CD4+ T cells by CRISPR-Cas9 for the identification and exploration of human-HIV, host-pathogen interactions (Fig. 1). crRNPs are synthesized in vitro by the incubation of target-specific guide RNA (gRNA), trans-activating CRISPR RNA (tracrRNA), and Cas9 protein14,15. These preformed crRNP complexes are then delivered to activated primary T cells by nucleofection for editing14,15. This approach can be used with minimal manipulation to screen a wide array of genes for phenotypic influence, toxicity, or off-target effects. These reactions can be carried out in arrayed, 96-well format and can be readily multiplexed for the generation of double-knockout pools. Although we have used this platform for the identification of human-HIV host-pathogen interactions, and the Procedure reported below focuses on this specific application, we envision that this general strategy can be adapted more broadly to other primary cell types for the interrogation of a wide range of complex biological phenomena directly in patient and healthy donor samples.
Fig. 1 |. Overview of primary T-cell editing using CRISPR-Cas9 RNPs. Primary CD4+ T cells are isolated from donor blood and activated.
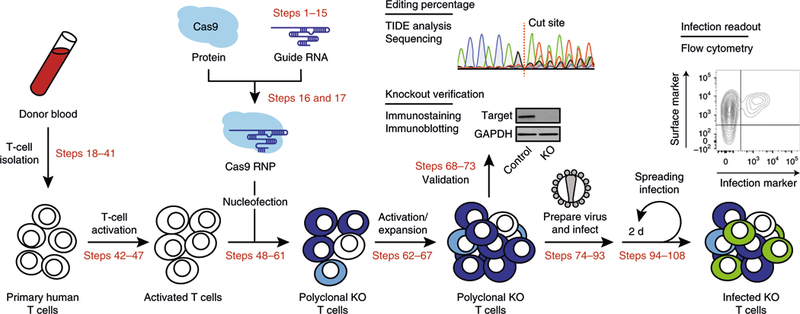
crRNPs are synthesized in vitro and delivered to the activated T cells by nucleofection. These cells are expanded for molecular validation of gene editing and downstream phenotypic assays. HIV-1 virus stocks are prepared and used to infect the cell pools, which are monitored for infection by flow cytometry over several days. Genes whose editing significantly alters the percentage of infected cells relative to non-targeting controls are candidate host factors for further mechanistic studies. KO, knockout. Adapted with permission from Hultquist et al.14, Elsevier.
Applications of crRNP-mediated primary cell gene editing
We have previously demonstrated that crRNPs can be successfully used to screen for and identify new HIV host factors in primary CD4+ T cells14. We have furthermore demonstrated that this technology can be used to validate the findings of hypothesis-driven and discovery-driven studies conducted in cell line models8,14. Given its scalability and ease of use, we believe that this approach is well suited for broad application, not only to the study of host-pathogen interactions, but to the investigation of other processes in which T cells are involved, such as autoimmunity, immune regulation, pathogen sensing, and immunologic memory. Furthermore, crRNP multiplexing, i.e., the use of multiple crRNPs at once to make double- or triple-knockout cell populations, enables the study of epistatic relationships and functional redundancy among gene families. Delivery of these same crRNPs to other primary cell types has the potential to transform many such cells into tractable genetic models with numerous applications across basic, translational, and clinical research.
This protocol has been employed successfully to screen potential host factors that interact with HIV integrase to influence HIV infection14. We are currently expanding these efforts to screen thousands of genes in parallel. Simultaneously, we are working to enhance multiplexing efficiency to perform systematic epistasis mapping directly in primary cell types. We are furthermore optimizing delivery to different primary cell types in an effort to study genetic programs in different cellular subsets from the same donor. Lessons gleaned from these efforts have been included in the text where applicable to assist in the optimization of these approaches to additional fields of study.
Limitations of crRNP-mediated primary cell gene editing
Although broadly applicable, scalable, and tractable, this approach has a number of distinct limitations. First, delivery of in vitro-synthesized crRNPs does not innately include a genetic selection marker. The resulting lack of selection and the imperfect repair of the generated double-strand breaks after cutting results in a polyclonal population of cells that includes unedited, heterozygous knockout, and homozygous knockout cells with heterogeneous sequences at the target locus. In other words, unlike RNAi, with which a given reagent results in a partial knockdown of protein expression across a population of cells, CRISPR-Cas9 editing will result in complete ablation of protein expression in a subset of cells while other cells remain unchanged. Phenotypic readout, therefore, is directly linked to the efficiency of the gRNA and the resultant frequencies of these genotypic populations in the final pool14,15. Successful knockout will prevent all protein expression from the targeted locus, allowing the detection of phenotypes for host factors that are required at only a small fraction of their normal cellular levels14,15. For example, if a cellular protein is required at only 10% of its endogenous level to be functionally active, an RNAi reagent that attains even 80% knockdown would be insufficient to yield a phenotype. A crRNP-edited pool, however, even at much lower efficacy, should result in some cells completely deficient in the protein of interest and can therefore yield a phenotype even at much lower editing efficiencies, depending on phenotypic assay output and sensitivity. In addition, approaches with single-cell readouts, such as flow cytometry or some next-generation sequencing methods, can be used to simultaneously monitor editing efficacy and phenotype in a single heterogeneous population of edited cells, providing robust internal controls. Fortunately, most loci are amenable to efficient CRISPR-Cas9 editing, and the gRNA design protocols detailed here will typically yield at least one guide out of every three with an average editing efficiency of ~75%14,37,41. Nevertheless, there are some loci that appear to be refractory to CRISPR-Cas9 editing, regardless of guide design. Although we hypothesize that this may be due to the genome architecture or chromatin structure at the locus, the reasons for it are currently unclear42,43. Similarly, certain cell types appear generally more resistant to CRISPR-Cas9 editing than activated T cells with the currently available technology. Regardless of the cause, the variability in editing efficiency across genomic loci and cell types underscores the importance of validating this parameter in each experiment and with every donor.
Second, although delivery of preformed crRNPs has yielded high-efficiency editing in primary T cells, the transient lifespan of these complexes limits the potential of this delivery method for applications beyond gene editing. Notably, CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) rely on continual occupancy of target loci by the Cas9 RNP complex44–47. Therefore, one-time delivery of preformed CRISPRi or CRISPRa RNPs may not cause long-term perturbations to gene expression, even though transient perturbations may be achievable, depending on the cell system.
Third, as gene ablation is complete in successfully edited cells, the editing of any genes essential to cell growth or cell survival may lead to cell cycle arrest or death and the preferential expansion of unedited cells. Conversely, editing of some cell cycle regulators may lead to preferential expansion of the edited population. Thus, verification of editing efficiency in the population should be performed on cellular lysates harvested at the time of phenotypic analysis. Concurrent monitoring of cell number and viability is also highly recommended to ensure that the observation of particular phenotypes is not simply a reflection of cell death or a cellular stress response. Below, we describe optional methods of verifying editing at the protein level by immunoblotting or immunostaining and at the genetic level by DNA sequencing.
Experimental design
Considerations for guide design
Since the advent of CRISPR-Cas9 editing, several different protocols and algorithms for the design of gRNAs have been described37,48,49, and a review of them has been published50. The performance of these algorithms has improved markedly over time, resulting in higher on-target hit rates with fewer off-target effects50,51. At the time of writing, the best such algorithm is arguably Rule Set 2, which is the basis for the Procedure we lay out below37. A number of resources exist to identify the best gRNA sequences for a given gene based on these rules, a few of which are described below. Guides can be custom-designed and ordered for synthesis by contract (as described in the Procedure), generated by in vitro transcription, or ordered pre-designed from a number of commercial providers14,15. If planning to use pre-designed guides, we recommend checking the specificity of each guide in silico before purchase, in order to ensure it targets the proper locus with minimal affinity for other genomic sequences. Stability-enhancing modifications of the RNA guides (such as 2’-O-methyl modifications) can be implemented with no loss of editing efficiency (see, e.g., Hendel A. et al.52).
For the generation of gene knockouts, we recommend ordering three to five distinct gRNAs per gene. For most loci, this approach will yield at least one guide that exhibits high-efficiency editing. Although any part of the gene can be targeted for editing, it is essential to take the gene structure into account during the design process to avoid intron-exon boundaries and alternatively spliced sites. In addition, naturally occurring human genetic variants could disrupt target sites or even create new off-target sites, so regions of high variation frequency should be avoided when possible. Many genome browsers provide isoform and variant information to assist in targeting key regions of the gene. We describe Ensembl as one example below, but we have had success using a variety of online tools for guide design. Benchling, for example, is free for academic use, and provides an online tutorial for guide design at https://benchling.com/tutorials/21/designing-and-analyzing-gRNAs. Recent publications have furthermore described pre-designed guides for human and mouse genomes37, many of which are available through Addgene.
Considerations for primary cell isolation, activation, and culture
Unlike cell lines, which can be expanded indefinitely, primary cells have limited longevity and expansion potential, so isolation of a sufficient number of cells to carry out the protocol is of paramount importance. The Procedure detailed below routinely involves the use of 500,000 to 1,000,000 cells per nucleofection reaction, but it may be applicable to cell counts both below and above this range, with some loss of viability or editing efficiency. Any scalable T-cell isolation protocol can be used to procure the required number of cells from whole blood or blood product, and many robust methods exist that are compatible with this protocol (e.g., positive-versus negative-selection protocols, selection of pan-T cells versus naive T cells, or magnetic versus filter-based separation methods). In the Procedure, we describe the isolation of peripheral blood mononuclear cells (PBMCs) from whole blood and the subsequent isolation of CD4+ T cells by negative selection using the StemCell Technologies EasySep T cell Isolation Kit.
Regarding CD4+ T-cell activation, we have achieved successful editing after stimulation with plate-bound anti-CD3 and soluble anti-CD28, bead-bound anti-CD3/anti-CD28, bead-bound anti-CD2/ anti-CD3/anti-CD28, or PHA and IL-2 (refs 14,15). Although it is important to achieve full cell activation for efficient editing under these conditions, it is just as important to avoid over-stimulation and antigen-induced cell death. Therefore, for knockout experiments such as those described in the Procedure, we recommend implementing a stimulation approach with plate-bound anti-CD3 and soluble anti-CD28 for 72 h before nucleofection, followed by a second round of stimulation with bead-bound anti-CD2/anti-CD3/anti-CD28 immediately after nucleofection. Optimal conditions for homology-directed repair can vary from those just described, so stimulation conditions should be optimized for each unique downstream application (see Roth et al.53). Furthermore, as the cells from each donor may respond differently to stimulation, the purity and activation state of the CD4+ T cells should be monitored by CD4 and CD25 cell surface staining on the day of nucleofection (as in Hultquist et al.14).
Cells will continue to expand after editing and can be maintained in culture for several weeks post nucleofection. If it is anticipated that the knockout of targeted genes may alter cell growth phenotypes, we recommend performing the planned phenotypic assays 3–7 d post nucleofection. This time interval should be long enough for the cells to recover from the editing reaction and for the existing protein to degrade, but insufficient for unedited cells to outcompete or substantially dilute out edited cells in the culture.
Considerations for screening
Screen design is dependent on the number of genes being screened and the downstream phenotypic assays to be performed. For targeted validation of a few genes, we recommend validating the efficiency of gRNA knockout with a test batch of primary T cells before embarking on phenotypic analyses. Guide efficiency varies by sequence, target locus, and cell type, but it is relatively consistent between donors. Indeed, although there is donor-to-donor variation in the overall permissivity to CRISPR–Cas9-mediated gene editing, the relative efficiency of editing between different gRNAs is nearly always consistent between donors (J.F.H., J.H., A.M., and N.J.K., unpublished observations).
For medium- to large-scale screens with three to five guides per gene, it may be easier to embark on the phenotypic assays without prior guide validation. gRNAs can be ordered from commercial sources pre-arrayed in 96-well plates. Conveniently, this layout can be preserved for crRNP generation, nucleofection, HIV infection, and additional phenotypic interrogation. Genomic or protein lysates can also be stored in this format for downstream validation. Thus, it may be expedient to proceed with phenotypic assays after validation of the positive controls only, limiting the broad validation of gRNA editing efficiencies to only those guides associated with a statistically significant phenotype. One consequence of this strategy is that negative results in phenotypic assays are difficult to interpret; they may indicate that the target host factor has no role in the process under examination or they may be due to editing failures. Disambiguation requires validation of every guide, which may or may not be a worthwhile effort, depending on the application and the experimental design.
Controls
As with any experimental system, conducting the right control experiments is critical for the downstream interpretation of results. The importance of such controls grows in proportion to the size of the experiment, as variations in donors, plate handling, and environment must be robustly accounted for. Although the controls we recommend in the Procedure are appropriate for studies focusing on host-pathogen interactions within the context of HIV infection, we strongly recommend including additional controls that are appropriate to the specific downstream phenotypic assay. Generally, each experiment and each plate within a larger screen should have at least three types of controls: a negative, non-targeting control; a positive control with a strong effect on the phenotype being assayed; and a toxicity control.
An important negative control for all applications is nucleofection of a crRNP that bears a non-targeting gRNA. Several such sequences have been published, and still more are commercially available as proprietary reagents37,48,54. We recommend including at least two distinct non-targeting guides per experiment, randomly distributed if on a plate. Other negative controls, including unedited cells, cells nucleofected with nothing, or cells nucleofected with Cas9 protein alone, may or may not be appropriate for the assay at hand. In addition, inclusion of a negative control that targets a gene not associated with the downstream phenotypic readout may be advisable in the case that DNA repair pathways activated by gene editing may influence the readout.
We also recommend including at least two distinct positive controls that have predictable effects on the phenotype being assayed. For HIV replication, we have found that knockout of the dependency factors CXCR4 and LEDGF results in reproducible, consistent defects in HIV replication. CXCR4 knockout prevents HIV entry into cells in a manner dependent on virus tropism and consistently decreases infection rates to levels close to the limit of detection14. LEDGF knockout, on the other hand, partially inhibits proviral integration and inhibits infection by typically 50–75%, but in a tropism-independent manner14.
One final control we recommend including is the knockout of a gene essential to cell health and proliferation, such as CDK9. In most donors, successful knockout of such a gene results in the activation of cell stress responses and cell death. Comparison of this control’s cell count, cell viability, and phenotype with those of experimental wells is useful for setting thresholds for analysis and for identifying other potentially essential genes. Although the toxicity of gene knockout will vary depending on cell type, culture conditions, and timing, several lists of essential human genes have been previously published55–57; these lists can be used to guide the search for potentially toxic gene knockouts.
Considerations for viral replication
The virus strain(s) used for virus replication assays will be dependent on the experimental setup and downstream analyses. Although many approaches have been developed for monitoring HIV replication in ex vivo cultures (e.g., p24 ELISA, reverse transcription assays, luciferase reporter viruses, fluorescent p24 antibodies)58–60, in the example application of the Procedure, we direct readers to use the replication-competent HIV-1 clone NL4–3 with a GFP reporter expressed behind the nef open-reading frame (nef:IRES:GFP)61. This virus offers a number of advantages for large-scale screening. First, this virus is replication competent, and thus we can monitor events that occur both early and late in the infection cycle. Second, this strain expresses all viral open reading frames, as well as a GFP reporter, for easy quantification without any required additional processing. The use of a fluorescent reporter obviates the need for cell staining before flow cytometry analysis, saving reagents, time, and the multiple centrifugations that can quickly become overwhelming when a large number of plates are in culture. Third, the use of flow cytometry to detect GFP provides single-cell data (including cell size, shape, and GFP mean fluorescence intensity), which can inform downstream mechanistic assays. The collection of single-cell data also enables the simultaneous monitoring of cell viability, such that potential confounding effects in gene editing can be readily identified. Inclusion of a toxicity control, such as a CDK9 targeting guide, can assist in the identification of similarly essential genes.
Note that the susceptibility of each donor’s cells to infection will vary, and, therefore, live virus titers may vary substantially from one donor to the next. In the Procedure below, we recommend titering the prepared virus stock on cells for each new donor to ensure levels of infection fall within a linear range. Alternatively, virus stocks can be prepared in bulk, and consistent amounts of the same lots can be used across multiple donors. The data can then be analyzed relative to internal controls in order to normalize out donor-to-donor variability in basal susceptibility. If high rates of infection are required, spinoculation of cultures can also be considered to boost initial infection rates62.
Considerations for crRNP multiplexing
The amount of crRNP that can be delivered to each cell is limited by the concentration of crRNPs that can be maintained in solution and the volume of crRNPs that can be added to the nucleofection cuvette without detrimental dilution of the nucleofection buffer. The volume of crRNPs can be increased up to twofold from what is recommended in the Procedure without the nucleofection efficiency being diminished. Although this volume increase may enhance the editing efficiency of otherwise inefficient guides, most targets do not require this modification, and the volumes recommended in the Procedure provide a good balance of editing efficiency, convenience, and cost in large-scale experiments. Given that the volumes can be effectively doubled in the reaction, two independent crRNPs can be delivered to the same set of cells simultaneously, against either the same or different genes. This approach therefore could allow the generation of condensed screening platforms and of double-knockout populations for the study of epistatic relationships and functional redundancy14. When multiplexing, we recommend preparing each pool of crRNPs independently and mixing the pools at a 1:1 ratio before nucleofection. Delivery of more than two independent crRNPs per reaction is likely to come at a cost in terms of nucleofection efficiency, could introduce trans-locations or other complex DNA repair outcomes, and may require additional optimization.
Consideration for studies in other cell types
This protocol has been optimized for use with primary CD4+ T cells, but it may be applicable to other T-cell subsets, as well as other primary cell types, with additional optimization. The key hurdle to adapting this protocol for use with additional cell types is optimization of crRNP delivery, although additional variables may also influence successful editing, including chromatin architecture, cell viability, and activation state. Successful delivery of the crRNPs can be monitored by immunostaining for Cas9 after nucleofection or by nucleofection of a fluorescently labeled Cas9 protein, but this may not always correlate directly with editing efficiency. We therefore typically recommend optimizing nucleofection conditions by quantifying editing efficiency at the genomic and/or protein levels using previously validated guides. As consistency of guide efficiency between cell types is not guaranteed, we recommend conducting these optimization experiments with at least two independent gRNAs.
When optimizing delivery to a new cell subset, pay special attention to input cell stimulation and concentration, cell culture conditions, nucleofection buffer, and nucleofection pulse code. The confluence of these factors will affect both editing efficiency and cell viability, collectively determining the frequency of edited cells in your resultant population. Several nucleofection optimization kits designed to test a range of conditions and buffers are commercially available (see, e.g., the Lonza Cell Line Optimization 4D-Nucleofector X Kit), and new delivery strategies for various cell types are published regularly15,34,41,63. The development of these strategies for peripheral and tissue-localized hematopoietic cell subsets, epithelial tissues, and an array of progenitor cells, including induced pluripotent stem cells, promises to revolutionize genetic study of primary human samples for the advancement of research into a wide array of diseases.
Materials
Biological materials
Whole-human blood or leukoreduction chamber
 CAUTION All human blood samples must be handled according to approved biosafety protocols and regulations set by each individual lab and institution. Improper handling of samples may lead to exposure to blood-borne pathogens.
CAUTION All human blood samples and data obtained from them must be handled according to approved biosecurity protocols for the protection of patient confidentiality and health information in accordance with relevant institutional and national regulations.
CAUTION All human blood samples must be handled according to approved biosafety protocols and regulations set by each individual lab and institution. Improper handling of samples may lead to exposure to blood-borne pathogens.
CAUTION All human blood samples and data obtained from them must be handled according to approved biosecurity protocols for the protection of patient confidentiality and health information in accordance with relevant institutional and national regulations.HEK293T cells (ATCC, cat. no. CRL-3216) for packaging of lentivirus
CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Reagents
Ficoll-Paque (Fisher, cat. no. 17144003)
FBS (heat-inactivated; Life Technologies, cat. no. 26140-079)
DMEM (high glucose; Corning, cat. no. MT10-017-CV)
RPMI-1640 (high glucose; Corning, cat. no. MT10-040-CV)
Penicillin-streptomycin (5 mg/mL; Corning, cat. no. MT30-002-CI)
100× Sodium pyruvate (Corning, cat. no. MT25-000-CI)
100× HEPES (Hyclone; Fisher, cat. no. SH3023701)
1× PBS (Corning, cat. no. MT46-013-CM)
0.5 M EDTA (pH 8.0; Corning, cat. no. MT46-034-CI)
Bleach (VWR, cat. no. 89414-502 or equivalent)
70% (vol/vol) Ethanol (VWR, cat. no. 89125-172)
Nuclease-free H2O (sterile, tissue culture grade; VWR, cat. no. EM4.86505.0500 or equivalent)
RNAse Away (Sigma, cat. no. 83931-250ML)
IL-2 (research grade, lyophilized; Miltenyi Biotec, cat. no. 130-097-744)
T Cell Activation and Stimulation Kit (Miltenyi, cat. no. 130-091-441)
EasySep T Cell Isolation Kit (StemCell Technologies, cat. no. 19052)
NucleoSpin Gel and PCR Clean-up Kit (Macherey Nagel, cat. no. 740609)
QuickTiter HIV Lentivirus Quantitation Kit (Cell Biolabs, cat. no. VPK-108-H)
Anti-CD3, human clone UCHT1 (Tonbo, cat. no. 40-0038-U500)
Anti-CD28, human clone 28.2 (Tonbo, cat. no. 40-0289-U500)
Anti-CD4-PE, human clone M-T466 (Miltenyi Biotec, cat. no. 130-091-231)
Anti-CD25-APC, human clone 3G10 (Miltenyi Biotec, cat. no. 130-101-435)
Anti-CXCR4-APC, human clone 12G5 (Miltenyi Biotec, cat. no. 130-098-357)
0.05% (wt/vol) Trypsin-EDTA (Corning, cat. no. MT25-052-CI)
PolyJet (SignaGen Laboratories, cat. no. SL100688)
HIV-1 NL4-3 nef:IRES:GFP full molecular clone (NIH AIDS Reagent Program, cat. no. 11349)
50% (wt/vol) Sterile PEG 6000 (Fisher, cat. no. 528877-1kg)
4 M Sodium chloride (NaCl, sterile; Fisher, cat. no. BP358-212)
0.5 M Tris-HCl (pH 6.8; BioBasic, cat. no. SD8112)
1% (wt/vol) Bromophenol blue (Sigma, cat. no. B5525)
10% (wt/vol) SDS (Fisher, cat. no. BP2436200)
50% (vol/vol) Glycerol (BioVision, cat. no. B1012-100)
2-Mercaptoethanol (Sigma, cat. no. M-6250)
2 M Potassium chloride (KCl; Fisher cat. no. AAJ75896AE)
1 M Potassium hydroxide (KOH; VWR, cat. no. 35113-1L)
Synthetic guide RNA (Dharmacon, cat. no. crRNA-XXXXX)
Synthetic tracrRNA (Dharmacon, cat. no. U-002000-50)
Purified Cas9-NLS protein (MacroLab, cat. no. Cas9-NLS)
Primary Cell Nucleofection Kit P3 (Amaxa; Lonza, cat. no. V4SP-3096)
 CRITICAL The P3 Primary Cell Nucleofection Kit is required for efficient editing with the Amaxa 4D Nucleofector unit.
CRITICAL The P3 Primary Cell Nucleofection Kit is required for efficient editing with the Amaxa 4D Nucleofector unit.10 mM Tris-HCl (pH 7.4, RNAse-free; Corning, cat. no. MT46-030-CM)
10 mM Tris-HCl (0.1 mM EDTA, pH 8.0, RNAse-free; Thermo Fisher, 12090015)
10 mM dNTPs (NEB, cat. no. N0447L)
Phusion High-Fidelity (HF) PCR Kit with 5× HF buffer (NEB, cat. no. E0553L)
Oligonucleotides for TIDE analysis (IDT, target-specific sequences)
Formaldehyde (Sigma, cat. no. F8775-4×25ML)
2.5× Laemmli sample buffer (Sigma, cat. no. S3401-10VL, or prepare in house as discussed below)
QuickExtract (EpiCentre, cat. no. QE09050)
Dimethyl sulfoxide (DMSO; Sigma, cat. no. D2650-100ML)
Equipment
Amaxa 4D Nucleofector with X Unit (and optional 96-well shuttle; Amaxa, cat. no. V4XC-1032)
Tissue culture plates (15 cm; Fisher, cat. no. 12-565-100
Mr. Frosty freezing container (Fisher, cat. no. 15-350-50 or equivalent)
T150 flasks (Fisher, cat. no. 1012634)
Single- and multichannel pipettors (Rainin, cat. nos. 30386739 and 30386597 or equivalent)
Filter tips for pipettors (Rainin, cat. nos. 17014961, 17014963, and 17014967 or equivalent)
Hemocytometer (Fisher, cat. no. 02-671-6 or equivalent cell counter)
Pipet-Aid (Drummond, cat. no. DP-100 or equivalent)
Flow cytometer (Thermo Fisher, model Attune NxT or equivalent)
48-Well plates (Fisher, cat. no. 08-772-52)
Non-treated 48-well tissue culture plates (Fisher, cat. no. 08-772-1C)
Cryovials (Corning, cat. no. CLS430488-500EA or equivalent)
1.5-mL Microcentrifuge tubes (Sigma, cat. no. T9661)
0.5-mL LoBind tubes (Eppendorf, cat. no. Z666491-100EA)
PCR plates (96-well; Bio-Rad, cat. no. MLP-9601)
Low-bind 96-well plates (E&K Scientific, cat. no. 651261)
Aluminum plate seals (Bio-Rad, cat. no. MSF1001)
Falcon conical tubes (15 and 50 mL; Fisher, cat. nos. 14-959-53A and 14-432-22)
Benchtop centrifuge with spinning bucket rotor (Beckman Coulter, cat. no. 392932 or equivalent)
0.22-μm Steriflip filters (Fisher, cat. no. SCGP00525)
500-mL 0.22-μm PVDF filter unit (Millipore, cat. no. SCGPU05RE)
Thermal cycler (Bio-Rad, cat. no. 170-9713 or equivalent)
Computer
Reagent setup
DMEM medium
To 450 mL of high-glucose DMEM, add 50 mL of FBS (10% (vol/vol) final) and 5 mL of penicillin-streptomycin (50 μg/mL final). Store the medium at 4 °C for up to 3 months.
Freezing medium
To 45 mL of FBS, add 5 mL of DMSO (10% (vol/vol) final) and sterilize by filtration through a 0.22- μm Steriflip filter. Store the medium at 4 °C for up to 1 year.
Complete RPMI medium for primary CD4+ T cells (cRPMI)
To 450 mL of high-glucose RPMI-1640, add 50 mL of FBS (10% (vol/vol) final), 5 mL of penicillin-streptomycin (50 μg/mL final), 5 mL of sodium pyruvate (5 mM final), and 12.5 mL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 5 mM final). Store the medium at 4 °C for up to 6 months.
PBS-EDTA
To 498 mL of 1× PBS, add 2 mL of 0.5 M EDTA, pH 8.0 (2 mM final). Store at room temperature (18-22 °C) or at 4 °C for up to 1 year.
IL-2
Suspend lyophilized IL-2 in nuclease-free H2O to a final concentration of 20 units (U) per μL. Make 50-μL aliquots in 500-μL LoBind tubes and store at −20 °C for up to 1 year.
Magnetic-activated cell sorting (MACS) buffer
To 488 mL of 1× PBS, add 10 mL of FBS (2% (vol/vol) final) and 2 mL of 0.5 M EDTA, pH 8.0 (2 mM final). Filter-sterilize through a 500-mL 0.22-μm PVDF filter unit. Store the buffer at 4 °C for up to 1 year.
2.5× Laemmli sample buffer
Combine 1.87 mL of 0.5 M Tris-HCl, pH 6.8 (3.1 mM final), 6 mL of 50% glycerol (10% (vol/vol) final), 3 mL of 10% SDS (1% (vol/vol) final), 750 μL of 2-mercaptoethanol (2.5% (vol/vol) final), 150 μL of 1% bromophenol blue (0.5% (vol/vol) final), and 18.23 mL of 1× PBS in a 50-mL conical tube and mix by pipetting up and down. Store the buffer at room temperature for up to 1 year.
Guide RNA/tracrRNA suspension buffer
To 45.25 mL of Milli-Q purified H2O, add 1 mL of 0.5 M Tris-HCl, pH 6.8 (10 mM final), and 3.75 mL of 2 M KCl (150 mM final). Add 1 M KOH dropwise as needed to adjust the pH of the final solution to 7.4. Sterilize by filtration through a 0.22-μm filter. Store the buffer at room temperature for up to 1 year.
T-cell stimulation beads
Prepare beads and store in accordance with the manufacturer’s instructions as included in the T Cell Activation and Stimulation Kit. Prepared beads can be stored at 4 °C for up to 6 months.
Oligonucleotide working stocks for TIDE analysis
Add 10 mM Tris-HCl and 0.1 mM EDTA, pH 8.0, to each lyophilized oligonucleotide to achieve a final oligo concentration of 100 μM. In sterile 1.5-mL centrifuge tubes, mix 90 μL of 10 mM Tris-HCl and 0.1 mM EDTA, pH 8.0, with 10 μL of the 100 μM oligo suspension to generate 10 μM working stocks for each oligo. Suspended oligos can be stored at −20 °C for several years.
2% (wt/vol) Formaldehyde in PBS
Mix 2 mL of formaldehyde solution with 35 mL of 1 PBS in a 50-mL conical tube, and vortex to mix for a final concentration of 2% (wt/vol). This dilution can be stored at room temperature for up to 1month.
Equipment setup
Nucleofection parameters
On the Amaxa 4D Nucleofector, prepare a customized program for nucleofection of primary T cells, setting the pulse code to EH-115 and the buffer to P3. Details on creating customized programs the Amaxa 4D Nucleofector can be found in the manufacturer’s instruction manual.
Tissue culture incubator
Prepare a sterile tissue culture incubator set to 37 °C with 5% CO2. Ensure that the environment remains humidified by refilling the humidifier pan with sterile water every 3–4 d.
Procedure
Guide RNA design  Timing 1 d
Timing 1 d
-
1
Open up the Ensembl human genome browser (http://ensembl.org) and search for the gene symbol or Ensembl ID of your gene of interest.
-
2
Open the ‘Human Gene’ page associated with your gene. Near the top of the page, you will notice a list of known and predicted transcript variants. TSL:1 transcript variants are considered validated sequences. For knockout of all known isoforms, identify an exon shared by at least all TSL:1 variants.
-
3
Open the page for an isoform Transcript ID by clicking on the hyperlink.
-
4
‘About this transcript’ will list the number of exons as a hyperlink. Click on the link to display the exon sequences.
-
5
For the best results, we recommend targeting conserved exonic sequences immediately following the start codon. If there is limited exonic coding sequence before the next intron, skip to the next exon that contains at least 100 nt. Copy 100–250 nt of sequence.
-
6
Open the CRISPR design webtool hosted by MIT (http://crispr.mit.edu/). Paste the sequence in the ‘sequence’ box and select ‘unique genomic region’.
-
7
Enter your e-mail address and the gene name into the appropriate fields. Click ‘Submit Query’. Guide sequences will be displayed as they are searched, including specificity scores and off-target hits.
-
8
Copy the sequences of 3–5 of the top-scoring gRNAs to an Excel file, excluding the protospacer adjacent motif (PAM). We recommend also recording the name of the exon searched, the target gene, the guide score, and any additionally pertinent notes.
CRITICAL STEP We do not recommend selecting guide sequences that score <65. If many guides are to be ordered, it may be worth searching several exons for the best possible gRNA sequences. -
9
Place synthetic gRNA orders through Integrated DNA Technologies, Dharmacon, or an appropriate alternative vendor, supplying the sequences designed above. Some companies offer RNA-stabilizing modifications (i.e., 2xMS from Dharmacon), and we have found all of them to be compatible with this protocol. Alternatively, instructions for in vitro transcription of gRNA can be found in Schumann et al.15. We recommend ordering synthetic tracrRNA from the same vendor as the gRNA, as the two reagents may not be compatible across vendors.
CRITICAL STEP RNAs for CRISPR-Cas9 gene editing are sold either as a two-part system of gRNA and tracrRNA that must be pre-complexed together before use, or as a single RNA with combined functionality known as a single gRNA (sgRNA). Although we use a two-part system of tracrRNA and gRNA in the Procedure, sgRNA systems are also compatible with this protocol.
Guide RNA and tracrRNA suspension Timing 1 d
-
10
Prepare an RNAse-free biosafety cabinet by wiping down the work surface, vortex, pipettes, and racks first with 70% (vol/vol) ethanol and then with RNAse Away. Prepare a tray of ice for working with suspended RNA.
-
11
Briefly centrifuge the tubes or 96-well plate of lyophilized gRNA and tracrRNA at 800g for 30 s at room temperature to ensure the RNA is on the bottom of the tubes or wells.
-
12
Add sufficient gRNA/tracrRNA suspension buffer to each tube or well for a final concentration of 160 μM. For example, suspend 20 nmol gRNA in 125 μL of buffer.
-
13
Pipette up and down several times to mix. Alternatively, seal the tubes or plate and vortex briefly to aid suspension.
-
14
Briefly centrifuge the tubes or plate (800g, 4°C, 15 s) to return the suspended RNA to the bottom of the wells/tubes. Keep all plates/tubes containing suspended RNA on ice.
-
15
Aliquot 5 μL of suspended gRNA into 0.5-mL LoBind tubes, or sterile PCR plates if working in plate format. As tracrRNA is used for multiple reactions and is provided only in tube format, we recommend making 20- to 50-μL aliquots of suspended tracrRNA in 0.5-mL LoBind tubes. Seal each tube or plate securely and label it with the appropriate description, date of preparation, and concentration. Store aliquots at −80 °C or proceed immediately with crRNP synthesis (Step 16).
CRITICAL STEP If working in plate format, we recommend proceeding immediately with crRNP generation (Step 16).
CRITICAL STEP Suspended gRNA and tracrRNA should be subjected to no more than one freeze-thaw cycle. PAUSE POINT Suspended gRNA and tracrRNA are stable at −80 °C for at least 6 months.
PAUSE POINT Suspended gRNA and tracrRNA are stable at −80 °C for at least 6 months.
crRNP generation Timing 1 d
-
16
Set a heat block, incubator, or thermocycler to warm to 37 °C.
CRITICAL STEP Work in an RNAse-free biosafety cabinet. Ensure that all tubes and pipette tips are sterile and RNAse-free. -
17Generate crRNPs in either tube format (option A) or plate format (option B) as appropriate:
-
(A)Small-scale preparation of crRNPs in individual tubes
-
(i)A single reaction requires 1 μL of tracrRNA (160 μM) and 1 μL of gRNA (160 μM). Accordingly, bring a sufficient number of aliquots of frozen or freshly prepared gRNA and tracrRNA from Step 15 to room temperature.
-
(ii)For each reaction, mix 1 μL of tracrRNA with 1 μL of gRNA in a sterile, PCR tube. These will anneal to form 2 μL of gRNA:tracrRNA duplex at an effective concentration of 80 μM. Prepare appropriate positive and negative controls at the same time (Experimental design).
-
(iii)Incubate the mixtures at 37 °C for 30 min in the prewarmed heat block, incubator, or thermocycler from Step 16 to promote duplex formation.
CRITICAL STEP If you are using an incubator, it is advisable to increase this incubation time to 40 min to account for the slower temperature change achieved by convection versus conduction.
-
(iv)A single reaction requires 2 μL of Cas9-NLS protein (40 μM). According to your experimental needs, remove sufficient Cas9-NLS protein from the freezer and allow it to thaw at room temperature. Near the end of the gRNA:tracrRNA incubation, briefly warm the Cas9-NLS protein in an incubator or thermocycler to 37 °C.
-
(v)Add 2 μL of the Cas9-NLS protein to the RNA mixture by moving the pipette tip in small, rapid circles while ejecting slowly. If a precipitate forms, gently pipette up and down until it goes back into solution. Incubate at 37 °C for 15 min to form crRNPs. Providing gRNA: tracrRNA in molar excess to Cas9 promotes efficient crRNP formation, yielding a preparation of crRNPs at a 20 μM concentration.
CRITICAL STEP Subjecting crRNPs to repeated freeze-thaw cycles will diminish editing efficiency. If storing crRNPs at −80 °C, we recommend preparing and freezing them in 4-μL single-reaction aliquots.
PAUSE POINT The steps outlined here generate sufficient crRNPs for a single nucleofection reaction. crRNPs can be stored at 4 °C for up to 2 weeks or at −80 °C for at least 6 months with little-to-no loss of editing efficiency.
-
(i)
-
(B)Medium- or large-scale preparation of crRNPs in plate format
CRITICAL If working with gRNAs in plate format, we recommend generating crRNPs immediately following gRNA suspension (Step 15).
CRITICAL For ease of manipulation when working in plate format, we recommend preparing sufficient crRNPs for at least five reactions at one time, as shown in our example below. Any unused crRNPs can be frozen and stored at −80 °C for at least 6 months before use.
-
(i)Each well will require 5 μL of gRNA (160 μM) and 5 μL of tracrRNA (160 μM) as prepared in Step 15. Accordingly, bring a sufficient number of aliquots of frozen or freshly prepared tracrRNA and gRNA to room temperature.
-
(ii)Add 5 μL of 160 μM tracrRNA to each well of a PCR plate.
-
(iii)Using a multichannel pipettor, add 5 μL of gRNA to the plate containing tracrRNA, pipetting up and down four to five times to mix. Seal the plate and incubate for 30 min at 37 °C in the pre-warmed heat block, incubator, or thermocycler from Step 16 to form gRNA:tracrRNA duplexes at an effective concentration of 80 μM.
CRITICAL STEP If you are using an incubator, it is advisable to increase this incubation time to 40 min to account for the slower temperature change achieved by convection versus conduction.
-
(iv)Each well will require 10 μL of Cas9-NLS protein (40 μM). Remove sufficient Cas9-NLS protein from the freezer accordingly and allow it to thaw at room temperature. Near the end of the gRNA:tracrRNA incubation, briefly warm the Cas9-NLS protein to 37 °C in an incubator or thermocycler.
-
(v)Using a multichannel pipettor, add 10 μL of the Cas9-NLS protein to the RNA mixture by moving the pipette tips in small, rapid circles while ejecting slowly. If a precipitate forms, gently pipette up and down until it goes back into solution. Incubate at 37 °C for 15 min to form crRNPs. Providing gRNA:tracrRNA in molar excess to Cas9 promotes efficient crRNP formation, yielding a preparation of crRNPs at a 20 μM concentration.
-
(vi)Using a multichannel pipettor, aliquot 3.5 μL of crRNPs into five separate LoBind 96-well V-bottom plates. Seal the plates with foil, label as appropriate, and store them either at 4 °C for immediate use or at −80 °C until needed.
CRITICAL STEP We strongly recommend that all crRNPs used in a screen be treated consistently (i.e., they are either all frozen at −80 °C or all used immediately). Subjecting crRNPs to repeated freeze-thaw cycles will diminish editing efficiency and is not recommended.
PAUSE POINT Preparing crRNPs ahead of time markedly improves screening workflow. Plates of crRNPs can be stored at 4 °C for up to 2 weeks or at −80 °C for at least 6 months with little-to-no loss of editing efficiency.
-
(i)
-
(A)
PBMC isolation Timing 1 d
CAUTION All human blood samples must be handled in accordance with approved biosafety protocols and regulations as dictated by institutional and federal regulations. Improper handling of samples may lead to exposure to blood-borne pathogens.
CAUTION All human blood samples and data obtained from them must be handled according to approved biosecurity protocols for the protection of patient confidentiality and health information.
CRITICAL PBMC isolations are best performed on a Monday or a Tuesday to allow 72 h for activation before nucleofection. Isolations should be performed within 24 h of the blood draw.
CRITICAL A standard PBMC and T-cell isolation from one donor will typically take between 4 and 5 h. Make sure sufficient time is secured in the appropriate biosafety hood on the day of isolation.
-
18
Before PBMC isolation, coat the wells of a sterile, non-treated 48-well tissue culture plate with anti-CD3 antibody for T-cell stimulation (Step 42) as follows. Dilute anti-CD3 (clone UCHT1) to 10 μg/mL in 1× PBS and add 150 μL per well. Incubate the plate for 2 h at 37 °C. This incubation may also proceed for 8 h at room temperature or overnight at 4 °C. In the latter case, make sure to wrap the plate in Parafilm to prevent evaporation.
CRITICAL STEP Each well will hold 1.25 million cells during stimulation, and each nucleofection can tolerate as few as 200,000 and as many as 1 million cells. Be sure to coat enough wells to stimulate sufficient cells for your experimental needs.
CRITICAL STEP The stimulation and culture conditions described herein have been optimized for the longevity and viability of the culture, but plate-bound stimulation may be eschewed entirely in favor of bead-bound stimulation, provided the beads are removed before nucleofection. Other culture plates may be used if volumes are scaled appropriately. -
19
Once the donor samples have arrived, post any required biosafety notices around your workspace, record any donor information in your logs, and equip yourself with any required personal protective equipment, including appropriate gloves, safety glasses, and lab coat.
-
20
Prepare eight 15-mL conical tubes per donor, each containing 4 mL of Ficoll-Paque.
-
21Depending on whether the experimenter is working with a whole-blood sample or a leukoreduction chamber, implement either option A or B, respectively.
-
(A)Working with whole-blood samples
-
(i)Carefully cut the drainage and ventilation cords and drain the sample into a sterile 50-mL conical tube.
-
(ii)Dilute the blood sample 1:1 with 1× PBS and mix gently using a pipette (e.g., if working with 50 mL of blood, dilute with 50 mL of 1× PBS).
-
(i)
-
(B)Working with a leukoreduction chamber
-
(i)Carefully cut the top cord leading to the conical chamber. Cut the bottom cord from the opposite end and set the conical chamber on top of a 50-mL conical tube to drain by gravity flow. By this approach, 5–10 mL of blood containing concentrated leukocytes should be released.
-
(ii)Dilute the blood with 1× PBS to a final volume of 65 mL. Mix gently by pipetting.
-
(i)
-
(A)
-
22
Using a 10-mL pipette, slowly dispense 8 mL of blood-PBS mixture onto the Ficoll-Paque in the eight 15-mL conical tubes prepared in Step 20. A clear boundary should be evident. Additional blood can be either discarded after sterilization in 10% (vol/vol) bleach or layered over additional Ficoll tubes.
-
23
Centrifuge the 15-mL tubes at 400g for 30 min at room temperature to separate the blood components. Make sure that the brake is turned off or is at the minimum setting.
-
24
The blood will separate into the five layers as follows from top to bottom: plasma, PBMCs (buffy coat), Ficoll, granulocytes, and erythrocytes. Carefully aspirate off and discard the plasma-containing top layer, leaving only a half centimeter of liquid above the buffy coat.
-
25
Using a 5-mL pipette, slowly remove the buffy coat and transfer it to a 50-mL conical tube. Removal and transfer of some of the Ficoll and plasma layer above and below the buffy coat, respectively, will not harm the PBMCs in subsequent steps. Pool PBMCs from the same donor into a single tube. Discard the Ficoll tubes after sterilization in 10% (vol/vol) bleach.
-
26
Dilute the PBMCs with PBS-EDTA to a final volume of 50 mL. Centrifuge the tubes at 300g for 10 min at room temperature (the brake can now be reapplied).
-
27
Remove the supernatant and discard. Note that it will be cloudy due to suspended platelets.
-
28
Suspend cells in 50 mL of PBS-EDTA and centrifuge at 200g for 10 min at room temperature. Remove the supernatant and discard. Suspend the PBMCs in 50 mL of PBS-EDTA.
-
29
Count the PBMCs using a hemocytometer or cell counter. Note that you may have to dilute a sample of the PBMCs 1:10 in PBS-EDTA to obtain an accurate count.
-
30
After counting, centrifuge the tube at 300g for 10 min at room temperature to pellet the cells. Remove the supernatant and discard.
-
31
Suspend cells in MACS buffer to obtain a final concentration of 5 × 107 cells per mL.
-
32
From each donor’s PBMC sample, remove a 50-μL aliquot and transfer it to a 1.5-mL centrifuge tube. Centrifuge the tubes at 400g for 3 min at room temperature to pellet the cells. Remove the supernatant and discard, suspend the cell pellet in 200 μL of cRPMI medium, and transfer the mixture to a 96-well round-bottom plate. Move this plate to a sterile tissue culture incubator (37 °C with 5% CO2) for 48–72 h. This ‘PBMC’ population will be stained for CD4 and CD25 as enrichment and stimulation controls, respectively, alongside the CD4+-enriched population in Step 41 and the activated CD4+ T cells in Step 46 (see Step 47 and Fig. 2a).
Fig. 2 |. Results from primary T-cell isolation and editing.

a, Flow cytometry histograms depicting CD4 and CD25 levels on the cell surface of PBMCs (red), CD4+ T cells post enrichment (blue), and CD4+ T cells post stimulation with anti-CD3/anti-CD28/IL-2 (orange). After successful isolation and stimulation, the resultant cell population should be >95% CD4+ and >90% CD25+. Samples were run on an Attune NxT Flow cytometer and analyzed using FlowJo software v.10.1 (n > 100,000 events). b, Flow cytometry histogram depicting CXCR4 levels on the cell surface of primary T cells treated with non-targeting crRNPs (blue) or CXCR4-targeting crRNPs (red) relative to an unstained control (gray). After successful editing with CXCR4-targeting crRNPs, the resultant cell population should be <10% CXCR4+. Samples were run on an Attune NxT Flow cytometer and analyzed using FlowJo software v.10.1 (n > 100,000 events). c (Left), Chromatograms from Sanger sequencing of PCR amplicons over a target site in the CCNT1 locus in non-targeting treated cells (bottom) and in cells treated with CCNTI-targeting crRNPs (top). The gRNA target sequence and associated PAM are highlighted below. (Right) The TIDE output calculating the percentage of indels from these chromatograms. The total efficiency of indel generation provides a good estimate of knockout percentage in a cell population following crRNP treatment.
CD4+ T-cell enrichment Timing 1 d
-
33
Determine the number of CD4+ T cells required for your experimental needs and calculate the volume of PBMCs to be used for isolation. Consider that ~500,000 CD4+ T cells will be used per nucleofection reaction and that PBMCs from most healthy donors will contain 4–20% CD4+ T cells. On average, using the procedures detailed below, processing 5 mL of suspended PBMCs will yield 10–50 million CD4+ T cells.
-
34
Transfer up to 5 mL of the PBMC suspension to a sterile 14-mL polystyrene tube. Any remaining PBMCs can be used for additional T-cell isolations, cryopreserved for future use, disposed of in 10% (vol/vol) bleach, or used for other experimental purposes.
-
35
Add 50 μL of negative-selection antibody cocktail from the EasySep T Cell Isolation Kit per mL of suspended PBMCs to each tube. Mix gently, but completely, and incubate at room temperature for 5 min.
-
36
During the incubation, suspend the RapidSphere magnetic beads from the EasySep T Cell Isolation Kit by vortexing vigorously for 30 s.
-
37
Add 50 μL of RapidSphere magnetic beads per mL of suspended PBMCs to each tube. Mix gently, using a pipette, and incubate at room temperature for 5 min.
-
38
Place the sample (without the lid) onto the EasySep magnet and incubate at room temperature for 5 min.
-
39
Keeping the tube(s) on the magnet, pick up the entire apparatus and pour off the remaining cell suspension into a clean, labeled tube. These tubes will contain an enriched population of CD4+ T cells.
-
40
If multiple T-cell isolations have been performed from one donor, combine cells from the same donor and mix. Determine the cell concentration using a hemocytometer or cell counter.
-
41
From each donor’s CD4+ T-cell sample, remove a 100-μL aliquot and transfer it to a 1.5-mL centrifuge tube. Centrifuge the tubes at 400g for 3 min at room temperature to pellet the cells. Remove the supernatant and discard, suspend the cell pellet in 200 μL of cRPMI medium, and transfer the cells to a 96-well round-bottom plate. Move this plate to a sterile tissue culture incubator (37 °C with 5% CO2) for 48–72 h. This ‘CD4+-enriched’ population will be stained for CD4 and CD25 as enrichment and stimulation controls, respectively, alongside the PBMCs from Step 32 and the activated CD4+ T cells in Step 46 (see Step 47 and Fig. 2a).
T-cell activation and expansion Timing 2–3 d
-
42
Aspirate the PBS-antibody mixture from the anti-CD3-bound plate prepared in Step 18.
-
43
Pellet the enriched CD4+ T cells from Step 40 by centrifugation at 300g for 5 min at room temperature. Remove the supernatant and discard. Suspend the cell pellet in cRPMI plus 20 U/mL IL-2 and 5 μg/mL anti-CD28 (clone CD28.2) to a final concentration of 2.5 × 106 cells per mL.
-
44
Add 500 μL of suspended CD4+ T cells to each well of the anti-CD3-bound plate and incubate for 48–72 h in a sterile tissue culture incubator at 37 °C with 5% CO2 to allow for cell stimulation and activation.
-
45
After 48–72 h, suspend the cells, remove them from the 48-well plate, and place them into a 50-mL conical tube, combining cells from the same donor. Determine the concentration of activated CD4+ T cells, using a hemocytometer or cell counter. The cells should have begun to multiply and should have gone from being small and round to displaying the larger and irregularly shaped morphology characteristic of activated T cells. Pellet the cells by centrifugation at 400g for 3 min at room temperature. Remove the supernatant and discard. Suspend the cell pellets in cRPMI plus 20 U/mL IL-2 to a final concentration of 2.5 × 106 cells per mL. Move the cells to a flask or plate of an appropriate size for the given culture volume.
-
46
From each donor’s activated CD4+ T-cell population, remove a 100-μL aliquot and transfer it to a 1.5-mL centrifuge tube. Centrifuge the tubes at 400g for 3 min at room temperature to pellet the cells. Remove the supernatant and discard, suspend the cell pellet in 200 μL of cRPMI medium plus 20 U/mL IL-2, and transfer the mixture to a 96-well round-bottom plate. Move this plate to a sterile tissue culture incubator (37 °C with 5% CO2). This ‘activated CD4+ T cell’ population will be stained for CD4 and CD25 as enrichment and stimulation controls, respectively, alongside the PBMCs from Step 32 and the CD4+ enriched population from Step 41 (see Step 47 and Fig. 2a). Return the remaining cultures to the sterile tissue culture incubator (37 °C with 5% CO2).
-
47
Immunostain the PBMCs, the CD4+-enriched T-cell samples, and the activated CD4+ T-cell samples collected from Step 32, 41, and 46, respectively, with anti-CD4-PE and anti-CD25-APC per the manufacturers’ instructions. Quantify the percentage of CD4+ and the percentage of CD25+ cells in each population by flow cytometry (refer to Supplementary Fig. 1a for gating strategy). The activated T-cell population should be >90% CD4+/CD25+ (see Anticipated results and Fig. 2a).
CRITICAL STEP If the activated CD4+ T-cell population is <90% CD4+ or <90% CD25+, there may have been a problem with the T-cell isolation or activation, respectively. Impure or non-activated cell populations may not yield good editing efficiency or infection and so should not be used in further experiments. TROUBLESHOOTING
TROUBLESHOOTING
crRNP nucleofection Timing 1 d
CRITICAL If T-cell isolations are performed on Monday or Tuesday, nucleofections can be performed on Thursday or Friday of the same week, allowing 72 h for T-cell stimulation.
-
48
Prepare the stimulatory beads from the T Cell Activation and Expansion Kit according to the manufacturer’s instructions. Allow the beads to rotate at 4 °C for at least 2 h before use.
-
49
Mix the P3 nucleofection buffer with the required supplement from the P3 Primary Cell 4D- Nucleofector X Kit according to the manufacturer’s instructions. Allow the supplemented nucleofection buffer to come to room temperature before it comes into contact with the cells.
CRITICAL STEP Supplemented nucleofection buffer is stable at 4 °C for up to 3 months. Do not prepare more supplemented nucleofection buffer than will be used in this time frame. -
50
Set up the Amaxa 4D Nucleofection System according to the manufacturer’s instructions, specifying buffer P3 and pulse code EH-115.
-
51
Prepare cRPMI plus 20 U/mL IL-2 and pre-warm it to 37 °C.
-
52
Determine the concentration of activated CD4+ T cells from Step 45, using a hemocytometer or automated cell counter. Each nucleofection reaction will require 500,000 cells per well. Calculate the total number of cells required from each donor, remove the required volume of culture and place it into a 50-mL conical tube, and pellet the cells by centrifugation in a spinning bucket rotor at 400g for 5 min at room temperature. For example, pellet 2 million cells for four nucleofection reactions.
CRITICAL STEP This procedure is effective when using anywhere between 200,000 and 1 million cells per reaction. The cell number used per reaction should be optimized on the basis of the required cell numbers in downstream applications. -
53
Thaw crRNPs in plate format from Step 17 or, if crRNPs are not in plate format, array 3.5 μL of each crRNP in a 96-well, LoBind, V-Bottom plate in the exact layout to be used for nucleofection. Bring to room temperature.
CRITICAL STEP Supplemented nucleofection buffer is toxic to cells. To minimize the amount of time the cells spend in supplemented nucleofection buffer, collect and arrange the necessary reagents beforehand for efficient handling. -
54
Remove the supernatant from the cell pellet obtained in Step 52 and suspend the cells in 20 μL of supplemented nucleofection buffer per reaction. If working in plate format, transfer the cell suspension to an appropriate vessel for multichannel pipetting.
-
55
For each nucleofection reaction, add 20 μL of the cell suspension to 3.5 μL of crRNPs as laid out in Step 53. Mix gently three to four times by pipette. If working in plate format, this can be done using a multichannel pipette.
-
56
Immediately transfer 20-μL aliquots of the cell/crRNP mixture to the nucleofection strip tubes/plate, leaving any extra solution behind. To avoid arc errors during nucleofection, dispense the reaction at the bottom of the wells and avoid the formation of air bubbles.
CRITICAL STEP Although arc errors will not prevent the nucleofection from being successful, they may negatively impact editing efficiency and/or cell viability. -
57
Tap the nucleofection strip tubes/plate gently on the surface of the hood to release any air bubbles that may have formed.
-
58
Nucleofect the cells on the Amaxa 4D-Nucleofector System, using program EH-115 as set up in Step 50.
-
59
As soon as possible after nucleofection, add 80 μL of pre-warmed cRPMI plus 20 U/mL IL-2 from Step 51 to each well of the nucleofection strip tube/plate. Afterward, move the nucleofection strip tubes/plate to the sterile tissue culture incubator (37 °C with 5% CO2) for at least 15 min to allow for cell recovery.
-
60
As the cells recover, prepare a flat-bottom, 96-well plate containing 100 μL of pre-warmed cRPMI plus 20 U/mL IL-2 from Step 51 plus T Cell Stimulation Beads. Sufficient beads should be added to achieve a 1:1 bead/cell ratio in accordance with the manufacturer’s instructions. Store this plate in the 37 °C incubator to keep it warm until needed.
-
61
After allowing sufficient time to recover, transfer the entirety of each reaction mixture from the nucleofection cuvette to the appropriate wells of the pre-warmed flat-bottom plate (from Step 60) for expansion in culture.
Cell activation and expansion Timing 4–8 d
CRITICAL To allow time for cell recovery, gene editing, and protein turnover, at least 3 d should pass before harvesting protein and genomic DNA samples for validation.
CRITICAL Edited cells will continue to expand for ~10–14 d post treatment with stimulatory beads, after which time the cells will lose activation and permissivity to infection. Cells can be expanded further with a second round of stimulation if necessary. In this example application of the Procedure, the cells are divided into five sets: one to harvest protein, one to harvest genomic DNA, and three to provide technical triplicate samples for infection.
-
62
48–72 h after nucleofection, feed the cells by adding 100 μL of cRPMI plus 20 U/mL IL-2 to each well of the plate.
-
63
48–72 h later, use a pipette to mix a representative well of cells and determine the cell density using a hemocytometer or cell counter. If 500,000 cells were nucleofected originally, expect the cell concentration to be ~1–2 million cells per mL.
-
64
Using a multichannel pipette, mix the cells in their media to suspend and transfer 200,000 from each well to each of two U-bottom, 96-well plates. To the original plate, add back an equivalent volume of cRPMI + 20 U/mL IL-2 as was just removed and return these cells to the sterile tissue culture incubator (37 °C with 5% CO2). These cells can be either used directly for phenotypic assays (such as an HIV spreading infection, see Step 94) or cryopreserved for future use.
PAUSE POINT Instead of proceeding immediately with infection, edited primary CD4+ T cells can be frozen and stored in cryovials in liquid nitrogen for several years. To freeze, transfer the cells to a 15-mL conical tube and pellet in a spinning bucket rotor by centrifugation at 400g for 3 min at room temperature. Remove the supernatant and suspend the cell pellet in FBS + 10% DMSO at a final concentration of 1–5 million cells per mL. Transfer this suspension to a cryovial, and place the cryovial in a freezing container (i.e., Mr. Frosty or equivalent). These containers ensure a slow decrease in cryovial temperature (<1 °C per min) to maximize cell viability. Move the freezing container to a −80 °C freezer overnight. The following day, transfer the cryovials to liquid nitrogen for long-term storage. -
65
Centrifuge the two U-bottom plates at 400g for 3 min at room temperature to pellet the cells. Gently remove the supernatant from each well, using a multichannel pipette and discard.
-
66
Suspend the cell pellets in one U-bottom plate in 50 μL of 2.5× Laemmli sample buffer per well. Transfer the resultant lysates to PCR tubes or plates and heat them at 98 °C for 20 min. These lysates are stable for up to 2 years when stored at −20 °C and can be used for immunoblotting analysis to determine knockout efficiency at the protein level (Anticipated results).
TROUBLESHOOTING -
67
Suspend the cell pellets in the second U-bottom plate in 50 μL of QuickExtract buffer per well. Transfer the resultant lysates to PCR tubes or plates and heat them first at 65 °C for 20 min, followed by 98 °C for 5 min. These lysates are stable for up to 2 years when stored at −20 °C and contain PCR-ready genomic DNA for tracking of indels by decomposition (TIDE) analysis (Steps 68–73).
Tracking of indels by decomposition analysis Timing 3–4 d
CRITICAL Before proceeding with phenotypic analyses, validate the successful knockout of a representative positive control through immunostaining, immunoblotting, or TIDE analysis (Steps 68–73). In the case of CXCR4, extra non-targeting and CXCR4-targeted cells can be stained with anti- CXCR4-APC per the manufacturer’s instructions and analyzed by flow cytometry (Fig. 2b).
-
68
CRISPR-Cas9 editing will result in the formation of randomly assorted indels at the cut site35. PCR amplification over the cut site from the genomic DNA of a polyclonal pool of cells provides a template for Sanger sequencing, which can then be analyzed for indel percentage using the TIDE webtool64 (Fig. 2c). To perform TIDE, first design single-stranded DNA oligos to serve as PCR primers over the targeted cut site. Aim for a product size between 500 and 700 bp, with the gRNA target site at least 150 bp removed from either primer. Reference genome sequences for primer design can be found using the UCSC Genome Browser (http://genome.ucsc.edu) or Ensembl (http://www.ensembl.org), and primers can be designed using a variety of webtools, such as Primer3 (refs 65,66; http://bioinfo.ut.ee/primer3-0.4.0/primer3/) or Benchling (https://benchling.com/tutorials/25/creating-and-analyzing-primers). Each gRNA will require a unique TIDE primer pair, unless the target sites are in close proximity.
-
69Prepare 50-μL PCR reactions as follows (buffer and enzyme are from the Phusion High-Fidelity PCR Kit). For each primer pair, include a negative-control reaction designed to amplify the intact gene product from non-targeting, control genomic DNA template.
Reagent Amount (μL) [Final] 5× Buffer 10 1× dNTPs (10 mM) 2 0.4 mM Nuclease-free dH2O 31 — Forward primer (10 μM) 2 0.4 μM Reverse primer (10 μM) 2 0.4 μM Phusion enzyme 0.5 — Genomic DNA lysate (Step 67) 2.5 — Total 50 -
70Amplify over the cut site, using a touch-down PCR amplification strategy with annealing temperatures adjusted as appropriate for the designed primers. The touch-down annealing temperature should start 5 °C higher than the theoretical melting temperature of the primers and end 2 °C lower. The theoretical melting temperature of the primers in the below example is 60 °C.
Cycle no. Denature Anneal Extend 1 98 °C, 5 min 2–15 98 °C, 30 s 65 °C, −0.5 °C per cycle, 20 s 72 °C, 1 min 16–35 98 °C, 30 s 58 °C, 20 s 72 °C, 1 min 36 72 °C, 10 min End -
71
Purify the amplified DNA for sequencing, using a PCR purification kit (i.e., the NucleoSpin Gel and PCR Clean-up Kit) according to the manufacturer’s instructions.
-
72
Submit the purified PCR products for Sanger sequencing, using either the forward or reverse TIDE oligos originally used for amplification. Refer to your Sanger sequencing provider for specifications on setting up these reactions. Once the chromatograms are returned, sequence diversification will become evident directly adjacent to the PAM sequence of the guide, if the editing reaction was successful (Fig. 2c).
-
73
An estimated percentage of indels can be generated by uploading the experimental and control chromatograms to the TIDE webtool (http://tide.nki.nl). The best guides will reveal allelic editing percentages of ≥75%.
CRITICAL STEP TIDE data should not be considered absolute empirical knockout percentages, but rather rough estimates that afford a rapid validation of the editing process.
TROUBLESHOOTING
Generation of concentrated HIV-1 virus stocks Timing 5 d
CAUTION HIV is a blood-borne pathogen and must be handled in accordance with approved biosafety protocols and regulations as dictated by institutional and federal regulations.
CRITICAL To allow time for virus generation, precipitation, and titering, viruses should be made before or during the week of primary cell isolation (Steps 18–47). Once precipitated and stored, the viruses will be stable at −80 °C for several months. Virus generation takes 5 d, so Monday is the recommended start day (Fig. 3).
Fig. 3 |. Overview of HIV-1 virus preparation.

HEK293T cells are plated and transfected with an HIV-1 molecular clone, using appropriate biosafety precautions. The virus-containing supernatant is harvested 48 and 72 h after transfection. PEG precipitation, followed by centrifugation, is implemented to pellet the virus for subsequent concentration, titering, and storage.
-
74
Pre-warm DMEM medium to 37 °C.
-
75
Starting with a confluent T175 flask of HEK293T cells, remove the medium, rinse the cells gently with 10 mL of 1× PBS, and add 5 mL of 0.05% (wt/vol) trypsin-EDTA. Gently rock for 2–3 min until the cells detach from the flask.
CRITICAL STEP Refer to the ATCC website (https://www.atcc.org/products/all/CRL-3216.aspx#culturemethod) for more information on the culture and proper care of HEK293T cells. -
76
Add 5 mL of pre-warmed DMEM medium to the flask of detached cells and mix well to neutralize the trypsin. Transfer the cells to a 50-mL conical tube and centrifuge in a spinning bucket rotor at 400g for 3 min at room temperature to pellet. Remove the supernatant and discard. Suspend the cells in 50 mL of pre-warmed DMEM medium.
-
77
Calculate cell density using a hemocytometer or cell counter. Transfer 5 million cells to each of eight 15-cm plates. Add pre-warmed DMEM medium to a final volume of 27 mL per plate. Swirl the cells in the plate to distribute them evenly over the plate surface and allow them to adhere overnight in a sterile tissue culture incubator (37 °C with 5% CO2).
-
78
The following day, prepare eight 1.5-mL microcentrifuge tubes, one per 15-cm dish, each containing 10 μg of replication-competent proviral vector in accordance with the experimental design. In this example application of the Procedure, our vector of choice is an HIV-1 NL4–3 full molecular clone with an integrated GFP reporter driven off an internal ribosome entry site (IRES) sequence following the nef open reading frame (nef:IRES:GFP).
-
79
Remove serum-free DMEM and polyJET from the refrigerator and allow them to warm to room temperature for transfection.
-
80
Add 250 μL of serum-free DMEM medium (25 μL per μg of DNA) to each tube containing plasmid DNA form Step 78.
-
81
Prepare a master mix consisting of 250 μL of serum-free DMEM medium (25 μL per μg of DNA) and 30 μL of polyJET reagent (3 μL per μg of DNA) per tube. For example, for eight plates, the composition of the master mix will be 2 mL of serum-free DMEM medium and 240 μL of polyJet. Mix well and incubate for 2 min at room temperature.
-
82
Add 250 μL of the polyJET:serum-free DMEM master mix (Step 81) to each plasmid DNA-serum-free DMEM mixture (Step 80) and pipette to mix. Incubate at room temperature for 15 min.
-
83
After incubation, add each tube of transfection mixture (plasmid DNA:serum-free DMEM-polyJET from Step 82) dropwise to its respective plate of HEK293T cells (prepared in Step 77). Swirl each dish to mix and return the cells to the sterile tissue culture incubator (37 °C with 5% CO2) for 48 h to generate virus.
CAUTION As soon as the cells are transfected, they are considered HIV-positive and therefore biohazardous. These cells must be handled according to the appropriate biosafety standards of the researcher’s institution. -
84
After 48 h, collect all 27 mL of virus-containing supernatant from each plate and place it in 50-mL conical tubes at 4 °C. Immediately replace the supernatant with 25 mL of fresh DMEM medium and return the cells to the tissue culture incubator overnight to generate additional virus.
-
85
The following day, collect the second batch of virus-containing supernatant and combine it with the first for a total yield of ~400 mL of virus supernatant. Inactivate the virus-producing cells with 10% (vol/vol) bleach and dispose of them.
-
86
Remove cell debris from the virus-containing supernatant by vacuum filtration through a 0.22-μm PVDF filter unit (e.g., a 0.22-μm Steriflip filter or similar).
-
87
Meanwhile, prepare sixteen 50-mL conical tubes for virus precipitation. To each tube, add 5.5 mL of 50% (vol/vol) PEG-6000 and 2.4 mL of 4 M NaCl. The PEG-6000-NaCl solutions can be prepared as much as 24 h in advance.
-
88
To each of these conical tubes, add 24 mL of filtered virus supernatant from Step 86 to achieve a final concentration of 8.5% (vol/vol) PEG-6000 and 0.3 M NaCl. Invert each tube to mix and allow the virions to precipitate at 4 °C for 4–8 h.
CRITICAL STEP Incubation times <4 h may result in incomplete precipitation and decreased yields. Conversely, incubation times >8 h may result in decreased virus infectivity. -
89
Pellet the virions by centrifugation in a spinning bucket rotor at 2,850g for 20 min at 4 °C.
CAUTION Avoid the formation and release of aerosols by performing all centrifugation steps with appropriate biosafety measures, including the use of biosafety seals and caps on all swinging buckets. Decontaminate the inside of the centrifugation buckets and caps thoroughly before returning them to the centrifuge. -
90
Immediately following centrifugation, decant or aspirate off the supernatant and discard. Add 250 μL of 1× PBS to the virus precipitate in each tube.
-
91
Pipette up and down to suspend the precipitated virus and combine all fractions. The expected yield is ~6 mL of virus stock at a 100-fold concentration.
CAUTION Concentrated, replication-competent HIV-1 stocks must be handled with extreme care according to the appropriate biosafety standards at the researcher’s institution. -
92
Make aliquots by dividing the concentrated virus stocks among 1.5-mL screw-top, microcentrifuge tubes for storage at −80 °C. Plan for roughly 24–36 aliquots of 50, 100, or 250 μL of virus per vial.
PAUSE POINT Concentrated virus stocks can be stored at −80 °C for at least 6 months without any loss of infectivity. -
93
(Optional) Owing to inherent donor variability in their susceptibility to virus infection (Anticipated results), we recommend virus stocks be quantified either by donor-matched live titer or by calculating p24 equivalents. Live titers can be estimated by infecting activated CD4+ T cells over a virus dilution series, whereas p24 levels in the virus stock can be quantified using a variety of commercially available kits (e.g., QuickTiter HIV Lentivirus Quantitation Kit).
Primary cell HIV spreading infection Timing 7–9 d
CRITICAL Although spreading infection time points may be recorded with some flexibility, it is generally advantageous to record at least three time points between 3 and 7 d post infection. If the initial infection occurs on Friday, the sampling can be performed on the Monday, Wednesday, and Friday of the following week. Three time points in technical triplicate will ultimately result in nine flow samples per nucleofected population.
CRITICAL To ensure successful infection, the cells must be activated at the time of virus challenge. Therefore, stimulation should be performed at least 2–7 d before infection. In this example, 4–6 d pass between stimulation and infection.
CRITICAL Although we use a GFP reporter virus for easy analysis of infection in this example application ofthe Procedure, alternative HIV molecular clones without integrated reporters can be used. Viruses without an engineered reporter gene, however, will require additional steps for virus detection, such as p24 immunostaining or monitoring of p24 in the culture supernatant by ELISA (see, e.g., Wehrly et al.67).
-
94
The edited cells in the original plate from Step 64 can now be prepared for HIV spreading infection. Suspend a representative well of cells and determine its cell density, using a hemocytometer or cell counter.
-
95
Using a multichannel pipette, suspend the cells in each well and transfer 50,000–100,000 cells per well to each of three U-bottom, 96-well plates. These plates will be used as technical replicates for the infection. Plate a few extra wells of cells to serve as uninfected controls.
-
96
Add additional cRMPI plus 20 U/mL IL-2 to each well of the U-bottom plates to achieve a total volume of 150 μL per well. Any cells remaining in the original plate can be discarded, cryopreserved, or used for additional experiments as necessary.
-
97
Calculate the amount of cRPMI plus 20 U/mL IL-2 required to add 50 μL to each well of edited cells and add this to a 50-mL conical tube.
-
98
Calculate the amount of virus per well required to achieve a ~1–2% initial infection after 72 h. For most donors and viruses, this amount will be ~2–5 μL of concentrated virus per well. If a live titer was calculated in Step 93, this can be used to determine the optimal volume of virus to be added.
-
99
Thaw the total amount of concentrated virus stock required for all infections and add this to the conical tube of cRPMI plus 20 U/mL IL-2 from Step 97 to generate the virus inoculate. Mix gently by pipetting.
-
100
Add 50 μL of virus inoculate to each well across each plate from Step 99. Add 50 μL of cRPMI plus 20 U/mL IL-2 without virus to any additional, uninfected control wells. Return all plates to the sterile tissue culture incubator (37 °C with 5% CO2) for 72 h.
-
101
Label three 96-well, U-bottom plates to be used for time point collection/cell fixation and add 75 μL of 2% (wt/vol) formaldehyde in 1× PBS to each well.
-
102
Gently mix each well of infected cells with a multichannel pipette. To each well of formaldehyde, add 75 μL of the mixed culture for a final concentration of 1% (wt/vol) formaldehyde. Pipette up and down to mix and prevent clumping.
-
103
Allow 30 min for complete fixation and inactivation of the virus. Clean the outside of the plates with 70% (vol/vol) ethanol and wrap them in aluminum foil. These cells are now ready for analysis by flow cytometry.
PAUSE POINT Fixed cells can be stored wrapped in aluminum foil at 4 °C for up to 1 month before flow cytometry. -
104
To each well of infected cells from Step 102, add 75 μL of fresh cRPMI plus 20 U/mL IL-2. Return the plate to the tissue culture incubator for 48 h.
-
105
Repeat steps 101–104 to obtain cells for the second time point (typically 5 d post infection).
-
106
Repeat steps 101–103 to obtain cells for the third and final time point (typically 7 d post infection).
-
107
Any remaining infected cells not collected for flow cytometry analysis can be disposed of after inactivation with 10% (vol/vol) bleach.
-
108
Run fixed cells from each time point (collected in Steps 103, 105, and 106) on a flow cytometer (i.e., a Thermo Fisher Attune NxT instrument or equivalent), calculating the percentage of live, infected cells over the time course of infection (i.e., Fig. 4b; see gating strategy in Supplementary Fig. 1b).
TROUBLESHOOTING
Fig. 4 |. HIV spreading infection results over a time course of infection in primary CD4+ T cells.
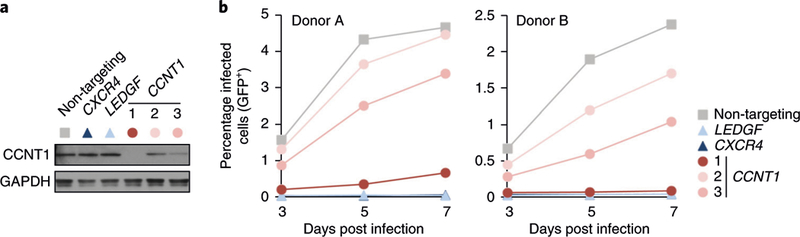
a, An immunoblot of CCNT1 levels in primary T cells treated with six different crRNPs: non-targeting (gray square), CXCR4-targeting (dark blue triangle), LEDGF-targeting (light blue triangle), and three unique crRNPs targeting CCNT1 (red circles). Cell lysates are probed for GAPDH as a loading control. b, Average percentage of HIV-infected (GFP+) cells at days 3, 5, and 7 post infection for six pools of edited cells originating from two donors (n = 3 technical triplicates). Relative to the non-targeting control, knockout of the CXCR4 and LEDGF control loci results in severe inhibition of infection (note that some data points overlap). Knockout of CCNT1 similarly results in decreased infection over time, with the replication defect correlating to protein knockout as depicted in a. Samples were run on an Attune NxT Flow cytometer and analyzed using FlowJo software v.10.1 (n > 10,000 events collected per data point).
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 47 | Poor enrichment of CD4+ T cells | Inefficient isolation of PBMCs from whole blood or impure PBMCs | Perform isolations within 24 h of blood draw Increase dilution of blood samples in Step 21 Handle centrifuged samples with care and turn off the centrifuge brake to ensure that a clean PBMC buffy coat forms in Step 23 Wash the PBMCs with PBS-EDTA one extra time after Step 28 |
| Inefficient binding and removal of CD4− cells from PBMCs | Recount the PBMCs to ensure a correct concentration of input cells in Step 31 Ensure complete mixing of antibody and beads during Steps 35–37 |
||
| Donor has low T-cell counts Inefficient CD4 staining | Start over with fresh blood from a new donor Increase antibody concentration during incubation or stain fewer cells in Step 47 Wash twice with MACS buffer before staining Incorporate a compatible live-dead cell stain to remove confounding signals |
||
| Inefficient T-cell activation | Poor CD3 adherence to the surface of the plate | Use non-treated plates and increase either CD3 concentration or incubation time in Step 18 Switch to either bead-based or small molecule-based activation protocols |
|
| Donor T cells are exhausted and cannot be activated | Start over with fresh blood from a new donor | ||
| Inefficient CD25 staining | Increase antibody concentration during incubation or stain fewer cells in Step 47 Wash cells twice with MACS buffer before staining Incorporate a compatible live-dead cell stain to remove confounding signals |
||
| 66, 73 | CRISPR-Cas9 RNPs show little activity in positive controls | Donor cells are not sufficiently activated | Cells should be nucleofected 48–72 h after activation and should be >90% CD25+ in Step 47. If not, reactivate or start over with fresh blood from a new donor |
| Guide RNA stocks were suspended >6 months earlier or frozen-thawed multiple times | Reorder guide RNA | ||
| crRNPs were generated >6 months earlier or frozen-thawed multiple times | Resynthesize crRNPs | ||
| Cas9 precipitated during synthesis | Preheat Cas9, tracrRNA, and guide RNA to 37 °C before mixing in Step 17 If a precipitate is visible, mix gently by pipette or flicking until precipitant re-enters solution in Step 17 |
||
| Too many cells or too few crRNPs were added to the nucleofection mixture | Repeat nucleofections with fewer cells or more crRNPs per reaction | ||
| Arc error during nucleofection of the positive=control well | Repeat nucleofections, making sure to avoid air bubbles, to pipette to the bottom of the cuvette, and to add no more than 20 μL of suspension per well in Step 56 | ||
| Cells were harvested for validation before protein turnover | Wait 72–96 h before harvesting protein or genomic DNA in Step 67 | ||
| Positive controls are validated, but experimental CRISPR-Cas9 RNPs do not work | Guide RNA does not target a translated exon | Double-check the intron-exon structure of the targeted gene, with special attention to alternative isoforms in Steps 1–9 | |
| Guide RNA does not cut the target site | Not all guides will be successful. Redesign and order an additional guide RNA targeting the desired locus | ||
| Targeted locus is not accessible to editing | Some loci appear resistant to editing in some cell types. Try multiple guide RNAs or alternative cell types or lines | ||
| 73 | TIDE analysis does not work | Unclear chromatograms prevent deconvolution | Adjust PCR thermocycler conditions to amplify a single amplicon in Step 70. Verify the success of adjustments by gel electrophoresis Design and order alternative primers in Step 68 if necessary Ensure clean DNA preparations after the purification performed in Step 71 by spectrophotometry |
| 74–108 | Low infection rate (<1%) | Poor yield during virus preparation | Check transfection efficiency in Steps 78–83 with a GFP or reporter plasmid under identical conditions and adjust if necessary Ensure purity and correct sequence of the HIV molecular clone by full-vector sequencing and spectrophotometry During concentration, make sure the mixture is cloudy after 4 °C incubation in Step 88, before pelleting of virions. If it is not, add more PEG-6000 and incubate for 2 h to promote precipitation Verify virus titer by p24 ELISA or infection of an easy-to-infect cell line as described in Step 93 |
| The molecular clone used for virological assays is not appropriate for replication in primary cells | Choose an alternative molecular clone | ||
| The cells are not fully activated | Stimulate again and repeat infection | ||
| The donor is not susceptible to HIV infection | Start over with a new blood sample from a different donor | ||
| Donor cells are only partially susceptible to infection | Infect with more virus in Step 100 or increase infection rates with alternative infection techniques such as spinoculation Allow more time for infection to spread between cells in Steps 100–106 |
||
Timing
Steps 1–9, gRNA design: 1 d
Steps 10–15, gRNA and tracrRNA suspension: 1 d
Steps 16 and 17, crRNP generation: 1 d
Steps 18–32, PBMC isolation: 1 d
Steps 33–41, CD4+ T-cell enrichment: 1 d
Steps 42–47, T-cell activation and expansion: 2–3 d
Steps 48–61, crRNP nucleofection: 1 d
Steps 62–67, cell activation and expansion: 4–8 d
Steps 68–73, TIDE analysis: 3–4 d
Steps 74–93, generation of concentrated HIV-1 virus stocks: 5 d
Steps 94–108, primary cell HIV spreading infection: 7–9 d
Anticipated results
Following implementation of the Procedure detailed above, three critical pieces of data will be generated: quality control staining for CD4+ T-cell isolation and stimulation, molecular validation of gene knockout (by immunostaining, immunoblotting, or TIDE), and HIV spreading infection time courses in each edited T-cell population.
CD4+ T-cell isolation and stimulation
Three primary cell samples are collected during isolation and stimulation at distinct stages: after Ficoll centrifugation (PBMCs, Step 32), after CD4+ T-cell isolation (CD4+ enriched, Step 41), and 72 h after stimulation (post stimulation, Step 46). These samples are then stained for CD4 and CD25 to ensure proper isolation and stimulation, respectively (Step 47, i.e., Fig. 2a; see Supplementary Fig. 1a for gating strategy). PBMCs from the whole blood of a typical donor under normal conditions will contain 5–25% CD4+ T cells, only a fraction of which will be activated (5–20% CD25+). Most enrichment techniques, including those used in the Procedure, enable researchers to isolate CD4+ T cells to >95% purity. Negative-selection approaches, including those used in the Procedure, rely on the binding and removal of all undesired cells from the PBMCs and result in a predominantly naive, unstimulated population of cells (<5% CD25+). Positive-selection approaches, on the other hand, rely on the selective binding and enrichment of CD4+ cells and result in a ratio of stimulated/unstimulated cells similar to what is observed in the PBMC fraction. If the post-enrichment sample contains <95% CD4+ cells or shows abnormally high numbers of activated (>25% CD25+) cells, the editing reaction or infection rates may be negatively impacted (refer to Troubleshooting). Post stimulation, all isolated cells should retain CD4 expression and may even show an increase in CD4 cell surface levels due to their larger overall cell size. Most stimulation approaches (such as treatment with anti-CD3/anti-CD28/IL-2 as used in the Procedure) will yield near-complete activation (>90% CD25+). As the efficient editing of most loci is dependent on T-cell activation using this protocol, lower percentages of activated cells in the population may adversely impact editing efficiency (refer to Troubleshooting).
Validation of gene editing
A variety of molecular biology techniques can be used to verify the successful knockout of target genes, including DNA sequencing, immunoblotting, and immunostaining. Although edits at the DNA level can be observed within 24 h of nucleofection, we recommend waiting at least 3–4 d post nucleofection to perform validation. This not only ensures that editing has been completed but also allows time for the recovery of cells post nucleofection, the resolution of persistent DNA breaks, and the turnover of protein pools.
For large numbers of samples with unique molecular readouts, it may be advantageous to complete phenotypic analysis before validation of each condition, depending on the experimental setup (refer to Experimental design). Regardless of the design, however, we strongly recommend validating the CXCR4 positive control before proceeding with phenotypic analyses. The CXCR4 locus has proven to be highly susceptible to editing in primary T cells and serves as a surrogate marker of efficiency. Relative to non-targeting controls, which should be >90% CXCR4+ by immunostaining, we expect cell populations treated with CXCR4-targeting crRNPs to be nearly devoid of CXCR4 (<10% CXCR4+, Fig. 2b). Inefficient knockout of CXCR4 probably indicates experimental error at the stage of cell isolation, cell stimulation, crRNP synthesis, or nucleofection (refer to Troubleshooting).
For proteins that lack appropriate antibodies for immunostaining or immunoblotting, DNA sequencing of the targeted cut site provides a good indication of successful editing. As the break sites within the polyclonal pool are unique to each locus and cell, PCR amplification over the targeted region will result in many unique fragments of differing length and sequence. These fragments can be precisely quantified by a number of deep-sequencing approaches, but the easiest way to quickly determine a rough editing percentage is through TIDE analysis (Steps 68–73). This approach relies on computational methods to deconvolute the Sanger sequencing of related, yet diverse, PCR amplicons. For example, Fig. 2c (right) shows the Sanger sequencing results from PCR amplicons over a targeted region of the CCNT1 locus in both experimental (top) and non-targeting control (bottom) samples. All amplicons in the non-targeting control display the same sequence. In the experimental sample, however, the sequences diverge at the targeted cut site, indicating successful editing and the presence of multiple indels. Uploading these chromatograms into the TIDE software provides an estimated knockout efficiency, including predicted frameshift percentages (Fig. 2c, right). Keep in mind, however, that although TIDE is a quick and cost-effective method of estimating indel percentage and validating site-specific editing, it does not reveal the exact nature of the repair. Deep sequencing of amplicons over the cut site is required for additional, detailed information on the diversity of repaired alleles in an edited population.
Editing efficiency will vary by locus, gRNA, cell type, and donor, so it is vital to validate knockout efficiency in each unique sample. Owing to the variability inherent in working with these primary cell types, it is critical to validate any pertinent results with multiple gRNAs in multiple donors.
HIV spreading infection
We recommend collecting data from at least three time points post infection, including one within the first 2–3 d. At this first time point, the virus will not yet have completed multiple rounds of replication, such that genes influencing early versus late events of the viral life cycle can be distinguished. In principle, at day 2 or 3, the proportion of infected cells should be between 1 and 3%, a low enough value that the virus can spread effectively without inducing massive cytotoxic effects in the culture. The percentage of infected cells in the culture will increase as the infection spreads. Eventually, as the virus infects and kills more cells and as the cells return to a resting state in which they are unsusceptible to infection, the percentage of infected cells in culture will decrease, with peak infectivity typically occurring between 5 and 9 d post infection.
Even before editing, cells from different donors will display different susceptibilities to infection, and baseline infection rates will subsequently vary. Therefore, each assay should be interpreted relative to donor-matched controls. For example, the results of an HIV spreading infection assay conducted on cell samples from two different donors are reported in Fig. 4b. The baseline infection rate in the non-targeting control is roughly twofold lower for the cells from donor B than for those from donor A, even though the two cell samples were treated identically and in parallel. Nevertheless, the results relative to the matched controls are consistent between the two donors. As expected, knocking out CXCR4 (dark blue triangles) and LEDGF (light blue triangles) markedly repressed replication relative to the non-targeting control (gray squares). Three crRNPs, all targeting CCNT1, were also delivered in parallel (red circles). These guides appear differentially effective in inhibiting HIV replication, with one guide (dark red circles, no. 1) inhibiting virus replication to near-control levels. Immunoblots of these samples (i.e., from lysates collected in Step 66 of the Procedure) probed for CCNT1 protein levels (Fig. 4a) confirm that the most effective crRNP gave the strongest HIV replication phenotype.
Correlating guide strength to phenotype strength and confirming the phenotype in multiple donors are critical to the interpretation of the results. For screens with large numbers of samples, we recommend performing these assays in at least two donors and normalizing the infectivity relative to the median infectivity across the plate at a given time point for a given donor. Assuming most guides will not impact replication, the distribution about the median will enable researchers to determine statistical cutoffs. If a large number of guides are anticipated to have an impact on the observed results, normalization relative to the average of the non-targeting controls is another option. Significant findings should be repeated in multiple donors after the screening is completed, with consideration of phenotypic variability, strength, and statistical robustness.
Supplementary Material
Acknowledgements
We thank L. Pache, E. Battivelli, and members of the Marson and Krogan labs for critical feedback and testing of the protocol. This research was supported by amfAR grant 109504–61-RKRL, using funds raised by generationCURE (J.F.H.); a fellowship of the Deutsche Forschungsgemeinschaft (SCHU3020/2–1; K.S.); the UCSF Sandler Fellowship (A.M.), a gift from Jake Aronov (A.M.); NIH/NIGMS funding for the HIV Accessory & Regulatory Complexes (HARC) Center (P50 GM082250; A.M., J.D., and N.J.K.); NIH funding for the FluOMICs cooperative agreement (U19 AI106754; J.F.H. and N.J.K.); NIH/NIAID funding for the HIV Immune Networks Team (P01 AI090935; N.J.K.); NIH funding for the Dengue Human Immunology Project Consortium (DHIPC, U19 AI118610; N.J.K.); NIH funding for the study of innate immune responses to intracellular pathogens (R01 AI120694 and P01 AI063302; N.J.K.); NIH funding for the UCSF-Gladstone Institute of Virology & Immunology Center for AIDS Research (CFAR, P30 AI027763); and an NIH/NIDA grant (DP2 DA042423–01; A.M.). A.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund, is a Chan Zuckerberg Biohub Investigator, and has received funding from the Innovative Genomics Institute (IGI). Special thanks to E. Brookes, M. Hall, and O. Cantada at Lonza Bioscience for their support in regard to the nucleofection transfection technology; D. Chow at Dharmacon for his support in regard to gRNA synthesis; T. Brown at Thermo Fisher Scientific for his support in regard to the Attune NxT Flow Cytometer, and C. Jeans at the University of California, Berkeley Macrolab for the production of Cas9 protein.
Footnotes
Competing interests
An intellectual property patent application has been filed for the use of CRISPR-Cas9 RNPs to edit the genome of human primary hematopoietic cells. A.M. is a cofounder of Spotlight Therapeutics, serves on the scientific advisory board of PACT Pharma, and was previously an adviser to Juno Therapeutics. The Marson lab has received sponsored research funding from Juno Therapeutics, Epinomics, and Sanofi, and a gift from Gilead. The remaining authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-018-0069-7.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Schumann, K. et al. Proc. Natl. Acad. Sci. USA 112, 10437–10442 (2015): https://doi.org/10.1073/pnas.1512503112 Hultquist, J. F. et al. Cell Rep. 17, 1438–1452 (2016): https://doi.org/10.1016Zj.celrep.2016.09.080 Park, R. J. et al. Nat. Genet. 49, 193–203 (2017): https://doi.org/10.1038/ng.3741
References
- 1.Harris RS, Hultquist JF & Evans DT The restriction factors of human immunodeficiency virus. J. Biol. Chem. 287, 40875–40883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goff SP Host factors exploited by retroviruses. Nat. Rev. Microbiol. 5, 253–263 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Hsu TH & Spindler KR Identifying host factors that regulate viral infection. PLoS Pathog. 8, e1002772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorr P et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49, 4721–4732 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gulick RM et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359, 1429–1441 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bushman FD et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 5, e1000437 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeung ML, Houzet L, Yedavalli VS & Jeang KT A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284, 19463–19473 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park RJ et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 49, 193–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan C, Kumar C, Bohl S, Klingmueller U & Mann M Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol. Cell. Proteomics 8, 443–450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alge CS, Hauck SM, Priglinger SG, Kampik A & Ueffing M Differential protein profiling of primary versus immortalized human RPE cells identifies expression patterns associated with cytoskeletal remodeling and cell survival. J. Proteome Res. 5, 862–878 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Lorsch JR, Collins FS & Lippincott-Schwartz J Cell biology. Fixing problems with cell lines. Science 346, 1452–1453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes P, Marshall D, Reid Y, Parkes H & Gelber C The costs of using unauthenticated, over-passaged cell lines: how much more data do we need? Biotechniques 43, 575 (2007). [DOI] [PubMed] [Google Scholar]
- 13.American Type Culture Collection Standards Development Organization Workgroup ASN-0002. Cell line misidentification: the beginning of the end. Nat. Rev. Cancer 10, 441–448 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Hultquist JF et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep. 17, 1438–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schumann K et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl Acad. Sci. USA 112, 10437–10442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brass AL et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Konig R et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou H et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4, 495–504 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Pache L, Konig R & Chanda SK Identifying HIV-1 host cell factors by genome-scale RNAi screening. Methods 53, 3–12 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Llano M et al. An essential role for LEDGF/p75 in HIV integration. Science 314, 461–464 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Llano M et al. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 78, 9524–9537 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fadel HJ et al. TALEN knockout of the PSIP1 gene in human cells: analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J. Virol. 88, 9704–9717 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shun MC et al. LEDGF/p75 functions downstream from preintegration complex formation to effect gene- specific HIV-1 integration. Genes Dev. 21, 1767–1778 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson AL & Linsley PS Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 9, 57–67 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Jackson AL et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12, 1179–1187 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tripathi S et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pichlmair A et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487, 486–490 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Carette JE et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Deans RM et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 12, 361–366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heaton NS et al. Targeting viral proteostasis limits influenza virus, HIV, and dengue virus infection. Immunity 44, 46–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin S, Staahl B, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal PK et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/ Cas9. Cell Stem Cell 15, 643–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Overbeek M et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol. Cell 63, 633–646 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Kim S, Kim D, Cho SW, Kim J & Kim JS Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR- Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleinstiver BP et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slaymaker IM et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu BX, Hansen LL, Artiles KL, Nonet ML & Fire AZ Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res. 42, 13778–13787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horlbeck MA et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horlbeck MA et al. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5, e12677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Isaac RS et al. Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. Elife 5, e13450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larson MH et al. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilbert LA et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perez-Pinera P et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maeder ML et al. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doench JG et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heigwer F, Kerr G & Boutros M E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Haeussler M et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haeussler M & Concordet JP Genome editing with CRISPR-Cas9: can it get any better? J. Genetics Genomics 43, 239–250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendel A et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blomen VA et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Hart T et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Healey DS, Jowett JB, Beaton F, Maskill WJ & Gust ID Comparison of enzyme immunoassay and reverse transcriptase assay for detection of HIV in culture supernates. J. Virol. Methods 17, 237–245 (1987). [DOI] [PubMed] [Google Scholar]
- 59.Healey DS, Maskill WJ, Neate EV, Beaton F & Gust ID A preliminary evaluation of five HIV antigen detection assays. J. Virol. Methods 20, 115–125 (1988). [DOI] [PubMed] [Google Scholar]
- 60.Gupta P, Balachandran R, Grovit K, Webster D & Rinaldo C Jr. Detection of human immunodeficiency virus by reverse transcriptase assay, antigen capture assay, and radioimmunoassay. J. Clin. Microbiol. 25, 1122–1125 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munch J et al. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J. Virol. 81, 13852–13864 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Doherty U, Swiggard WJ & Malim MH Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74, 10074–10080 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mandegar MA et al. CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18, 541–553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brinkman EK, Chen T, Amendola M & van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koressaar T & Remm M Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Untergasser A et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wehrly K & Chesebro B p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12, 288–293 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.