

Abstract
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a familial cancer syndrome associated with the development of cutaneous and uterine leiomyomas, and an aggressive form of type 2 papillary kidney cancer. HLRCC is characterized by germline mutation of the FH gene. This study evaluated the prevalence and clinical phenotype of FH deletions in HLRCC patients. Patients with phenotypic manifestations consistent with HLRCC who lacked detectable germline FH intragenic mutations were investigated for FH deletion. A series of 28 patients from 13 families were evaluated using a combination of a comparative genomic hybridization (CGH) array and/or CLIA-approved FH deletion/duplication analyses. Thirteen distinct germline deletions were identified in the 13 UOB families, including 11 complete FH gene deletions and 2 partial FH gene deletions. The size of eight evaluated complete FH deletions varied from ~4.74 Mb to 249 kb, with all deletions resulting in additional gene losses. Two partial FH gene deletions were identified, with one resulting in loss of exon 1 and the upstream region of the FH gene only. Kidney cancer was diagnosed in 9 (32%) of 28 patients and 7 (54%) of 13 families possessing either complete or partial FH deletions. Cutaneous and uterine leiomyomas were observed at similar rates to those in FH point mutation families. Complete or partial FH gene alterations in HLRCC families are associated with all of the canonical HLRCC manifestations, including type 2 papillary kidney cancer and should be screened for in any patient at-risk for this disorder.
1 |. INTRODUCTION
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is a familial cancer syndrome associated with the development of cutaneous and uterine leiomyomas and a highly aggressive form of type 2 papillary kidney cancer.1–4 Cutaneous and uterine leiomyomas are common clinical features of HLRCC;5–7 kidney cancer presents in a minority of HLRCC patients.6,7 Notably, HLRCC renal tumors can be aggressive, with patients at risk to develop locally advanced or disseminated disease even when associated with small primary tumors 1-2 cm in size.3 Due to the propensity for HLRCC-associated kidney cancer to spread when the primary tumors are small, it is important to screen at risk individuals periodically from a relatively young age, and HLRCC-associated kidney cancer has been diagnosed in children as young as 11 years old.1,8 Therefore, accurate identification of “at risk” individuals is needed.
HLRCC is an autosomal dominant inherited cancer syndrome characterized by a germline mutation of the fumarate hydratase (FH) gene in chromosome band 1p42.1 that encodes the enzyme responsible for conversion of fumarate to malate in the Krebs cycle.6,7,9 Cells in which there is somatic loss of the remaining wild-type copy of the FH gene undergo a metabolic shift to aerobic glycolysis with impaired oxidative phosphorylation. Loss of FH is associated with increased levels of fumarate.10,11 Increased intracellular fumarate can inhibit several α-ketoglutarate dependent dioxygenases including the prolyl hydroxylase domain enzymes leading to increased levels of hypoxia inducible factor (HIF) and activation of the HIF pathway.10,12 Additionally, increased intracellular fumarate induces succination of KEAP1, resulting in loss of NRF2 transcription factor inhibition and upregulation of the antioxidant response pathway that can combat the increased levels of reactive oxygen species associated with FH-deficient RCC.13,14
In our previous experience, germline FH gene mutation detection was positive in nearly 90% of patients with phenotypic manifestations consistent with HLRCC.7 The remaining individuals could lack point mutations within the FH gene for several reasons including inactivating mutations lying within the promoter or enhancer regions not assessed by the FH mutation test or by deletion of the FH gene. To date, no mutations outside of the FH coding region or splice junctions have been identified, and no other gene has been associated with HLRCC. Several studies have reported germline complete deletions of the FH gene,5,9,15–17 and a few have estimated the extents of these deletions and identified the additional genes that would be lost in conjunction with FH.9,16 Several reports have demonstrated that germline deletion of FH is associated with cutaneous and uterine leiomyomas. Conversely, there has only been a single report of a germline complete deletion of FH associated with kidney cancer and a single report of a germline partial deletion of FH, resulting in the loss of exon 1, in a family with kidney cancer.18,19
In another autosomal dominant inherited kidney cancer syndrome, von Hippel-Lindau, patients can present with germline mutation, partial deletion, or complete deletion of the VHL disease gene.20,21 It has been shown that the size of the deletion can affect the overall incidence of kidney cancer with the additional loss of the BRK1 (HSPC300) gene significantly lowering the rate of kidney cancer.22,23
Within our cohort of kindreds, we have a number of suspected or clinically confirmed HLRCC patients, both with and without kidney cancer, for which no FH point mutation had been identified. This study was conducted to search for complete or partial FH gene deletion in these kindreds and to characterize the phenotypic manifestations of these families and represents the largest case study reported to date.
2 |. METHODS
2.1 |. Patients and DNA samples
Patients with phenotypic manifestations consistent with HLRCC who tested negative for mutations within the 10 coding exons and exon-intron boundaries of FH by bidirectional DNA sequencing were selected for this study. Patients and, in some cases, additional family members were seen at the Urologic Oncology Branch (UOB) of the National Cancer Institute (NCI), National Institutes of Health (NIH) for clinical assessment, and peripheral blood samples were obtained for DNA extraction. This study was approved by the Institutional Review Board of the National Cancer Institute, and all patients provided written informed consent.
2.2 |. Fluorescence in situ hybridization analysis
The metaphase chromosomes were prepared from patient blood samples. A bacterial artificial chromosome-based human genomic DNA, 409K12 (RPC1-11), covering exons 1-6 of the FH gene (144 kb) was used as gene specific probe for fluorescence in situ hybridization (FISH) analysis (shown in red). Commercial probes for the centromere of chromosome 1 were used to highlight chromosome 1 (shown in green).
2.3 |. Array-based comparative genomic hybridization
An Agilent custom high-definition comparative genomic hybridization (CGH) array (Agilent, Santa Clara, CA) had been previously designed to assess copy number aberrations in several selected kidney cancer-associated genes.24 Included within this array were 74 probes selected from the Agilent HD-CGH database from within the 22.2 kb genomic region containing FH that were computationally preselected to provide an average probe density of ~3 probes per kb. Within the 50 kb flanking regions 5′ and 3′ to FH, a fade-out design achieved an average density of ~1 probe per kb diminishing to an average of ~1 probe per 40 kb over the entire genome. The custom designed arrays were printed on an Agilent 4x44K customer array and processed according to the manufacturer’s protocol. Briefly, 0.5 μg of patient genomic DNA and 0.5 μg of normal human reference DNA (Promega, Madison, WI) were fragmented by Alu I/Rsa I digestion, labeled with Cy3/Cy5 fluorescent dyes and hybridized at 65°C for 24 h. Following hybridization and washing, the arrays were scanned using an Agilent Microarray Scanner. Data were extracted with Agilent Feature Extraction Software (v10.7.1.1) and analyzed with Agilent DNA Analytics 4.0 software (v4.0.85). Deletions were calculated as the distance between the first and last probes that lost ~50% of their signal in comparison to the normal signal.
2.4 |. CLIA-Approved FH deletion/duplication analysis
Several commercial companies provide a service for the evaluation of deletions or duplications of the FH gene. All patients from the seven families with germline deletions identified by the Agilent custom high-definition CGH array were confirmed by CLIA-approved FH deletion/duplication analysis provided by either GeneDx, the Children’s Hospital of Philadelphia (CHOP), or Emory Genetics Laboratory. A further six patients were directly evaluated using CLIA-approved FH deletion/duplication analysis provided by the same companies.
2.5 |. Real-time PCR-based deletion analysis
The CLIA deletion analysis report by GeneDx for the patient representing UOB-4838 was inconclusive for exon 1 of FH and only confirmed loss of part of the region upstream of the gene. To demonstrate that important elements of the gene were lost a series of genomic primer pairs were designed to amplify ~150 bp sized DNA fragments in the region within and flanking the predicted deletion region including exon 1 and intron 1 of FH. Genomic primer pairs to exons 2 and 10 of FH were designed as normal copy number controls. Real-Time polymerase chain reaction (PCR) analysis was performed with these primer pairs using the SYBR Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA) on 10 ng of patient genomic DNA and analyzed using an ViiA 7 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA) using standard reverse transcriptase PCR (RT-PCR) conditions. A normal blood control was used to represent normal copy number and the genomic DNA from a member of the UOB-1815 complete FH deletion family was used to represent single copy number loss at these loci. All reactions were normalized to a primer pair for the ALB gene on chromosome 4 as a control for normal copy number.
2.6 |. PCR
Primers were designed adjacent to the estimated deleted region boundaries, and a Qiagen Taq PCR Core Kit (Germantown, MD) was used to amplify the deletion boundaries. DNA fragments were gel purified using E-Gel SizeSelect Gels (Life Technologies, Grand Island, NY).
2.7 |. Sequencing
All purified DNA products were sequenced bidirectionally using the Big Dye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems) according to manufacturer’s specifications and run on an ABI 3130xl Genetic Analyzer.
3 |. RESULTS
Our initial family (UOB-1815) with clinical features suggestive of HLRCC and extensive kidney cancer disease was investigated by FISH analysis using a probe that encompassed the first six exons of the FH gene (409K12 (RPC1-11)). This demonstrated a germline deletion of FH in multiple members of this family including patients with kidney cancer but did not assess the extent of the deletion beyond the extent of the probe (Figure 1A,B). This result was confirmed by CLIA-approved FH deletion/duplication analysis that demonstrated complete loss of the FH gene but did not reveal the extent of the deletion or whether or not additional genes were deleted.
FIGURE 1.
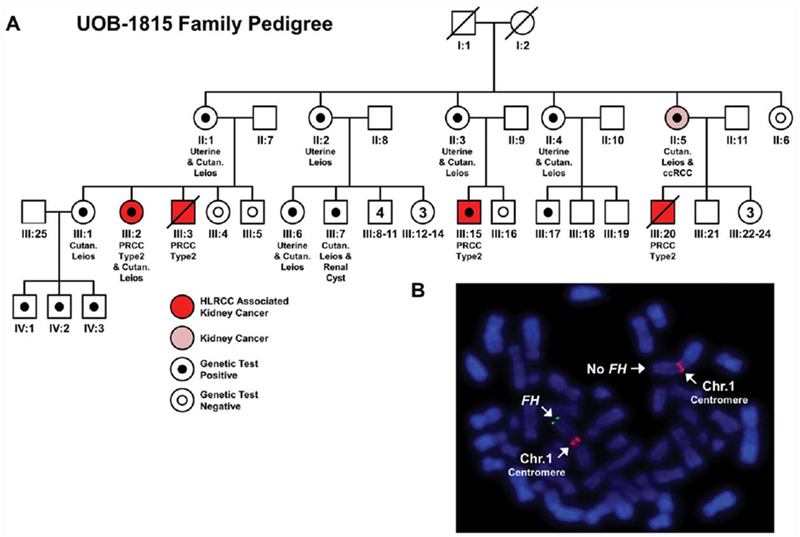
Analysis of a large HLRCC family (UOB-1815) with a complete germline deletion of the FH gene. (A) A pedigree for a large HLRCC family that demonstrated several individuals with HLRCC associated type 2 papillary kidney cancer (patients highlighted in red). One individual (highlighted in pink) presented with clear cell kidney cancer that is currently not associated with HLRCC but is the most common sporadic kidney cancer. Additional features of HLRCC are listed under each individual if known. Solid black dots indicate complete loss of FH that has been confirmed by CLIA testing, while white dots with black outlines indicate testing confirmed no loss of FH . (B) An example of the FISH analysis of blood cells for a member of this pedigree that demonstrated germline loss of FH (green) near the q telomere of one copy of chromosome 1. Chromosome 1 was identified using a chromosome 1 centromere specific probe (red). [Color figure can be viewed at wileyonlinelibrary.com]
To further investigate UOB-1815 and six other potential HLRCC families (UOB-4428, UOB-4366, UOB-3952, UOB-1126, UOB-4087, UOB-4391), a custom high-definition CGH array that had been designed to assess copy number aberrations in several selected kidney cancer associated genes was used.24 The array contained a high concentration of probes across the entirety of the FH gene and its surrounding DNA sequence with further probes representing the entire genome at a lower density. This array had the capability to detect the extents of both partial and complete FH gene deletions. This assay identified the complete germline deletion of a single copy of FH in all seven families, including the UOB-1815 HLRCC family previously identified by FISH analysis (Table 1). The minimum ranges for the deletions based on the probe positions varied greatly from ~4.74 Mb (UOB-4428) to ~249 kb (UOB-4391) and in all cases resulted in the loss of additional genes with minimal loss of KMO, OPN3, CHML, and WDR64 (Figure 2 and Table 1). The deletion breakpoints for two families were mapped to the exact nucleotide, and this slightly increased the actual size of the deletions to 487,769 bp and 251,982 bp for UOB-1815 and UOB-4391, respectively. Neither pair of breakpoints demonstrated any evidence for the involvement of Alu repeats (Figure 3).
TABLE 1.
Size and range of complete germline deletion of FH in HLRCC patients
| UOB family | FH deletion | Size of deletion | Additional deleted/partially deleted genes | Minimum range of deletion (GRCh37/hg19) |
|
|---|---|---|---|---|---|
| Starta | Finisha | ||||
| UOB-4428 | Complete | ~4.74 Mb | RYR2, ZP4, CHRM3, FMN2, GREM2, RGS7, KMO, OPN3, CHML, WDR64, EXO1, PLD5 | Chr1:237,941,801 | Chr1:242,677,983 |
| UOB-4366 | Complete | ~2.40 Mb | CHRM3, FMN2, GREM2, RGS7, KMO, OPN3, CHML, WDR64, EXO1, PLD5 | Chr1:239,927,943 | Chr1:242,322,958 |
| UOB-3952 | Complete | ~2.25 Mb | CHRM3, FMN2, GREM2, RGS7, KMO, OPN3, CHML, WDR64, EXO1, PLD5 | Chr1:240,071,458 | Chr1:242,322,958 |
| UOB-1126 | Complete | ~1.83 Mb | RGS7, KMO, OPN 3, CHML, WDR64, EXO1, PLD5 | Chr1:240,845,554 | Chr1:242,677,983 |
| UOB-4636 | Complete | ~1.25 Mb | RGS7, KMO, OPN3, CHML, WDR64, EXO1 | Chr1:240,849,498 | Chr1:242,094,956 |
| UOB-4087 | Complete | ~981 kb | RGS7, KMO, OPN3, CHML, WDR64 | Chr1:240,918,766 | Chr1:241,900,239 |
| UOB-1815 | Complete | 484 kbb | RGS7, KMO, OPN3, CHML, WDR64 | Chr1:241,465,096b | Chr1:241,949,083b |
| UOB-4391 | Complete | 249 kbb | KMO, OPN3, CHML, WDR64 | Chr1:241,650,802b | Chr1:241,900,239b |
| UOB-4515 | Complete | Unknown | N/A | N/A | N/A |
| UOB-4431 | Complete | Unknown | N/A | N/A | N/A |
| UOB-4861 | Complete | Unknown | N/A | N/A | N/A |
| UOB-4862 | Partial | Unknown | N/A | Exon 7 | Downstream of Exon 10 |
| UOB-4838 | Partial | 4.01 kbb | None | Chr1: 241,683,108b | Chr1: 241,684,038b |
Start and Finish coordinates represent the position of the first and last CGH array probes lost respectively and demonstrate the minimum range of the deletion based on CGH array probes.
Deletion mapped to the exact base pair breakpoint.
FIGURE 2.

Size and range of germline FH deletions identified within HLRCC patients. An Agilent custom high-definition CGH array was used to identify the presence and boundaries of FH deletions in a series of HLRCC patients. The boundaries were defined by the first and last probe with ~50% loss in the patient compared to a normal control. The genes lost due to each deletion are mapped underneath with a labeled arrow that indicates the direction of transcription. A red dotted line indicates the location of the FH gene. [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 3.
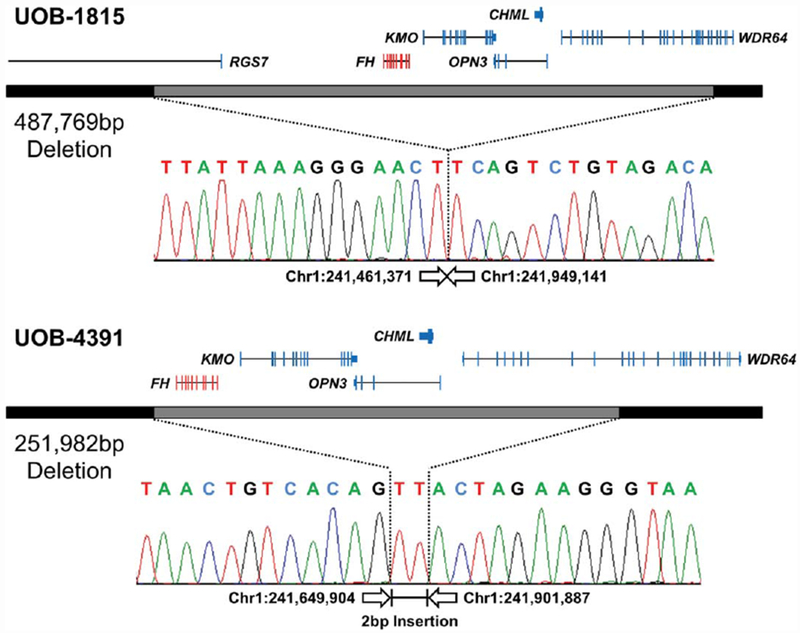
Breakpoint mapping of two CGH array-based FH complete deletions in HLRCC patients. The Agilent custom high-definition CGH array refined the breakpoints down to the region between the first and last probes that demonstrated no loss or 50% loss. A combination of primers was designed to sit within the potentially retained regions near each end of the deletion and used to PCR amplify across the deleted region. Sequencing of these PCR products demonstrated the breakpoint at the nucleotide level for two deletions with one containing a 2 bp insertion. The completely or partially lost genes are indicated above the deletion with the target FH gene in red and the additionally deleted genes in blue. All positions and size calculations are based on the GRCh37/hg19 build. [Color figure can be viewed at wileyonlinelibrary.com]
As with the UOB-1815 deletion, the six newly identified complete FH germline deletions were confirmed using CLIA-approved FH deletion/duplication analysis. An additional six potential HLRCC families were also thus evaluated. Four additional families with complete FH germline deletion (UOB-4636, UOB-4515, UOB-4431, UOB-4861) were identified. Mapping of family UOB-4636 demonstrated a deletion of ~1.25 Mb resulting in loss of RGS7, KMO, OPN3, CHML, WDR64, EXO1 (Table 1). In addition, two families (UOB-4862 and UOB-4838) were found to have partial FH germline deletions. While UOB-4862 was shown to have a germline deletion of the terminal four exons of FH (exons 7-10), additional gene loss downstream of the FH gene was not assessed (Table 1). UOB-4838 was found to have a partial germline deletion in a region upstream of the 5′ untranslated region (UTR) of the FH gene, but the deletion status of exon 1 was not evaluated and exon 2 did not exhibit loss. Thus, the deletion was reported as having “uncertain significance” due to no observation of loss within the coding region. To further evaluate the extent of this deletion a series of PCR probes were designed within the FH gene and upstream (UpStr) of transcription start site, including the predicted deletion region, exon 1, and intron 1 (Figure 4A). Real-Time PCR analysis using these probes demonstrated the expected normal copy number in exons 2 and 10 and the loss of exon 1 and three upstream probes, further refining the minimal deletion and confirming loss within the coding region (Figure 4A). Using these data, the deletion breakpoints were mapped to the nucleotide level revealing a 4088 bp deletion which encompassed the entirety of exon 1 of the FH gene (Table 1, Figure 4B). One of the two breakpoints was in an L2 repeat sequence, and the other was not in or near a repeat element.
FIGURE 4.
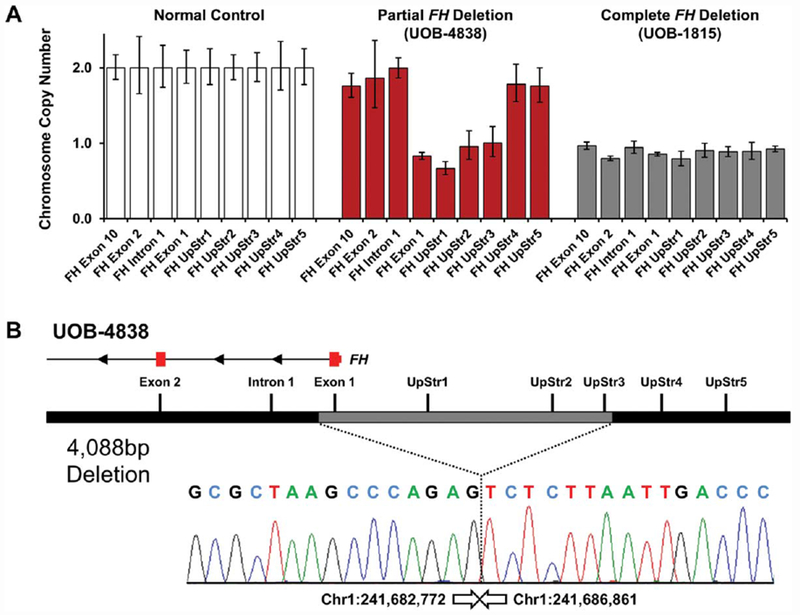
Breakpoint mapping of a partial FH deletion in a HLRCC patient. (A) The GeneDx aCGH identified loss of a region upstream of the FH gene and provided inconclusive results for exon 1. A series of PCR primer pairs was designed within the FH gene and upstream (UpStr) of transcription start site, including the suggested deleted region. Real-Time PCR analysis of these loci demonstrated loss of exon 1 and three upstream probes. (B) Sequencing of the PCR fragment produced using the primers designed for the RT-PCR analysis demonstrated the exact breakpoint and the loss of 4088 bp including all of exon 1. The lost and retained exons within the FH gene are shown in red, and all positions and size calculations are based on the GRCh37/hg19 build. [Color figure can be viewed at wileyonlinelibrary.com]
Clinical data from 28 patients with germline FH deletions from the 13 UOB families were investigated. Uterine leiomyomas were present in 13/13 (100%) female patients from seven different families (Table 2). Uterine leiomyomas were observed in patients with both the largest and second smallest complete FH germline deletions. None of female patients had the partial FH germline deletions (Tables 1 and 2). Cutaneous leiomyomas were present in 18/20 (90%) of patients from 11 families (Table 2). Both patients who were negative for cutaneous leiomyomas were members of the UOB-1815 family in which other family members were found to have cutaneous leiomyomas. Cutaneous leiomyomas were observed in patients with both the largest and smallest complete FH germline deletions and in the patients with both of the partial FH germline deletions (Tables 1 and 2). Kidney cancer was present in 9 (32%) of 28 patients that were evaluated and was found in 7 different families (Table 2). Eight out of the nine kidney cancers demonstrated the distinct type 2 papillary histology associated with HLRCC tumors; one patient had a clear cell renal cell carcinoma and was a member of a family in which other family members have demonstrated the classic HLRCC renal tumor histology. One additional patient reported a family history of kidney cancer; however, the tumor histology data was not available. Kidney cancer was present in patients with both the second largest and smallest complete FH germline deletions and in both patients with partial FH germline deletions (Tables 1 and 2).
TABLE 2.
Clinical features of HLRCC patients with confirmed complete/partial deletion of FH
| Clinical features |
||||||
|---|---|---|---|---|---|---|
| Family designation | Status | Gender | Uterine leiomyomas | Cutaneous leiomyomas | Kidney cancer | |
| UOB-4428 | Alive | F | Yes | Yes | No | |
| UOB-4428 | Alive | F | Yes | Yes | No | |
| UOB-4366 | Dead | M | – | n/a | Yes | |
| UOB-3952 | Alive | F | Yes | Yes | Family history | |
| UOB-1126 | Alive | M | – | Yes | No | |
| UOB-4636 | Alive | F | Yes | Yes | No | |
| UOB-4087 | Alive | F | Yes | Yes | No | |
| UOB-1815 | II:1 | Alive | F | Yes | Yes | No |
| UOB-1815 | II:2 | Alive | F | Yes | n/a | No |
| UOB-1815 | II:3 | Alive | F | Yes | Yes | No |
| UOB-1815 | II:4 | Alive | F | Yes | Yes | No |
| UOB-1815 | II:5 | Alive | F | Yes | n/a | Yes |
| UOB-1815 | III:1 | Alive | F | n/a | Yes | No |
| UOB-1815 | III:2 | Alive | F | n/a | Yes | Yes |
| UOB-1815 | III:6 | Alive | F | Yes | Yes | No |
| UOB-1815 | III:7 | Alive | M | – | Yes | No |
| UOB-1815 | III:15 | Alive | M | – | No | Yes |
| UOB-1815 | III:17 | Alive | M | – | No | No |
| UOB-1815 | IV:1 | Alive | M | – | n/a | No |
| UOB-1815 | IV:2 | Alive | M | – | n/a | No |
| UOB-1815 | IV:3 | Alive | M | – | n/a | No |
| UOB-4391 | Dead | M | – | Yes | Yes | |
| UOB-4391 | Alive | M | – | n/a | No | |
| UOB-4515 | Dead | M | – | Yes | Yes | |
| UOB-4431 | Alive | F | Yes | Yes | No | |
| UOB-4861 | Dead | F | Yes | n/a | Yes | |
| UOB-4862 | Dead | M | – | Suspected | Yes | |
| UOB-4838 | Alive | M | – | Yes | Yes | |
n/a: not assessed.
4 |. DISCUSSION
HLRCC is a relatively rare syndrome with most HLRCC patients demonstrating a point mutation within FH (~90%).7 The remaining suspected HLRCC patients without detectable point mutations represent an appreciable percentage of patients. In this study we report the comprehensive genomic and phenotypic characterization of complete or partial FH gene deletion in 28 patients from 13 HLRCC families.
Thirteen separate germline deletions were identified, with 11 families found to have complete germline FH gene deletion and two families demonstrating partial germline loss of the FH gene. There was notable variation in the sizes of the eight complete FH gene deletions that were evaluated, ranging from approximately 4.74 Mb to only 249 kb and included larger deletions than any previously reported.9,16 All evaluated complete deletions resulted in loss of additional genes, with a minimum of four other genes, KMO, OPN3, CHML, and WDR64, always lost. While several previous studies have shown one or two cases of complete deletion within their cohorts,5,9,15–19 this study more than doubles the number of individual deletions reported, reports complete genomic characterization of the germline deletions, highlighting the loss of additional genes, and detailed phenotypic characterization of the families. Two partial FH gene deletions were identified with one resulting in the loss of the last four exons of FH plus an unknown amount of downstream sequence and the other resulting only in the loss of exon 1 and the region immediately upstream of the FH gene. This second partial deletion represents the first confirmed deletion alteration that was limited to loss of the FH gene alone. Both partial deletions could result in the production of truncated versions of the FH protein. Future studies are planned to investigate whether any truncated protein is present and their potential for any dominant negative activity. With two partial FH deletions in our cohort and only one case previously reported, this suggests partial deletion is a rarer event.15 The mapping of deletion breakpoints revealed no evidence for the involvement of Alu repeats, as is observed in deletions of the VHL kidney cancer risk gene.23
Patients with either complete or partial FH gene deletions appear to have a similar rate of cutaneous (90%) and uterine leiomyomas (100%) as HLRCC patients with FH point mutations, although no female patients with partial FH gene deletions were identified and thus could not be evaluated for uterine leiomyomas. Previous studies involving HLRCC patient cohorts in which the majority of patients had point mutations of FH demonstrated a rate of cutaneous leiomyomas ranging from 76% to 100% and a rate of uterine leiomyomas in female patients also ranging from 76% to 100%.5–7
Kidney cancer was found in 9 (32%) of 28 patients and 7 (54%) of 13 families and was associated with both complete and partial FH gene deletions. In the majority of cases (8/9), the kidney cancers were type 2 papillary renal cell carcinoma, which has been associated with HLRCC. The large HLRCC pedigree (UOB-1815) effectively demonstrates that the presence of the same germline FH deletion can result in varying clinical presentation in different members of the same family. In comparison, only a single previous report identified FH deletion patients with evidence of HLRCC associated type 2 papillary kidney cancer.19 This could have been due to the small number of reported cases of germline FH deletion in combination with the lower penetrance of kidney cancer in HLRCC.22,23 These findings demonstrate that HLRCC associated type 2 papillary kidney cancer can occur in patients with either complete or partial germline FH deletions, ranging from the smallest partial deletion, resulting in only the loss of the first exon of the FH gene (UOB-4838), to the second largest deletion (UOB-4366) that resulted in a larger loss than any previously mapped.9,16 The prevalence of kidney cancer in HLRCC patients in reported cohorts has varied greatly from 2% to 43% and this is partially dependent upon whether the selection criteria for patient acquisition was based on the presence of cutaneous leiomyomas or the combination of leiomyomas and kidney cancer.5–7 This study is comparable to the studies that acquired patients due to a combination of leiomyomas and kidney cancer that demonstrated a rate of kidney cancer ranging from 16% to 43% in comparison to the 32% reported in this study.6,7
These findings demonstrate that deletion analysis of FH should be performed in patients who are suspected to be at risk for HLRCC. The identification of genomic alteration of FH in patients suspected of being affected with HLRCC will be very useful for both confirming the diagnosis and for effectively screening family members.25 Testing for the presence of all FH alterations is particularly important due to the highly aggressive nature of the HLRCC-associated type 2 papillary kidney cancers and its potential to manifest in patients at a very early age.1,8 Thus, the identification of deletions in these patients and families is important for identifying the relevant at risk individuals and, in particular, ensuring that the appropriate screening and surveillance for all related clinical features can be instigated at the earliest time point to provide the optimal clinical management of these individuals.
Acknowledgments
Funding information
Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, Grant numbers: ZIA BC011028, ZIA BC011038, ZIA BC011089, and ZIC BC011044
REFERENCES
- [1].Grubb RL III, Franks ME, Toro J, et al. 2007. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 177:2074–2080. [DOI] [PubMed] [Google Scholar]
- [2].Launonen V, Vierimaa O, Kiuru M, et al. 2001. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA. 98:3387–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Merino MJ, Torres-Cabala C, Pinto PA, Linehan WM. 2007. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 31:1578–1585. [DOI] [PubMed] [Google Scholar]
- [4].Schmidt LS, Linehan WM. 2014. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 7:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alam NA, Olpin S, Leigh IM. 2005. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol. 153:11–17. [DOI] [PubMed] [Google Scholar]
- [6].Toro JR, Nickerson ML, Wei MH, et al. 2003. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 73:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wei MH, Toure O, Glenn GM, et al. 2006. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 43:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alrashdi I, Levine S, Paterson J, et al. 2010. Hereditary leiomyomatosis and renal cell carcinoma: very early diagnosis of renal cancer in a paediatric patient. Fam Cancer. 9:239–243. [DOI] [PubMed] [Google Scholar]
- [9].Tomlinson IP, Alam NA, Rowan AJ, et al. 2002. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 30: 406–410. [DOI] [PubMed] [Google Scholar]
- [10].Tong WH, Sourbier C, Kovtunovych G, et al. 2011. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 20:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yang Y, Lane AN, Ricketts CJ, et al. 2013. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS One. 8:e72179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Isaacs JS, Jung YJ, Mole DR, et al. 2005. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer. Cell 8:143–153. [DOI] [PubMed] [Google Scholar]
- [13].Adam J, Hatipoglu E, O’Flaherty L, et al. 2011. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 20:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ooi A, Wong JC, Petillo D, et al. 2011. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 20:511–523. [DOI] [PubMed] [Google Scholar]
- [15].Ahvenainen T, Lehtonen HJ, Lehtonen R, et al. 2008. Mutation screening of fumarate hydratase by multiplex ligation-dependent probe amplification: detection of exonic deletion in a patient with leiomyomatosis and renal cell cancer. Cancer Genet Cytogenet. 183: 83–88. [DOI] [PubMed] [Google Scholar]
- [16].Alam NA, Rowan AJ, Wortham NC, et al. 2003. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 12: 1241–1252. [DOI] [PubMed] [Google Scholar]
- [17].Gardie B, Remenieras A, Kattygnarath D, et al. 2011. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet. 48:226–234. [DOI] [PubMed] [Google Scholar]
- [18].Smit DL, Mensenkamp AR, Badeloe S, et al. 2011. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 79:49–59. [DOI] [PubMed] [Google Scholar]
- [19].Trpkov K, Hes O, Agaimy A, et al. 2016. Fumarate hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol. 40:865–875. [DOI] [PubMed] [Google Scholar]
- [20].Stolle C, Glenn GM, Zbar B, et al. 1998. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 12:417–423. [DOI] [PubMed] [Google Scholar]
- [21].Zbar B, Kishida T, Chen F, et al. 1996. Germline mutations in the von Hippel-Lindau disease (VHL) gene in families from North America, Europe and Japan. Hum Mutat. 8:348–357. [DOI] [PubMed] [Google Scholar]
- [22].Cascon A, Escobar B, Montero-Conde C, et al. 2007. Loss of the actin regulator HSPC300 results in clear cell renal cell carcinoma protection in Von Hippel-Lindau patients. Hum Mutat. 28:613–621. [DOI] [PubMed] [Google Scholar]
- [23].Maranchie JK, Afonso A, Albert PS, et al. 2004. Solid renal tumor severity in von Hippel Lindau disease is related to germline deletion length and location. Hum Mutat. 23:40–46. [DOI] [PubMed] [Google Scholar]
- [24].Benhammou JN, Vocke CD, Santani A, et al. 2011. Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dube syndrome. Genes Chromosomes Cancer. 50:466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Menko FH, Maher ER, Schmidt LS, et al. 2014. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 13:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]