

Summary
The ascomycete Neofusicoccum parvum, one of the causal agents of Botryosphaeria dieback, is a destructive wood‐infecting fungus and a serious threat to grape production worldwide. The capability to colonize woody tissue, combined with the secretion of phytotoxic compounds, is thought to underlie its pathogenicity and virulence. Here, we describe the repertoire of virulence factors and their transcriptional dynamics as the fungus feeds on different substrates and colonizes the woody stem. We assembled and annotated a highly contiguous genome using single‐molecule real‐time DNA sequencing. Transcriptome profiling by RNA sequencing determined the genome‐wide patterns of expression of virulence factors both in vitro (potato dextrose agar or medium amended with grape wood as substrate) and in planta. Pairwise statistical testing of differential expression, followed by co‐expression network analysis, revealed that physically clustered genes coding for putative virulence functions were induced depending on the substrate or stage of plant infection. Co‐expressed gene clusters were significantly enriched not only in genes associated with secondary metabolism, but also in those associated with cell wall degradation, suggesting that dynamic co‐regulation of transcriptional networks contributes to multiple aspects of N. parvum virulence. In most of the co‐expressed clusters, all genes shared at least a common motif in their promoter region, indicative of co‐regulation by the same transcription factor. Co‐expression analysis also identified chromatin regulators with correlated expression with inducible clusters of virulence factors, suggesting a complex, multi‐layered regulation of the virulence repertoire of N. parvum.
Keywords: Botryosphaeria dieback, CAZymes, cell wall degradation, RNA‐seq, secondary metabolism, single‐molecule real‐time (SMRT) sequencing, weighted co‐expression network analysis
Introduction
Over 20 species of fungi in the family Botryosphaeriaceae (Botryosphaeriales, Ascomycota) cause Botryosphaeria dieback of grape (Vitis vinifera L.) and similar diseases of other woody plants (Slippers and Wingfield, 2007; Úrbez‐Torres, 2011). Neofusicoccum parvum (Pennycook & Samuels) is one of the most aggressive Botryosphaeriaceae species on grape (Úrbez‐Torres and Gubler, 2009). Neofusicoccum parvum colonizes the woody tissue through wounds, killing spurs and adjacent shoots, and causes necrosis of dormant buds (Fig. 1) (Úrbez‐Torres and Gubler, 2009, 2011). Wedge‐shaped cankers in grapevine spurs, cordons and trunks are typical symptoms of the disease (Fig. 1B). Botryosphaeria dieback is part of the grapevine trunk disease complex which, together with Eutypa dieback, Phomopsis dieback and Esca, causes chronic wood infections that accumulate over time (Bertsch et al., 2013). Because no practices are available to eradicate the causative fungi (aka trunk pathogens), short of physically cutting out infected wood, a practice known as ‘vine surgery’ (Sosnowski et al., 2011), disease management relies on preventative practices of delayed pruning or application of pruning wound protectants (Petzoldt et al., 1981; Rolshausen et al., 2010; Weber et al., 2007).
Figure 1.

Culture of Neofusicoccum parvum isolate UCD646So on potato dextrose agar (PDA) (A) and symptoms of Botryosphaeria dieback on grapevine wood (B, C): wedge‐shaped canker (B) and dead spurs (arrows) (C).
During wood colonization, the growth and survival of the invading microorganism depend on its ability to decompose secondary cell walls to access extra‐ and intracellular nutrients whilst overcoming host immune responses (Bruno and Sparapano, 2006; Gómez et al., 2016). Woody tissue decomposition is a result of the activities of secreted enzymes that disassemble structural polymers of the primary and secondary cell walls (King et al., 2011; Rolshausen et al., 2008). Plant cell walls are not only an important barrier to be overcome by pathogens to access intercellular material, but are also a carbon‐rich substrate (Cantu et al., 2008). In addition to the enzymatic decomposition of cell wall components, trunk disease development has also been associated with the activities of toxins secreted by the invading pathogens (Andolfi et al., 2011). These structurally heterogeneous, low‐molecular‐mass compounds are products of fungal secondary metabolism and are mostly derived from either polyketides or non‐ribosomal peptides. Toxins can injure host cells by interfering with enzymatic reactions and cellular transport, and damaging cell membranes. Previous experiments have shown that N. parvum produces a broad variety of phytotoxins in vitro, such as dihydrotoluquinones, epoxylactones, dihydroisocoumarins, hydroxybenzoic acids and fatty esters (Abou‐Mansour et al., 2015; Evidente et al., 2010; Martos et al., 2008; Uranga et al., 2016). Purified hydroxymellein and sphaeropsidone produced in vitro by N. parvum have been shown to cause severe foliar symptoms, whereas terremutin and mellein have been identified in grape wood with Botryosphaeria dieback symptoms (Abou‐Mansour et al., 2015).
In fungal genomes, genes involved in secondary metabolism are often organized into clusters, where a central biosynthesis gene is surrounded by genes encoding transporters and enzymes involved in the post‐synthesis modification of the metabolites, such as cytochrome P450 monooxygenases (P450s), dehydrogenases and FAD‐binding domain proteins (Brakhage, 2013). Common transcriptional regulation is thought to be the major evolutionary driver of this genomic organization (Brakhage, 2013). Shared regulatory mechanisms, including transcription factors, as well as chromatin remodelling, can in fact lead to the co‐expression of gene clusters. As a result, a complex and multi‐layered regulatory system, involving global as well as pathway‐specific regulators, modulates the production of secondary metabolites in response to environmental stimuli, including carbon and nitrogen sources, light, pH, reactive oxygen species and hypoxic conditions (Brakhage, 2013; Bruns et al., 2010; Vödisch et al., 2011; Yin and Keller, 2011). Using short‐read shotgun sequencing, we have recently assembled the first draft genome of N. parvum (Blanco‐Ulate et al., 2013) and have evaluated its repertoire of potential virulence factors (Morales‐Cruz et al., 2015). Despite the fragmentary assembly and gene prediction solely based on ab initio methods, we were able to identify a large number of genes encoding cell wall‐degrading enzymes (CWDEs), as well as several genes involved in secondary metabolism. Comparative analysis with genomes of other trunk pathogens revealed a significant expansion in N. parvum of gene families associated with specific oxidative functions, which may contribute to lignin degradation and secondary metabolic products, such as non‐ribosomal peptides and amino acid‐derived compounds (Morales‐Cruz et al., 2015). Lineage‐specific expansion of such gene families in N. parvum supports their potential role in pathogenesis and adaptation.
The objective of this study was to determine the genome‐wide expression dynamics of potential virulence functions of N. parvum in response to different growth substrates in vitro and during colonization of grapevine woody stems in planta. We first applied single‐molecule real‐time (SMRT) DNA sequencing to generate a more complete and contiguous reference genome. Using a combination of evidence‐based and ab initio prediction methods, we then expanded and refined the predicted transcriptome. The improved reference was used to determine genome‐wide patterns of expression of putative virulence factors. Pairwise statistical testing of differential expression, followed by Weighted Gene Co‐expression Network Analysis (WGCNA), revealed a condition‐dependent regulation of physically clustered genes. The analysis of promoter regions showed that all adjacent genes composing each genomic cluster shared at least one common motif, indicating that the expression of adjacent genes might be co‐regulated by a common transcription factor. Interestingly, co‐regulated gene clusters were significantly enriched not only in genes associated with secondary metabolism, but also in those associated with cell wall degradation, suggesting that dynamic co‐regulation of transcriptional networks contributes to multiple aspects of N. parvum virulence.
Results and Discussion
SMRT DNA sequencing of the N. parvum genome and gene discovery
The genome of N. parvum isolate UCD646So was sequenced using the PacBio P6‐C4 chemistry (Appendix S1, see Supporting Information: Table S1). Sequencing reads were assembled into 28 contigs, providing a significant improvement in sequence contiguity compared with the 1877 contigs (1287 scaffolds) generated previously using short‐read technology for the genome of isolate UCR‐NP2 (Appendix S1: Fig. S1A and Table S2; Blanco‐Ulate et al., 2013). One of the 28 contigs of isolate UCD646So corresponded to the entire mitochondrial genome with a length of 115.8 kb (Appendix S2, see Supporting Information). The other 27 contigs formed the nuclear genome with a total assembly size of 43.7 Mb and a remarkable N90 of 2.0 Mb (Table 1; Appendix S2). The total assembly size of the genome was close to the genome size estimate based on k‐mer frequency (44.2 Mb), suggesting that the shotgun sequencing approach yielded the complete N. parvum genome. The completeness of the genome was further supported by the analysis of terminal contig sequences. We identified TTAGGG telomeric repeats (Podlevsky et al., 2008; Appendix S1: Fig. S2) at both ends of 16 contigs, suggesting that these contigs encompass complete chromosomes, telomere to telomere. For two contigs, telomeric repeats were found only at one end. The 18 contigs with terminal telomeric repeats represented 99.1% of the total assembly and had a similar mean GC content of 56.5 ± 1.0% (Fig. 2). The remaining nine contigs displayed a significantly different GC content from the rest of the genome (41.6 ± 10.1%; P = 1.3 × 10−6) and did not carry any detectable protein‐coding genes, suggesting that these much shorter fragments are derived from intergenic and repetitive regions of the genome. We cannot rule out the possibility that some of these contigs represent whole or fragments of dispensable chromosomes. Resequencing of multiple isolates is necessary to shed light on the relevance of dispensable chromosomes in N. parvum virulence and host adaptation.
Table 1.
General assembly and annotation statistics of the nuclear genome of Neofusicoccum parvum UCD646So.
| Number of contigs | 27 |
| Total assembly size (bp) | 43 674 556 |
| N50 (bp) | 2 555 423 |
| N90 (bp) | 2 020 352 |
| Total interspersed repeats (bp) | 2 055 934 |
| Predicted complete protein‐coding genes | 13 124 |
Figure 2.
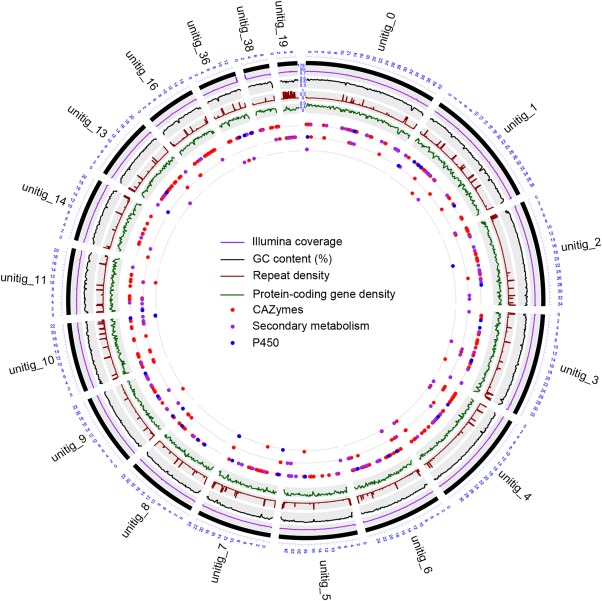
Circular representation of the Neofusicoccum parvum genome. Different tracks denote (moving inwards): (i) mapping coverage of short Illumina reads; (ii) percentage GC content; (iii) repeat density; (iv) protein‐coding gene density; (v–vii) localization of genes encoding virulence factors: (v) induced in presence of wood, (vi) expressed exclusively in planta, and (vii) differentially regulated in planta. Figure was prepared using the OmicCircos Bioconductor package (Hu et al., 2014).
To determine the rate of potential sequence errors not corrected by Quiver, we sequenced the genome of the same isolate using an Illumina HiSeq2500 platform. Mapping of the Illumina reads, followed by the GATK variant‐discovery pipeline, detected 103 sites in which Illumina and PacBio sequences were in discordance. Assuming that the Illumina reads were correct, we concluded that the genome sequence obtained using SMRT technology is 99.99976% accurate. Gene space completeness was assessed using CEGMA (Parra et al., 2009) and BUSCO (Simão et al., 2015). Both methods identified in the assemblies approximately 97% of the complete eukaryotic conserved protein‐coding sequences, providing further evidence of the completeness of the genome (Appendix S1: Table S4).
We applied nucmer (Kurtz et al., 2004) and Assemblytics (Nattestad and Schatz, 2016) to compare this assembly with the contigs previously generated using short reads of isolate UCR‐NP2 (Blanco‐Ulate et al., 2013; Appendix S1: Fig. S1B). A total of 474.84 kb was affected by structural variation (≥50 bp). Fifty per cent of the structural variation detected was caused by shorter repeats in UCR‐NP2, probably as a result of the failure to reconstruct repeats using short reads. In agreement with this observation, the total repeat content in the PacBio assemblies was higher than in the assembly generated using short reads (6.20% vs. 3.60%; Appendix S1: Table S3). Interestingly, 180 sites (≥50 bp), for a total of 113 kb, were absent in the UCR‐NP2 genome, possibly because they were inaccessible to Illumina sequencing.
To identify the protein‐coding genes in the assembly, we applied the BRAKER gene discovery pipeline (Hoff et al., 2015), which combines GeneMark‐ET (Lomsadze et al., 2014) and Augustus (Stanke and Waack, 2003) for unsupervised RNA‐sequencing (RNA‐seq)‐based genome annotation. BRAKER identified 13 124 complete protein‐coding genes (Appendix S2), representing 2654 additional genes in comparison with our previous transcriptome prediction of isolate UCR‐NP2, based on short‐read assembly and ab initio gene discovery (Blanco‐Ulate et al., 2013; Appendix S1: Table S3). The mean peptide length was also greater in this new predicted proteome, probably as a result of a more accurate prediction. Approximately 97% of the 1438 conserved BUSCO orthologues were found in the predicted proteome, supporting its completeness. Gene density was generally uniform across the contigs with 3.2 ± 2.3 genes every 10 kb (Fig. 2). These results show that shotgun sequencing using SMRT technology resulted in a more complete and contiguous assembly which, in combination with evidence‐based gene prediction, provided the most comprehensive view to date of the protein‐coding genes of N. parvum.
Annotation of putative virulence factors
Annotation of the predicted transcriptome focused on functions potentially associated with pathogenesis and virulence, including: (i) wood degradation and host colonization, such as carbohydrate‐active enzymes (CAZymes), peroxidases and P450s; (ii) cellular transporters; and (iii) secondary metabolism, including toxin production. Fifty‐two per cent of the total transcriptome (6805 genes) was assigned to these functional groups. We found 567 genes belonging to 52 CAZyme families potentially involved in the degradation of all components of primary and secondary plant cell walls (Fig. 3; Appendix S1: Table S5; Appendix S3, see Supporting Information). A diagram summarizing the CAZymes and their hypothetical targets in the plant host is provided in Fig. 3A. Glycoside hydrolases (GHs) accounted for 50% of the total CWDE repertoire. The most abundant families were involved in hemicellulose breakdown and included 42 β‐glucosidases (GH3s), 23 endo‐β‐1,4‐cellulases (GH5s), 32 xyloglucan endo‐β‐1,4‐glucanases (GH16s) and 31 GH43s.
Figure 3.
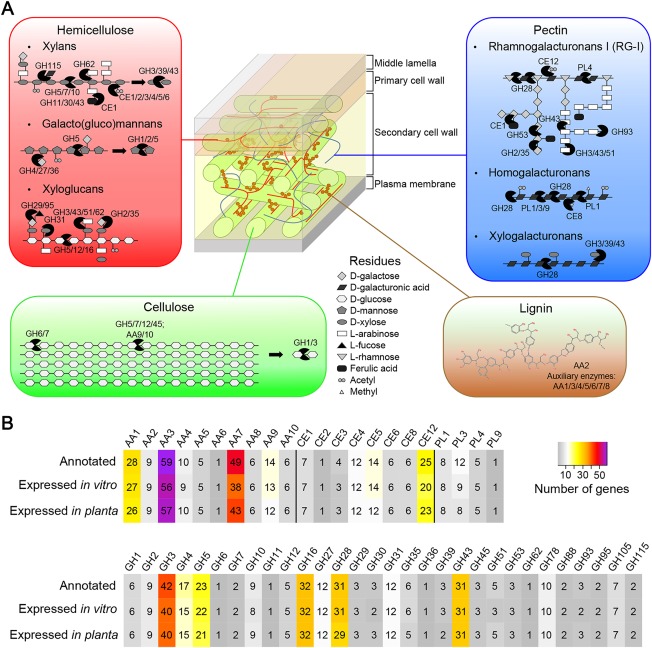
Repertoire of putative plant cell wall‐degrading enzymes (CWDEs) encoded in the Neofusicoccum parvum genome. (A) List of carbohydrate‐active enzymes (CAZymes), grouped by substrate, showing a schematic representation of their enzymatic activities. Adapted from van den Brink and de Vries (2011). (B) Total number of protein‐coding genes annotated as lignocellulolytic CAZymes and detected as expressed in the in vitro and in planta experiments.
The ability of N. parvum to decompose lignin is suggested by the abundant presence of genes belonging to the Auxiliary Activities (AA) families, namely 28 AA1s, 59 AA3s and 49 AA7s. AA1s are multicopper oxidases involved in lignin degradation; laccases, for example, oxidize the phenolic moiety of lignin (Kawai et al., 1987; Levasseur et al., 2013). Both AA3s and AA7s generate hydrogen peroxide, which can be used by peroxidases (AA2s) during lignin decomposition (van Hellemond et al., 2006; Hernández‐Ortega et al., 2012). Two hundred and ninety‐four of the CAZyme‐coding genes targeting host cell walls carried a secretion signal peptide, suggesting their potential role in extracellular activities.
Biosynthetic gene clusters (BGCs) potentially involved in secondary metabolism and toxin production were identified using antiSMASH (Weber et al., 2015). In total, 160 BGCs were found, corresponding to 4501 genes (Appendix S3). Among these, 52 BGCs belonged to known classes, such as polyketide synthase (PKS), non‐ribosomal peptide synthetase (NRPS) and terpene synthase classes (543, 221 and 222 genes, respectively). Nevertheless, most of the detected BGCs were annotated as unknown biosynthetic types (108 BGCs corresponding to 2858 genes).
We extended the functional annotation to peroxidases and P450s, which are involved in a broad array of primary, secondary and xenobiotic metabolic reactions that have been associated with fungal adaptation to various ecological niches (Chen et al., 2014; Črešnar and Petrič, 2011; Durairaj et al., 2015; Moktali et al., 2012). We identified 65 peroxidase‐ and 265 P450‐encoding genes. The latter belonged to 95 different clans, based on the classification of Moktali et al. (2012; Appendix S3). These included 39 genes encoding CYP65s, shown to catalyse the epoxidation reaction during the biosynthesis of the mycotoxin trichothecene in Fusarium spp. (Ward et al., 2002), as well as during radicicol biosynthesis in Aspergillus parasiticus (Wang et al., 2008).
Cellular transporters can also contribute to pathogenesis and virulence, for example those involved in the secretion of virulence factors and in the exclusion of antimicrobial compounds produced by the host plant (Denny and VanEtten, 1983; Denny et al., 1987). In total, we identified 1983 transporters (Appendix S3), 41% more than in the previous annotation (Morales‐Cruz et al., 2015). Among them, the largest transporter family was the Major Facilitator Superfamily (MFS, TCBD code 2.A.1) with 494 members, followed by the Peroxisomal Protein Importer (PPI) family (TCDB code 3.A.20) and the ATP‐binding cassette (ABC) superfamily (TCBD code 3.A.1) with 115 and 68 genes, respectively. MFS and ABC family transporters, which are both involved in efflux, have been shown to contribute to pathogenesis in other fungi (Coleman and Mylonakis, 2009).
Change in growth substrate triggers major transcriptional reprogramming of virulence‐associated genes
A genome‐wide expression profiling experiment, using RNA‐seq, was carried out to determine the portion of the N. parvum transcriptome that is induced when grapevine wood is the carbon source. Neofusicoccum parvum mycelia were collected after two cycles of growth either on agar mixed with ground wood of V. vinifera ‘Cabernet Sauvignon’ or ‘Merlot’, or on potato dextrose agar (PDA). An average of 9 786 825 ± 1 142 788 reads from three biological replicates were mapped onto the transcriptome of N. parvum UCD646So. By statistical testing, we identified 1349 genes up‐regulated in wood agar (adjusted P ≤ 0.05; Appendix S4C, see Supporting Information). Among these differentially expressed (DE) genes, 73 were putative CAZymes involved in host cell wall degradation, mostly targeting pectin and hemicellulose. Wood from both cultivars promoted the up‐regulation of 12 GMC oxidoreductase genes (AA3s), which may contribute to the degradation of lignocellulosic components, whereas the three ligninase‐encoding genes (AA2s) identified in the genome were constitutively expressed at similar levels on all media.
The presence of wood as the sole nutrient promoted not only the expression of genes involved in wood decomposition, but also a large set of transporters, which were the functional category with the most prominent induction with wood in the growth media. Among these were 92 genes coding for MFS transporters, including 29 sugar transporters possibly involved in the transport of products of cell wall degradation, as seen in Trichoderma reesei (Ivanova et al., 2013): these included six galactose‐H+ symporters (TCBD code 2.A.1.1.9), five xylose facilitators (TCBD code 2.A.1.1.40) and two glucose/xylose‐H+ symporters (TCBD code 2.A.1.1.51). Five quinate‐H+ symporters (TCBD code 2.A.1.1.7) and eight high‐affinity nicotinate permeases (TCBD code 2.A.1.14.11) were also found, indicating an extensive reprogramming of the transporter repertoire in wood agar.
These results show that the addition of wood as a substrate in vitro affected the expression of approximately 10% of the predicted genes in the genome. A similar phenomenon was observed in Phanerochaete carnosa, a white‐rot basidiomycete fungus, in which a wood agar mixture, with wood from soft‐ and hardwood hosts, induced the expression of lignocellulolytic CAZyme‐coding genes (MacDonald et al., 2011). The induction of genes encoding CWDEs, such as cellulase and hemicellulase, has been shown to involve a positive feedback regulation by the respective hydrolysis and/or transglycosylation products of cellulose and/or xylan, such as, for example, gentiobiose in Penicillium purpurogenum (Kurasawa et al., 1992) and sophorose in Aspergillus terreus and T. reesei (Amore et al., 2013; Hrmová et al., 1991; Mandels et al., 1962). In addition, genes encoding CWDEs in filamentous fungi have also been shown to be repressed when preferred carbon sources (e.g. glucose) are present (Aro et al., 2005; Kubicek et al., 2009; Ruijter and Visser, 1997; de Vries and Visser, 2001).
Interestingly, we also observed that genes sharing a common pattern of up‐regulation in response to the type of medium often clustered physically. These clusters of potentially co‐regulated genes included genes belonging to BGCs, but also genes with functions other than secondary metabolism, such as cell wall degradation (e.g. acetyl xylan esterases and GMC oxidoreductases) and transcription factors (e.g. C2H2 and C6 zinc finger domain transcription factors). However, we also found that, within BGCs, not all genes were co‐expressed. For example, in cluster BGC_114 (Fig. 4), the five central genes of the cluster were constitutively expressed, whereas the rest of the genes of the cluster were all significantly induced in wood agar.
Figure 4.
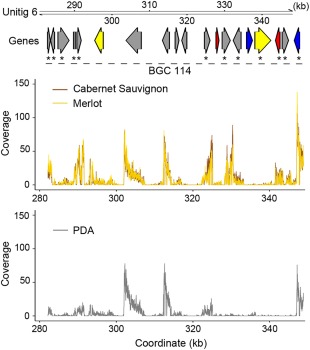
Example of Neofusicoccum parvum gene clusters modulated as a function of in vitro substrate. Arrows represent genes coding for carbohydrate‐active enzymes (CAZymes) (red), P450s (blue) and transporters (yellow). Grey arrows correspond to genes predicted to be part of a secondary metabolism cluster, but with other annotations. Asterisks highlight genes with significant up‐regulation in the presence of wood in vitro. Line plots show the RNA‐sequencing mapping coverage in samples grown on minimal medium amended with finely ground grape wood of either ‘Cabernet Sauvignon’ or ‘Merlot’ or on potato dextrose agar (PDA) without wood.
In planta RNA‐seq experiment reveals clusters of virulence factors differentially regulated during grapevine woody stem infection
In order to determine the patterns of expression of potential virulence functions as N. parvum colonizes viable host tissues, we profiled its transcriptome during a time course of infection of the woody stems of potted grape plants. Total mRNA was sequenced from three biological replicates at seven time points after inoculation. On average, 14 806 320 ± 2 636 797 reads were mapped onto a combined reference that included the predicted transcriptomes of N. parvum UCD646So and grape (V. vinifera var. ‘PN40024’). The number of N. parvum reads was low at 0 and 3 h post‐inoculation (hpi) (830 ± 362 and 367 ± 198, respectively), increased from 24 hpi (11 439 ± 900) to 6 weeks post‐inoculation (wpi) (1 071 368 ± 816 298), and finally declined until 12 wpi (14 423 ± 5087) (Appendix S1: Fig. S3). The overall increase in N. parvum transcripts in the sample probably reflected the accumulation of fungal biomass during wood colonization. Despite the lower sequencing coverage of the in planta experiment, a similar number of genes were detected in the two experiments: 12 513 (95.3%) in vitro compared with 12 211 (93%) in planta.
After application of multiple statistical cut‐offs to exclude constitutively expressed genes, as described in the Experimental Procedures section, we identified 78 genes that were DE in planta (adjusted P ≤ 0.05; Appendix S4E). K‐means and hierarchical clustering were used to divide the DE genes based on their pattern of expression (Appendix S1: Fig. S4). Both clustering methods divided the 78 DE genes into four groups (Fig. 5; Appendix S4E). Groups 1 and 2 were composed of genes with peaks of expression at 24 hpi and 2 wpi, respectively.
Figure 5.
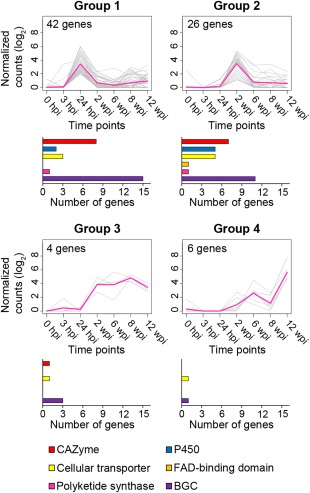
Expression profiles of four groups of genes sharing a similar expression pattern during in planta colonization of grape stems. Groups were obtained by performing a K‐means clustering analysis using the 78 differentially expressed (DE) genes expressed in planta. Each single line represents the log2‐transformed average of the normalized counts for an individual transcript. Histograms show the counts of genes belonging to each virulence factor category [i.e. carbohydrate‐active enzyme (CAZyme), P450, cellular transporter, FAD‐binding domain, polyketide synthase (PKS) and part of biosynthetic gene cluster (BGC)].
Group 1 included five CWDE‐coding genes: two pectate lyases (PL1 and PL3) (Benen et al., 2000), a GH7 and a GH16 involved in cellulose and xylan/xyloglucan backbone disassembly (Cantarel et al., 2009; Gielkens et al., 1999) and a GMC oxidoreductase (AA3), possibly contributing to cellulose, hemicellulose and lignin degradation (Kremer and Wood, 1992). In Group 1, there was also a cluster of five genes belonging to BGC_115, which included a PKS, an MFS transporter (TCBD code 2.A.1.2.17), a salicylate 1‐monooxygenase, a P450 and a cupin‐like protein (Fig. 6A). Early up‐regulation of a salicylate 1‐monooxygenase suggests a mechanism for N. parvum to overcome host immunity. In the corn smut pathogen Ustilago maydis, a salicylate 1‐monooxygenase has been shown to be transcriptionally induced by salicylic acid and to be required during the biotrophic stages of development (Rabe et al., 2013).
Figure 6.
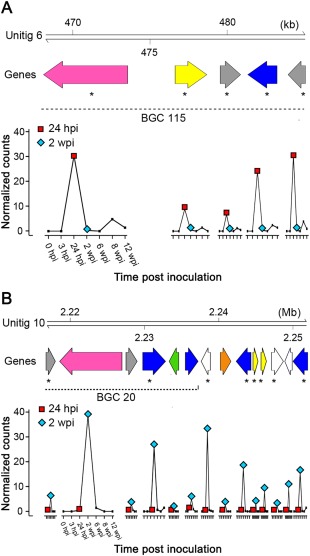
Examples of gene clusters associated with secondary metabolism among the co‐expressed gene Groups 1 (A) and 2 (B) shown in Fig. 5. Arrows represent genes coding for polyketide synthases (PKSs) (pink), P450s (blue), transporters (yellow), transcription factors (green) and FAD‐binding proteins (orange). Grey arrows correspond to genes predicted to be part of a secondary metabolism cluster, but with other annotations. White arrows represent genes outside of the biosynthetic gene clusters (BGCs). Asterisks highlight genes differentially regulated at 24 h post‐inoculation (hpi) (A) and 2 weeks post‐inoculation (wpi) (B). For each transcript, the gene expression profile is represented by the average of the normalized counts at each time point of woody stem colonization.
Although the gene composition of Group 1 suggests that genes involved in the degradation of the plant cell wall and in nutrient storage contribute to the early development of N. parvum, the genes of Group 2 suggest that biosynthetic metabolism is the predominant process during later stages of infection. Indeed, Group 2 was mostly composed of genes associated with secondary metabolism. These included loci encompassing BGCs 20 and 149 on unitigs 10 and 7, respectively (Fig. 6B). The co‐expressed cluster on unitig 10 was composed of the epoxide hydrolase, the PKS, the transferase and the two P450 genes of BGC_20, as well as genes downstream to BGC_20, an aristolochene synthase gene, a FAD‐binding protein gene, two P450 genes, two MFS transporter genes (TCBD codes 2.A.1.3.33 and 2.A.1.48.4) and a hydroxymethylglutaryl‐CoA synthase gene (Fig. 6B). The latter catalyses the production of hydroxymethylglutaryl‐CoA from acetyl‐CoA and acetoacetyl‐CoA, and participates in mevalonate biosynthesis, a precursor required for the biosynthesis of sterol and terpenoids (Wawrzyn et al., 2012; Withers and Keasling, 2007). Epoxide hydrolases convert epoxides arising from aromatic compound degradation into dihydrodiols. Fungal epoxide hydrolases can play a role in the synthesis of toxins (Bradshaw and Zhang, 2006; Morisseau et al., 1999), although they can also be involved in the detoxification of plant aromatic compounds (Harris et al., 2016). Gene composition suggests that BGC_149 may be involved in toxin biosynthesis. Its central biosynthesis gene encodes a PKS, whereas the other genes in the cluster are a FAD‐binding domain protein, two hypothetical proteins and an aldo‐keto reductase, followed by two S‐adenosylmethionine‐dependent methyltransferases, a MFS transporter (TCBD code 2.A.1.3.27), an alcohol dehydrogenase and a P450. In both gene clusters, genes that were not considered statistically as DE nonetheless showed the same expression pattern (i.e. up‐regulation at 2 wpi) as their DE neighbours. Interestingly, within BGC_20 and in close proximity to BGC_149, we found putative transcription factors containing a GAL4‐like Zn(II)2Cys6 DNA‐binding domain, which are potentially involved in the regulation of the respective BGCs, as shown previously in Fusarium graminearum (Lawler et al., 2013).
The comparison between N. parvum genes detected in wood agar in vitro and in planta revealed 304 genes expressed exclusively during the colonization of woody grape stems (Appendix S4D). Table S6 (Appendix S1) provides a summary of the functional annotations of these genes, as well as the 1342 genes up‐regulated in wood agar and detected in planta. These transcriptional differences may be caused by the complexity of both tissue and plant cell wall structures in planta, as the finely ground grape wood added to the agar had no tissue structure. More challenging growth conditions in the woody tissue, including partial anoxia and dehydration, as well as the accumulation of fungitoxic metabolites produced by the host, may also contribute to the transcriptional differences. Among the 304 genes, 38 genes were located on unitig 19, including 12 of the 22 genes that form BGC_60. This biosynthetic cluster spanned 96 kb on unitig 19 and comprised three genes coding for putative short‐chain dehydrogenase/reductases (SDRs): one classical SDR, one aldehyde reductase and one NmrA‐like protein. SDRs constitute a large family of NAD(P)(H)‐dependent oxidoreductases (Jörnvall et al., 1999). In fungi, some members of the SDR family have been found to be key enzymes in fungal melanin and toxin biosynthesis (Butchko et al., 2003; Thompson et al., 1997). An SDR has been shown to be required for pathogenicity of the rice pathogen Magnaporthe oryzae (Kwon et al., 2010). In total, 89 genes belonging to BGCs, as well as seven P450‐ and 20 CWDE‐encoding genes, were found to be expressed exclusively in planta. These results suggest that N. parvum expresses a repertoire of virulence factor genes during colonization of living woody tissue, which is unique when compared with that of non‐viable woody tissue.
Co‐expression network analysis identifies genomic clusters of co‐regulated virulence factors
As described above, transcriptional profiling, followed by pairwise statistical testing, revealed that different sets of putative virulence functions are induced depending on the substrate or the stage of woody stem colonization. In addition, we observed that genes with a shared pattern of up‐regulation were often physically clustered on the fungal genome, suggesting that they are co‐regulated. Therefore, we extended the analysis of co‐expression and co‐regulation to the entire transcriptome by constructing a gene expression correlation network. The objective of this analysis was to define groups of co‐expressed genes (i.e. modules) and to identify, within these groups, clusters of adjacent genes with shared patterns of expression and putative promoter motifs, suggestive of co‐regulation by common transcription factors. Co‐expression analysis was carried out using WGCNA (Langfelder and Horvath, 2008). Both RNA‐seq datasets were processed together. An undirected weighted network with scale‐free topology composed of 40 modules of genes with correlated expression (scale‐free model fitting index R 2 > 0.882) was obtained (Fig. 7A; Appendix S4F). For each module, a unique colour label was assigned as specific module identifier. The modules were composed, on average, of 319.8 genes, with the largest module (‘turquoise’) containing 3252 genes. Twenty‐four genes were not grouped in any of the 40 modules and were added to the ‘grey’ (or improper) module.
Figure 7.
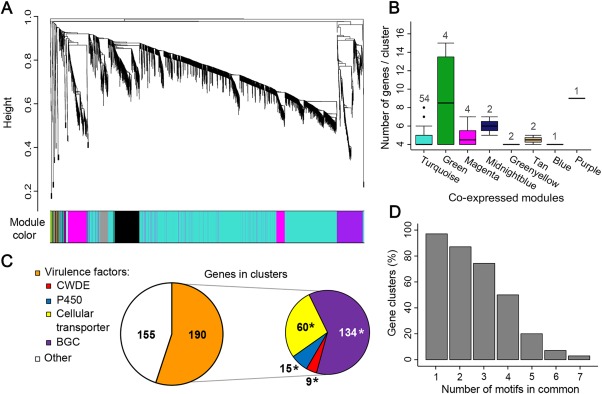
Graphical visualization of the Neofusicoccum parvum co‐expression network. (A) Dendrogram of all transcripts clustered based on a dissimilarity measure (1 – TOM; TOM, topological overlap mapping metric). Each line of the dendrogram corresponds to a gene. The multi‐colour bar below the dendrogram shows the 40 modules with each gene colour coded based on module assignment. (B) Boxplots showing the number of adjacent co‐expressed genes in each genomic cluster (≥4 adjacent genes per cluster) identified within the co‐expression modules. Numbers above the boxplots represent the total number of genomic clusters in each co‐expression module. (C) Functional category distribution of the genes organized in co‐expressed genomic clusters. Asterisks point to virulence categories significantly over represented (Fisher's exact test; adjusted P < 0.01) in the genomic clusters. BGC, biosynthetic gene cluster; CWDE, cell wall‐degrading enzyme. (D) Bar plot representing the percentage of genomic clusters that contain at least one common promoter motif in all genes that compose the cluster.
Within the WGCNA modules, we identified 70 genomic clusters containing a minimum of four co‐expressed adjacent genes. These clusters comprised a total of 345 genes, including 29 genes up‐regulated in wood‐agar and nine genes up‐regulated at 2 wpi in planta. The greatest number of these clusters was found in the ‘turquoise’ module (54 clusters; Fig. 7B; Appendix S4G). The two largest clusters, found in the ‘green’ module, corresponded to genes belonging to BGCs 20 and 149, which were composed of 15 and 13 genes, respectively. These two genomic clusters were described above as part of the set of genes significantly up‐regulated at 2 wpi (Fig. 6B). Approximately 55% of the genes in co‐expressed clusters were annotated as potential virulence factors (Fig. 7C). Importantly, all virulence categories were significantly overrepresented (Fisher's exact test; adjusted P < 0.01), suggesting that virulence functions in N. parvum tend to be organized as physically clustered and co‐expressed genes.
We then explored the possibility that co‐expressed and clustered genes are regulated by common transcriptional regulators. We applied the MEME motif discovery analysis (Bailey et al., 2009) to identify recurrent DNA motifs in the promoter regions (600 bp upstream of the coding sequences) of all gene clusters. Interestingly, in almost all clusters (97.1%), the promoter regions of all genes in each cluster shared at least one common motif. A maximum of seven shared motifs was found in two clusters in the ‘green yellow module’ (Fig. 7D). Shared motifs showing similarity to Saccharomyces cerevisiae motifs and motif‐associated proteins (ScAPs), together with their putative N. parvum homologues of ScAPs, are provided in Appendix S5 (see Supporting Information). Network topology was analysed to identify highly connected genes (i.e. intramodular hubs), which potentially include major transcriptional regulators of the co‐expressed gene modules in which they are located (Ma et al., 2013). The weighted network was converted into an unweighted network preserving all connections with topological overlap mapping metric (TOM) > 0.1 (Appendix S4H). Twenty‐one transcription factors, listed in Appendix S4I, were among the most highly connected nodes (top 5%). Interestingly, we also found six gene hubs coding for PHD finger domain‐containing proteins, which are involved in chromatin‐mediated gene regulation (Bienz, 2006; Appendix S4I). Chromatin modification has been shown to play an important role in the transcriptional regulation of the tightly linked genes involved with the mycelial pigment aurofusarin in F. graminearum (Reyes‐Dominguez et al., 2012). Although chromatin remodelling can prime the response and set the basal expression of a broad set of genes, transcription factors can activate smaller subsets of environment‐responsive genes, as proposed in F. graminearum (Lawler et al., 2013) and in mammalian systems (Garber et al., 2012).
Conclusions
In this study, we show that the expression of potential virulence factors of N. parvum, including functions involved in cell wall degradation and secondary metabolism, is influenced by the presence of woody substrate in vitro and even more so during colonization of woody stem tissue in planta. Our finding of this condition‐dependent regulation was primarily a result of the co‐expression of physically clustered genes. Most of the co‐expressed gene clusters shared at least one common promoter motif, suggesting that they are co‐regulated by common transcription factors. Co‐expression analysis revealed not only transcription factors, but also chromatin regulators, whose expression was correlated with inducible clusters of virulence factors. Such multi‐layered regulation suggests a complex range of potential virulence factors in N. parvum. Our findings serve as a basis for further studies on how N. parvum is able to colonize woody tissues or adapt to this environment. An understanding of the functions that lead to the colonization of certain cell types/tissues, and the corresponding fungal genes activated during subsequent degradation of such host tissues, may help us to understand the mechanism(s) of cultivar resistance and interactions within the trunk pathogen community.
Experimental Procedures
Biological material
Neofusicoccum parvum isolate UCD646So (ATCC number: MYA‐3706) was isolated from V. vinifera ‘Sauvignon blanc’ in Sonoma County (California, USA; Úrbez‐Torres et al., 2006). For transcriptome profiling of fungal cultures on solid medium (i.e. in vitro experiments), isolate UCD646So was inoculated onto PDA (Difco, Detroit, MI, USA) or SuperPure agar (BTS) containing finely ground grape wood (V. vinifera ‘Cabernet Sauvignon’ FPS 08 and ‘Merlot’ FPS 03), prepared as described in Morales‐Cruz et al. (2015). Cultures were incubated at 25 ºC for 7 days, and then subcultured to the same medium and incubated again for 7 days. Subculturing was carried out three times before collection of RNA. Hyphae were harvested from the surface of the agar for RNA extraction. For in planta transcriptome profiling, 1‐year‐old potted V. vinifera ‘Cabernet Sauvignon’ FPS 19 plants were inoculated with isolate UCD646So mycelia, as described in Travadon et al. (2013). Woody stems were collected at seven time points: 0, 3 and 24 hpi, and 2, 6, 8 and 12 wpi. Wood samples from 1 cm below the inoculation site were collected using flame‐sterilized forceps, immediately placed in liquid nitrogen and then stored at −80°C for RNA extraction. Infections were confirmed by positive recovery of the pathogen after 5 days of growth on PDA.
DNA extraction, library preparation and sequencing
High‐molecular‐weight DNA was extracted following the protocol described in Stoffel et al. (2012). Four grams of frozen ground mycelia were combined with 15 mL of 2 × extraction buffer. DNA purity was measured with a spectrophotometer, and integrity was evaluated with pulsed field electrophoresis. For PacBio sequencing library preparation, 15 µg of high‐quality genomic DNA was sheared using a Megaruptor (Diagenode, Denville, NJ, USA). After shearing, fragmented DNA was processed using a SMRTbell Template Prep Kit 1.0 (Pacific Biosciences, Menlo Park, CA, USA). Libraries were size selected at 12–50‐kb cut‐offs using a Blue Pippin (Sage Science, Beverly, MA, USA), submitted to a final DNA repair treatment (SMRTbell Damage Repair Kit, Pacific Biosciences) and then sequenced using a PacBio RS II platform (DNA Technologies Core Facility, University of California, Davis, CA, USA). Nine SMRT cells yielded a total of 7.2 Gb of sequence data: 698 022 reads were generated with median and maximum lengths of 10 and 52.5 kb, respectively (Appendix S1: Table S1). Sequencing coverage was approximately 160 times the expected genome size of 44.2 Mb, which was estimated based on DNA k‐mer distribution using SGA v.0.10.6 (Simpson, 2014). For Illumina sequencing, DNA libraries were prepared as described in Jones et al. (2014). Paired‐end, 150‐bp reads were sequenced on an Illumina HiSeq2500 (DNA Technologies Core Facility, University of California). A total of 2.36 Gb was generated, achieving a median coverage of 50 times the N. parvum genome. Genomic sequences can be retrieved from the National Center for Biotechnology Information (NCBI, Bioproject PRJNA321421).
RNA extraction, library preparation and sequencing
Mycelia from cultures on solid medium were collected in a 2‐mL micro‐centrifuge tube, which was immediately frozen in liquid nitrogen, and then ground to a powder with a TissueLyser II (Qiagen, Hilden, Germany) at 30 Hz for 30 s. One millilitre of TRIzol reagent (Ambion, Austin, TX, USA) was added to the ground mycelia; extraction of total RNA was performed following the manufacturer's protocol. For the in planta experiment, stem fragments 1 cm below the point of inoculation were placed into liquid nitrogen in pre‐cooled, 25‐mL stainless‐steel jars (Retsch, Haan, Germany) and ground to a fine powder, as described above. Total RNA was then isolated using a cetyltrimethylammonium bromide (CTAB)‐based extraction protocol, as described in Blanco‐Ulate et al. (2013). RNA‐seq libraries were prepared using the Illumina TruSeq RNA sample preparation kit v.2 (Illumina, San Diego, CA, USA). Libraries were sequenced on an Illumina HiSeq3000 sequencer (DNA Technologies Core Facility, University of California). Libraries from cultures and infected wood were sequenced in paired‐end, 150‐bp mode and single‐end, 50‐bp mode, respectively. Sequences were deposited to GEO (NCBI; accession: GSE85079).
Neofusicoccum parvum genome assembly and gene prediction
PacBio reads were assembled using HGAP v.3.0 (Chin et al., 2013). Reads were filtered with the parameters: minimum sub‐read length = 1000; minimum polymerase read quality = 0.8; minimum polymerase read length = 500. The minimum seed read length for the assembly was set at 6000. Assembly polishing was carried out with Quiver using only unambiguously mapped reads. To estimate the error rate, Illumina paired‐end reads were mapped using Bowtie2 v.2.2.327 (Langmead and Salzberg, 2012), polymerase chain reaction (PCR) and optical duplicates were removed with Picard tools (v.1.119; http://broadinstitute.github.io/picard/) and HaplotypeCaller (GATK v.3.3.0; McKenna et al., 2010) was used to call sequence variants with the following parameters: –ploidy 1 –min_base_quality_score 20.
Gene prediction was carried out using BRAKER v.1.8 (Hoff et al., 2015). Paired‐ and single‐end RNA‐seq reads from in vitro and in planta expression studies, respectively, were mapped onto the genome assemblies using TopHat v.2.0.9 (Trapnell et al., 2009). Prior to gene prediction, repetitive regions were masked using a combination of ab initio and homology‐based approaches, as described in Jones et al. (2014). BRAKER prediction was carried out on the soft‐masked contigs applying GeneMark‐ET with the branch point model, which was shown to be useful for fungal genomes. Only complete protein‐coding sequences without internal stop codons were retained for further analysis. Functional annotations were carried out as described in Morales‐Cruz et al. (2015).
Transcriptome mapping and statistical analysis
Forward reads from the dataset of in vitro cultures were trimmed to 50 bp using the FASTX‐Toolkit v.0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) in order to obtain results comparable with those obtained in the in planta experiment. Quality trimming (Q > 20) and adapter contamination removal were carried out using sickle (v.1.2.1; Joshi and Fass, 2011) and scythe (v.0.991; Buffalo, 2011), respectively. Single‐end reads were mapped onto the isolate UCD646So transcriptome (CDSs) using Bowtie2 v.2.2.327 with the following parameters: ‐q ‐end‐to‐end ‐sensitive ‐no‐unal. For the in planta experiment, the predicted transcriptome of isolate UCD646So was combined with the predicted transcriptome of V. vinifera ‘PN40024’ (v.V1 from http://genomes.cribi.unipd.it/grape/) and used as a reference for read mapping. Counts of uniquely mapped reads (mapping quality Q > 30) were extracted using sam2counts.py v.0.91 (https://github.com/vsbuffalo/sam2counts). Details on the results of data trimming and mapping are reported in Appendix S1 (Table S7). The Bioconductor package DESeq2 (Love et al., 2014) was used for normalization and for statistical testing of differential expression for both experiments (Appendix S4A,B). As described in the Results section, sequencing coverage varied greatly between time points and biological replicates, as a result of fungal biomass accumulation and the variable tissue colonization rate, commonly observed in experiments with trunk pathogens (e.g. Czemmel et al., 2015). In order to avoid false discovery of DE genes as a result of the changes in coverage between time points not corrected completely by DESeq2, we applied two additional cut‐offs that removed potentially constitutively expressed genes from the analysis. Fungal RNA‐seq read counts provide a good estimation of fungal biomass accumulation in the infected tissue (Blanco‐Ulate et al., 2015). Therefore, constitutive genes should: (i) increase their transcript abundance (as raw reads) linearly as the fungal biomass accumulates (as total fungal raw reads); and (ii) their normalized reads should display small variation between time points. To identify constitutive genes, we first calculated a Pearson correlation coefficient (R) for each gene by comparing the raw read count of the gene with the total number of raw read counts in each biological replicate. We imposed a second filter based on gene expression variability across time points as in Massa et al. (2011). A coefficient of variation (CV) was calculated for each gene using DESeq2‐normalized count means of all time points. Finally, we considered genes with R > 0.7 and/or CV < 50% as expressed constitutively (Appendix S1: Fig. S5). Therefore, genes were considered DE not only if they had a significant differential expression (adjusted P ≤ 0.05) between each subsequent pair of time points (3 hpi vs. 0 hpi, 24 hpi vs. 3 hpi, 2 wpi vs. 24 hpi, etc), but also if they had both R < 0.7 and CV > 50%. K‐means and hierarchical clustering analyses were performed on the genes DE at least at one time point during the in planta experiment using MeV v.4.9 (Saeed et al., 2003). K‐means cluster analysis was performed with a maximum number of 100 iterations and a number of random sets equal to four. The number of clusters was established using a plot of figure of merit (FOM) vs. the number of clusters (1–20 clusters, 100 iterations) (Appendix S1: Fig. S6).
Genome‐wide co‐expression analysis
Co‐expression analysis was conducted using WGCNA v.1.51 (Langfelder and Horvath, 2008). RNA‐seq counts from both experiments were combined into one dataset and normalized using DESeq2 (Love et al., 2014). A soft‐thresholding power of 7 with a scale‐free model fitting index R 2 > 0.882 was applied with a minimum module size of 20. Within the co‐expression modules identified by WGCNA, genomic clusters were defined as a minimum of four adjacent co‐expressed genes. Fisher's exact test was used to calculate functional enrichments in the genomic clusters. Following Lawler et al. (2013), the promoter region was defined as up to 600 bp upstream of each CDS. Upstream regions of adjacent genes were analysed using the MEME SUITE (Bailey et al., 2009). MEME v.4.11.1 was run with the following parameters: ‐dna ‐mod zoops ‐nmotifs 10 ‐maxw 9. Motifs were annotated by comparison with MacIsaac_v1 and SCPD databases (MacIsaac et al., 2006; Zhu and Zhang, 1999), using tomtom v.4.11.1 with the following parameters: ‐evalue ‐no‐ssc. Neofusicoccum parvum protein homologues of ScAPs were identified using BLASTp (e‐value < 10−6 and matched regions covering ≥30% of the query or target sequence). For network topological analyses, the unweighted network was extracted using a hard threshold of TOM > 0.1 and imported into Cytoscape v.3.4 (Shannon et al., 2003).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Appendix S1 Supplementary tables and figures.
Appendix S2 Genome assemblies and protein‐coding gene coordinates.
Appendix S3 Functional annotations.
Appendix S4 Normalized RNA‐sequencing counts in the in vitro (A) and in planta (B) experiments, list of genes up‐regulated in the presence of wood (C) and exclusively expressed in planta (D), and groups of co‐expressed genes during Neofusicoccum parvum colonization obtained by both K‐means and hierarchical clustering analysis (E). Gene co‐expression modules obtained from Weighted Gene Co‐expression Network Analysis (WGCNA) and the corresponding degree of connectivity in the unweighted network (F), genomic clusters identified among the gene co‐expression modules (G), network properties of the gene co‐expression modules (H) and transcription factor‐coding genes and PHD finger domain‐containing protein genes identified among the most highly connected genes (5%) (I).
Appendix S5 Shared motifs showing similarity to yeast motifs (MacIsaac_v1 database) and Saccharomyces cerevisiae motifs and motif‐associated proteins (ScAPs) (SCPD database) (E < 1 and motif length ≤ 9) (A) and Neofusicoccum parvum protein homologues of ScAPs (B).
Acknowledgements
This research was funded by the US Department of Agriculture (USDA), National Institute of Food and Agriculture, Specialty Crop Research Initiative (grant number 2012‐51181‐19954).
References
- Abou‐Mansour, E. , Débieux, J.L. , Ramírez‐Suero, M. , Bénard‐Gellon, M. , Magnin‐Robert, M. , Spagnolo, A. , Chong, J. , Farine, S. , Bertsch, C. , L'haridon, F. , Serrano, M. , Fontaine, F. , Rego, C. and Larignon, P. (2015) Phytotoxic metabolites from Neofusicoccum parvum, a pathogen of Botryosphaeria dieback of grapevine. Phytochemistry, 115, 207–215. [DOI] [PubMed] [Google Scholar]
- Amore, A. , Giacobbe, S. and Faraco, V. (2013) Regulation of cellulase and hemicellulase gene expression in fungi. Curr. Genomics, 14, 230–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfi, A. , Mugnai, L. , Luque, J. , Surico, G. , Cimmino, A. and Evidente, A. (2011) Phytotoxins produced by fungi associated with grapevine trunk diseases. Toxins, 3, 1569–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aro, N. , Pakula, T. and Penttilä, M. (2005) Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol. Rev. 29, 719–739. [DOI] [PubMed] [Google Scholar]
- Bailey, T.L. , Boden, M. , Buske, F.A. , Frith, M. , Grant, C.E. , Clementi, L. , Ren, J. , Li, W.W. and Noble, W.S. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benen, J.A.E. , Kester, H.C. , Parenicová, L. and Visser, J. (2000) Characterization of Aspergillus niger pectate lyase A. Biochemistry, 39, 15 563–15 569. [DOI] [PubMed] [Google Scholar]
- Bertsch, C. , Ramírez‐Suero, M. , Magnin‐Robert, M. , Larignon, P. , Chong, J. , Abou‐Mansour, E. , Spagnolo, A. , Clément, C. and Fontaine, F. (2013) Grapevine trunk diseases: complex and still poorly understood. Plant Pathol. 62, 243–265. [Google Scholar]
- Bienz, M. (2006) The PHD finger, a nuclear protein‐interaction domain. Trends Biochem. Sci. 31, 35–40. [DOI] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Rolshausen, P. and Cantu, D. (2013) Draft genome sequence of Neofusicoccum parvum isolate UCR‐NP2, a fungal vascular pathogen associated with grapevine cankers. Genome Announc. 1, e00339–e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco‐Ulate, B. , Amrine, K.C. , Collins, T.S. , Rivero, R.M. , Vicente, A.R. , Morales‐Cruz, A. , Doyle, C.L. , Ye, Z. , Allen, G. , Heymann, H. , Ebeler, S.E. and Cantu, D. (2015) Developmental and metabolic plasticity of white‐skinned grape berries in response to Botrytis cinerea during noble rot. Plant Physiol. 169, 2422–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw, R.E. and Zhang, S. (2006) Biosynthesis of dothistromin. Mycopathologia, 162, 201–213. [DOI] [PubMed] [Google Scholar]
- Brakhage, A.A. (2013) Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 11, 21–32. [DOI] [PubMed] [Google Scholar]
- van den Brink, J. and de Vries, R.P. (2011) Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 91, 1477–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno, G. and Sparapano, L. (2006) Effects of three esca‐associated fungi on Vitis vinifera L.: III. Enzymes produced by the pathogens and their role in fungus‐to‐plant or in fungus‐to‐fungus interactions. Physiol. Mol. Plant Pathol. 69, 182–194. [Google Scholar]
- Bruns, S. , Seidler, M. , Albrecht, D. , Salvenmoser, S. , Remme, N. , Hertweck, C. , Brakhage, A.A. , Kniemeyer, O. and Müller, F.M. (2010) Functional genomic profiling of Aspergillus fumigatus biofilm reveals enhanced production of the mycotoxin gliotoxin. Proteomics, 10, 3097–3107. [DOI] [PubMed] [Google Scholar]
- Buffalo, V. (2011) Scythe: a Bayesian adapter trimmer. Available at https://github.com/vsbuffalo/scythe [accessed 08 August 2013].
- Butchko, R.A.E. , Plattner, R.D. and Proctor, R.H. (2003) FUM13 encodes a short chain dehydrogenase/reductase required for C‐3 carbonyl reduction during fumonisin biosynthesis in Gibberella moniliformis . J. Agric. Food Chem. 51, 3000–3006. [DOI] [PubMed] [Google Scholar]
- Cantarel, B.L. , Coutinho, P.M. , Rancurel, C. , Bernard, T. , Lombard, V. and Henrissat, B. (2009) The Carbohydrate‐Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu, D. , Vicente, A.R. , Labavitch, J.M. , Bennett, A.B. and Powell, A.L. (2008) Strangers in the matrix: plant cell walls and pathogen susceptibility. Trends Plant Sci. 13, 610–617. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Lee, M.K. , Jefcoate, C. , Kim, S.C. , Chen, F. and Yu, J.H. (2014) Fungal cytochrome P450 monooxygenases: their distribution, structure, functions, family expansion, and evolutionary origin. Genome Biol. Evol. 6, 1620–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.S. , Alexander, D.H. , Marks, P. , Klammer, A.A. , Drake, J. , Heiner, C. , Clum, A. , Copeland, A. , Huddleston, J. , Eichler, E.E. , Turner, S.W. and Korlach, J. (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Coleman, J.J. and Mylonakis, E. (2009) Efflux in fungi: La Piece de Resistance. PLoS Pathog. 5, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Črešnar, B. and Petrič, Š. (2011) Cytochrome P450 enzymes in the fungal kingdom. Biochim . Biophys. Acta, 1814, 29–35. [DOI] [PubMed] [Google Scholar]
- Czemmel, S. , Galarneau, E.R. , Travadon, R. , McElrone, A.J. , Cramer, G.R. and Baumgartner, K. (2015) Genes expressed in grapevine leaves reveal latent wood infection by the fungal pathogen Neofusicoccum parvum . PLoS One, 10, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny, T.P. and VanEtten, H.D. (1983) Characterization of an inducible, nondegradative tolerance of Nectria haematococca MPVI to phytoalexins. J. Gen. Microbiol. 12, 2903–2913. [Google Scholar]
- Denny, T.P. , Matthews, P.S. and VanEtten, H.D. (1987) A possible mechanism of nondegradative tolerance of pisatin in Nectria haematococca MPVI. Physiol. Mol. Plant Pathol. 30, 93–107. [Google Scholar]
- Durairaj, P. , Malla, S. , Nadarajan, S.P. , Lee, P.G. , Jung, E. , Park, H.H. , Kim, B.G. and Yun, H. (2015) Fungal cytochrome P450 monooxygenases of Fusarium oxysporum for the synthesis of ω‐hydroxy fatty acids in engineered Saccharomyces cerevisiae . Microb. Cell Fact. 14, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evidente, A. , Punzo, B. , Andolfi, A. , Cimmino, A. , Melck, D. and Luque, J. (2010) Lipophilic phytotoxins produced by Neofusicoccum parvum, a grapevine canker agent. Phytopathol. Medit. 49, 74–79. [Google Scholar]
- Garber, M. , Yosef, N. , Goren, A. , Raychowdhury, R. , Thielke, A. , Guttman, M. , Robinson, J. , Minie, B. , Chevrier, N. , Itzhaki, Z. , Blecher‐Gonen, R. , Bornstein, C. , Amann‐Zalcenstein, D. , Weiner, A. , Friedrich, D. , Meldrim, J. , Ram, O. , Cheng, C. , Gnirke, A. , Fisher, S. , Friedman, N. , Wong, B. , Bernstein, B.E. , Nusbaum, C. , Hacohen, N. , Regev, A. and Amit, I. (2012) A high‐throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell, 47, 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielkens, M.M.C. , Dekkers, E. , Visser, J. and de Graaff, L.H. (1999) Two cellobiohydrolase‐encoding genes from Aspergillus niger require D‐xylose and the xylanolytic transcriptional activator XlnR for their expression. Appl. Environ. Microbiol. 65, 4340–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez, P. , Báidez, A.G. , Ortuño, A. and Del Río, J.A. (2016) Grapevine xylem response to fungi involved in trunk diseases. Ann. Appl. Biol. 169, 116–124. [Google Scholar]
- Harris, L.J. , Balcerzak, M. , Johnston, A. , Schneiderman, D. and Ouellet, T. (2016) Host‐preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 120, 111–123. [DOI] [PubMed] [Google Scholar]
- van Hellemond, E.W. , Leferink, N.G. , Heuts, D.P. , Fraaije, M.W. and van Berkel, W.J. (2006) Occurrence and biocatalytic potential of carbohydrate oxidases. Adv. Appl. Microbiol. 60, 17–54. [DOI] [PubMed] [Google Scholar]
- Hernández‐Ortega, A. , Ferreira, P. and Martínez, A.T. (2012) Fungal aryl‐alcohol oxidase: a peroxide‐producing flavoenzyme involved in lignin degradation. Appl. Microbiol. Biotechnol. 93, 1395–1410. [DOI] [PubMed] [Google Scholar]
- Hoff, K.J. , Lange, S. , Lomsadze, A. , Borodovsky, M. and Stanke, M. (2015) BRAKER1: unsupervised RNA‐Seq‐based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics, 32, 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrmová, M. , Petráková, E. and Biely, P. (1991) Induction of cellulose‐ and xylan‐degrading enzyme systems in Aspergillus terreus by homo‐ and heterodisaccharides composed of glucose and xylose. J. Gen. Microbiol. 137, 541–547. [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Yan, C. , Hsu, C.H. , Chen, Q.R. , Niu, K. , Komatsoulis, G.A. and Meerzaman, D. (2014) OmicCircos: a simple‐to‐use R Package for the circular visualization of multidimensional omics data. Cancer Inform. 13, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova, C. , Bååth, J.A. , Seiboth, B. and Kubicek, C.P. (2013) Systems analysis of lactose metabolism in Trichoderma reesei identifies a lactose permease that is essential for cellulase induction. PLoS One, 8, e62631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, L. , Riaz, S. , Morales‐Cruz, A. , Amrine, K.C. , McGuire, B. , Gubler, W.D. , Walker, M.A. and Cantu, D. (2014) Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator . BMC Genomics, 15, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörnvall, H. , Höög, J.O. and Persson, B. (1999) SDR and MDR: completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett. 445, 261–264. [DOI] [PubMed] [Google Scholar]
- Joshi, N. and Fass, J. (2011) Sickle: a sliding‐window, adaptive, quality‐based trimming tool for FastQ files (Version 1.33) [Software]. Available at https://github.com/najoshi/sickle [accessed 08 August 2013].
- Kawai, S. , Umezawa, T. , Shimada, M. , Higushi, T. , Koide, K. , Nishida, T. , Morohoshi, N. and Haraguchi, T. (1987) Ca‐Cb cleavage of phenolic b‐1 lignin substructure model compound by laccase of Coriolus versicolor . Mokuzai Gakkaishi, 33, 792–797. [Google Scholar]
- King, B.C. , Waxman, K.D. , Nenni, N.V. , Walker, L.P. , Bergstrom, G.C. and Gibson, D.M. (2011) Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol. Biofuels, 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer, S.M. and Wood, P.M. (1992) Production of Fenton's reagent by cellobiose oxidase from cellulolytic cultures of Phanerochaete chrysosporium . Eur. J. Biochem. 208, 807–814. [DOI] [PubMed] [Google Scholar]
- Kubicek, C.P. , Mikus, M. , Schuster, A. , Schmoll, M. and Seiboth, B. (2009) Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina . Biotechnol. Biofuels, 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasawa, T. , Yachi, M. , Suto, M. , Kamagata, Y. , Takao, S. and Tomita, F. (1992) Induction of cellulase by gentiobiose and its sulfur‐containing analog in Penicillium purpurogenum . Appl. Environ. Microbiol. 58, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S. , Phillippy, A. , Delcher, A.L. , Smoot, M. , Shumway, M. , Antonescu, C. and Salzberg, S.L. (2004) Versatile and open software for comparing large genomes. Genome Biol. 5, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, M.J. , Kim, K.S. and Lee, Y.H. (2010) A short‐chain dehydrogenase/reductase gene is required for infection‐related development and pathogenicity in Magnaporthe oryzae . Plant Pathol. J. 26, 8–16. [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler, K. , Hammond‐Kosack, K. , Brazma, A. and Coulson, R.M. (2013) Genomic clustering and co‐regulation of transcriptional networks in the pathogenic fungus Fusarium graminearum . BMC Syst. Biol. 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur, A. , Drula, E. , Lombard, V. , Coutinho, P.M. and Henrissat, B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels, 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze, A. , Burns, P.D. and Borodovsky, M. (2014) Integration of mapped RNA‐Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, S. , Shah, S. , Bohnert, H.J. , Snyder, M. and Dinesh‐Kumar, S.P. (2013) Incorporating motif analysis into gene co‐expression networks reveals novel modular expression pattern and new signaling pathways. PLoS Genet. 9, e1003840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald, J. , Doering, M. , Canam, T. , Gong, Y. , Guttman, D.S. , Campbell, M.M. and Master, E.R. (2011) Transcriptomic responses of the softwood‐degrading white‐rot fungus Phanerochaete carnosa during growth on coniferous and deciduous wood. Appl. Environ. Microbiol. 77, 3211–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac, K.D. , Wang, T. , Gordon, D.B. , Gifford, D.K. , Stormo, G.D. and Fraenkel, E. (2006) An improved map of conserved regulatory sites for Saccharomyces cerevisiae . BMC Bioinformatics, 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandels, M. , Parrish, F.W. and Reese, E.T. (1962) Sophorose as an inducer of cellulase in Trichoderma viride . J. Bacteriol. 83, 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martos, S. , Andolfi, A. , Luque, J. , Mugnai, L. , Surico, G. and Evidente, A. (2008) Production of phytotoxic metabolites by five species of Botryosphaeriaceae causing decline on grapevines, with special interest in the species Neofusicoccum luteum and N. parvum . Eur. J. Plant Pathol. 121, 451–461. [Google Scholar]
- Massa, A.N. , Childs, K.L. , Lin, H. , Bryan, G.J. , Giuliano, G. and Buell, C.R. (2011) The transcriptome of the reference potato genome Solanum tuberosum Group Phureja Clone DM1‐3 516R44. PLoS One, 6, e26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. and DePristo, M.A. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moktali, V. , Park, J. , Fedorova‐Abrams, N.D. , Park, B. , Choi, J. , Lee, Y.H. and Kang, S. (2012) Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genomics, 13, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales‐Cruz, A. , Amrine, K.C. , Blanco‐Ulate, B. , Lawrence, D.P. , Travadon, R. , Rolshausen, P.E. , Baumgartner, K. and Cantu, D. (2015) Distinctive expansion of gene families associated with plant cell wall degradation, secondary metabolism, and nutrient uptake in the genomes of grapevine trunk pathogens. BMC Genomics, 16, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau, C. , Ward, B.L. , Gilchrist, D.G. and Hammock, B.D. (1999) Multiple epoxide hydrolases in Alternaria alternata f. sp. lycopersici and their relationship to medium composition and host‐specific toxin production. Appl. Environ. Microbiol. 65, 2388–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattestad, M. and Schatz, M.C. (2016) Assemblytics: a web analytics tool for the detection of assembly‐based variants. Bioinformatics, 32, 3021–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. , Ning, Z. , Keane, T. and Korf, I. (2009) Assessing the gene space in draft genomes. Nucleic Acids Res. 37, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzoldt, C.H. , Moller, W.J. and Sall, M.A. (1981) Eutypa dieback of grapevine: seasonal differences in infection and duration of susceptibility of pruning wounds. Phytopathology, 71, 540–543. [Google Scholar]
- Podlevsky, J.D. , Bley, C.J. , Omana, R.V. , Qi, X. and Chen, J.J.L. (2008) The telomerase database. Nucleic Acids Res. 36, D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe, F. , Ajami‐Rashidi, Z. , Doehlemann, G. , Kahmann, R. and Djamei, A. (2013) Degradation of the plant defence hormone salicylic acid by the biotrophic fungus Ustilago maydis . Mol. Microbiol. 89, 179–188. [DOI] [PubMed] [Google Scholar]
- Reyes‐Dominguez, Y. , Boedi, S. , Sulyok, M. , Wiesenberger, G. , Stoppacher, N. , Krska, R. and Strauss, J. (2012) Heterochromatin influences the secondary metabolite profile in the plant pathogen Fusarium graminearum . Fungal Genet. Biol. 49, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolshausen, P.E. , Greve, L.C. , Labavitch, J.M. , Mahoney, N.E. , Molyneux, R.J. and Gubler, W.D. (2008) Pathogenesis of Eutypa lata in grapevine: identification of virulence factors and biochemical characterization of cordon dieback. Phytopathology, 98, 222–229. [DOI] [PubMed] [Google Scholar]
- Rolshausen, P.E. , Urbez‐Torres, J.R. , Rooney‐Latham, S. , Eskalen, A. , Smith, R.J. and Gubler, W.D. (2010) Evaluation of pruning wound susceptibility and protection against fungi associated with grapevine trunk diseases. Am. J. Enol. Vitic. 61, 113–119. [Google Scholar]
- Ruijter, G.J.G. and Visser, J. (1997) Carbon repression in Aspergilli. FEMS Microbiol. Lett. 151, 103–114. [DOI] [PubMed] [Google Scholar]
- Saeed, A.I. , Sharov, V. , White, J. , Li, J. , Liang, W. , Bhagabati, N. , Braisted, J. , Klapa, M. , Currier, T. , Thiagarajan, M. , Sturn, A. , Snuffin, M. , Rezantsev, A. , Popov, D. , Ryltsov, A. , Kostukovich, E. , Borisovsky, I. , Liu, Z. , Vinsavich, A. , Trush, V. and Quackenbush, J. (2003) TM4: a free, open‐source system for microarray data management and analysis. Biotechniques, 34, 374–378. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , Amin, N. , Schwikowski, B. and Ideker, T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson, J.T. (2014) Exploring genome characteristics and sequence quality without a reference. Bioinformatics, 30, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slippers, B. and Wingfield, M.J. (2007) Botryosphaeriaceae as endophytes and latent pathogens of woody plants: diversity, ecology and impact. Fungal Biol. Rev. 21, 90–106. [Google Scholar]
- Sosnowski, M.R. , Wicks, T.J. and Scott, E.S. (2011) Control of Eutypa dieback in grapevines using remedial surgery. Phytopathol. Medit. 50, S277–S284. [Google Scholar]
- Stanke, M. and Waack, S. (2003) Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics, 19, 215–225. [DOI] [PubMed] [Google Scholar]
- Stoffel, K. , van Leeuwen, H. , Kozik, A. , Caldwell, D. , Ashrafi, H. , Cui, X. , Tan, X. , Hill, T. , Reyes‐Chin‐Wo, S. , Truco, M.J. , Michelmore, R.W. and Van Deynze, A. (2012) Development and application of a 6.5 million feature Affymetrix Genechip® for massively parallel discovery of single position polymorphisms in lettuce (Lactuca spp.). BMC Genomics, 13, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.E. , Basarab, G.S. , Andersson, A. , Lindqvist, Y. and Jordan, D.B. (1997) Trihydroxynaphthalene reductase from Magnaporthe grisea: realization of an active center inhibitor and elucidation of the kinetic mechanism. Biochemistry, 36, 1852–1860. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travadon, R. , Rolshausen, P.E. , Gubler, W.D. , Cadle‐Davidson, L. and Baumgartner, K. (2013) Susceptibility of cultivated and wild Vitis spp. to wood infection by fungal trunk pathogens. Plant Dis. 97, 1529–1536. [DOI] [PubMed] [Google Scholar]
- Uranga, C.C. , Beld, J. , Mrse, A. , Córdova‐Guerrero, I. , Burkart, M.D. and Hernández‐Martínez, R. (2016) Fatty acid esters produced by Lasiodiplodia theobromae function as growth regulators in tobacco seedlings. Biochem. Biophys. Res. Commun. 472, 339–345. [DOI] [PubMed] [Google Scholar]
- Úrbez‐Torres, J.R. (2011) The status of Botryosphaeriaceae species infecting grapevines. Phytopathol. Medit. 50, 5–45. [Google Scholar]
- Úrbez‐Torres, J.R. and Gubler, W.D. (2009) Pathogenicity of Botryosphaeriaceae species isolated from grapevine cankers in California. Plant Dis. 93, 584–592. [DOI] [PubMed] [Google Scholar]
- Úrbez‐Torres, J.R. and Gubler, W.D. (2011) Susceptibility of grapevine pruning wounds to infection by Lasiodiplodia theobromae and Neofusicoccum parvum . Plant Pathol. 60, 261–270. [Google Scholar]
- Úrbez‐Torres, J.R. , Leavitt, G.M. , Voegel, T.M. and Gubler, W.D. (2006) Identification and distribution of Botryosphaeria spp. associated with grapevine cankers in California. Plant Dis. 90, 1490–1503. [DOI] [PubMed] [Google Scholar]
- Vödisch, M. , Scherlach, K. , Winkler, R. , Hertweck, C. , Braun, H.P. , Roth, M. , Haas, H. , Werner, E.R. , Brakhage, A.A. and Kniemeyer, O. (2011) Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin A biosynthesis gene cluster in response to hypoxia. J. Proteome Res. 10, 2508–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries, R.P. and Visser, J. (2001) Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol. Mol. Biol. Rev. 65, 497–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Xu, Y. , Maine, E.A. , Wijeratne, E.M. , Espinosa‐Artiles, P. , Gunatilaka, A.A. and Molnár, I. (2008) Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii . Chem. Biol. 15, 1328–1338. [DOI] [PubMed] [Google Scholar]
- Ward, T.J. , Bielawski, J.P. , Kistler, H.C. , Sullivan, E. and O'donnell, K. (2002) Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc . Natl. Acad. Sci. USA, 99, 9278–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrzyn, G.T. , Bloch, S.E. and Schmidt‐Dannert, C. (2012) Discovery and characterization of terpenoid biosynthetic pathways of fungi. Methods Enzymol. 515, 83–105. [DOI] [PubMed] [Google Scholar]
- Weber, E.A. , Trouillas, F.P. and Gubler, W.D. (2007) Double pruning of grapevines: a cultural practice to reduce infections by Eutypa lata . Am. J. Enol. Vitic. 58, 61–66. [Google Scholar]
- Weber, T. , Blin, K. , Duddela, S. , Krug, D. , Kim, H.U. , Bruccoleri, R. , Lee, S.Y. , Fischbach, M.A. , Müller, R. , Wohlleben, W. , Breitling, R. , Takano, E. and Medema, M.H. (2015) antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers, S.T. and Keasling, J.D. (2007) Biosynthesis and engineering of isoprenoid small molecules. Appl. Microbiol. Biotechnol. 73, 980–990. [DOI] [PubMed] [Google Scholar]
- Yin, W. and Keller, N.P. (2011) Transcriptional regulatory elements in fungal secondary metabolism. J. Microbiol. 49, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J. and Zhang, M.Q. (1999) SCPD: a promoter database of the yeast Saccharomyces cerevisiae . Bioinformatics, 15, 607–611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Appendix S1 Supplementary tables and figures.
Appendix S2 Genome assemblies and protein‐coding gene coordinates.
Appendix S3 Functional annotations.
Appendix S4 Normalized RNA‐sequencing counts in the in vitro (A) and in planta (B) experiments, list of genes up‐regulated in the presence of wood (C) and exclusively expressed in planta (D), and groups of co‐expressed genes during Neofusicoccum parvum colonization obtained by both K‐means and hierarchical clustering analysis (E). Gene co‐expression modules obtained from Weighted Gene Co‐expression Network Analysis (WGCNA) and the corresponding degree of connectivity in the unweighted network (F), genomic clusters identified among the gene co‐expression modules (G), network properties of the gene co‐expression modules (H) and transcription factor‐coding genes and PHD finger domain‐containing protein genes identified among the most highly connected genes (5%) (I).
Appendix S5 Shared motifs showing similarity to yeast motifs (MacIsaac_v1 database) and Saccharomyces cerevisiae motifs and motif‐associated proteins (ScAPs) (SCPD database) (E < 1 and motif length ≤ 9) (A) and Neofusicoccum parvum protein homologues of ScAPs (B).