

Summary
The fungus Sclerotinia sclerotiorum is a necrotrophic plant pathogen causing significant damage on a broad range of crops. This fungus produces sclerotia that serve as the long‐term survival structures in the life cycle and the primary inoculum in the disease cycle. Melanin plays an important role in protecting mycelia and sclerotia from ultraviolet radiation and other adverse environmental conditions. In this study, two genes, SCD1 encoding a scytalone dehydratase and THR1 encoding a trihydroxynaphthalene reductase, were disrupted by target gene replacement, and their roles in mycelial growth, sclerotial development and fungal pathogenicity were investigated. Phylogenetic analyses indicated that the deduced amino acid sequences of SCD1 and THR1 were similar to the orthologues of Botrytis cinerea. Expression of SCD1 was at higher levels in sclerotia relative to mycelia. THR1 was expressed at similar levels in mycelia and sclerotia at early stages, but was up‐regulated in sclerotia at the maturation stage. Disruption of SCD1 or THR1 did not change the pathogenicity of the fungus, but resulted in slower radial growth, less biomass, wider angled hyphal branches, impaired sclerotial development and decreased resistance to ultraviolet light.
Keywords: hyphal branch, pathogenicity, sclerotium, scytalone dehydratase, trihydroxynaphthalene reductase, UV light sensitivity
Introduction
Sclerotinia sclerotiorum (Lib.) de Bary is a necrotrophic fungal pathogen attacking a broad range of plants worldwide and causing significant crop losses (Boland and Hall, 1994). This fungus produces sclerotia, which are long‐term survival structures against adverse environments in the soil and play a crucial role in the life cycle of the fungus and in disease progression (Bolton et al., 2006). Germination of sclerotia is affected by environmental conditions, resulting in carpogenic or myceliogenic germination (Bardin and Huang, 2001). Both types of germination can initiate disease, although host infection is mostly caused by the former via saprophytic hyphae developed from ascospores (Bolton et al., 2006). Carpogenic germination normally forms apothecia and releases ascospores that infect the senescent tissue and aboveground plant parts (Huang, 1985; Turkington and Morrall, 1993).
Sclerotia are multihyphal aggregates with pigmentation and consist of three distinct layers: an outer black rind containing melanin, a thin‐walled cortex and an inner large central medulla (Kosasih and Willetts, 1975). The formation of sclerotia has been described as three distinct stages: initiation, development and maturation (Townsend and Willetts, 1954; Willetts and Bullock, 1992). In another description, the development stage was further partitioned into four stages: condensation, enlargement, consolidation and pigmentation (Li and Rollins, 2009). All of these stages involve specific morphological, physiological, biochemical and metabolic changes, and are affected by endogenous and exogenous challenges, such as temperature, toxic metals, ultraviolet (UV) and visible irradiation, microbial antagonists and competitors (Bell and Wheeler, 1986; Chet and Henis, 1975). Pigmentation achieved by melanin accumulation is an important feature of sclerotial formation because it can increase the resistance of the fungus to adverse environmental conditions (Butler and Day, 1998).
Fungal melanin is a dark poly‐aromatic complex polymer composed of various types of phenolic or indolic monomers, as well as carbohydrates and proteins (Butler and Day, 1998; Butler et al., 2005). Melanin is important to the fungus because it can improve its survival and competitive abilities against certain adverse environments or microbial degradation (Bell and Wheeler, 1986; Butler and Day, 1998). Furthermore, melanin contributes to the pathogenicity of various fungal species by influencing the development of penetration structures (Bolton et al., 2006; Butler and Day, 1998). For example, Magnaporthe grisea and Colletotrichum lagenarium mutants unable to form melanized appressoria are unable to penetrate host plants (Howard and Valent, 1996; Kubo et al., 1987). In other fungal species, such as Alternaria alternata and Venturia inaequalis, melanin production may not be essential for fungal pathogenicity (Fitzgerald et al., 2004; Thomma, 2003). Melanin is normally observed in the cell walls of sclerotia or spores in some fungi, but is also found in the cell wall of hyphae of many fungal species (Butler et al., 2005). The melanogenesis of dihydroxynaphthalene (DHN) is one of the most prevalent and extensively studied fungal melanin pathways. It is synthesized through the pentaketide pathway, which has been identified previously in S. sclerotiorum (Butler et al., 2005, 2009).
Some genes and proteins involved in sclerotial development in S. sclerotiorum have been identified and functionally characterized (Li and Rollins, 2009, 2010; Liang et al., 2010; Russo et al., 1982). Among them, two melanin biosynthesis‐related proteins have been found to be up‐regulated between the initiation and developmental stages of sclerotium formation, and the abundance of these two proteins is maintained at high levels after the initiation stage (Liang et al., 2010). As the effect of sclerotial melanin on the biology and pathogenicity of S. sclerotiorum is barely understood, in this study, we investigated the biological and pathogenicity function of the genes encoding these two proteins through a target gene replacement approach.
Results
Sequence and phylogenetic analysis
Two proteins with the GenBank accession numbers SS1G_13314 and SS1G_13315 have been identified (Liang et al., 2010). The deduced amino acid sequences of these two proteins are annotated in GenBank with the hypothetical functions of scytalone dehydratase and trihydroxynaphthalene reductase (1,3,8‐naphthalenetriol reductase), respectively. Both proteins have been reported to be functional in the process of melanogenesis in some fungal species (Langfelder et al., 2003). We named these two proteins Scd1 and Thr1, and the corresponding genes SCD1 and THR1, representing scytalone dehydratase and trihydroxynaphthalene reductase, respectively.
SCD1 consists of a coding sequence of 612 nucleotides (nt) in length and a 67‐nt intron, whereas THR1 consists of a coding sequence of 840 nt and a 308‐nt intron. These features were further supported by sequencing of the cDNA fragments of the two genes (data not shown). Both Scd1 and Thr1 lack a putative signal peptide. The three‐dimensional (3D) structural models of the hypothetical proteins were constructed on the basis of domain homology of the deduced amino acid sequence, which primarily provided the detailed structural analysis models representing the secondary structure, oligo‐state without any ligands (Fig. 1A). Sequences of orthologues of Scd1 and Thr1 in other fungi were retrieved from the National Center for Biotechnology Information (NCBI) database. Phylogenetic analyses revealed that both Scd1 and Thr1 sequences of S. sclerotiorum and Botrytis cinerea formed a monophyletic group (Fig. 1B).
Figure 1.
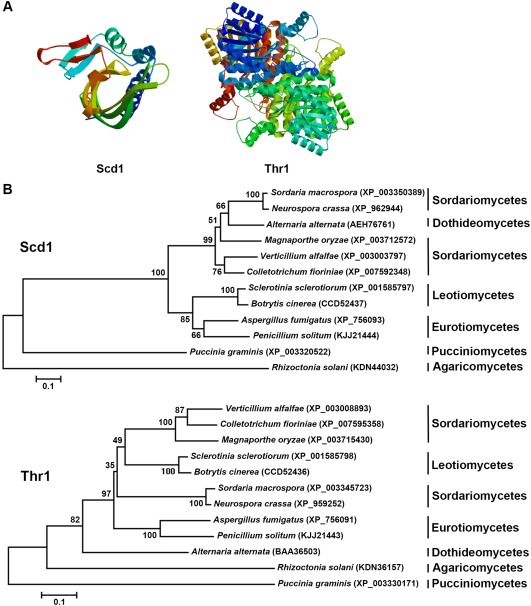
Sequence and phylogenetic analysis of scytalone dehydratase (Scd1) and trihydroxynaphthalene reductase (Thr1). (A) The three‐dimensional (3D) model of Scd1 and Thr1 predicted by SWISS‐MODEL. (B) Phylogenetic trees of Scd1 and Thr1 orthologues constructed by sequence alignment.
Expression of SCD1 and THR1
Expression levels of SCD1 and THR1 were investigated by semi‐quantitative reverse transcription‐polymerase chain reaction (RT‐PCR). Mycelial and sclerotial tissues were collected from four different stages: vegetative [3 days post‐inoculation (dpi)], initial (4 dpi), developmental (5 dpi) and matured (8 dpi). The expression level of SCD1 was significantly (P < 0.05) higher in sclerotia than in mycelia at all developmental stages, whereas the expression in mycelia did not change between stages (Fig. 2A). Moreover, SCD1 expression in sclerotia was significantly up‐regulated relative to that in mycelia in the matured stage (Fig. 2A). The expression levels of THR1 in both sclerotia and mycelia were significantly enhanced at the matured stage relative to the developing sclerotia and mycelia (Fig. 2B).
Figure 2.
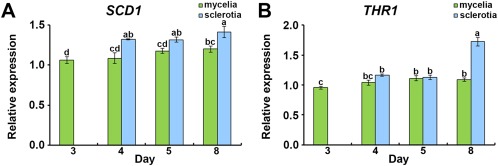
Expression analysis of SCD1 and THR1. Relative expression of SCD1 (A) and THR1 (B) in different stages of mycelia and sclerotia. Mycelia and sclerotia were sampled from three wild‐type strains as biological replicates in different stages. The bars in the plot represent the standard error. Means in the plot topped by the same letter do not differ on the basis of Duncan's multiple range test at P < 0.05.
Knockout of SCD1 and THR1
Protoplast transformation resulted in six and eight positive transformants for SCD1 and THR1, respectively. The transformants were screened on selection medium with hygromycin B and verified by PCR and RT‐PCR. From the SCD1 transformants, a 3.7‐kb single band was amplified by PCR, in contrast with a 1.3‐kb amplicon from the wild‐type. RT‐PCR indicated that the 0.6‐kb transcript of SCD1 is present only in the wild‐type (Fig. 3A). From the THR1 transformants, a 3.8‐kb single band was amplified by PCR, in contrast with a 1.1‐kb amplicon from the wild‐type. The 0.8‐kb THR1 transcript was present in the wild‐type, but absent in the transformants (Fig. 3B). The presence of the selection marker gene hygromycin B phosphotransferase (hph) in the SCD1 and THR1 transformants was confirmed by the amplification of a 3.3‐kb product (Fig. 3C). The reference control, ACT1, was amplified from both wild‐type and transformants (Fig. 3D). In addition, PCR products of the wild‐type and transformants were sequenced and the gene knockout events were confirmed (data not shown). Three SCD1 transformants and five THR1 transformants were verified as the gene knockout strains, indicating a gene knockout efficiency of 50% for SCD1 and 63% for THR1. From the confirmed gene knockout strains, three transformants were selected for each gene, named scd1 or thr1, and subjected to further analyses.
Figure 3.
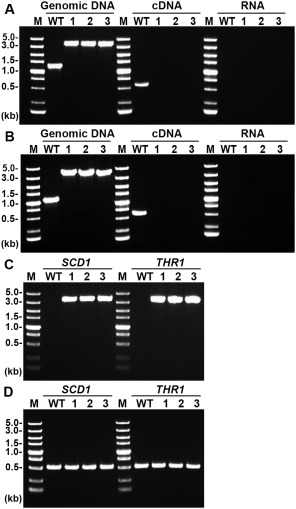
Polymerase chain reaction (PCR) confirmation of gene knockout events in three SCD1 and three THR1 transformants. (A) A 1.3‐kb band representing SCD1 and a 3.7‐kb band representing the transforming DNA were amplified from genomic DNA of the wild‐type (WT) and the three SCD1 transformants, respectively; a 0.6‐kb band representing the SCD1 transcript was amplified from cDNA of the WT only; no band was amplified from RNA samples. (B) A 1.1‐kb band representing THR1 and a 3.8‐kb band representing the transforming DNA were amplified from genomic DNA of the WT and the three THR1 transformants, respectively; a 0.8‐kb band representing the THR1 transcript was amplified from cDNA of the WT only. No band was amplified from RNA samples. (C) A 3.3‐kb band representing the hph gene cassette was amplified from all transformants, but not from the WT. (D) A 0.6‐kb band representing ACT1 was amplified from the WT and all transformants.
Morphological characterization of the gene knockout strains
The radial growth of the wild‐type and the knockout strains on potato dextrose agar (PDA) plates was investigated with a time course study. On PDA plates, the scd1 and thr1 strains exhibited approximately 30% reduction in growth compared with the wild‐type at 24 and 48 h post‐inoculation (hpi). Compared with 24 and 48 hpi, the radial growth rate of knockout strains was increased slightly at 72 and 96 hpi. However, the mycelial colonies of scd1 and thr1 were still significantly smaller than that of the wild‐type at each time point (Fig. 4A). In liquid media, the knockout strains produced significantly less biomass (P < 0.05) than the wild‐type after 3 days of culture (Fig. 4B). The pH values of the media used for the culture of the knockout strains were pH 5.45 ± 0.02 for scd1 and pH 5.61 ± 0.03 for thr1, which were higher than that used for the wild‐type (pH 3.62 ± 0.05), but lower than that used for the blank (pH 7.03 ± 0.02). Calculation of the production of H+ protons per milligram of fungal biomass indicated that the gene knockout strains produced less H+ than the wild‐type (Fig. 4C).
Figure 4.
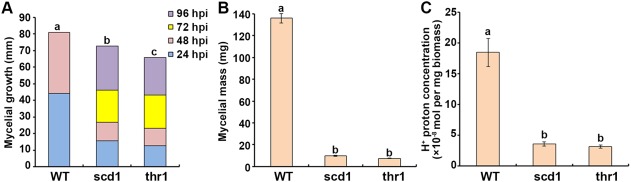
Biological characterization of the wild‐type (WT) and gene knockout strains (scd1 and thr1). (A) Mycelial growth for a period of 96 h with 24‐h time intervals. hpi, hours post‐inoculation. Mycelial mass (B) and H+ proton production (C) after incubation for 3 days in liquid medium. Three WT strains and three knockout strains for each gene were assayed. The bar in the plot represents the standard error. Means in the plot topped by the same letter do not differ on the basis of Duncan's multiple range test at P < 0.05.
Morphological changes of the apical hyphae were observed in which the angles of the hyphal branches in the knockout strains were wider than those in the wild‐type (Fig. 5A,B). This was observed in all confirmed gene knockout strains. Compared with the wild‐type, scd1 and thr1 formed sparse and less pigmented colonies and showed delayed melanization (Fig. 6A). Sclerotial formation of all gene knockout strains was delayed and with fewer numbers compared with the wild‐type (Fig. 6B). Generally, the wild‐type produced 20–24 matured and fully melanized sclerotia in each PDA plate, whereas the knockout strains produced only one or two smaller sized sclerotia with limited melanization and were covered by a cottony white mycelium (Fig. 6A,B).
Figure 5.
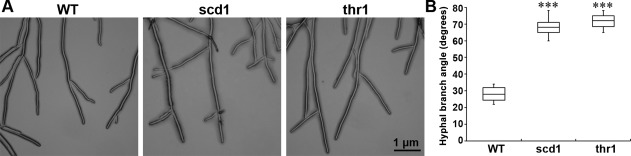
Hyphal morphology and branching of the wild‐type (WT) and gene knockout strains (scd1 and thr1). (A) Hyphal branching and extension. (B) Box‐and‐whisker plots (median and interquartile ranges) of hyphal branching angle of the WT and gene knockout strains. Three WT strains and three knockout strains for each gene were used. The bar in the plot represents the standard error. Asterisks indicate significance (***P < 0.001) with Student's t‐test.
Figure 6.
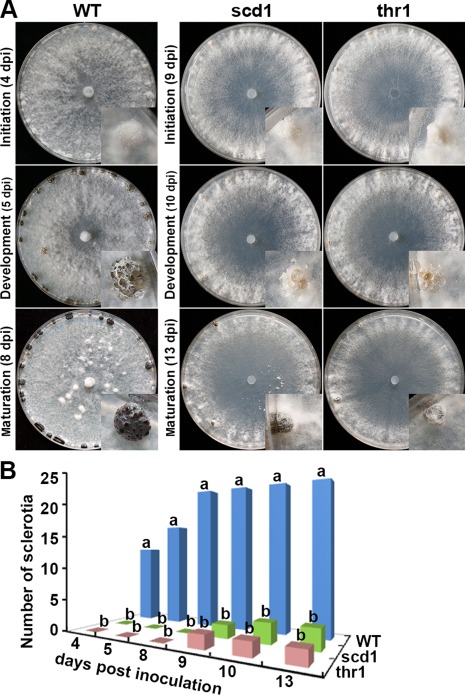
Development of sclerotia of the wild‐type (WT) and the gene knockout strains (scd1 and thr1). (A) Morphologies of colonies and sclerotia. The insets show enlarged views of a sclerotium. dpi, days post‐inoculation. (B) Numbers of sclerotia formed from 4 to 13 days after incubation. Three wild‐type (WT) strains and three knockout strains for each gene were assayed. The bar in the plot represents the standard error. For each time point, means in the plot topped by the same letter do not differ on the basis of Duncan's multiple range test at P < 0.05.
UV irradiation sensitivity of the gene knockout strains
The sensitivity to UV light of the knockout strains was assessed by measuring the radial growth of mycelial colonies after different durations of UV irradiation. After 10 and 30 min of UV irradiation, the knockout strains and wild‐type produced similar colonies. However, the growth of the knockout strains declined significantly (P < 0.05) if UV irradiation lasted for more than 30 min. In contrast, the wild‐type grew steadily even after 120 min of UV irradiation treatment (Fig. 7).
Figure 7.
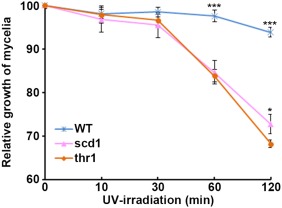
The sensitivity to ultraviolet (UV) irradiation of the wild‐type (WT) and gene knockout strains (scd1 and thr1). Asterisk indicates significance (*P < 0.05; ***P < 0.001) with Student's t‐test.
Pathogenicity assay of the gene knockout strains
Pathogenicity assays were conducted on wounded and unwounded leaves of rapeseed seedlings. All gene knockout strains caused symptoms similar to those of the wild‐type with both inoculation methods. Typical disease symptoms (e.g. necrosis and water‐soaked lesions) were observed on all inoculated leaves at 24 hpi (Fig. 8A). The necrotic area consistently developed at 48 hpi, resulting in fungal colonization (data not shown). For either the wounded or unwounded inoculation method, no difference was found on the basis of the lesion areas between the gene knockout strains and the wild‐type at both 24 and 48 hpi (Fig. 8B).
Figure 8.
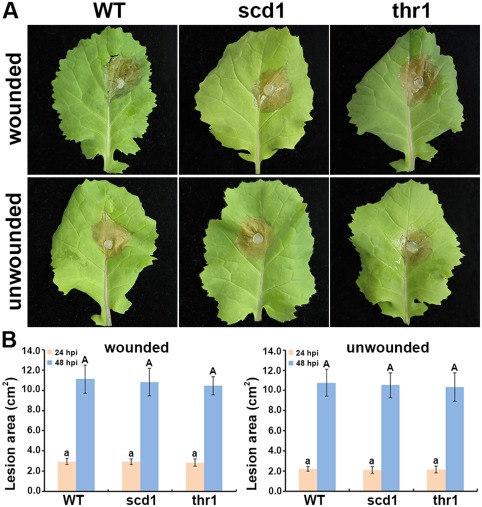
Pathogenicity assay for the wild‐type (WT) and gene knockout strains (scd1 and thr1) on intact rapeseed leaves. (A) Representative photographs taken at 24 h post‐inoculation (hpi). (B) Lesion areas measured from inoculated leaves. Three WT strains and three knockout strains for each gene were assayed. The bar in the plot represents the standard error. For each time point, means in the plot topped by the same letter do not differ on the basis of Duncan's multiple range test at P < 0.05.
Discussion
Scytalone dehydratase and hydroxynaphthalene reductase in S. sclerotiorum participate in the pentaketide pathway through which the fungal melanin DHN is produced (Butler et al., 2009). In the present study, we provided evidence to demonstrate that SCD1 and THR1 contribute to the development of sclerotia in S. sclerotiorum. Expression of SCD1 was detected at the initiation stage of sclerotia, with a similar level in the development and maturation stages, suggesting that SCD1 contributes to the initial formation of sclerotia. In contrast, expression levels of THR1 did not change between the early stages of fungal development, but increased significantly during sclerotial maturation, suggesting that THR1 may be involved in sclerotial maturation. These results are in agreement with a previous proteomic study (Liang et al., 2010), which indicated the induced up‐regulation of proteins Scd1 and Thr1 involved in melanin biosynthesis during sclerotial development.
In this study, a split‐marker gene replacement strategy was applied to generate the transforming DNA construct. The efficiency of gene replacement in fungi has been shown to be improved using a split‐marker approach (Feng et al., 2014). For split‐marker gene replacement, three crossovers occur: both flanking regions in the genome crossover with their complementary sequences in the two constructs and the overlapping regions of partial marker segments crossover to form the complete selectable marker. Thus, the efficiency of gene replacement will be improved because only transformants that contain the two successfully integrated overlapping marker fragments will grow on selective media. In this study, we obtained a gene knockout efficiency of 50% for SCD1 and 63% for THR1, similar to the efficiencies obtained from other studies (Feng et al., 2014; Yajima et al., 2009).
Disruption of SCD1 and THR1 in S. sclerotiorum did not affect the pathogenicity of the fungus. This was expected because reports on the pathogenicity function of melanin in fungal phytopathogens have been inconsistent. The production of melanin has been reported to aid the appressorium‐mediated penetration of C. lagenarium and M. grisea, whereas the penetration in A. alternata was not affected by melanin deficiency (Thomma, 2003; Tucker and Talbot, 2001). In Gaeumannomyces graminis, melanized hyphae or hyphopodia were not required for plant infection, whereas, in a Curvularia species, melanization improved hyphal development in planta (Butler et al., 2001). In another study, mutation of genes encoding polyketide synthase and tetrahydroxynaphthalene reductases, involved in melanin biosynthesis, enhanced the mycelial growth and virulence of B. cinerea (Zhang et al., 2015).
Compared with the wild‐type, the gene knockout strains produced hyphae with wider angled branches, which is associated with a reduction in hyphal growth and colony size. The NADPH oxidase‐deficient mutants of S. sclerotiorum remained fully pathogenic and exhibited radial growth, but hyphae were more branched than those of the wild‐type (Kim et al., 2011). In another study, inactivation of catalase in S. sclerotiorum not only compromised radial growth and pathogenic development, but also induced hyphal hyperbranching and changes in colony morphology (Yarden et al., 2014). In a recent study, knockdown of an atypial forkhead‐containing transcription factor in S. sclerotiorum reduced mycelial growth and pathogenicity, but did not change the hyphal branching pattern (Fan et al., 2016).
The initiation of sclerotia in the gene knockout strains was delayed compared with the wild‐type. In addition, the number, size and pigmentation of sclerotia produced by the gene knockout strains were much less than those of the wild‐type, indicating that both genes are important for sclerotial formation. Slower hyphal growth and less biomass of the gene knockout strains may cause the fungus to require more time to form sclerotia, because a certain amount of mycelial accumulation and aggression are necessary. The fewer sclerotia observed in the gene knockout strains compared with the wild‐type, even in the late developmental stage (13 dpi), indicated that these two genes may have functions in the regulation of the number of sclerotia in the fungus. In addition, sclerotial pigmentation was not fully inhibited by the disruption of either gene, probably because an alternative melanogenesis pathway is functional in this fungal species.
In both sclerotia‐forming and non‐forming fungal species, melanin helps fungi survive in adverse environments (Butler et al., 2001). The protective function of melanin is because it can resist UV irradiation (Brenner and Hearing, 2008). In the present study, disruption of either gene resulted in increased sensitivity to UV light, indicating that these two genes, as well as melanin, are important for the survival and completion capacity of the fungus. In field conditions, overwintering sclerotia are probably insolated in sunlight for at least a certain period of time. Without SCD1 or THR1, or if either of these genes are silenced or interfered with, the fungus would find it difficult to survive.
In conclusion, we have characterized SCD1 and THR1 which participate in the melanin biosynthetic pathway of S. sclerotiorum. Phylogenetic analysis indicated that proteins Scd1 and Thr1 are closely related to their orthologues in B. cinerea. The biological functions of these two genes were studied by gene knockout. Investigation of the gene knockout strains indicated that these two genes play important roles in mycelial growth, ambient pH, hyphal branching, sclerotial formation and resistance to irradiation. This study provides novel insights into a better understanding of sclerotial biology, as well as pathogenesis, of S. sclerotiorum and other sclerotia‐forming fungi.
Experimental Procedures
Fungal and plant materials
A strain of S. sclerotiorum was isolated from diseased rapeseed (Brassica napus) in a field and maintained on PDA at 25 °C as the wild‐type. Rapeseed cv. Westar was sown and grown in a glasshouse at 24 °C/18 °C (day/night), 16‐h photoperiod and 80% relative humidity. The true leaves of 28‐day‐old seedlings were subjected to a pathogenicity assay.
Sequence analysis
The nucleic acid sequences and deduced amino acid sequences of GenBank accession numbers SS1G_13314 and SS1G_13315 were retrieved from the NCBI database. Based on the nucleic acid sequences, primers (Table 1) were designed with Prime Premier 5.0 software (Premier Biosoft, Palo Alto, CA, USA) and synthesized by Invitrogen (Beijing, China). Similarity searches were conducted with the basic local alignment search tool (blast) against the NCBI database. The presence of signal peptides in the preproproteins was predicted using the SignalP 4.1 platform (http://www.cbs.dut.dk/services/SignalP). The 3D structural models of the proteins were constructed based on domain homology using the SWISS‐MODEL server (www.expasy.ch). Phylogenetic trees were constructed using mega 5.0 software (www.megasoftware.net) with the neighbour‐joining algorithm based on multiple sequence alignment of the deduced amino acid sequences.
Table 1.
Primers used in this study.
| Name | Sequence (5′–3′) |
|---|---|
| SCD1F | CACTCCCGAACCAAAT |
| SCD1R | TTCCTCCTACTTCTTACA |
| THR1F | GTCACCAGCTCCATTT |
| THR1R | AACCACTCGTTTTTCTG |
| ActinF | CACCAGAGGAGCACCCA |
| ActinR | TCAAGACCCAAGACAGA |
| SCD1qF | CCACCATCGGCAAATCCC |
| SCD1qR | CAAACCCGCGAACTT |
| THR1qF | GCTGAGCAGGTCGTTC |
| THR1qR | GCCTTAGCCTCTTGGA |
| ActinqF | TTCGTGTAGCACCAGAG |
| ActinqR | GGAAGCGTAAAGGGAGA |
| SCD1sF | AGGCTGATTCGTTCTT |
| SCD1sR | ATGGCCCACGCCAAGATCTGGGATTTGCCGATGGTGGT1 |
| SCD1asF | GAAAGCTTGGATCCCCGGGTTTTTGCGTCATCATCAGTT2 |
| SCD1asR | ATGGATTCCCTTTGTAT |
| THR1sF | GCATTTCGCAGTCGCAT |
| THR1sR | ATGGCCCACGCCAAGATCTGGGGTCCAGCGGAGTTGGCA1 |
| THR1asF | GAAAGCTTGGATCCCCGGGTTTTCTGTTCGCTGGGACG2 |
| THR1asR | TCGGCATCAAGTGGTA |
| hphsF | CCCAGATCTTGGCGTGGGCCATTGTGCATGG3 |
| hphsR | GCGGTTACCATTGTCCGTCAGGACATTGTTG |
| hphasF | GCGGTTACCAGATCGTTATGTTTATCGGCAC |
| hphasR | AAACCCGGGGATCCAAGCTTTCGAGTGGAGAT4 |
Italic sequences labelled with 1 and 2 are reverse complemented to the italic sequences labelled with 3 and 4, respectively.
Nucleic acid manipulation
Mycelia and sclerotia at different developmental stages were collected from PDA plates and ground in liquid nitrogen. The samples were subjected to genomic DNA extraction following the protocol described by Feng et al. (2010). A DNA pellet was dried in a vacuum evaporator system (Eppendorf, Hamburg, Germany) and dissolved in 30 μL of sterile distilled water. The concentration and quality of the extracted DNA were determined by a NanoDrop 1000 spectrophotometer (ThermoFisher, Waltham, MA, USA) and the samples were stored at −20 °C for further study. Total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized with 200 ng of template RNA by an iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA) as recommended by the manufacturer.
Amplification and expression analyses
To verify the presence and investigate the expression levels of SCD1 and THR1, primers were designed using a strategy such that the 1.3‐kb fragment of SCD1 would be amplified using the primer pair SCD1F/SCD1R, whereas the 1.1‐kb fragment of THR1 would be amplified using the primer pair THR1F/THR1R. The primer pair ACT1F/ACT1R was used to amplify a 0.5‐kb fragment of the actin gene ACT1 (SS1G_08733) as the endogenous reference (Table 1). PCR was conducted using genomic DNA from mycelia as a template with an initial heat denaturation step of 95 °C for 4 min, followed by 35 cycles of 95 °C for 30 s, 55 °C for 40 s and 72 °C for 1 min, with a final extension at 72 °C for 10 min. Aliquots of the PCR products were visualized on 1.0% agarose gels and the leftovers were purified with an EasyPure Quick Gel Extraction Kit (TransGen, Beijing, China). The purified products were ligated into the pMD19‐T vector (TaKaRa, Dalian, China) according to the manufacturer's instructions. Then, competent Escherichia coli DH5α cells were transformed and the plasmid DNA of PCR‐verified colonies was isolated for sequencing (Invitrogen).
The transcript accumulation of the two genes was preliminarily investigated by RT‐PCR with the primer pairs given in Table 1 against the same amounts of cDNA derived from mycelia, sclerotia, rapeseed leaves infected by wild‐type S. sclerotiorum at 24 hpi or uninoculated leaves (mock control). The amplicons of SCD1, THR1 and ACT1 from cDNA were 0.6, 0.8 and 0.5 kb in size, respectively.
To further investigate the expression of the two genes during fungal development, semi‐quantitative RT‐PCR was conducted using the primer pairs SCD1qF/SCD1qR, THR1qF/THR1qR and ACT1qF/ACT1qR (Table 1). cDNA templates were derived from mycelia at 3, 4, 5 or 8 dpi and from sclerotia at 4, 5 or 8 dpi from PDA. The PCR program consisted of an initial cycle at 94 °C for 4 min, 35 cycles of 94 °C for 30 s, 55 °C for 40 s and 72 °C for 30 s, followed by a final extension at 72 °C for 10 min. A 0.3‐μL cDNA template synthesized from the same amount of RNA was used in a total PCR of 20 μL. The amplified products were visualized on 1.0% agarose gels and the grey values of electrophoresis were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Construction of transforming DNA and fungal transformation
The construction of transforming DNA for gene knockout of SCD1 and THR1 followed the split‐marker gene replacement strategy (Feng et al., 2014). The up‐hph (2.1 kb) and down‐hph (1.7 kb) fragments were amplified from vector pGC1‐1 with the primer pairs hphsF/hphsR and hphasF/hphasR (Table 1), respectively. The 5′ franking up‐SCD1 (1.2 kb) was amplified with the primer pair SCD1sF/SCD1sR, whereas the 3′ franking down‐SCD1 (1.0 kb) was amplified with the primer pair SCD1asF/SCD1asR. The PCR products of up‐SCD1 and up‐hph were mixed and used as a template to amplify a 3.2‐kb up‐fusion fragment with the primer pair SCD1sR/hphsF. The PCR products of down‐hph and down‐SCD1 were mixed and used as a template to amplify a 2.7‐kb down‐fusion fragment with the primer pair SCD1asF/hphasR. The two amplified fusion fragments (up‐ and down‐fusion) were purified and stored at –20 °C for fungal transformation. Using the same strategy, up‐THR1 (1.1 kb) and down‐THR1 (1.4 kb) were generated and fused with up‐hph and down‐hph to amplify a 3.2‐kb up‐fusion and a 3.1‐kb down‐fusion fragment for transformation.
Fungal protoplasts were prepared (Liang et al., 2013) and followed by polyethylene glycol (PEG)‐mediated transformation to improve the efficiency of homologous recombination (Feng et al., 2014). After transformation, the hyphal tips growing on selection medium containing hygromycin B (600 µg/mL) were transferred onto PDA plates (containing 100 µg/mL of hygromycin B) and subsequently regenerated four more times on PDA containing 100 µg/mL of hygromycin B with 3‐day intervals.
Confirmation of gene knockout
To verify the gene knockout event in the transformants, genomic DNA was extracted from mycelia of the wild‐type and transformants. PCR was conducted against DNA using primer pairs SCD1F/SCD1R and THR1F/THR1R. From the gene knockout strains, the expected sizes of the amplicons generated by the two primer pairs were 3.7 and 3.8 kb, respectively, larger than the 1.2 and 1.1 kb from the wild‐type. In addition, the amplicons were purified and sequenced. Based on the genomic PCR results, the gene knockout event was further confirmed by RT‐PCR using cDNA synthesized from total RNA of the wild‐type and knockout strains. The primer pair ACT1F/ACT1R was used for Actin amplification as the reference control. In addition, extracted total RNA was also used directly as a template to avoid the false positive caused by DNA contamination (Yajima et al., 2009).
Phenotypic characterization of gene knockout strains
Colony radial growth was investigated in the wild‐type and knockout strains. A 5‐mm mycelial plug from the edge of an actively growing colony was excised and transferred to a fresh PDA plate (90 mm). The wild‐type and three gene knockout strains for each gene were assayed with three replicates. At 24‐h intervals in a 96‐h time course, the diameter of the colonies was measured. Furthermore, the changes in hyphal morphology were investigated using an Eclipse E100‐LED microscope equipped with a DS‐Vi1 digital camera and NIS‐Elements software (Nikon, Tokyo, Japan). Moreover, the biomass accumulation of the gene knockout strains and the wild‐type was investigated in liquid cultures. Three 5‐mm mycelial plugs excised from each culture of the wild‐type and gene knockout strains were inoculated into a volume of 100 mL potato dextrose broth (20 g potato extract and 20 g glucose) medium in a 250‐mL Erlenmeyer flask and incubated with 130 rpm agitation at 25 ºC for 3 days. Mycelia were collected by centrifugation at 3000 g for 10 min. The resulting mycelial pellets were washed and lyophilized using a SpeedVac concentrator (ThermoFisher). The dry weight of mycelia was measured. Meanwhile, the pH values of the supernatant (liquid medium after the fungal culture was removed) were measured. Based on the dry weight and pH data, the production of H+ protons per unit of fungal biomass was calculated using the formula [H+] = (10–pH × V – 10–pH of the blank × V)/dry weight, where [H+] is the number of moles of H+ protons produced per milligram of fungal biomass and V is the volume (L) of the medium. The wild‐type and three gene knockout strains for each gene were assayed in triplicate.
The sclerotial development of the wild‐type and knockout strains was investigated (Liang et al., 2010). Initial time points of sclerotial production and total number of sclerotia (i.e. no more sclerotia occurred) were assessed with triplicates of the wild‐type and three knockout strains of the two genes.
UV irradiation assay
A 5‐mm mycelial plug was inoculated onto the centre of a PDA plate and cultured until the colony grew to approximately one‐half of the plate. The growing colonies were irradiated at a distance of 40 cm under UV light (wavelength, 254 nm) for 10, 30, 60 and 120 min (Wlaschek et al., 1997). The treated cultures were incubated for a second 24 h and the radial diameter of the colonies was measured. The effect of UV irradiation was calculated using the formula R t (%) = (D 1 – D 0)/ΔD, where t is the different time duration of UV irradiation, R is the relative growth at each time interval, D 0 is the colony diameter before UV irradiation, D 1 is the colony diameter at a second 24‐h culture under UV irradiation treatment and ΔD is the difference in the colony diameter of the corresponding controls within a second 24‐h culture without UV irritation. The wild‐type and three gene knockout strains for each gene were assayed with three replicates.
Pathogenicity assay
Fully expanded leaves of rapeseed were inoculated by the wild‐type and gene knockout strains. Before wounded inoculation, the fully expanded leaves were wounded by a pair of forceps. Mycelial plugs of the wild‐type and knockout strains were inoculated on wounded and non‐wounded leaves. The inoculated plants were incubated at 22 °C and photographed at 24 and 48 hpi. The lesion (necrotic) area was quantified using APS Assess: Image Analysis Software for Plant Disease Quantification (APS Press, St. Paul, MN, USA), which was normalized against a square area (1 cm2) photographed in the same image of each leaf (Liang et al., 2013). The wild‐type and three knockout strains for each gene were used in the inoculation assays with three replicates.
Statistical analysis
Data were analysed for statistical significance using the general linear model (GLM) procedure of the Statistical Analysis System (SAS Institute, Cary, NC, USA). All of the data were subjected to Student's t‐test or analysis of variance (ANOVA, P < 0.05) followed by Duncan's multiple range test.
Acknowledgements
This work was supported by the Scientific Research Foundation for the Introduced Talents of Shenyang Agricultural University (20153040) and the Natural Science Foundation Project of CQ CSTC (2011BB1101). The authors declare no conflicts of interest.
References
- Bardin, S.D. and Huang, H.C. (2001) Research on biology and control of Sclerotinia diseases in Canada. Can. J. Plant Pathol. 23, 88–98. [Google Scholar]
- Bell, A.A. and Wheeler, M.H. (1986) Biosynthesis and functions of fungal melanins. Annu. Rev. Phytopathol. 24, 411–451. [Google Scholar]
- Boland, G.J. and Hall, R. (1994) Index of plant hosts of Sclerotinia sclerotiorum . Can. J. Plant Pathol. 16, 93–108. [Google Scholar]
- Bolton, M.D. , Thomma, B.P.H.J. and Nelson, B.D. (2006) Sclerotinia sclerotiorum (Lib.) de Bary: biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 7, 1–16. [DOI] [PubMed] [Google Scholar]
- Brenner, M. and Hearing, V.J. (2008) The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 84, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, M.J. and Day, A.W. (1998) Fungal melanins: a review. Can. J. Microbiol. 44, 1115–1136. [Google Scholar]
- Butler, M.J. , Day, A.W. , Henson, J.M. and Money, N.P. (2001) Pathogenic properties of fungal melanins. Mycologia, 93, 1–8. [Google Scholar]
- Butler, M.J. , Gardiner, R.B. and Day, A.W. (2005) Degradation of melanin or inhibition of its synthesis: are these a significant approach as a biological control of phytopathogenic fungi? Biol. Control. 32, 326–336. [Google Scholar]
- Butler, M.J. , Gardiner, R.B. and Day, A.W. (2009) Melanin synthesis by Sclerotinia sclerotiorum . Mycologia, 101, 296–304. [DOI] [PubMed] [Google Scholar]
- Chet, I. and Henis, Y. (1975) Sclerotial morphogenesis in fungi. Annu. Rev. Phytopathol. 13, 169–192. [Google Scholar]
- Fan, H. , Yu, G. , Liu, Y. , Zhang, X. , Liu, J. , Zhang, Y. , Rollins, J.A. , Sun, F. and Pan, H. (2016) An atypical forkhead‐containing transcription factor SsFKH1 is involved in sclerotial formation and is essential for pathogenicity in Sclerotinia sclerotiorum . Mol. Plant Pathol. 18, 963–975 doi: 10.1111/mpp.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, J. , Hwang, R. , Chang, K.F. , Hwang, S.F. , Strelkov, S.E. , Gossen, B.D. and Zhou, Q. (2010) An inexpensive method for extraction of genomic DNA from fungal mycelia. Can. J. Plant Pathol. 32, 396–401. [Google Scholar]
- Feng, J. , Zhang, H. , Strelkov, S.E. and Hwang, S.F. (2014) The LmSNF1 gene is required for pathogenicity in the canola blackleg pathogen Leptosphaeria maculans . PLoS One, 9, e92503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, A. , van Kan, J.A.L. and Plummer, K.M. (2004) Simultaneous silencing of multiple genes in the apple scab fungus, Venturia inaequalis, by expression of RNA with chimeric inverted repeats. Fungal Genet. Biol. 41, 963–971. [DOI] [PubMed] [Google Scholar]
- Howard, R.J. and Valent, B. (1996) Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea . Annu. Rev. Microbiol. 50, 491–512. [DOI] [PubMed] [Google Scholar]
- Huang, H.C. (1985) Factors affecting myceliogenic germination of sclerotia of Sclerotinia sclerotiorum . Phytopathology, 75, 433–437. [Google Scholar]
- Kim, H.J. , Chen, C. , Kabbage, M. and Dickman, M.B. (2011) Identification and characterization of Sclerotinia sclerotiorum NADPH oxidases. Appl. Environ. Microbiol. 77, 7721–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosasih, B.D. and Willetts, H.J. (1975) Ontogenetic and histochemical studies of the apothecium of Sclerotinia sclerotiorum . Ann. Bot. 39, 185–191. [Google Scholar]
- Kubo, Y. , Furusawa, I. and Shishiyama, J. (1987) Relationship between pigment intensity and penetrating ability in appressoria of Colletotrichum lagenarium . Can. J. Microbiol. 33, 870–873. [Google Scholar]
- Langfelder, K. , Streibel, M. , Jahn, B. , Haase, G. and Brakhage, A.A. (2003) Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet. Biol. 38, 143–158. [DOI] [PubMed] [Google Scholar]
- Li, M. and Rollins, J.A. (2009) The development‐specific protein (Ssp1) from Sclerotinia sclerotiorum is encoded by a novel gene expressed exclusively in sclerotium tissues. Mycologia, 101, 34–43. [DOI] [PubMed] [Google Scholar]
- Li, M. and Rollins, J.A. (2010) The development‐specific p1 and p2 genes of Sclerotinia sclerotiorum encode lectins with distinct yet compensatory regulation. Fungal Genet. Biol. 47, 531–538. [DOI] [PubMed] [Google Scholar]
- Liang, Y. , Rahman, M.H. , Strelkov, S.E. and Kav, N.N.V. (2010) Developmentally induced changes in the sclerotial proteome of Sclerotinia sclerotiorum . Fungal Biol. 114, 619–627. [DOI] [PubMed] [Google Scholar]
- Liang, Y. , Yajima, W. , Davis, M.R. , Kav, N.N.V. and Strelkov, S.E. (2013) Disruption of a gene encoding a hypothetical secreted protein from Sclerotinia sclerotiorum reduces its virulence on Brassica napus canola. Can. J. Plant Pathol. 35, 46–55. [Google Scholar]
- Russo, G.M. , Dahlberg, K.R. and van Etten, J.L. (1982) Identification of a development‐specific protein in sclerotia of Sclerotinia sclerotiorum . Exp. Mycol. 6, 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomma, B.P.H.J. (2003) Alternaria spp. from general saprophyte to specific parasite. Mol. Plant Pathol. 4, 225–236. [DOI] [PubMed] [Google Scholar]
- Townsend, B.B. and Willetts, H.J. (1954) The development of sclerotia in certain fungi. Trans. Br. Mycol. Soc. 37, 213–221. [Google Scholar]
- Tucker, S.L. and Talbot, N.J. (2001) Surface attachment and pre‐penetration stage development by plant pathogenic fungi. Annu. Rev. Phytopathol. 39, 385–417. [DOI] [PubMed] [Google Scholar]
- Turkington, T.K. and Morrall, R.A.A. (1993) Use of petal infestation to forecast Sclerotinia stem rot of canola: the influence of inoculum variation over the flowering period and canopy density. Phytopathology, 83, 682–689. [Google Scholar]
- Willetts, H.J. and Bullock, S. (1992) Developmental biology of sclerotia. Mycol. Res. 96, 801–816. [Google Scholar]
- Wlaschek, M. , Wenk, J. , Brenneisen, P. , Briviba, K. , Schwarz, A. , Sies, H. and Scharffetter‐Kochanek, K. (1997) Singlet oxygen is an early intermediate in cytokine‐dependent ultraviolet‐A induction of interstitial collagenase in human dermal fibroblasts in vitro. FEBS Lett. 413, 239–242. [DOI] [PubMed] [Google Scholar]
- Yajima, W. , Liang, Y. and Kav, N.N.V. (2009) Gene disruption of an arabinofuranosidase/β‐xylosidase precursor decreases Sclerotinia sclerotiorum virulence on canola tissue. Mol. Plant–Microbe Interact. 22, 783–789. [DOI] [PubMed] [Google Scholar]
- Yarden, O. , Veluchamy, S. , Dickman, M.B. and Kabbage, M. (2014) Sclerotinia sclerotiorum catalase SCAT1 affects oxidative stress tolerance, regulates ergosterol levels and controls pathogenic development. Physiol. Mol. Plant Pathol. 85, 34–41. [Google Scholar]
- Zhang, C. , He, Y. , Zhu, P. , Chen, L. , Wang, Y. , Ni, B. and Xu, L. (2015) Loss of bcbrn1 and bcpks13 in Botrytis cinerea not only blocks melanization but also increases vegetative growth and virulence. Mol. Plant–Microbe Interact. 28, 1091–1101. [DOI] [PubMed] [Google Scholar]