

Summary
Recombination is a major evolutionary force, increasing genetic diversity and permitting efficient coevolution of fungal pathogen(s) with their host(s). The ascomycete Fusarium graminearum is a devastating pathogen of cereal crops, and can contaminate food and feed with harmful mycotoxins. Previous studies have suggested a high adaptive potential of this pathogen, illustrated by an increase in pathogenicity and resistance to fungicides. In this study, we provide the first detailed picture of the crossover events occurring during meiosis and discuss the role of recombination in pathogen evolution. An experimental recombinant population (n = 88) was created and genotyped using 1306 polymorphic markers obtained from restriction site‐associated DNA sequencing (RAD‐seq) and aligned to the reference genome. The construction of a high‐density linkage map, anchoring 99% of the total length of the reference genome, allowed the identification of 1451 putative crossovers, positioned at a median resolution of 24 kb. The majority of crossovers (87.2%) occurred in a relatively small portion of the genome (30%). All chromosomes demonstrated recombination‐active sections, which had a near 15‐fold higher crossover rate than non‐active recombinant sections. The recombination rate showed a strong positive correlation with nucleotide diversity, and recombination‐active regions were enriched for genes with a putative role in host–pathogen interaction, as well as putative diversifying genes. Our results confirm the preliminary analysis observed in other F. graminearum strains and suggest a conserved ‘two‐speed’ recombination landscape. The consequences with regard to the evolutionary potential of this major fungal pathogen are also discussed.
Keywords: crossover rate, dense genetic linkage map, genotyping‐by‐sequencing, Gibberella zeae, meiosis, RAD‐seq
Introduction
Fusarium graminearum sensu stricto is one of the main causal agents of Fusarium head blight (FHB), a disease of cereal crops that constitutes a limiting factor for global production (Trail, 2009). The most damaging aspect of FHB infection is the contamination of grains with stable and harmful mycotoxins, such as deoxynivalenol (DON), whose presence in food and feed is widely regulated throughout the word (Waskiewicz and Golinski, 2013). Therefore, research targeting improved understanding of this pathogen and its control is critical to food security.
In addition to homothallic reproduction, the genetic diversity observed between field isolates suggests that F. graminearum outcrosses frequently (Chen and Zhou, 2009; Liang et al., 2014; Talas and McDonald, 2015a). Fungal pathogens that exhibit mixed reproduction can evolve rapidly, thereby endangering the efficiency and durability of control strategies (McDonald and Linde, 2002; Pariaud et al., 2009). With regard to F. graminearum, a shift towards more aggressive endogenic populations has been reported (Ward et al., 2008), as well as the emergence of isolates resistant to fungicides (Chen and Zhou, 2009; Talas and McDonald, 2015b). Furthermore, experimental studies have revealed more aggressive strains following sexual outcrossing (Cumagun and Miedaner, 2004; Voss et al., 2010), providing a role of sexual reproduction and recombination in the maintenance or increase in aggressiveness. A better characterization of recombination in F. graminearum is needed to understand pathogen evolution.
Fusarium graminearum is haploid for most of its life cycle, but meiosis can occur in the fruiting body (perithecium) formed after the fusion of two haploid hyphae (Trail, 2009). Meiosis is a highly conserved mechanism in eukaryotic genomes, where double‐strand breaks are formed between homologous chromosomes, ensuring their correct transmission and leading to either crossover or non‐crossover events (Mezard et al., 2015). Both events can have direct mutagenic effects as a result of gene conversion (Mezard et al., 2015). However, only crossovers will break parental haplotypes, limiting the hitchhiking of large chromosomal segments during selection (Charlesworth and Campos, 2014). The crossover rate has been shown to be highly variable along eukaryotic genomes (Bensasson, 2011; Bhakta et al., 2015; Brachet et al., 2012; Croll et al., 2015; Mezard, 2006; Petes, 2001; Sonnenberg et al., 2016; Tsai et al., 2016; Yelina et al., 2015). Furthermore, recent insights from several fungal plant pathogen genomes have revealed a bipartite distribution of recombination activity, diversity and gene function, often referred to as the ‘two‐speed’ genome hypothesis (Croll and McDonald, 2012; Dong et al., 2015; Raffaele et al., 2010).
In F. graminearum, the first draft of the genome assembly revealed a relatively condensed genome (36 Mb), 13 718 genes of which were annotated using an automated approach (Cuomo et al., 2007; Wong et al., 2011), whereas genome mining analysis suggested a significant number of effectors as secreted proteins or secondary metabolites (Brown et al., 2012; Sieber et al., 2014). Alignment of the reference genome with a linkage map suggested an increase in recombination rate within telomeric/subtelomeric regions, in addition to interstitial regions (Cuomo et al., 2007; Gale et al., 2005). However, as both the linkage map and genome assembly were incomplete, the interpretation of these results is limited. Strikingly, the genes hypothesized to be induced in host–pathogen interactions were more polymorphic and often located in regions of high recombination (Cuomo et al., 2007). Despite the recent revision and improvement of the reference genome assembly (38 Mb, 14 164 protein‐coding genes; King et al., 2015), the linkage maps currently available lack the required resolution to provide better insight into the patterns of recombination across the genome (Gale et al., 2005; Lee et al., 2008). In addition, questions arise about the organization of polymorphism in different genetic backgrounds, and the role of recombination in the distribution of polymorphisms. Recently, sequencing‐based genotyping strategies have revolutionized the process of obtaining genome‐wide marker data (Davey et al., 2011). Of these techniques, restriction site‐associated DNA sequencing (RAD‐seq) has been widely employed for the generation of high‐density linkage maps for several species (e.g. Baird et al., 2008; Davey et al., 2013; Gonen et al., 2014; Lendenmann et al., 2014; Palaiokostas et al., 2013).
Our working hypothesis was that we would be able to detect regions with high and low levels of recombination, consistent with a ‘two‐speed’ genome architecture in F. graminearum. In order to test this hypothesis, we assessed the distribution of meiotic recombination events in F. graminearum by constructing an accurate, high‐resolution genetic map based on RAD‐seq data from a progeny set of 88 strains. Finally, these results provided new clues about the role of recombination in the evolution of this pathogen.
Results
Strains and genotyping
The parental strains used for this analysis, namely INRA‐156 and INRA‐171, were isolated from wheat in France. We re‐sequenced their genomes to help with downstream analysis, and to enable an accurate reference‐based RAD‐seq approach to genotyping. In total, 63 486 single nucleotide polymorphisms (SNPs) were discovered. Using parental genomes, we simulated PstI enzyme digestion of the INRA‐156 and INRA‐171 genomes, which predicted 12 610 and 12 634 fragments with a mean size of 3016 and 3010 base pairs (bp), respectively. The large majority of cutting sites were common between the two genomes (12 489). Cutting site distribution for PstI was predicted to be random (KS test, P > 0.01) avoiding the introduction of subsequent bias (Fig. 1, track B), suggesting that the use of PstI was appropriate.
Figure 1.
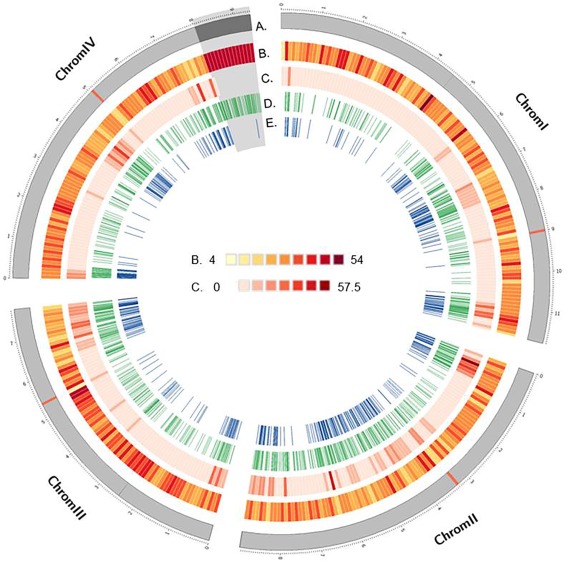
Circos plot of the distribution of several genomic attributes and genotyping features along the four chromosomes of Fusarium graminearum. The shaded region at the end of chromosome IV shows the highly repetitive rRNA encoding region proposed by King et al. (2015). (A) Representation (in Mb) of the four chromosomes of F. graminearum, with the positions of the predicted centromeres in red. (B) Predicted PstI cutting sites based on parental sequences. (C) Single nucleotide polymorphism frequency between whole genome sequences of parental strains. (D) Frequency of restriction site‐associated DNA sequencing (RAD‐seq) polymorphic marker sites. (E) Position of markers with non‐redundant segregation profiles.
The digestion of genomic DNA from the 88 recombinant progeny and two parental strains using the PstI enzyme was followed by RAD‐seq library preparation and Illumina paired‐end sequencing. A total of 401 726 418 paired‐end reads of 125 bp was produced, resulting in a total of 50.2 Gbp of sequence data. Filtering and demultiplexing resulted in a variable number of reads assigned per strain (ranging from 308 147 to 10 818 740), the majority of which were successfully aligned with the reference genome (97%). A catalogue of sequenced loci was constructed, recovering 17% of the total length of the reference genome. Strain‐specific sequences matching the catalogue resulted in the identification of 1866 polymorphic loci, 1306 of which passed the quality control requirements (Fig. 1, track D). Segregation profiles of 31 randomly chosen RAD loci SNPs were matched to corresponding profiles obtained from cleaved amplified polymorphic sequence (CAPS) assays. Only 15 mismatches were observed from the 2714 combinations with known genotypes, giving an overall correspondence of 99.4% (data not shown).
The dataset of markers used for downstream analysis was composed of 1306 RAD markers, 21 markers genotyped during the validation of recombinant strain isolation, together with three additional markers named KSNP100, KSNP101 and KSNP102 (File S1, see Supporting Information). Chromosomes I and II were under‐represented (chi‐squared test, P < 0.001) and over‐represented (chi‐squared test, P < 0.001) in markers, respectively, whereas no significant tendencies were observed for the other two chromosomes. The distribution of markers along chromosomes was not random (Fig. 1, track D, P < 0.01), with a maximal distance between pairs of markers of 498.8 kb (chromosome I at the position 4.0–4.5 Mb). The median physical distance between markers was 12.9 kb, ranging from 11.6 kb for chromosome II to 15.1 kb for chromosome IV (Table 1). Marker density was highest in subtelomeric and interstitial regions within chromosomes (Fig. 1, track D), and correlated with the density of SNPs detected between parental genomes (Fig. 1, track C, Rho = 0.76, P < 2.2E‐16). An exception to this pattern was observed at the end of chromosome IV (∼1.3 Mb), where highly repetitive rRNA‐encoding DNA content resulted in difficulty in read alignment and subsequent variant calling. As a result of the repetitive nature of the DNA in this region, there were very few informative polymorphic markers, despite an apparent abundance of PstI restriction sites (Fig. 1). For the remainder of the genome, the pattern of polymorphic marker distribution along the chromosomes probably reflects the polymorphism rate rather than the restriction site frequency, because of the random distribution of PstI sites.
Table 1.
General features of genotyping procedure and linkage map construction.
| Linkage group/chromosome | Marker number | Average marker number per 100 kb | Median distance between markers (kb) | Maximum distance between markers (kb) | Marker used for linkage map construction* | Average spacing (cM) | Linkage group size (cM) | Chromosome size (Mb) | Recombination rate (cM/Mb) | Percentage anchored | Average number of crossovers per strain |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG‐1/chromosome I | 342 | 2.9 | 11.7 | 499 | 130 | 3.4 | 435 | 11.76 | 37.0 | 98.0 | 4.5 |
| LG‐2/chromosome II | 387 | 4.3 | 11.6 | 351.7 | 164 | 3.2 | 522.9 | 9.00 | 58.1 | 99.4 | 5.5 |
| LG‐3/chromosome III | 278 | 3.6 | 12.8 | 301.1 | 92 | 2.8 | 250.8 | 7.79 | 32.2 | 99.2 | 2.8 |
| LG‐4/chromosome IV | 323 | 3.4 | 15.1 | 398.7 | 97 | 3 | 288.4 | 9.41 | 30.7 | 99.4 | 3.1 |
| Overall | 1330 | 3.5 | 12.9 | 499 | 483 | 3.1 | 1497.1 | 37.96 | 39.4 | 99.0 | 15.9 |
Segregation analysis, linkage map construction and alignment to the reference genome
Only five markers, sparsely located on the genome, were found to exhibit segregation distortion (P < 0.01, Table S1, see Supporting Information). With regard to the segregation pattern of these markers, we assumed that the observed bias of segregation could be reasonably attributed to missing data or genotyping errors. In contrast with the segregation of chromosomal markers, segregation analysis of mitochondrial markers and the KSNP102 marker (File S1) revealed uniparental inheritance from the INRA‐156Δmat strain of mitochondria and of the HG970330 sequence, respectively.
On investigation of marker information across the progeny, 483 different profiles of segregations were identified. Only one representative marker per profile was used to construct the framework linkage map (n = 483; Fig. 1, Table 1). Four linkage groups, named ‘LG‐1’, ‘LG2’, ‘LG‐3’ and ‘LG‐4’, were constructed [logarithm of the odds (LOD) threshold, 6], corresponding to chromosomes I, II, III and IV, respectively, described in the reference genome assembly (version 4.0). Markers were then ordered and genetic distances were calculated using the Kosambi function (Table 1). Alignment of the linkage map to the reference genome revealed a remarkable collinearity (Fig. 2, File S1C). Only 13 pairs of markers exhibited inverted order out of a possible 479 pairs of successive markers (2.7%). Manual investigation of these pairs revealed that the order of seven could be inverted and resulted from incertitude as a result of the map resolution. The six other pairs remained inverted, and may reflect errors in the linkage map or reference genome assembly order (Fig. 2, asterisks). The final map length was 1497.1 cM with an average genetic distance between markers of 3.1 cM (Table 1).
Figure 2.
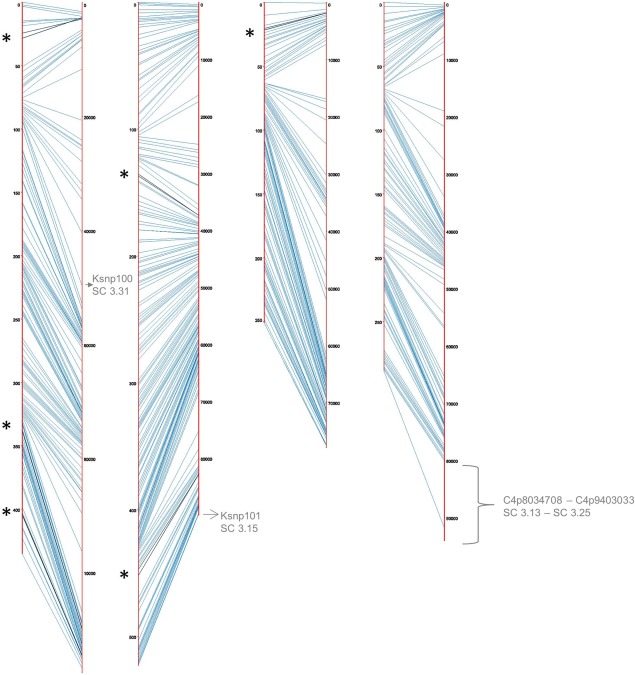
Alignment of the linkage map (left side) to the reference genome RRES v4.0 (right side). The asterisks and black lines represent the six inversions remaining in the linkage map. The location of the sequence placed by King et al. (2015) in the assembly and anchored by the linkage map is indicated in grey.
Marker orders and distances given by this linkage map were then used to add the 847 co‐segregating markers (those showing similar patterns of segregation compared with the representative markers), anchoring a total of 99.0% of the markers to the reference genome (Table 1). By doing so, one additional pair of inverted markers was detected at the end of chromosome IV (File S1C). The markers KSNP100 and KSNP101, designed to align the supercontigs 3.31 and 3.15 of the reference genome FGDG v3.1, have been successfully anchored to chromosomes I and II, respectively (Fig. 2), and this is in agreement with the location proposed in the RRes V4.0 assembly (Table S2, see Supporting Information). Similarly, the RAD‐seq markers C4p8034708 to C4p9403033, constituting a single recombination block, aligned the end of chromosome IV previously proposed in the RRes V4.0 assembly (Fig. 2, Table S2).
Recombination landscape
By using the segregation information of the 1330 markers ordered on the linkage map, 1451 putative crossovers were mapped, within intervals of a median length of 24 kb. The average recombination rate across the entire genome was high at 39.4 cM/Mb (±12.1 cM/Mb, Table 1). For each strain, 4.4 crossovers per chromosome were detected, giving an average of one crossover every 2.4 Mb (Table 1). However, the recombination rate was variable between chromosomes, and ranged between 30.7 cM/Mb for chromosome IV to 58.1 cM/Mb for chromosome II (Table 1). As a result of the limited number of chromosomes, the correlation between recombination rate and chromosome length could not be tested statistically, but no obvious pattern was evident.
A high level of variation in the recombination rate within chromosomes was typically observed, as shown in Fig. 3, which represents the alignment of the linkage map (y‐axis) with the physical map (x‐axis). Recombination‐active sections, corresponding to regions with positive slope in the curve and with at least two‐fold greater recombination than the genome‐wide average of 78.8 cM/Mb, and recombination deserts, corresponding to flat regions in the curve, could be identified for each chromosome. Twelve chromosomal segments greater than 0.5 Mb in length were identified as recombination active (Table S3, see Supporting Information). Nine recombination hotspots, i.e. with more than four crossovers in a 20‐kb region, were detected on all chromosomes (Table S4, see Supporting Information). Isolated recombination hotspots did not always result in recombination‐active sections, as illustrated by the hotspot on chromosome III at 3.4 Mb, which was not associated with any recombination‐active section (Fig. 4). The recombination‐active sections cover 30% of the physical genome, 56% of the total number of markers and 87% of the length of the linkage map. Chromosome I contained two distal recombination‐active sections, and two proximal and central sections, each flanked by a recombination desert of 1 Mb in size. Chromosome II contained three recombination‐active sections, with the intermediate one alone encompassing more than one‐half of the recombination activity recorded in the entire chromosome. Chromosome III contained two recombination‐active regions spanning more than one‐half of the linkage group. Chromosome IV contained three recombination‐active sections, but in contrast with other chromosomes, one subtelomeric region contained a non‐recombinant chromosomal segment, corresponding to an rRNA‐rich encoding region (Figs 3 and 4, track A in dark grey). The marker density was greater in active sections and varied by a factor of 2.4‐fold on average between sections. The average recombination rate was drastically different between non‐active and active sections, and varied overall by a factor of 15.3‐fold on average (Fig. 5A). The recombination rate in the defined recombination‐active sections ranged from 103.9 to 175.8 cM/Mb (Fig. 5A, Table S3), whereas the recombinant rate of recombination deserts ranged from 3.7 to 25.0 cM/Mb (Fig. 5A). The total physical length of recombination‐active sections per chromosome was positively correlated with the overall chromosome recombination rate (Fig. 5B).
Figure 3.
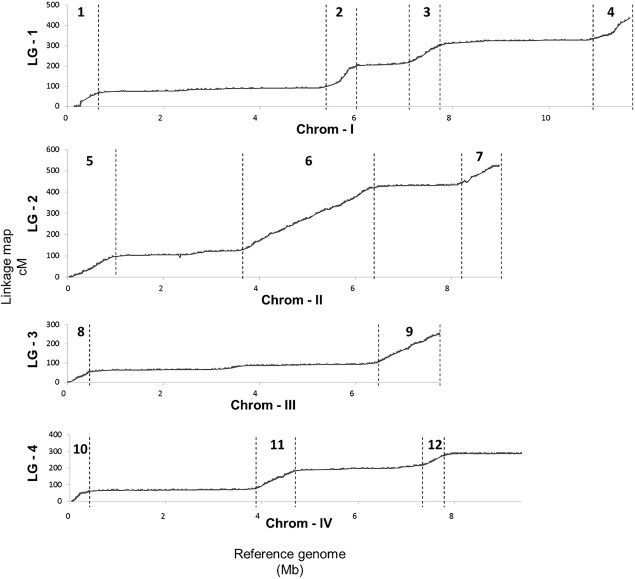
Scatter plot showing the linkage map position (y‐axis) and physical position (x‐axis). The dotted lines delimit the recombination‐active sections (numbered with arabic numerals) from recombinant desert sections.
Figure 4.
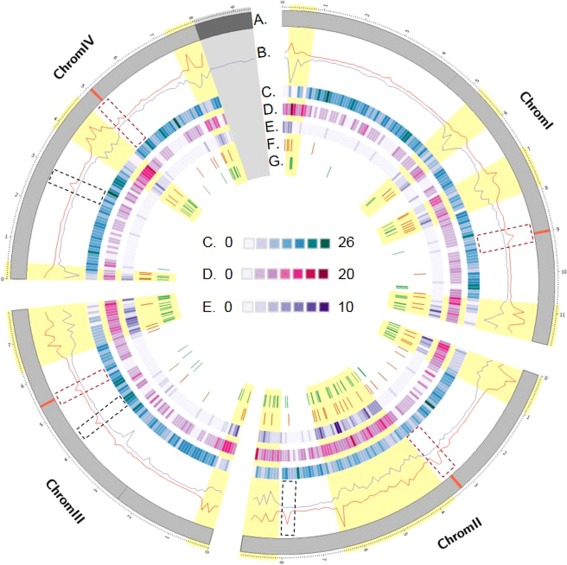
Circos plot of the distribution of several genomic sequence features along the four chromosomes of Fusarium graminearum. (A) Representation of the four chromosomes of F. graminearum (in Mb); the red segments delimit the positions of the predicted centromeres. (B) In red, the single nucleotide polymorphism (SNP) density (SNP/kb) is calculated in windows of 100‐kb bins along the parental genomes. In blue, the number of COs is calculated in separate windows of 100‐kb bins across the progeny. The dashed rectangular boxes show the typical patterns observed at centromere positions; the dashed black boxes show the centromere‐like patterns. Recombination‐active sections are highlighted in yellow. (C) Protein‐coding genes expressed constitutively in all in planta conditions tested conditions (Harris et al., 2016). (D) Protein‐coding genes expressed in host‐specific conditions (Harris et al., 2016). For (C) and (D), the gene density was calculated in 100‐kb bin windows. (E) Location of genes predicted to code for secreted proteins (Brown et al., 2012; King et al., 2015). (F) Location of predicted secondary metabolite clusters (Sieber et al., 2014). (G) Location of genes showing evidence of diversification.
Figure 5.
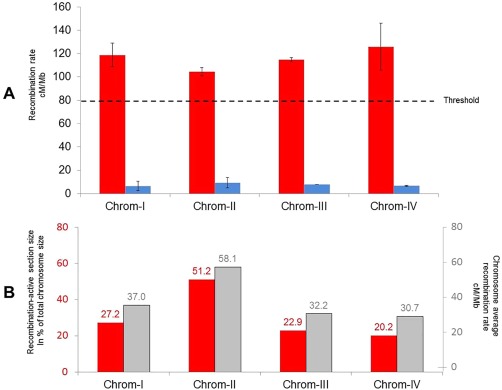
(A) Recombination rate distribution according to chromosomal section. Recombination‐active sections are given in red, and recombinant desert sections are shown in blue. The dashed line shows the recombination rate threshold considered to be the recombination‐active section (two‐fold increase in genome‐wide recombination rate). (B) The percentage of the total size of the chromosome allocated to recombination‐active sections is given in red, and the chromosome average recombination rate (cM/Mb) is shown in grey.
Functional and sequence enrichment analysis of highly recombinant regions
Overall, the crossover density was positively correlated with the polymorphism rate (measured in SNP/kb) along the genome (Fig. 4, track B, Rho = 0.67, P < 2.20E‐16). The crossover density was also positively correlated with the gene density along the genome (Rho = 0.50, P < 2.20E‐16), and negatively with GC content (Rho = −0.56, P < 2.20E‐16). No correlation was observed between the non‐synonymous/synonymous ratio and crossover count, as calculated in 100‐kb bins. However, the majority of genes showing an excess of mutation with non‐synonymous effects identified in this analysis (92 of the 121 genes identified), and assumed to represent genes under diversifying selection, were located in recombination‐active sections (Fig. 4, track G).
Several distinct local regions showed high levels of polymorphism, whereas no recombination was detected (Fig. 4). Four were located within the predicted centromeres of the chromosomes (Fig. 4, red segments on track A). Three other regions, dispersed throughout the genome, showed similar patterns (Fig. 4, track B). As reported previously, the regions located at the predicted centromere positions always show large decreases in GC content, and this feature was not observed for the other three regions (Table S5, see Supporting Information).
The 12 recombination‐active regions contained a total of 4947 genes, representing 35% of the total number of protein‐encoding genes annotated in the reference genome (File S2A, see Supporting Information). Gene ontology (GO) analysis of these genes revealed several enriched categories (File S2B). The highest significant enrichment was recorded for amino acid and transmembrane transport, oxidation–reduction and carbohydrate metabolic processes, as well as the regulation of several cellular processes, such as transcription or nitrogen compound metabolism (1.97‐fold to 1.27‐fold, File S2B). At the other end of the spectrum, recombinant desert sections were enriched for gene categories arguably associated with basal mechanisms, such as localization and protein transport and metabolism or translation (File S2B). Nonetheless, it is worth noting that 50.4% and 42.8% of the protein‐coding genes located in active and desert sections, respectively, had no predicted GO code.
Following the general GO approach described above, a more specific enrichment analysis was performed using specific datasets of F. graminearum retrieved from the literature. For example, genes predicted to encode for secreted proteins, and suggested to be putative effectors, were enriched by two‐fold in recombination‐active regions (P < 0.001, Fig. 4, track E and File S2C), as were genes predicted to act in secondary metabolite biosynthesis (Fig. 4, track F, 1.4‐fold, P < 0.001), or those previously reported to show host specificity of expression (2.0‐fold, P < 0.001, Fig. 4, track D and File S2C). In addition, genes previously shown to be expressed non‐specifically during the infection of a panel of hosts were over‐represented in the non‐recombinant section, and under‐represented by 1.5‐fold in recombination‐active regions (P < 0.001, Fig. 4, track C and File S2C).
To go further, motif enrichment analysis of genes located in recombination‐active sections (including 500‐bp upstream and downstream sequences) revealed a significant over‐representation of motifs similar to C2H2 zing finger factors of Saccharomyces cerevisiae (P < 1E‐14), as well as motifs similar to the High Mobility Group of Mus musculus (Table S6, see Supporting Information, P < 1E‐13).
Discussion
In this study, we estimated the recombination activity and its distribution across the genome of F. graminearum using the first high‐density linkage map of the species based on RAD‐seq. The linkage map is almost fully integrated with the reference genome sequence (99%). It also improves the resolution of the previously published genetic map of F. graminearum by six‐fold (Gale et al., 2005; Lee et al., 2008), allowing the first thorough characterization of the recombination landscape of the species.
Crossover mapping reveals the chromosome‐specific landscape of recombination
The recombination rate and its distribution are key components of genome biology, and can vary tremendously across organisms. In sexually reproducing fungi, the recombination rate has been reported to vary from ∼11 cM/Mb in the edible mushroom Agaricus bisporus var. bisporus (Sonnenberg et al., 2016) to ∼600 cM/Mb in S. cerevisiae (Mancera et al., 2008). The recombination rate estimate in this analysis for F. graminearum (∼39 cM/Mb) is substantially lower overall than that for S. cerevisiae and other pathogenic fungi, such as the causal agent of wheat Septoria tritici blotch, Zymoseptoria tritici (130 cM/Mb; Lendenmann et al., 2014). However, this recombination rate is consistent with a previously reported estimate for F. graminearum (∼34 cM/Mb; Gale et al., 2005), and may be higher as a result of the increased power to detect crossovers using high‐resolution genomic tools (Sonnenberg et al., 2016). Overall, it suggests that F. graminearum does not have a particularly high genome‐wide recombination rate in comparison with other pathogenic fungi. Furthermore, genome‐wide average recombination rates do not reflect the variation observed along chromosomes.
In many eukaryotic species, an increase in recombination rate is observed in subtelomeric regions, and a decrease near centromeres (Bhakta et al., 2015; Croll and McDonald, 2012; Cuomo et al., 2007; Jensen‐Seaman et al., 2004; Limborg et al., 2016; Mancera et al., 2008; Sonnenberg et al., 2016; Tsai et al., 2016). As highlighted previously (Cuomo et al., 2007; Gale et al., 2005), F. graminearum is no exception to this rule. However, additional regions of the genome with high recombination rate have also been described in interstitial areas and corresponding to previously suggested ancestral chromosomal fusion sites (Cuomo et al., 2007; Ma et al., 2010). Markers were not randomly distributed in the genome, and we suggest that this result may arise from the non‐random distribution of polymorphism in the genome. The variation in marker density along the genome may have affected the accuracy of crossover detection in some regions relative to others (Posada et al., 2002). However, the difference in magnitude between recombination rate variation and marker density variation along the genome supports the existence of true biological explanations rather than experimental bias. The recombination patterns identified in this study are strikingly consistent with previous reports that used different strains (Cuomo et al., 2007; Gale et al., 2005), albeit at a higher resolution, suggesting foundational control of recombination in F. graminearum. The downstream analysis discussed below provides some insight into the potential explanations of this phenomenon.
This high‐density linkage map was also used to test the latest version of the F. graminearum reference genome assembly, which is based solely on short read alignment (King et al., 2015). The supercontigs not positioned in the genome reference FGDB v3.1 (Cuomo et al., 2007; Wong et al., 2011), that were assembled in version RRES v4.0, were consistent with our linkage map assignments. The last remaining unassigned sequence contig (HG970330) in RRES v4.0 was found to show cytoplasmic inheritance, in line with the hypothesis of phage DNA given by King et al. (2015). It is surprising that these DNA sequences were conserved in non‐related strains, even though the four genes encoded on this sequence (King et al., 2015) may have important cellular function(s).
The predicted positions of centromeres proposed by King et al. (2015) were also consistent with the typical recombination pattern of centromeres identified in the current study. Centromeric regions typically show adenosine‐ and thymine‐rich sequences and have high DNA polymorphism, whereas recombination is suppressed as a result of physical constraints (Bensasson et al., 2008; Henikoff et al., 2001; King et al., 2015). Interestingly, other genomic positions demonstrate similar characteristics according to polymorphism and recombination rate, and could correspond to ancestral centromeres following chromosome fusions. Nevertheless, the base compositions of these putative centromeres were not enriched in adenosine and thymine, suggesting either that they have lost their functions or that the patterns observed for these regions have different origins. The DNA sequences of these regions (File S4, see Supporting Information) may help to identify their roles and origins.
Characterization of recombination‐active genome regions
Overall, the recombination landscape of F. graminearum and its close relative F. pseudograminearum (Gardiner et al., 2016) is rather unique amongst the fungal species studied to date. There are clearly defined peripheral and central genomic regions in which the recombination activity differs by approximately 15‐fold in comparison with recombinant desert regions. The fact that chromosomes contain specific recombination‐active regions in interstitial DNA supports the theory of distinct chromosome fusion events (Ma et al., 2010) that have retained their recombination characteristics, possibly related to conserved molecular mechanisms.
Tremendous advances have been made in model species to understand the molecular mechanisms controlling meiotic recombination, including the important role of epigenetics (Brachet et al., 2012; Galazka and Freitag, 2014; Mezard et al., 2015). For example, euchromatin seems to favour crossover formation compared with heterochromatin. However, a paradox remains in F. graminearum as the epigenetic mark associated with ‘recombinophobic’ chromatin in most organisms is enriched in recombination‐active regions (Connolly et al., 2013).
Variations in recombination rate can also be attributed to the action of several genes, or specific sequences, for which the absence or presence can control the recombination rate locally or globally on a genome (Catcheside, 1981, Mercier et al., 2015; Wahls and Davidson, 2010; Yeadon et al., 2002, 2004). An example is the case of the mating‐type genes, associated in some species with a suppression of recombination in their vicinity, whereas recombination seems to be induced nearby in other species (Idnurm et al., 2015). In this analysis, the Mat locus of F. graminearum (at position 3.0 Mb of chromosome II) was found in a region in which recombination was suppressed hundreds of kilobases away. At a broader scale, consensus sequences are known to be linked to recombination hotspots in several eukaryotes (Wahls and Davidson, 2010), such as, for example, the M26 motif (5′‐ATGACTG‐3′). Preliminary results made using our data suggest that the presence of such motifs is also linked with the global recombination activity observed herein for F. graminearum (data not shown). Furthermore, an interesting perspective would be to identify homologous proteins of model species known to be implicated in double‐strand break formation or repair (for a review, see Mercier et al., 2015) in F. graminearum, and to test their role in the organization of recombination activity. For example, the Spo11 protein (probably corresponding to FGRRES_05949), implicated in double‐strand break formation, or the FANCM‐like protein (probably corresponding to FGRRES_17603), involved in the repair of double‐strand breaks in non‐crossovers rather than in crossovers (Girard et al., 2015; Lorenz et al., 2012), are interesting targets.
The in‐depth recombination pattern proposed by our work marks an important milestone in the study of recombination in F. graminearum and opens up great perspectives for the investigation of its control in this pathogenic species.
Recombination‐active sections seem to be linked to several crossover hotspots
Relatively small genomic regions (1–5 kb) with high crossover rates have been identified in a wide range of organisms (mammals, plants, yeasts), and are commonly referred to as ‘hotspots’ (Mancera et al., 2008; Mezard, 2006; Mezard et al., 2015; Paigen and Petkov, 2010). Hotspots are often located in promoters of genes, leading to the hypothesis that they are associated with chromatin accessibility (Brachet et al., 2012; Comeron et al., 2012; Goodstadt, 2011). High recombination levels can be defined in two ways: tightly defined smaller regions corresponding to the precise length of crossover hotspots, and a broader scale genomic region corresponding to recombination‐active sections (Comeron et al., 2012; Duret and Arndt, 2008; Myers et al., 2005, Simchen and Stamberg, 1969), as analysed in the current study. Enrichment of motifs similar to the yeast C2H2 zinc finger or to High Mobility Group motifs in genes located in recombination‐active regions is interesting, because these motifs have previously been implicated in hotspot formation (Baudat et al., 2010; Bergeron et al., 2005; Goodstadt, 2011; Panday and Grove, 2016) and could be directly correlated to hotspot presence in F. graminearum. We found such limited hotspots in the F. graminearum genome, and the frequency of these hotspots may help to define larger recombination‐active regions of the genome. In Z. tritici, it has been demonstrated that hotspots are not always consistent between crosses (Croll et al., 2015). Although the data in the current study do not exclude the possibility of transient hotspots in F. graminearum, the general conservation of the recombination pattern across multiple genetic backgrounds (Cuomo et al., 2007) suggests that their locations are conserved along the genome, as observed in human chromosomes (Myers et al., 2005).
Potential role of recombination landscape in pathogen evolution
As observed here and as reported previously (Cuomo et al., 2007), the distribution of recombination events and polymorphisms is strikingly similar across several different F. graminearum genomes, a common feature of other eukaryotic genomes (Charlesworth and Campos, 2014; Manzano‐Winkler et al., 2013; Noor, 2008; Roselius et al., 2005; Spencer et al., 2006). Sexual recombination with different partners, suggested to occur at high frequency in F. graminearum populations (Talas and McDonald, 2015a), can play a role in evolution by increasing genotypic diversity and enabling the selection of favourable alleles and haplotypes in selective sweeps (Goddard et al., 2005).
Linkage disequilibrium, resulting from the lack of recombination in recombination deserts, coupled with the presence of genes under positive and purifying selection, should reduce nucleotide diversity over time via selective sweeps (Smith and Haigh, 1974). In F. graminearum, genes expressed independently in the infected host, which may correspond to conserved biological functions (Harris et al., 2016), are over‐represented in regions with low observed levels of recombination, whereas genes expressed in a host‐specific manner are over‐represented in regions with high observed levels of recombination. The diversifying selection acting on host‐specific effectors (Sperschneider et al., 2015), such as secreted proteins or genes implicated in secondary metabolite production (Brown et al., 2012; Harris et al., 2016; King et al., 2015; Sieber et al., 2014), is probably made efficient by the frequent break‐up and reshuffling of haplotypes in these regions and favours genotypic diversity.
Under the hypothesis that recombination is mutagenic during DNA repair, as demonstrated in yeast (Strathern et al., 1995), the higher rate of recombination observed in active sections of the F. graminearum genome may create genetic diversity. This latter point would thus make both heterothallic and homothallic reproduction an additional source of diversity and a driver for evolution.
Overall, the distribution and characterization of observed recombination events in F. graminearum are consistent with the emerging concept of a bipartite architecture of genome evolution in pathogenic fungi, also referred to as a ‘two‐speed’ genome. Under this concept, genes that are critical to host–pathogen interaction cluster in genomic regions associated with high recombination to facilitate a more rapid evolutionary response (Croll et al., 2015; Dong et al., 2015). However, the recombination pattern in F. graminearum differs from that of other fungal pathogens in at least two aspects: (i) F. graminearum has an unusual chromosome architecture, and (ii) the epigenetic marks associated with recombination activity are not always located in areas in which they are predicted to be. The uniqueness of the F. graminearum recombination patterns and genome organization makes this pathogen a significant exception to some of the emerging patterns of fungal genome organization, and suggests that genome organization is a dynamic process that remains in flux rather than a static arrangement. Overall, these results shed light on the high potential of adaptation inherent to the recombination landscape of this pathogen and alert us to the risk of the appearance of more aggressive populations.
Experimental Procedures
Progeny construction and culture conditions
The INRA‐156 and INRA‐171 strains were isolated from wheat in 2001 and 2002 in different fields in central and south‐west France, respectively. The strains were characterized as Fusarium graminearum sensu stricto and are both pathogenic and toxinogenic on wheat. Eighty‐eight recombinant strains were produced from a cross between polymorphic INRA‐156Δmat strain and INRA‐171 strain. Recombinant strains were further validated using 21 targeted marker assays (see section on ‘Targeted marker genotyping’ and Files S1, S4). Potato dextrose agar (PDA, Difco™ BD, Sparks, MD, USA) was used throughout vegetative growth. INRA‐156Δmat was obtained from INRA‐156 strain by replacing the mat1‐2‐1 coding sequence (FGREES_08893) by a hygromycin resistance cassette using the split marker method (Goswami, 2012; File S3, see Supporting Information). The crossing procedure was adapted from Lee et al. (2003) and Leslie and Summerell (2006), except for incubation, which was conducted under continuous white light (Osram T8 L 36W 840 G13, Lumilux). The plate was incubated at 25°C under continuous white light until perithecia reached maturity (Cavinder et al., 2012). Visible cirrhi were collected in sterile water, spread and incubated on PDA plates. Single germinating spores were isolated twice, as described previously in Leslie and Summerell (2006).
Genomic resources
The F. graminearum genome version RRes V4.0 (King et al., 2015), used in this study, is available at EMBL‐EBI (HG970331, HG970332, HG970333, HG970334, HG970335). Genes predicted to code for the secreted proteins of F. graminearum were retrieved from King et al. (2015). Genes previously described as expressed in wheat, barley and/or maize were retrieved from Harris et al. (2016), and genes predicted to belong to secondary metabolite pathway clusters were retrieved from Sieber et al. (2014).
Genomic DNA extraction
Genomic DNA was extracted from lyophilized mycelium previously grown on PDA (39 g/L, Difco™). Mycelia were lysed in a buffer containing 100 mm Tris‐HCl (pH 9.0), 10 mm ethylenediaminetetraacetic acid (EDTA), 1% sarkosyl and 200 μg/mL proteinase K for 2 h at 65°C. After centrifugation (10 min at 10 000 g), the supernatant was extracted successively with phenol, phenol–chloroform (50 : 50) and, finally, chloroform. Nucleic acids were precipitated with cold ammonium acetate (3 m) and isopropanol, washed in 70% ethanol and dissolved in 100 μL of nuclease‐free water.
Whole genome sequencing of parental strains and analysis
Whole nuclear DNA from the parental isolates was sequenced by the MGX platform in Montpellier, France (http://www.mgx.cnrs.fr/) using Illumina sequencing technology (HiSeq™ Sequencing Systems, Illumina, Inc. San Diego, CA, USA). The reads were filtered and trimmed using Prinseq software (Schmieder and Edwards, 2011). They were then aligned to the reference genome RRes V4.0 (King et al., 2015) using BWA (v 0.7.8) and the BWA‐MEM algorithm with standard parameters, and a seed size of 15 nucleotides (Li and Durbin, 2009). SNP calling was performed using GATK (v 2.4) and the Unified Genotyper walker in haploid mode (DePristo et al., 2011). Parental consensus sequences were constructed using the VcfToFasta tool, and by correcting the reference genome with variants identified in each parental genome. In silico digestion of the genomes by the PstI enzyme was conducted using EMBOSS and the ‘Restrict’ tool. Genes showing an excess of mutations were identified and defined as exhibiting more than 75% of non‐synonymous mutations compared with the total number of mutations in the genic sequence (upper third quartile of genome‐wide distribution) and at least four genic mutations (genome‐wide median mutation number per gene × 2).
RAD‐seq library preparation and sequencing
The library and sequencing were prepared and conducted by the MGX platform team in Montpellier, France (http://www.mgx.cnrs.fr). The libraries were prepared according to Baird et al. (2008) using the PstI restriction enzyme. The main modification was the use of Ampure XP beads for the different purification steps. A detailed version of the RAD‐seq procedure is available in File S3. Sequence data are retrievable from the GenBank database under SRA accession SRP083578.
RAD‐seq SNP discovery and genotyping analysis
The quality of the 125‐nucleotide‐long paired‐end reads was analysed using FastQC v0.11.2. Reads were demultiplexed according to barcode sequences using the ‘process_radtags’ program from the Stacks software pipeline v1.32 with the following parameters: PstI enzyme cutting site recognition, removal of reads with at least one uncalled base and/or an average quality score below the phred score of 20. For each strain, reads were then mapped onto the reference genome RRes V4.0 (King et al., 2015) using Bowtie2 v0.12.9 with default parameters. SAM files were then sorted and transformed to BAM files using Samtools v1.1 (Li and Durbin, 2009). The stacks software (Catchen et al., 2011) was used to identify SNP and InDel markers. The ref_map.pl program was used: (i) to compare both parental BAM files and the 88 BAM files from the progeny in order to build RAD loci and call SNPs; (ii) to create a catalogue of all loci; and (iii) to match each sample against the catalogue. The minimum depth of coverage to call a stack was ‘3’; one mismatch was allowed between loci when building the catalogue. Markers were retained if they were bi‐allelic, contained up to 10 SNPs compared with the consensus sequences, and at least 80% of the progeny were genotyped.
Targeted marker genotyping
SNP‐derived markers genotyped using either CAPS or Kompetitive Allele‐Specific PCR (KASP) techniques were designed from the whole genome polymorphism information between the two parental sequences. Microsatellite (simple sequence repeat, SSR) markers were retrieved from the literature (Brygoo and Gautier, 2007; Giraud et al., 2002; Naef and Défago, 2006; Naef et al., 2006; Vogelgsang et al., 2009). Ten SSR and 11 CAPS markers were used to confirm the recombinant progeny. Three KASP markers were designed to align supercontig 3.31, supercontig 3.15 and supercontig 3.12, respectively. These sequences corresponded to supercontigs that were not anchored during initial assembly (Cuomo et al., 2007). The first two supercontigs have been anchored in the version RRes V4.0. Supercontig 3.12 (HG970330) remains unanchored. Details on the primers and experimental conditions are available in File S5 (see Supporting Information).
Genetic map construction
The linkage map was constructed using R/qtl (Broman et al., 2003). First, markers exhibiting redundant segregation patterns in the population were set aside from the mapping procedure. Linkage groups were formed using a maximum recombination fraction of 0.35 and a decimal LOD score threshold of 6. Markers were ordered and the genetic distance was calculated using the Kosambi function. Groups of co‐segregating markers were repositioned manually at the marker position (that used for linkage map construction) and following reference genome order. Genotyping errors were investigated using the calc.errorlod function of R/qtl and the identified genotypes were replaced by missing data. Inversions were manually investigated. When the likelihoods were similar for the two alternative orders, the physical positions were preferred.
Alignment of genetic and physical maps and analysis of recombination
The linkage map was aligned on the reference genome (RRES v4.0). For RAD tags containing several genetic variants, only the position of the first variant was considered. ArkMap software was used to construct the illustration showing the alignment of the linkage map to the reference genome (http://www.bioinformatics.roslin.ed.ac.uk/arkmap/). The recombination rates of the overall chromosome and the recombination‐active and recombinant desert sections were calculated by dividing the linkage group size by the chromosome or section size. The detection of crossovers was performed manually using the inheritance of markers along the chromosome according to the linkage map order. The resolution of the crossover position was obtained by calculating the median size spanning two markers adjacent to the corresponding crossover. Recombination‐active sections were defined when they contained a two‐fold increase in recombination rate compared with the genome‐wide average over a region larger than 0.5 Mb. Recombination hotspots were defined as the genomic loci delimited by markers for which more than four crossovers were recorded and presenting an estimated recombination rate greater than a 10‐fold increase in the genome‐wide recombination rate (> 394 cM/Mb).
Statistical analysis
The Poisson distribution was used to test the distribution pattern of the predicted PstI cutting sites and marker sites in the genome. Marker distribution analysis was carried out as described in Bhakta et al. (2015) using an interval of 100 kb. Significant differences between the observed and expected frequencies of the number of markers per interval were tested through Kolmogorov–Smirnov tests, with P > 0.01 suggesting that the observed frequency does not show a statistically significant difference from the expected frequency. Enrichment of functional categories was calculated using the Gene Ontology Enrichment tools proposed online by the EuPathDB project (http://fungidb.org) using Biological Ontology and InterPro predictions. Enrichments were defined for P < 0.001. A chi‐squared test was used to compare the observed distribution of genes located in recombination‐active regions compared with the theoretical distribution under the hypothesis of a random distribution in the genome. Over‐ and under‐representations were defined for P < 0.001. Gene density, GC content and observed crossovers were calculated using different windows of 100‐kb bins. Correlations were calculated using Spearman rank order test correlation and accepted for P < 0.001. Motif search was conducted using Homer software (Heinz et al., 2016) employing the findMotifs.pl script with standard parameters.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
File S1 Dataset of markers. (A) Lab markers. (B) Restriction site‐associated DNA (RAD) markers. (C) Alignment of the genetic map with the reference genome.
File S2 Recombination and gene functions. (A) All protein‐coding genes described in the reference genome (RRES v4.0) with features. (B) Gene ontology enrichment of recombination‐active and desert sections. (C) Representation of specific gene lists within recombination‐active and desert sections
File S3 Supporting procedures.
File S4 Fasta sequences of the putative centromeres.
File S5 Development of targeted markers.
Table S1 List of markers exhibiting non‐Mendelian segregation.
Table S2 Overview of the supercontigs anchored during the RRes4.0 assembly proposal, with information concerning the marker used to anchor these supercontigs.
Table S3 List of the chromosomal segments classified as recombinant active.
Table S4 List of the hypothetical crossover hotspots identified in this analysis.
Table S5 Positions and GC content of peaks showing typical centromeric‐like patterns of recombination and polymorphisms. The regions containing the predicted positions of centromeres proposed by King et al. (2015) are given in yellow.
Table S6 Motifs significantly over‐represented in genes located in recombination‐active sections (gene sequence in addition to 500‐bp downstream and upstream sequences).
Acknowledgements
The authors would like to acknowledge the Genotoul Bioinformatic Platform Toulouse Midi‐Pyrénées and the Sigenae group for providing help with the bioinformatic analysis and storage resources (http://bioinfo.genotoul.fr/). B.L. received a PhD fellowship from the French Research Ministry. Collaboration between the French National Institute for Agricultural Research (INRA) and The Roslin Institute was made possible by a grant provided by the Plant Health and Environment INRA division. R.D.H. and C.P. were supported by the Bioscience and Biotechnology Research Council (BBSRC) funds BB/J004235/1 and BB/J004324/1.
References
- Baird, N.A. , Etter, P.D. , Atwood, T.S. , Currey, M.C. , Shiver, A.L. , Lewis, Z.A. , Selker, E.U. , Cresko, W.A. and Johnson, E.A. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One, 3, e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat, F. , Buard, J. , Grey, C. , Fledel‐Alon, A. , Ober, C. , Przeworski, M. , Coop, G. and Massy, B.D. (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science, 327, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensasson, D. (2011) Evidence for a high mutation rate at rapidly evolving yeast centromeres. BMC Evol. Biol. 11, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron, S. , Madathiparambil, T. and Swanson, P.C. (2005) Both high mobility group (HMG)‐boxes and the acidic tail of HMGB1 regulate recombination‐activating gene (RAG)‐mediated recombination signal synapsis and cleavage in vitro . J. Biol. Chem. 280, 31 314–31 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakta, M.S. , Jones, V.A. and Vallejos, C.E. (2015) Punctuated distribution of recombination hotspots and demarcation of pericentromeric regions in Phaseolus vulgaris L.. PLoS One, 10, e0116822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachet, E. , Sommermeyer, V. and Borde, V. (2012) Interplay between modifications of chromatin and meiotic recombination hotspots. Biol. Cell, 104, 51–69. [DOI] [PubMed] [Google Scholar]
- Broman, K.W. , Wu, H. , Sen, S. and Churchill, G.A. (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics, 19, 889–890. [DOI] [PubMed] [Google Scholar]
- Brown, N.A. , Antoniw, J. and Hammond‐Kosack, K.E. (2012) The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis. PLoS One, 7, e33731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brygoo, Y. and Gautier, A. (2007) Molecular polymorphism of Fusarium strains isolated from wheat and corn ears in France Collects 2003 and 2004. Paper presented at the Conférence sur les Progrès et Perspectives de la Recherche sur les Mycotoxines de Fusarium dans les Céréales, Arcachon, France.
- Catchen, J.M. , Amores, A. , Hohenlohe, P. , Cresko, W. and Postlethwait, J.H. (2011) Stacks: building and genotyping loci de novo from short‐read sequences. G3 Genes Genomes Genet. 1, 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catcheside, D.E.A. (1981) Genes in Neurospora that suppress recombination when they are heterozygous. Genetics, 98, 55–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavinder, B. , Sikhakolli, U. , Fellows, K.M. and Trail, F. (2012) Sexual development and ascospore discharge in Fusarium graminearum . J. Vis. Exp. 61 , e3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B. and Campos, J.L. (2014) The relations between recombination rate and patterns of molecular variation and evolution in Drosophila . Annu. Rev. Genet. 48, 383–403. [DOI] [PubMed] [Google Scholar]
- Chen, Y. and Zhou, M.G. (2009) Sexual recombination of carbendazim resistance in Fusarium graminearum under field conditions. Pest Manag. Sci. 65, 398–403. [DOI] [PubMed] [Google Scholar]
- Comeron, J.M. , Ratnappan, R. and Bailin, S. (2012) The many landscapes of recombination in Drosophila melanogaster . PLoS Genet. 8, e1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, L.R. , Smith, K.M. and Freitag, M. (2013) The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet. 9, e1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll, D. and McDonald, B.A. (2012) The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 8, e1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll, D. , Lendenmann, M.H. , Stewart, E. and McDonald, B.A. (2015) The impact of recombination hotspots on genome evolution of a fungal plant pathogen. Genetics, 201, 1213–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumagun, C.J.R. and Miedaner, T. (2004) Segregation for aggressiveness and deoxynivalenol production of a population of Gibberella zeae causing head blight of wheat. Eur. J. Plant Pathol. 110, 789–799. [Google Scholar]
- Cuomo, C.A. , Guldener, U. , Xu, J.R. , Trail, F. , Turgeon, B.G. , Di Pietro, A. , Walton, J.D. , Ma, L.J. , Baker, S.E. , Rep, M. , Adam, G. , Antoniw, J. , Baldwin, T. , Calvo, S. , Chang, Y.L. , Decaprio, D. , Gale, L.R. , Gnerre, S. , Goswami, R.S. , Hammond‐Kosack, K. , Harris, L.J. , Hilburn, K. , Kennell, J.C. , Kroken, S. , Magnuson, J.K. , Mannhaupt, G. , Mauceli, E. , Mewes, H.W. , Mitterbauer, R. , Muehlbauer, G. , Münsterkötter, M. , Nelson, D. , O'Donnell, K. , Ouellet, T. , Qi, W. , Quesneville, H. , Roncero, M.I. , Seong, K.Y. , Tetko, I.V. , Urban, M. , Waalwijk, C. , Ward, T.J. , Yao, J. , Birren, B.W. and Kistler, H.C. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science, 317, 1400–1402. [DOI] [PubMed] [Google Scholar]
- Davey, J.W. , Hohenlohe, P.A. , Etter, P.D. , Boone, J.Q. , Catchen, J.M. and Blaxter, M.L. (2011) Genome‐wide genetic marker discovery and genotyping using next‐generation sequencing. Nat. Rev. Genet. 12, 499–510. [DOI] [PubMed] [Google Scholar]
- Davey, J.W. , Cezard, T. , Fuentes‐Utrilla, P. , Eland, C. , Gharbi, K. and Blaxter, M.L. (2013) Special features of RAD sequencing data: implications for genotyping. Mol. Ecol. 22, 3151–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. , del Angel, G. , Rivas, M.A. , Hanna, M. , McKenna, A. , Fennell, T.J. , Kernytsky, A.M. , Sivachenko, A.Y. , Cibulskis, K. , Gabriel, S.B. , Altshuler, D. and Daly, M.J. (2011) Additional data: a framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat. Genet. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S. , Raffaele, S. and Kamoun, S. (2015) The two‐speed genomes of filamentous pathogens: waltz with plants. Curr. Opin. Genet. Dev. 35, 57–65. [DOI] [PubMed] [Google Scholar]
- Duret, L. and Arndt, P.F. (2008) The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 4, e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galazka, J.M. and Freitag, M. (2014) Variability of chromosome structure in pathogenic fungi–of “ends and odds.” Curr. Opin. Microbiol. 20, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale, L.R. , Bryant, J.D. , Calvo, S. , Giese, H., Katan, T., O'Donnell, K., Suga, H., Taga, M., Usgaard, T.R., Ward, T.J. and Kistler, H.C. (2005) Chromosome complement of the fungal plant pathogen Fusarium graminearum based on genetic and physical mapping and cytological observations. Genetics, 171, 985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner, D.M. , Benfield, A.H. , Stiller, J. , Stephen, S. , Aitken, K. , Chunji, L. and Kazan, K. (2016) A high resolution genetic map of the wheat crown rot pathogen Fusarium pseudograminearum provides a near complete genome assembly. Mol. Plant Pathol. doi: 10.1111/mpp.12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard, C. , Chelysheva, L. , Choinard, S. , Froger, N. , Macaisne, N. , Lemhemdi, A. , Mazel, J. , Crismani, W. and Mercier, R. (2015) AAA‐ATPase FIDGETIN‐LIKE 1 and helicase FANCM antagonize meiotic crossovers by distinct mechanisms. PLoS Genet. 11, e1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, T. , Fournier, E. , Vautrin, D. , Solignac, M. , Vercken, E. , Bakan, B. and Brygoo, Y. (2002) Isolation of eight polymorphic microsatellite loci, using an enrichment protocol, in the phytopathogenic fungus Fusarium culmorum . Mol. Ecol. Notes, 2, 121–123. [Google Scholar]
- Goddard, M.R. , Godfray, H.C.J. and Burt, A. (2005) Sex increases the efficacy of natural selection in experimental yeast populations. Nature, 434, 636–640. [DOI] [PubMed] [Google Scholar]
- Gonen, S. , Lowe, N.R. , Cezard, T. , Gharbi, K. , Bishop, S.C. and Houston, R.D. (2014) Linkage maps of the Atlantic salmon (Salmo salar) genome derived from RAD sequencing. BMC Genomics, 15, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstadt, C.P.P. (2011) Is the control of recombination conserved among diverse eukaryotes? Heredity (Edinb.), 106, 710–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami, R.S. (2012) Targeted gene replacement in fungi using a split‐marker approach. Methods Mol. Biol. 835, 255–269. [DOI] [PubMed] [Google Scholar]
- Harris, L.J. , Balcerzak, M. , Johnston, A. , Schneiderman, D. and Ouellet, T. (2016) Host‐preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 120, 111–123. [DOI] [PubMed] [Google Scholar]
- Heinz, S. , Benner, C. , Spann, N. , Bertolino, E. , Lin, Y.C. , Laslo, P. , Cheng, J.X. , Murre, C. , Singh, H. and Glass, C.K. (2016) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff, S. , Ahmad, K. and Malik, H.S. (2001) The centromere paradox: stable inheritance with rapidly evolving DNA. Science, 293, 1098–1102. [DOI] [PubMed] [Google Scholar]
- Idnurm, A. , Hood, M.E. , Johannesson, H. and Giraud, T. (2015) Contrasted patterns in mating‐type chromosomes in fungi: hotspots versus coldspots of recombination. Fungal Biol. Rev. 29, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen‐Seaman, M.I. , Furey, T.S. , Payseur, B.A. , Lu, Y. , Roskin, K.M. , Chen, C.F. , Thomas, M.A. , Haussler, D. and Jacob, H.J. (2004) Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, R. , Urban, M. , Hammond‐Kosack, M. , Hassani‐Pak, K. and Hammond‐Kosack, K. (2015) The completed genome sequence of the pathogenic ascomycete fungus Fusarium graminearum . BMC Genomics, 16, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Lee, T. , Lee, Y.W. , Yun, S.H. and Turgeon, B.G. (2003) Shifting fungal reproductive mode by manipulation of mating type genes: obligatory heterothallism of Gibberella zeae . Mol. Microbiol. 50, 145–152. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Jurgenson, J.E. , Leslie, J.F. and Bowden, R.L. (2008) Alignment of genetic and physical maps of Gibberella zeae . Appl. Environ. Microbiol. 74, 2349–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendenmann, M.H. , Croll, D. , Stewart, E.L. and McDonald, B.A. (2014) Quantitative trait locus mapping of melanization in the plant pathogenic fungus Zymoseptoria tritici . G3 Genes Genomes Genetics, 4, 2519–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, J.F. and Summerell, B.A. (2006) The Fusarium Laboratory Manual. Ames, IA: Blackwell Publications. [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J.M. , Xayamongkhon, H. , Broz, K. , Dong, Y. , McCormick, S.P. , Abramova, S. , Ward, T.J. , Ma, Z.H. and Kistler, H.C. (2014) Temporal dynamics and population genetic structure of Fusarium graminearum in the upper Midwestern United States. Fungal Genet. Biol. 73, 83–92. [DOI] [PubMed] [Google Scholar]
- Limborg, M.T. , McKinney, G.J. , Seeb, L.W. and Seeb, J.E. (2016) Recombination patterns reveal information about centromere location on linkage maps. Mol. Ecol. Resour. 16, 655–661. [DOI] [PubMed] [Google Scholar]
- Lorenz, A. , Osman, F. , Sun, W. , Nandi, S. , Steinacher, R. and Whitby, M.C. (2012) The fission yeast FANCM ortholog directs non‐crossover recombination during meiosis. Science, 336, 1585–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L.J. , Van Der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.B. , Woloshuk, C. , Xie, X. , Xu, J.R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera, E. , Bourgon, R. , Brozzi, A. , Huber, W. and Steinmetz, L.M. (2008) High‐resolution mapping of meiotic crossovers and non‐crossovers in yeast. Nature, 454, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzano‐Winkler, B. , McGaugh, S.E. and Noor, M.A.F. (2013) How hot are Drosophila hotspots? Examining recombination rate variation and associations with nucleotide diversity, divergence, and maternal age in Drosophila pseudoobscura . PLoS One, 8, e71582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, B. and Linde, C. (2002) The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica, 124, 163–180. [Google Scholar]
- Mercier, R. , Mézard, C. , Jenczewski, E. , Macaisne, N. and Grelon, M. (2015) The molecular biology of meiosis in plants. Annu. Rev. Plant Biol. 66, 297–327. [DOI] [PubMed] [Google Scholar]
- Mezard, C. (2006) Meiotic recombination hotspots in plants. Biochem. Soc. Trans. 34, 531–534. [DOI] [PubMed] [Google Scholar]
- Mezard, C. , Jahns, M.T. and Grelon, M. (2015) Where to cross? New insights into the location of meiotic crossovers. Trends Genet. 31, 393–401. [DOI] [PubMed] [Google Scholar]
- Myers, S. , Bottolo, L. , Freeman, C. , McVean, G. and Donnelly, P. (2005) A fine‐scale map of recombination rates and hotspots across the human genome. Science, 310, 321–324. [DOI] [PubMed] [Google Scholar]
- Naef, A. and Défago, G. (2006) Population structure of plant‐pathogenic Fusarium species in overwintered stalk residues from Bt‐transformed and non‐transformed maize crops. Eur. J. Plant Pathol. 116, 129–143. [Google Scholar]
- Naef, A. , Senatore, M. and Défago, G. (2006) A microsatellite based method for quantification of fungi in decomposing plant material elucidates the role of Fusarium graminearum DON production in the saprophytic competition with Trichoderma atroviride in maize tissue microcosms. FEMS Microbiol. Ecol. 55, 211–220. [DOI] [PubMed] [Google Scholar]
- Noor, M.A.F. (2008) Connecting recombination, nucleotide diversity and species divergence in Drosophila. Fly, 2, 255–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen, K. and Petkov, P. (2010) Mammalian recombination hot spots: properties, control and evolution. Nat. Rev. Genet. 11, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaiokostas, C. , Bekaert, M. , Khan, M.G.Q. , Taggart, J.B. , Gharbi, K. , McAndrew, B.J. and Penman, D.J. (2013) Mapping and validation of the major sex‐determining region in Nile Tilapia (Oreochromis niloticus L.) using RAD Sequencing. PLoS One, 8, e68389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panday, A. and Grove, A. (2016) The high mobility group protein HMO1 functions as a linker histone in yeast. Epigenetics Chromatin, 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariaud, B. , Ravigné, V. , Halkett, F. , Goyeau, H. , Carlier, J. and Lannou, C. (2009) Aggressiveness and its role in the adaptation of plant pathogens. Plant Pathol. 58, 409–424. [Google Scholar]
- Petes, T.D. (2001) Meiotic recombination hot spots and cold spots. Nat. Rev. Genet. 2, 360–369. [DOI] [PubMed] [Google Scholar]
- Posada, D. , Crandall, K.A. and Holmes, E.C. (2002) Recombination in evolutionary genomics. Annu. Rev. Genet. 36, 75–97. [DOI] [PubMed] [Google Scholar]
- Raffaele, S. , Farrer, R.A. , Cano, L.M. , Studholme, D.J. , MacLean, D. , Thines, M. , Jiang, R.H. , Zody, M.C. , Kunjeti, S.G. , Donofrio, N.M. , Meyers, B.C. , Nusbaum, C. and Kamoun, S. (2010) Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science, 330, 1540–1543. [DOI] [PubMed] [Google Scholar]
- Roselius, K. , Stephan, W. and Städler, T. (2005) The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics, 171, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder, R. and Edwards, R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber, C.M.K. , Lee, W. , Wong, P. , Münsterkötter, M. , Mewes, H.W. , Schmeitzl, C. , Varga, E. , Berthiller, F. , Adam, G. and Güldener, U. (2014) The Fusarium graminearum genome reveals more secondary metabolite gene clusters and hints of horizontal gene transfer. PLoS One, 9, e110311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J.M. and Haigh, J. (1974) The hitch‐hiking effect of a favourable gene. Genet. Res. 23, 23. [PubMed] [Google Scholar]
- Sonnenberg, A.S.M. , Gao, W. , Lavrijssen, B. , Hendrickx, P. , Sedaghat‐Tellgerd, N. , Foulongne‐Oriol, M. , Kong, W.S. , Schijlen, E.G. , Baars, J.J. and Visser, R.G. (2016) A detailed analysis of the recombination landscape of the button mushroom Agaricus bisporus var. bisporus . Fungal Genet. Biol. 93, 35–45. [DOI] [PubMed] [Google Scholar]
- Spencer, C.C.A. , Deloukas, P. , Hunt, S. , Mullikin, J. , Myers, S. , Silverman, B. , Donnelly, P. , Bentley, D. and McVean, G. (2006) The influence of recombination on human genetic diversity. PLoS Genet. 2, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider, J. , Gardiner, D.M. , Thatcher, L.F. , Lyons, R. , Singh, K.B. , Manners, J.M. and Taylor, J.M. (2015) Genome‐wide analysis in three Fusarium pathogens identifies rapidly evolving chromosomes and genes associated with pathogenicity. Genome Biol. Evol. 7, 1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern, J.N. , Shafer, B.K. and McGill, C.B. (1995) DNA synthesis errors associated with double‐strand‐break repair. Genetics, 140, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talas, F. and McDonald, B. (2015a) Genome‐wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genomics, 16, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talas, F. and McDonald, B.A. (2015b) Significant variation in sensitivity to a DMI fungicide in field populations of Fusarium graminearum . Plant Pathol. 64, 664–670. [Google Scholar]
- Trail, F. (2009) For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 149, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, H.Y. , Robledo, D. , Lowe, N.R. , Bekaert, M. , Taggart, J.B. , Bron, J.E. and Houston, R.D. (2016) Construction and annotation of a high density SNP linkage map of the Atlantic salmon (Salmo salar) genome. G3 Genes Genomes Genetics, 6, 2173–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelgsang, S. , Widmer, F. , Jenny, E. and Enkerli, J. (2009) Characterisation of novel Fusarium graminearum microsatellite markers in different Fusarium species from various countries. Eur. J. Plant Pathol. 123, 477–482. [Google Scholar]
- Voss, H.H. , Bowden, R.L. , Leslie, J.F. and Miedaner, T. (2010) Variation and transgression of aggressiveness among two Gibberella zeae crosses developed from highly aggressive parental isolates. Phytopathology, 100, 904–912. [DOI] [PubMed] [Google Scholar]
- Ward, T.J. , Clear, R.M. , Rooney, A.P. , O'Donnell, K. , Gaba, D. , Patrick, S. , Starkey, D.E. , Gilbert, J. , Geiser, D.M. and Nowicki, T.W. (2008) An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet. Biol. 45, 473–484. [DOI] [PubMed] [Google Scholar]
- Waskiewicz, A. and Golinski, P. (2013) Mycotoxins in foods, feeds and their components. Krmiva, 55, 35–45. [Google Scholar]
- Wahls, W.P. and Davidson, M.K. (2010) Discrete DNA sites regulate global distribution of meiotic recombination. Trends Genet. 26, 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, P. , Walter, M. , Lee, W. , Mannhaupt, G. , Münsterkötter, M. , Mewes, H.W. , Adam, G. and Güldener, U. (2011) FGDB: revisiting the genome annotation of the plant pathogen Fusarium graminearum . Nucleic Acids Res. 39, D637–D639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeadon, P.J. , Koh, L.Y. , Bowring, F.J. , Rasmussen, J.P. and Catcheside, D.E.A. (2002) Recombination at his‐3 in Neurospora declines exponentially with distance from the initiator, cog. Genetics, 162, 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeadon, P.J. , Bowring, F.J. and Catcheside, D.E.A. (2004) Alleles of the hotspot cog are codominant in effect on recombination in the his‐3 region of Neurospora . Genetics, 167, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelina, N. , Diaz, P. , Lambing, C. and Henderson, I.R. (2015) Epigenetic control of meiotic recombination in plants. Sci. China‐Life Sci. 58, 223–231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
File S1 Dataset of markers. (A) Lab markers. (B) Restriction site‐associated DNA (RAD) markers. (C) Alignment of the genetic map with the reference genome.
File S2 Recombination and gene functions. (A) All protein‐coding genes described in the reference genome (RRES v4.0) with features. (B) Gene ontology enrichment of recombination‐active and desert sections. (C) Representation of specific gene lists within recombination‐active and desert sections
File S3 Supporting procedures.
File S4 Fasta sequences of the putative centromeres.
File S5 Development of targeted markers.
Table S1 List of markers exhibiting non‐Mendelian segregation.
Table S2 Overview of the supercontigs anchored during the RRes4.0 assembly proposal, with information concerning the marker used to anchor these supercontigs.
Table S3 List of the chromosomal segments classified as recombinant active.
Table S4 List of the hypothetical crossover hotspots identified in this analysis.
Table S5 Positions and GC content of peaks showing typical centromeric‐like patterns of recombination and polymorphisms. The regions containing the predicted positions of centromeres proposed by King et al. (2015) are given in yellow.
Table S6 Motifs significantly over‐represented in genes located in recombination‐active sections (gene sequence in addition to 500‐bp downstream and upstream sequences).