

Summary
We have developed EumicrobeDBLite—a lightweight comprehensive genome resource and sequence analysis platform for oomycete organisms. EumicrobeDBLite is a successor of the VBI Microbial Database (VMD) that was built using the Genome Unified Schema (GUS). In this version, GUS has been greatly simplified with the removal of many obsolete modules and the redesign of others to incorporate contemporary data. Several dependences, such as perl object layers used for data loading in VMD, have been replaced with independent lightweight scripts. EumicrobeDBLite now runs on a powerful annotation engine developed at our laboratory, called ‘Genome Annotator Lite’. Currently, this database has 26 publicly available genomes and 10 expressed sequence tag (EST) datasets of oomycete organisms. The browser page has dynamic tracks presenting comparative genomics analyses, coding and non‐coding data, tRNA genes, repeats and EST alignments. In addition, we have defined 44 777 core conserved proteins from 12 oomycete organisms which form 2974 clusters. Synteny viewing is enabled by the incorporation of the Genome Synteny Viewer (GSV) tool. The user interface has undergone major changes for ease of browsing. Queryable comparative genomics information, conserved orthologous genes and pathways are among the new key features updated in this database. The browser has been upgraded to enable user upload of GFF files for quick view of genome annotation comparisons. The toolkit page integrates the EMBOSS package and has a gene prediction tool. Annotations for the organisms are updated once every 6 months to ensure quality. The database resource is available at www.eumicrobedb.org.
Keywords: comparative genomics, database, genome browser, oomycetes, orthologous genes, toolkit
Introduction
Many oomycetes are destructive pathogens against crop plants, animals and humans, and pose a major threat to global food security (Dong et al., 2014; Pennisi, 2010). These pathogens were earlier believed to be fungi, mostly because of their morphology, but were later grouped under stramenopiles (Adhikari et al., 2013). The early progenitors of oomycetes have been proposed to be phototrophic brown algae which lost their ability to photosynthesize and became parasites (Tyler et al., 2006). Although many pathogens and parasites have undergone genome reduction, some oomycetes have undergone substantial genome expansion (Raffaele and Kamoun, 2012). There is significant lifestyle diversity among these pathogens, with some being obligatory biotrophs (e.g. Hyaloperonospora sp.; Baxter et al., 2010), some necrotrophs (e.g. many members of the family Pythiaceae), some hemibiotrophs (e.g. many Phytophthora species) and some saprophytes, exhibiting significant environmental adaptability. The genome sizes of oomycete pathogens vary substantially, with the smallest having a size of 37 Mb (Albugo laibachii) and the largest having a size of 240 Mb (Phytophthora infestans) (Pais et al., 2013).
Several oomycete pathogen genomes have been sequenced at different genome centres. However, most of the genome centres have created their own databases for dissemination of data, such as the Joint Genome Institute (JGI), Pythium Genome Database, Broad Institute, etc. Some of these existing databases are on the verge of retirement and also do not contain all the available oomycete genomes. For example, the Broad Institute's resources recently closed. FungiDB hosts many fungal and oomycete genomes, but is a very extensive resource, more appropriate for complete genomes having exhaustive functional annotation data. EumicrobeDB, however, is well suited for draft genomes that are still undergoing changes in terms of genome assembly and annotation. Changes made to a genome can be quickly and easily incorporated into EumicrobeDB. The data in this database have been integrated from different sources, and so the nomenclature followed by the different centres had to be unified. We have adopted a standard system of nomenclature that is applicable to all of the genomes. This system includes different assembly versions, annotations and the nomenclature of the features. The entire database package comprises ∼180 000 lines of code. The database resource is publicly available at www.eumicrobedb.org.
Results and Discussion
EumicrobeDB is publicly available at www.eumicrobedb.org. It has been significantly upgraded from its earlier version, VMD (VBI Microbial Database), in terms of functionality and content. Some of the advanced features are discussed below.
EumicrobeDB runs on Genome Annotator Lite
EumicrobeDBLite is an advanced version of VMD (Tripathy et al., 2006), with major changes in its architecture and functionality. VMD was built on the Genome Unified Schema (GUS), which was based on an Oracle framework and had many interdependent Bioperl modules for data integration and analysis. EumicrobeDBLite, however, is independent of proprietary software and external modules. It runs on a powerful genome analysis virtual machine, Genome Annotator Lite (GAL), developed at our laboratory at the Indian Institute of Chemical Biology, Kolkata, India (A. Panda et al., unpublished data). GAL is a powerful, yet lightweight, virtual machine with most of the open‐source genome annotation tools embedded within it (A. Panda et al., unpublished data). In addition, the data parsing scripts in GAL do not require Bioperl, as inclusion of Bioperl makes the installation of the package cumbersome. The workflow of GAL is illustrated in Fig. 1. The entire process of uploading a completely annotated genome takes about 4 h (for a genome with approximately 20 000 predicted genes). Alternatively, unannotated draft assemblies can be used as inputs that will be subsequently annotated, parsed and uploaded to the database by GAL; this process takes slightly longer, depending on the genome size and the amount of analysis needed.
Figure 1.
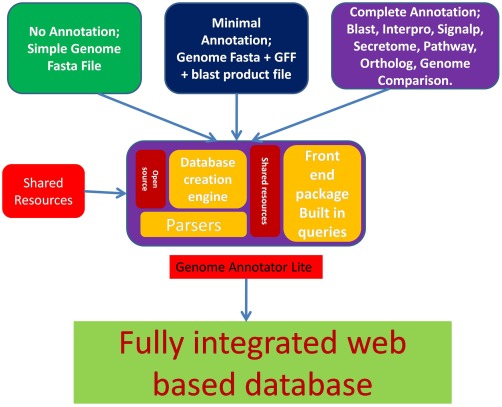
An overview of the GAL (Genome Annotator Lite) workflow. Three different types of data can be provided as input to GAL: unannotated, partially annotated and fully annotated. Depending on the input type, GAL determines the type of analysis needing to be run on the data. GAL has the unique capability of creating a database schema if it is not present already. It can also download shared resources from public databases, parse the data and upload them into EumicrobeDB.
Although genome sequencing technologies have been much improved, their annotation and visualization still remain a challenge (Yandell and Ence, 2012). When a genome is sequenced for the first time, the first and foremost step is to clean the reads and assemble them, and this is usually the most computationally intensive procedure. Currently, GAL does not handle genome assembly, as genome assembly can be extremely compute intensive for many genomes. However, it takes care of the downstream data annotation. Users can submit data to GAL either as a draft assembly in a partially annotated form or as a fully annotated assembly. GAL will automatically recognize the data type and determine the type of analysis that is required. Currently, EumicrobeDBLite contains analysed genomes of 26 oomycete pathogens, including one near‐complete genome of Phytophthora sojae (V5). Of the 26 listed genomes in EumicrobeDBLite, 21 genomes have been published, whereas five are unpublished genomes that have some restrictions on use, namely Phytophthora parasitica, Saprolegnia diclina, Aphanomyces astaci, Aphanomyces invadans and Phytophthora cinnamomi. These 26 organisms are from different orders of the phylum Oomycota, e.g. Albuginales (Albugo sp., white rusts), Peronosporales (including plant pathogens, such as Phytophthora sp. and downy mildews), Pythiales (Pythium sp., water moulds) and Saprolegniales (Saprolegnia sp., freshwater moulds; many animal pathogens). Some organisms, such as P. sojae, have several different assembly versions and one of the earlier versions (version 1) is still widely used by researchers. Therefore, we have included both genome versions of P. sojae (i.e. version 1 and version 5).
The details of the number of scaffolds, genes, genome size, organism prefix, etc. are available in the statistics link of the page (Table S1, see Supporting Information). All the publicly available expressed sequence tag (EST) sequences of oomycete pathogens with their genome alignment data are also integrated into the database.
EMBOSS analysis package is integrated into the database
The sequence analysis package EMBOSS is a powerful tool comprising 245 lightweight sequence analysis programs (Rice et al., 2000). Of these, we found 92 programs to be unsuitable or redundant in nature for web‐based applications. Therefore, we have incorporated the remaining 153 useful programs from the EMBOSS package into the toolkit section of this database. Several packages extremely useful for sequence analysis have been integrated into the oomycete genomes present in the database, so that the users can select the genomes of interest through a dropdown menu and perform the desired analysis on them. A few examples of these are listed in Table 1.
Table 1.
List of EMBOSS programs available as dropdown menus built on oomycete genomes that are listed in the gene detail page.
| Tool | Function | Dropdown available | Listed in gene detail page |
|---|---|---|---|
| banana | Plot bending and curvature data for B‐DNA | N | Y |
| biosed | Replace or delete sequence sections | Y | N |
| btwisted | Calculate twisting of B‐DNA | N | Y |
| cusp | Create a codon usage table from nucleotide sequences | N | Y |
| cpgplot | Identify and plot cpg islands in a DNA sequence | N | Y |
| cutseq | Remove a section from the sequence | Y | Y |
| degapseq | Remove non‐alphabetic characters from the sequence | Y | Y |
| Descseq | Alter the description of a sequence | Y | N |
| einverted | Finding inverted repeats in a sequence | Y | Y |
| entret | Retrieve sequence data from flat files and databases | Y | Y |
| extractseq | Extract regions of a sequence | Y | N |
| extractfeat | Extract features from a sequence | Y | N |
| eprimer3 | Pick PCR primers and hybridization oligos | N | Y |
| geecee | Calculate fractional GC content of nucleotide sequences | N | Y |
| listor | List of logical OR of two sequences | Y | N |
| maskambignuc | Mask ambiguous characters in a sequence | Y | N |
| maskseq | Mask a region of a sequence | Y | N |
| pasteseq | Insert one sequence into another | Y | N |
| prettyseq | Write a nucleotide sequence and its translation to a file | N | Y |
| plotorf | Plot open reading frames in a nucleotide sequence | N | Y |
| revseq | Reverse and complement a sequence | Y | Y |
| remap | Display restriction enzyme mapping sites in a nucleotide sequence | N | Y |
| seqcount | Count the number of sequences | Y | N |
| showpep | Show peptide of a sequence | Y | Y |
| trimseq | Remove unwanted characters from a sequence | Y | N |
| vectorstrip | Strip vectors | Y | N |
Many sequence utility programs can be accessed from the gene detail page (Table 1). These are linked to the gene models, so that, with a single click, users can run an analysis using the nucleotide or the protein sequence as automatic input. If the user chooses to run more than one analytical tool from this page, the outputs are arranged in a tab separated menu. A ‘clear all’ option is available to remove all analysis results from the page.
The analytical interfaces used in EumicrobeDB provide a very simple and intuitive way to quickly run a variety of sequence analysis programs. The existing oomycete databases, such as FungiDB (Stajich et al., 2012), JGI's Mycocosm (Grigoriev et al., 2014), the Pythium functional genomics database at Michigan State University (Hamilton et al., 2011), etc., disseminate pre‐computed genomics and comparative genomics data, but lack this feature for users to access a web‐based analysis platform.
Manually curated datasets for oomycete genomes are available in EumicrobeDB
In addition to the automated genome annotations, we have also carried out extensive semi‐automated annotation and data curation of the oomycete genomes. One such curated data resource is the Core Orthologue dataset. The Core Orthologue dataset was obtained after comparing the entire proteomes of 12 representative members belonging to four different orders, namely: Albugo laibachii Nc14 belonging to order Albuginales; P. sojae P6497, Phytophthora ramorum Pr102, P. infestans T30‐4, Phytophthora capsici LT1534, P. parasitica INRA‐310, Hyaloperonospora arabidopsidis Emoy2, Plasmopara halstedii, Phytopythium vexans DAOMBR484 (belonging to Peronosporales); Pythium ultimum DAOMBR144 (V1) (belonging to order Pythiales); and Saprolegnia parasitica CBS223.65 (V1) and Aphanomyces invadans (belonging to order Saprolegniales).
The Oomycete Molecular Genetics Network (OMGN at omgn.org) has conducted genome sequence jamborees for a number of newly released oomycete genomes. In these jamborees, community scientist members come together and manually annotate the data; the manual annotation effort often continues after the jamboree also. Over the last several years, we have collected these annotation data from community members and incorporated them into this database.
Core Orthologue dataset for oomycete pathogens
Although many oomycete genomes are available, an organized effort to generate a core proteome was needed. We generated a comprehensive core proteome by choosing 12 representative oomycetes. As all the genomes available currently are draft genomes, an attempt to include all 26 genomes for this study would have resulted in no core dataset.
Initially, we generated pairwise bidirectional best blast hits from these 12 proteomes (207 636 total protein sequences), followed by orthoMCL (Fischer et al., 2011), producing a set of core orthologues. We also performed an orthologue search using multiparanoid (Alexeyenko et al., 2006) for comparison. The total numbers of core orthologues generated by multiparanoid and orthoMCL were comparable with each other, i.e. 2862 and 2974, respectively. We used the core set generated by OrthoMCL as a number of clusters produced by multiparanoid were not very reliable. The clusters generated by OrthoMCL encompassed 44 777 protein sequences. The largest cluster had about 275 members (group 1) belonging to the ABC transporter superfamily (Tables S2, S3; Fig. S1, see Supporting Information).
Of the 2974 clusters generated using OrthoMCL, annotations of the individual clusters were obtained from the COG IDs of their members. Only about 1894 had COG annotations and the remaining 1080 were unique to this group of organisms. The greatest numbers of groups were annotated as hypothetical protein, unnamed protein product or conserved hypothetical protein. Among the annotated group members, the predominant classes of protein families were protein kinase (52 groups), ATP binding cassette (49 groups), transmembrane protein (49 groups), vacuolar associated proteins (31 groups) and serine protease family (31 groups) (Fig. S1). The orthologue clusters are available for search through the query page in eumicrobedb.org using two different options, namely: query by cluster_ID or by annotation of clusters. Cluster information and the tree structure for a protein are also available on the protein's gene detail page. A pre‐computed cluster analysis for core orthologues is a very valuable resource for inferring the biological role and phylogeny of a protein sequence (Yang et al., 2015).
We generated an HMM profile of each of the orthologue cores using HMMER 3.1 (Johnson et al., 2010). Then, the database containing the protein sequences of the remaining 14 organisms (222 582 sequences) was searched against the HMM profiles using hmmsearch with a cut‐off of 1e‐05. A matrix was generated from the output with present calls as ‘1’ and absent calls as ‘0’. The distance between a pair of gene clusters in two genomes was calculated using the Jaccard distance method, followed by single, complete and average clustering methods implemented in the Vegan Package in R (Scaria et al., 2015). The Jaccard distance has been used widely to examine genome fluidity. A value close to zero indicates no difference between two genomes. We computed Jaccard distances between the set of 12 organisms as one group and the single genomes of the remaining 14 organisms as the other (Table S4, see Supporting Information; Fig. 2). Phytophthora taxon totara diverged least from the cluster, whereas most of the Pythium sp. diverged the most. Some of these divergences may reflect variations in the quality of the respective assemblies and annotations. Phytophthora sojae (V1) recorded a very high distance (range of 0.04–0.05) compared with the others, indicating that it is the least complete genome (Table S4).
Figure 2.

Heat map of the Jaccard distance between the core group and the remaining 14 organisms using single, complete and average linkage clustering employing the Vegan package in R.
User‐annotated genes and effectors
Effectors are virulence proteins that enter host cells to promote infection. They undergo rapid evolution to evade detection by the host resistance machinery (Jiang et al., 2008). As their rate of adaptation is very high, the sequences undergo rapid changes in composition. Gene predictors are therefore often unable to successfully predict these genes in a draft genome. The prediction of effectors requires the use of HMM searches and manual intervention in many cases. Most of the oomycete Arg‐x‐Leu‐Arg (RXLR) effectors have been curated by community users manually in conjunction with a variety of prediction strategies (Jiang et al., 2008). For those species with manually curated effector sets (P. sojae, P. ramorum and H. arabidopsidis), we have replaced the electronically annotated effector sets with the manually curated gene models in this database version. Presently, there are about 125 curated RxLR effectors from H. arabidopsidis, 370 curated RxLR effectors from P. ramorum, 396 curated RxLR effectors from P. sojae (V1) and 385 curated RxLR effectors from P. sojae (V5) (Table 2). There are 1898 user‐annotated non‐effector gene models in the database. The user‐predicted gene models, as well as the gene models reviewed by the community reviewers, are colour coded in the browser (Fig. 3B). Earlier versions of EumicrobeDB (VMD) had a user annotation interface. However, users prefer to send bulk annotations in excel files rather than filling in gene details one by one in the web‐based forms. Therefore, we have taken off this feature in this version. Now, the researchers can send us the gene‐related information through the ‘contact us’ page.
Table 2.
Number of curated RxLR effectors in EumicrobeDB.
| Organism | Total |
|---|---|
| Phytophthora sojae P6497 (V1) | 396* |
| Phytophthora sojae P6497 (V5) | 385* |
| Hyaloperonospora arabidopsidis Emoy2 | 125*, † |
| Phytophthora ramorum Pr102 (V1) | 370* |
| Phytophthora cinnamomi CBS 144.22 | 8 |
| Phytophthora capsici LT1534 | 159 |
| Phytophthora infestans T30‐4 | 563 ‡ |
*Manually curated.
†Does not include 242 RxLR‐like (RxLL) proteins judged to be poor quality effector candidates or 22 crinkler genes with RxLR strings (RxLCRN genes).
‡From Haas et al. (2009).
Figure 3.
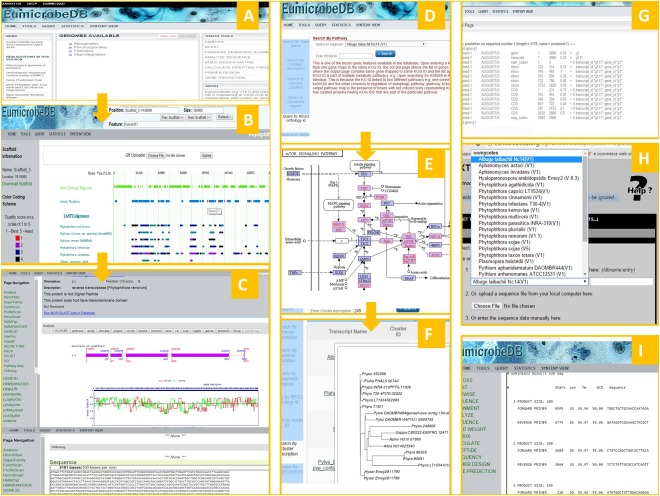
Graphical user interface of EumicrobeDB. Screenshots are shown in each case. The home page (A) opens to a tree view list of the genomes present in EumicrobeDB. The genome browser page (B) opens showing the default scaffold with all available tracks. The tracks are clickable and clicking on a coding region opens to the gene detail page (C). Query items include KEGG (Kyoto Encyclopedia of Genes and Genomes) ID (D), which leads to a pathway map (E), where the coloured members are the euKaryotic Ortholog Groups (KOG) of proteins present in the query organism for that pathway. The cluster query output (F) shows the phylogenetic relationship between the genes from the same cluster. The gene prediction output (G), extract sequence with pull down genome menu (H) and output of primer design (I) are all part of the new toolkit menu.
Data visualization interface has many additional features
The data visualization interface of the database has five major components, namely genome browser, gene detail page, genome synteny page, query page and toolkit page. Several other accessory components, such as statistics page, download page and tutorial pages, are also available. The gene detail page is the central part of the user interface in which detailed annotation and analytical information is available for a gene. All the other pages eventually link to the gene detail page. A brief overview of the user interface is summarized in Fig. 3.
Synteny viewer
The newly created synteny viewer is based on Genome Synteny Viewer (GSV) (Revanna et al., 2011). Genome synteny was computed by running an all‐versus‐all comparison among all pairs of oomycete genomes using Lastz (Harris, 2007). The user interface has been modified so that it is intuitive for new users. For example, users can choose to see the highly syntenic regions between a pair of organisms by just clicking on the ‘check synteny’ option. The scaffolds that display the most synteny between a pair of organisms will be listed on the page. Syntenic regions are displayed only if at least 10 000 bases are syntenic and the insertions and deletions cover less than 5% of the matching length (Fig. 4).
Figure 4.
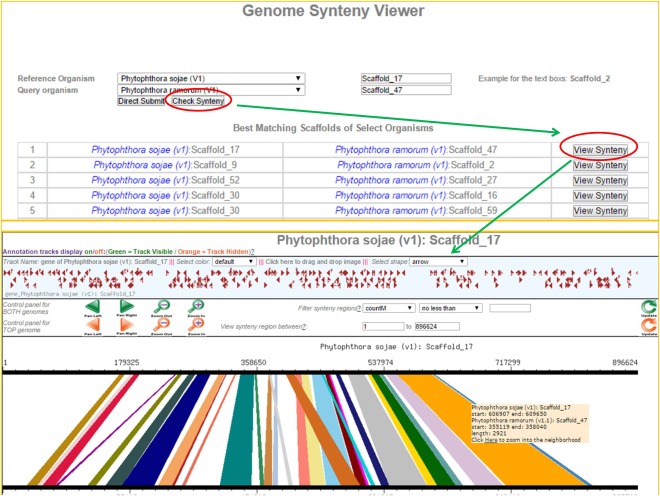
Scaffold synteny comparison. (A) Screenshot of the ‘check synteny option’ between two specified scaffolds, which is one of two synteny query options (the other is querying the best syntenic regions for a particular scaffold, see Fig. 4). A query is depicted between Scaffold_17 of Phytophthora sojae V1 and Scaffold_47 of Phytophthora ramorum V1. (B) All possible syntenic regions are listed for the user to choose to visualize synteny between a pair of scaffolds.
Genome browser
The genome browser serves as one of the entry points to this database from the main page. The organism list is arranged in a tree view format with taxonomic hierarchy, e.g. orders → genus → species → strains (if available) and in a list view format. On clicking an organism name, the browser page opens to the default scaffold page (largest scaffold) with default scaffold region (1–150 000). The uppermost green‐coloured track represents non‐coding DNA sequences. This track is very useful when users are interested in retrieving upstream or downstream regions of a coding sequence.
Non‐coding regions of the genomes are particularly interesting in the context of the ENCODE Project Consortium (2012) project. Natural selection plays a very important role in determining the virulence of a pathogen and may act on non‐coding as well as coding regions of the genome (Rech et al., 2014). By offering the clickable non‐coding track, researchers can quickly analyse the non‐coding regions.
The next blue‐coloured track identifies coding regions with introns and exons plotted as pointed rectangles indicating their orientation. The gene model is coloured red when a community researcher has either reviewed or curated it. There are other feature tracks, such as repeats and tRNA tracks, available currently. We have added a new feature into the browser tracks, namely the conserved region track. The pre‐computed comparative genomics regions between the genomes are quality sorted and represented in five different colours on these tracks. The best‐quality conserved regions are ranked as ‘1’ (colour coded in red) and the poorest quality conserved regions are ranked as ‘5’ (please refer to Experimental procedures for details of the scoring schemes). On ‘mouse‐over’ on the conserved region tracks, the scaffold location of the conserved region pops up in a text box. This track is clickable and opens to a page containing the list of coding transcripts present in that region. Next to these tracks are the EST BLAT alignment tracks showing regions mapping between the reference genome and the assembled ESTs.
The genome browser and genome synteny viewer offer a quick and easy way to explore regions of a genome in which genes are co‐localized or in which there is a repeat‐mediated expansion of the genome (Figs 5 and 6).
Figure 5.
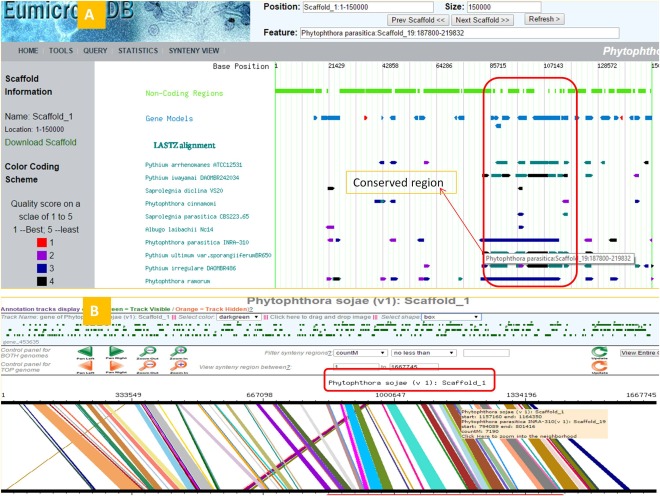
Scaffold genome‐wide synteny query. (A) Screenshot of browser page showing synteny between Scaffold_19 of Phytophthora parasitica with conserved regions of other oomycete genomes. (B) Phytophthora sojae Scaffold_1 shows significant synteny with Scaffold_19 of P. parasitica.
Figure 6.
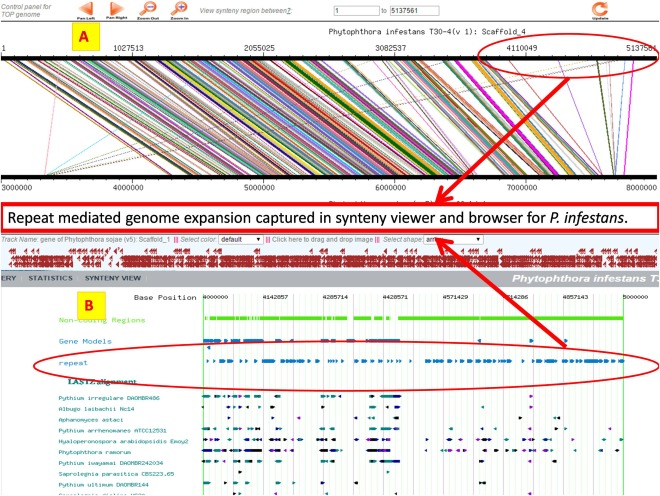
Repeat‐mediated genome expansion in Phytophthora infestans illustrated with the synteny viewer and browser. (A) Synteny view of P. infestans Scaffold_4 aligned with Phytophthora sojae V5 Scaffold_1. (B) Browser view of P. infestans showing region of Scaffold_4: 4 000 000–5 000 000.
Users can now upload data in GFF format into the browser for quick visualization (Template file provided in download page at http://www.eumicrobedb.org/uploadable_gff/). The coding tracks of the user‐uploaded GFF tracks are clickable.
Query page
The updated query page is frame based, where different kinds of queries can be carried out by clicking on the left menu item. Each query result is then stored in the browser cache for easy data browsing. The query options include: query by gene_id, query by primary and secondary annotation, query by gene ontology (GO) annotation, query by cluster_ID, query by cluster annotation, EST query, synteny query, query by KEGG orthology ID, etc. Bulk data download for the secretomeP package (Prop, SignalP, TMHMM, Psort) is available via this page. Query outputs for genes open into the relevant gene detail page. Query results that contain multiple transcripts or genes open as a list in which each item is linked to the relevant gene detail page. ‘Query by genome location’ retrieves a list of features that occur within the queried region, together with links to the relevant gene pages. ‘Query by conserved region’ is a new feature that is similar to ‘query by genome location’, except that the output is a set of features contained in regions of other genomes that are conserved with the query region. ‘Query by KEGG Orthology’ results in a list of the genes tagged to the KEGG ID query. On clicking the KEGG IDs, the pathway image maps appear with coloured EC numbers. These coloured EC numbers indicate members of the pathway present in the reference genome.
Gene detail page
The gene detail page, or main annotation page, contains detailed information about a gene. This page has a summary header containing brief information on the gene/transcript. As this page contains a long list of information, quick links to different features are provided in the left panel. Log‐likelihood (McLachlan et al., 1984) and Fickett statistics (Fickett, 1982) plots of the genes are computed on‐the‐fly using pre‐computed codon usage values. These plots help to validate the correctness of the predicted gene model. The gene model plot on the top is clickable and the page leads to the translated CDS and nucleotide sequences for the gene. A new analytical feature is incorporated in the top panel that runs some of the sequence analysis programs chosen from EMBOSS. The users can click on the tools and the gene sequence in the page will be used as the input for the program and output will be displayed on the same page.
Toolkit page
The toolkit page is the sequence analysis interface of EumicrobeDB with many useful open‐source as well as in‐house tools for sequence analysis. blast, pairwise sequence comparison and the EMBOSS interface are a few of the most useful packages that are part of this suite. From the EMBOSS package, 150 sequence analysis programs are integrated with the 26 oomycete genome sequences. The inputs to many of these programs can be selected from the dropdown menu box and analysis can be carried out directly. By integrating open‐source sequence analysis packages that normally exist as stand‐alone packages, EumicrobeDB provides a convenience that is invaluable for biologists.
Another very useful feature added to the toolkit is the integration of the gene prediction software Augustus (Stanke et al., 2008). We have refined training datasets for each of the 26 oomycete genomes, and users can choose the training dataset of their choice to predict coding regions from an unknown stretch. This is one of the most useful features for the research community.
Experimental Procedures
Genomes were downloaded from genome centre websites. The sources and origins of the genomes are listed on the statistics page of the EumicrobeDB website and EST datasets were downloaded from GenBank.
Data processing
Genome sequence files and gene names have been renamed with genus, species and strain prefixes for uniformity. For genomes such as P. sojae, in which more than one assembly version is available, we have appended the assembly version to the genus_species_strain prefix. For unification of scaffold nomenclature, we have size‐sorted scaffolds and numbered them in descending order from larger to smaller; thus, each largest scaffold is named as Scaffold_1. For some organisms, such as H. arabidopsidis and P. infestans, in which the scaffolds already followed this rule, we kept the old name. A map file is provided at www.eumicrobedb.org/ForEMBOSS/for comparing the old with the new names. Genes and genome prefixes are listed on the statistics page.
We analysed and annotated 406 500 protein coding genes from these 26 oomycete organisms. blastx (Altschul et al., 1990) against the National Center for Biotechnology Information's nonredundant (NCBI's nr) database was used to assign putative primary annotations to the genes. We ran interproscan (Zdobnov and Apweiler, 2001) annotation for predicting domains and GO features. Pathway and KOG prediction was performed using the KAAS annotation server from KEGG (Moriya et al., 2007). In addition, we created an entire secretome repertoire using SignalP 3.0 (Petersen et al., 2011) and the secretomeP (Bendtsen et al., 2004) package; the latter includes Prop (Duckert et al., 2004) (prediction of proprotein convertase sites), Psort (Horton and Nakai, 1997) (prediction of protein subcellular locations) and TMHMM (Krogh et al., 2001).
To create a core proteome of the oomycetes, we used 12 oomycete organisms as described in the Results and Discussion section. Clusters of orthologous proteins were extracted from 12 organisms using OrthoMCL version 2.0.9 with default parameters. A total of 207 636 proteins was clustered into 22 592 groups. Core orthologues were defined as the 2974 orthologous groups present in all 12 organisms. In order to detect core orthologues in the remaining 14 organisms (of a total of 26), we performed a profile search based on HMMER. Each of the 2974 clusters contained multiple proteins and were separated into individual clusters. Profiles were built from each of the 2974 clusters and an HMMSearch was carried out against these 2974 profiles for all the 14 other organisms that were not part of the cluster building. A matrix of ‘1s’ and ‘0s’ was constructed for the 25 organisms consisting of 2974 rows. This matrix was then converted into a distance matrix using the Jaccard distance implemented in the Vegan package in R. Hierarchical clustering was performed on the resulting distance matrix using ‘single’, ‘complete’ and ‘average’ linkage clustering as implemented in Vegan. Heat maps were drawn to visualize the presence/absence with the stats package in R. Annotations of the clusters were manually edited to fit into a broader category before uploading into tagcrowd.com for the generation of word clouds.
We built phylogenetic relationships among these proteins with ClustalW (Thompson et al., 2002) and mega (Tamura et al., 2013). The multiple sequence alignment and the tree features are available in the gene detail page.
We included secretomeP analysis for the prediction of non‐signal peptide secretory proteins.
Whole genome synteny analysis was performed by running all‐versus‐all whole genome comparisons using LastZ (Harris, 2007). EST datasets were cleaned using in‐house scripts, clustered and assembled using TGICL (Pertea et al., 2003). EST contig alignment to genome assemblies was performed using BLAT (Kent, 2002).
Comparative genomics module
We aligned all the existing 26 genomes against each other (624 runs) using Lastz, a package that handles pairwise sequence alignments (Harris, 2007). We performed chained, gapped alignments with the default mismatch count (<50 mismatches) for Lastz over windows of 1000 bases. The alignments were further filtered into five categories with the best being ‘1’ and the worst being ‘5’. The best matches have a matching region of over 10 000 bases with <5% mismatches and gaps. Second best matches have >1000 bases and <10 000 bases matching region with <5% mismatches and gaps. The third category is for matching regions over 1000 bases with mismatches and gaps >5% and <10%. The fourth category is for matching regions over 1000 bases with mismatches >10% and <15%. The remainder are category 5.
Future Directions
Many new oomycete genomes are being sequenced at several genome centres. We are on our way to collecting the publicly available genomes into this database in the next release.
Author Contributions
S.T. and B.M.T. designed the project and wrote the manuscript; S.T., M.M.C., D.S., A.P., G.P.M. and A.G. analysed the data; A.P., A.G. and S.T. uploaded the data; A.P. designed and created the front end; W.W.Y. and B.M.T. provided curated lists of RxLR effectors.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Fig. S1 Word cloud for 100 most frequent words in the annotation file for 2974 core groups computed using OrthoMCL with 12 representative members.
{kind=link}
Table S1 Gene statistics of all the members present in EumicrobeDB.
Table S2 2974 clusters generated with OrthoMCL with their COG IDs and annotations.
Table S3 Members of each of the 2974 clusters with organism IDs, gene_ids, cluster_id and annotation.
Table S4 Distance matrix showing Jaccard distance between the cluster of 12 organisms with other members.
Acknowledgements
Financial support from a DBT‐RLS fellowship, Department of Biotechnology, and CSIR‐Genesis, Government of India, to S.T. is gratefully acknowledged. This work was supported in part by grants to B.M.T. from the US Department of Agriculture, National Institute of Food and Agriculture (#2007‐35600‐18530 and #2011‐68004‐30104) and US National Science Foundation (#MCB‐0731969).
Contributor Information
Brett M. Tyler, Email: Brett.Tyler@oregonstate.edu
Sucheta Tripathy, Email: tsucheta@iicb.res.in, Email: tsucheta@gmail.com.
References
- Adhikari, B.N. , Hamilton, J.P. , Zerillo, M.M. , Tisserat, N. , Levesque, C.A. and Buell, C.R. (2013) Comparative genomics reveals insight into virulence strategies of plant pathogenic oomycetes. PLoS One, 8, e75072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyenko, A. , Tamas, I. , Liu, G. and Sonnhammer, E.L. (2006) Automatic clustering of orthologs and inparalogs shared by multiple proteomes. Bioinformatics, 22, e9–e15. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Baxter, L. , Tripathy, S. , Ishaque, N. , Boot, N. , Cabral, A. , Kemen, E. , Thines, M. , Ah‐Fong, A. , Anderson, R. , Badejoko, W. , Bittner‐Eddy, P. , Boore, J.L. , Chibucos, M.C. , Coates, M. , Dehal, P. , Delehaunty, K. , Dong, S. , Downton, P. , Dumas, B. , Fabro, G. , Fronick, C. , Fuerstenberg, S.I. , Fulton, L. , Gaulin, E. , Govers, F. , Hughes, L. , Humphray, S. , Jiang, R.H. , Judelson, H. , Kamoun, S. , Kyung, K. , Meijer, H. , Minx, P. , Morris, P. , Nelson, J. , Phuntumart, V. , Qutob, D. , Rehmany, A. , Rougon‐Cardoso, A. , Ryden, P. , Torto‐Alalibo, T. , Studholme, D. , Wang, Y. , Win, J. , Wood, J. , Clifton, S.W. , Rogers, J. , Van den Ackerveken, G. , Jones, J.D. , McDowell, J.M. , Beynon, J. and Tyler, B.M. (2010) Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science, 330, 1549–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen, J.D. , Nielsen, H. , Von Heijne, G. and Brunak, S. (2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. [DOI] [PubMed] [Google Scholar]
- Dong, S. , Stam, R. , Cano, L.M. , Song, J. , Sklenar, J. , Yoshida, K. , Bozkurt, T.O. , Oliva, R. , Liu, Z. , Tian, M. , Win, J. , Banfield, M.J. , Jones, A.M. , van der Hoorn, R.A. and Kamoun, S. (2014) Effector specialization in a lineage of the Irish potato famine pathogen. Science, 343, 552–555. [DOI] [PubMed] [Google Scholar]
- Duckert, P. , Brunak, S. and Blom, N. (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickett, J.W. (1982) Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 10, 5303–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, S. , Brunk, B.P. , Chen, F. , Gao, X. , Harb, O.S. , Iodice, J.B. , Shanmugam, D. , Roos, D.S. and Stoeckert, C.J. (2011) Using OrthoMCL to assign proteins to OrthoMCL‐DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics, Chapter 6, Unit 6 12 1‐9. Available at 10.1002/0471250953.bi0612s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev, I.V. , Nikitin, R. , Haridas, S. , Kuo, A. , Ohm, R. , Otillar, R. , Riley, R. , Salamov, A. , Zhao, X. , Korzeniewski, F. , Smirnova, T. , Nordberg, H. , Dubchak, I. and Shabalov, I. (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42, D699–D704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, J.P. , Neeno‐Eckwall, E.C. , Adhikari, B.N. , Perna, N.T. , Tisserat, N. , Leach, J.E. , Lévesque, C.A. and Buell, C.R. (2011) The Comprehensive Phytopathogen Genomics Resource: a web‐based resource for data‐mining plant pathogen genomes. Database (Oxford), 2011, bar053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, R.S. (2007) Improved pairwise alignment of genomic DNA. Ph.D Thesis, Pennsylvania State University, University Park, PA.
- Horton, P. and Nakai, K. (1997) Better prediction of protein cellular localization sites with the k nearest neighbors classifier. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 147–152. [PubMed] [Google Scholar]
- Jiang, R.H. , Tripathy, S. , Govers, F. and Tyler, B.M. (2008) RXLR effector reservoirin two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc. Natl. Acad. Sci. USA, 105, 4874–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, L.S. , Eddy, S.R. and Portugaly, E. (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics, 11, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J. (2002) BLAT–the BLAST‐like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , Von Heijne, G. and Sonnhammer, E.L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. [DOI] [PubMed] [Google Scholar]
- McLachlan, A.D. , Staden, R. and Boswell, D.R. (1984) A method for measuring the non‐random bias of a codon usage table. Nucleic Acids Res. 12, 9567–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya, Y. , Itoh, M. , Okuda, S. , Yoshizawa, A.C. and Kanehisa, M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais, M. , Win, J. , Yoshida, K. , Etherington, G.J. , Cano, L.M. , Raffaele, S. , Banfield, M.J. , Jones, A. , Kamoun, S. and Saunders, D.G. (2013) From pathogen genomes to host plant processes: the power of plant parasitic oomycetes. Genome Biol. 14, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi, E. (2010) Armed and dangerous. Science, 327, 804–805. [DOI] [PubMed] [Google Scholar]
- Pertea, G. , Huang, X. , Liang, F. , Antonescu, V. , Sultana, R. , Karamycheva, S. , Lee, Y. , White, J. , Cheung, F. , Parvizi, B. , Tsai, J. and Quackenbush, J. (2003) TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics, 19, 651–652. [DOI] [PubMed] [Google Scholar]
- Petersen, T.N. , Brunak, S. , Von Heijne, G. and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods, 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Raffaele, S. and Kamoun, S. (2012) Genome evolution in filamentous plant pathogens: why bigger can be better. Nat. Rev. Microbiol. 10, 417–430. [DOI] [PubMed] [Google Scholar]
- Rech, G.E. , Sanz‐Martín, J.M. , Anisimova, M. , Sukno, S.A. and Thon, M.R. (2014) Natural selection on coding and noncoding DNA sequences is associated with virulence genes in a plant pathogenic fungus. Genome Biol. Evol. 6, 2368–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revanna, K.V. , Chiu, C.C. , Bierschank, E. and Dong, Q. (2011) GSV: a web‐based genome synteny viewer for customized data. BMC Bioinformatics, 12, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, I. and Bleasby, A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Scaria, J. , Suzuki, H. , Ptak, C.P. , Chen, J.W. , Zhu, Y. , Guo, X.K. and Chang, Y.F. (2015) Comparative genomic and phenomic analysis of Clostridium difficile and Clostridium sordellii, two related pathogens with differing host tissue preference. BMC Genomics, 16, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich, J.E. , Harris, T. , Brunk, B.P. , Brestelli, J. , Fischer, S. , Harb, O.S. , Kissinger, J.C. , Li, W. , Nayak, V. , Pinney, D.F. , Stoeckert, C.J. and Roos, D.S. (2012) FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 40, D675–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Diekhans, M. , Baertsch, R. and Haussler, D. (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics, 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. and Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D. , Gibson, T.J. and Higgins, D.G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics, Chapter 2, Unit 2 3. Available at 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- Tripathy, S. , Pandey, V.N. , Fang, B. , Salas, F. and Tyler, B.M. (2006) VMD: a community annotation database for oomycetes and microbial genomes. Nucleic Acids Res. 34, D379–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler, B.M. , Tripathy, S. , Zhang, X. , Dehal, P. , Jiang, R.H. , Aerts, A. , Arredondo, F.D. , Baxter, L. , Bensasson, D. , Beynon, J.L. , Chapman, J. , Damasceno, C.M. , Dorrance, A.E. , Dou, D. , Dickerman, A.W. , Dubchak, I.L. , Garbelotto, M. , Gijzen, M. , Gordon, S.G. , Govers, F. , Grunwald, N.J. , Huang, W. , Ivors, K.L. , Jones, R.W. , Kamoun, S. , Krampis, K. , Lamour, K.H. , Lee, M.K. , McDonald, W.H. , Medina, M. , Meijer, H.J. , Nordberg, E.K. , Maclean, D.J. , Ospina‐Giraldo, M.D. , Morris, P.F. , Phuntumart, V. , Putnam, N.H. , Rash, S. , Rose, J.K. , Sakihama, Y. , Salamov, A.A. , Savidor, A. , Scheuring, C.F. , Smith, B.M. , Sobral, B.W. , Terry, A. , Torto‐Alalibo, T.A. , Win, J. , Xu, Z. , Zhang, H. , Grigoriev, I.V. , Rokhsar, D.S. and Boore, J.L. (2006) Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science, 313, 1261–1266. [DOI] [PubMed] [Google Scholar]
- Yandell, M. and Ence, D. (2012) A beginner's guide to eukaryotic genome annotation. Nat. Rev. Genet. 13, 329–342. [DOI] [PubMed] [Google Scholar]
- Yang, L. , Tan, J. , O'Brien, E.J. , Monk, J.M. , Kim, D. , Li, H.J. , Charusanti, P. , Ebrahim, A. , Lloyd, C.J. , Yurkovich, J.T. , Du, B. , Dräger, A. , Thomas, A. , Sun, Y. , Saunders, M.A. and Palsson, B.O. (2015) Systems biology definition of the core proteome of metabolism and expression is consistent with high‐throughput data. Proc. Natl. Acad. Sci. USA, 112, 10 810–10 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. (2001) InterProScan—an integration platform for the signature‐recognition methods in InterPro. Bioinformatics, 17, 847–848. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website.
Fig. S1 Word cloud for 100 most frequent words in the annotation file for 2974 core groups computed using OrthoMCL with 12 representative members.
Table S1 Gene statistics of all the members present in EumicrobeDB.
Table S2 2974 clusters generated with OrthoMCL with their COG IDs and annotations.
Table S3 Members of each of the 2974 clusters with organism IDs, gene_ids, cluster_id and annotation.
Table S4 Distance matrix showing Jaccard distance between the cluster of 12 organisms with other members.