

SUMMARY
Root‐knot nematodes (RKNs, Meloidogyne spp.) are sedentary biotrophic pathogens that establish within the vascular cylinder of plant roots, forming a gall and inducing several feeding cells, giant cells (GCs), essential for completion of their life cycle. GCs suffer gene expression changes, repeated mitosis and endoreduplication events. Transcriptomics has revealed that an extensive down‐regulation of transcripts, a molecular signature of early‐developing galls and GCs that is conserved in tomato and Arabidopsis, may be achieved through small RNA (sRNA) gene silencing pathways. The role of some microRNAs (miRNAs) in plant–RKN interactions has recently been addressed, but little is known about the regulatory roles of other sRNA types. Here, we perform a differential accumulation analysis to show which repeat‐associated small interfering RNAs (rasiRNAs) are distinctive or enriched in early Arabidopsis galls vs. uninfected roots. Those distinctive from galls are preferentially located in pericentromeric regions with predominant sizes of 24 and 22 nucleotides. Gall‐distinctive rasiRNAs target primarily GYPSY and COPIA retrotransposons, which show a marked repression in galls vs. uninfected roots. Infection tests and phenotypic studies of galls from Meloidogyne javanica in Arabidopsis mutants impaired in post‐transcriptional gene silencing and/or canonical RNA‐directed DNA methylation (RdDM) pathways, as well as quantitative polymerase chain reaction analysis, suggest the implication of canonical and non‐canonical RdDM pathways during gall formation, possibly through the regulation of retrotransposons. This process may be crucial for the maintenance of genome integrity during the reprogramming process of galls/GCs from their vascular precursor cells, and/or to ensure a faithful DNA replication during the repeated mitosis/endoreduplication that concurs with feeding site formation.
Keywords: galls, giant cells, Meloidogyne spp., retrotransposons, root‐knot nematode, small RNAs (sRNAs), transposable elements (TEs)
1. INTRODUCTION
Transposable elements (TEs) and their derived fragments represent around 15% of the 135‐MB Arabidopsis thaliana genome (Arabidopsis Genome Initiative, 2000). However, most TEs are in a transcriptionally repressed or silenced state, as they can generate mutations and instability in genomes (Slotkin and Martienssen, 2007). In plants, the regulatory mechanism that prevents TE expression is referred to as RNA‐directed DNA methylation (RdDM) and involves the biosynthesis of repeat‐associated small interfering RNAs (rasiRNAs) that subsequently guide the methylation of TEs and other repetitive sequences (Xie and Yu, 2015; Zhang et al., 2010). In the Arabidopsis canonical RdDM pathway (Matzke and Mosher, 2014), RNA polymerase IV (Pol IV) is recruited to previously silenced TEs through the histone H3 lysine 9 methylation mark (Law et al., 2013), which is linked to transcriptional repression. The TE transcript product of Pol IV is converted into double‐stranded RNA by RNA‐dependent RNA POLYMERASE 2 (RDR2) and subsequently cleaved into 24‐nucleotide rasiRNAs by DICER‐LIKE 3 (DCL3). Then, 24‐nucleotide rasiRNAs are loaded into ARGONAUTE 4 and 6 (AGO4 and AGO6) and these proteins move to nascent Pol V transcripts still associated with their chromatin template. These Pol V transcripts serve as an RNA scaffold on the chromatin that allows rasiRNA‐based silencing information to direct chromatin modification. Sometimes the transcriptional silencing is lost (for example, as a result of stress, during development, after TE horizontal transfer, etc.) and silencing must begin through post‐transcriptional gene silencing (PTGS) via the plant’s endogenous RNA interference (RNAi) mechanism (Cavrak et al., 2014; Diao et al., 2005; Dowen et al., 2012; El‐Baidouri et al., 2014; Sarkies et al., 2015), known as the non‐canonical RdDM pathway. In this pathway, RDR6, AGO1, DCL2 and DCL4 degrade TE mRNA into 21–22‐nucleotide rasiRNAs which are the first trigger to homology‐independent initiation of TE silencing (McCue et al., 2012; Nuthikattu et al., 2013).
Although it is well documented that RdDM represses plant defence‐related genes in biotrophic plant–pathogen interactions, such as bacteria and fungi (Dowen et al., 2012; López et al., 2011; Slaughter et al., 2012; Yu et al., 2013), little is known about its involvement in the plant–nematode interaction. Plant‐parasitic nematodes from the genus Meloidogyne spp. are obligate parasites that penetrate through the root elongation zone, migrate intercellularly through the cortex and establish in the vascular cylinder where they form feeding sites (Escobar et al., 2015; Wyss et al., 1992). From five to eight feeding cells, called giant cells (GCs), develop in the vascular cylinder and constitute the sole nutrient source for the nematodes during their life cycle (Bird, 1962; Escobar et al., 2015). Around the GCs, the vascular cells proliferate profusely and the cortex cells hypertrophy, giving rise to a swelling or knot in the roots, called a gall (Dropkin, 1972), which gives the generic name to Meloidogyne spp. nematodes as root‐knot nematodes (RKNs). Genes differentially expressed during plant–nematode interactions have been identified through transcriptomic analyses of GCs, galls or infected roots (reviewed in Ali et al., 2015 and Cabrera et al., 2016a). Most of the functional studies have centred their attention on the genes that are induced during the interaction. However, a distinctive signature of early‐developing galls and GCs, conserved in tomato and Arabidopsis, is a large down‐regulation of transcripts (Barcala et al., 2010; Portillo et al., 2013). A putative mechanism to establish massive gene repression is the activation of small RNA (sRNA) pathways that repress gene expression at the transcriptional or translational level (Ruiz‐Ferrer and Voinnet, 2009). In line with this, the roles of different microRNAs (miRNAs) on the plant–RKN interaction have been addressed recently (Cabrera et al., 2016b; Díaz‐Manzano et al., 2016a, 2018; Kaur et al, 2017; Medina et al., 2017; Subramanian et al., 2016; Zhang et al., 2016; Zhao et al., 2015). However, little is known about the regulatory role of other types of siRNA in this interaction.
Recently, Hewezi et al. (2017) have reported that 24‐nucleotide rasiRNAs are highly abundant in the syncytia formed by the cyst nematode Heterodera schachtii and are accumulated in hypermethylated regions of the genome (Hewezi et al., 2017). A similar analysis of Meloidogyne javanica galls is still lacking. In previous work by Cabrera et al. (2016b), we described an enrichment of 24‐nucleotide sequences (independent of their origin as rasiRNAs or other types of siRNA) in the galls. In addition, we reported that the group of rasiRNAs was accordingly more abundant in galls than in control roots, and that they preferentially targeted TEs. However, we did not conduct a more comprehensive statistical analysis to select the rasiRNA sequences differentially accumulated in the galls at 3 days post‐infection (dpi) relative to control roots, or to study their length and distribution along the Arabidopsis genome with the aim to hypothesize the significance of their predominance in galls.
In this study, we selected the rasiRNAs statistically enriched or distinctive in Arabidopsis galls vs. uninfected roots. This extensive analysis showed that the rasiRNAs distinctive from galls, with predominant sizes of 24 and 22 nucleotides, are preferentially located in pericentromeric regions, where type I transposons (retrotransposons) are the major targets. Moreover, retrotransposon (COPIA, GYPSY, LINE and SINE) superfamilies are drastically repressed in galls at early times of infection relative to those in control roots. Further experimental results obtained from quantitative polymerase chain reaction (qPCR) analysis, as well as infection tests with M. javanica in single, double and triple Arabidopsis mutants defective in key functions from RdDM pathways, strongly suggest the implication of canonical (through 24‐nucleotide) and non‐canonical (through 22‐ and 21‐nucleotide) RdDM pathways during gall formation, possibly through the regulation of retrotransposons in galls.
2. RESULTS
2.1. rasiRNAs complementing repetitive sequences of the genome are enriched in Arabidopsis galls vs. uninfected roots
We have previously performed a global analysis of three sRNA libraries from galls at 3 dpi, induced by M. javanica in Arabidopsis roots, compared with three sRNA libraries from uninfected root segments (Cabrera et al., 2016b). We showed that the total accumulation of rasiRNAs was higher in galls than in control roots (Fig. 1; unique rasiRNAs: G1 = 561 968, G2 = 511 928 and G3 = 534 338; RC1 = 329 316, RC2 = 350 914 and RC3 = 381 262). In the present work, we deepen and expand the analysis of this subset of rasiRNAs, selected because they fully match with repetitive Arabidopsis DNA sequences deposited in the repetitive elements database Repbase (Fig. 1; Bao et al., 2015). Repbase includes TEs and non‐TE repetitive sequences, such as satellite sequences, microsatellites and multi‐copy RNA genes (rRNA, tRNA, snRNA). First, we normalized the rasiRNA sequences with respect to the total number of sRNA sequences found in each independent library after cleaning low‐quality reads, 5′ primer contaminants, and those without 3′ primer, without the insert tag, with poly A and/or shorter than 18 nucleotides (clean reads; Cabrera et al., 2016b), to be able to compare between replicates. Then, we performed a differential abundance analysis between galls and control roots. We found 4147 rasiRNAs differentially accumulated in galls relative to control roots (Fig. 1; Table S1a, see Supporting Information) after statistical analysis (t‐tests, P < 0.05) followed by a Benjamini–Hochberg correction with a false discovery rate of 5%. Hence, differences in the accumulation of rasiRNAs in galls relative to control uninfected Arabidopsis roots were significant (G1 = 3162, G2 = 3161 and G3 = 3161) in each of the three replicates (RC1 = 1272, RC2 = 1276 and RC3 = 1282; Fig. 1; Table S1a). The 4147 rasiRNAs differentially accumulated were single sequences, but they also matched with other loci (Table S1b–d) in multiple regions as the promoter, exons, introns, 3′ untranslated region (UTR), 5′ UTR, etc. (Table S1b–d), i.e. 17 933 hits were found in the genome for these rasiRNAs (Table S1e). Only 1.3% (52) of the 4147 differentially accumulated rasiRNAs were not present in the three replicates of either gall or control rasiRNAs; thus, they were not considered for further studies (Table S1a). The remaining 4095 differentially accumulated rasiRNAs were further classified into three groups (Fig. 1): (i) 2847 rasiRNAs (69.5%) present in galls, but not in control roots (dGall‐rasiRNAs, i.e. rasiRNAs distinctive from galls; Table S1b); (ii) 982 rasiRNAs (24%) present in control roots, but not in galls (dRC‐rasiRNAs, i.e. rasiRNAs distinctive from roots; Table S1c); and (iii) 266 rasiRNAs (6.5%) that were differentially accumulated in galls, but present in both (common‐rasiRNAs). Amongst these common‐rasiRNAs, 146 sequences were significantly up‐regulated and 120 down‐regulated in galls, relative to uninfected Arabidopsis roots (Fig. 1; Table S1d).
Figure 1.
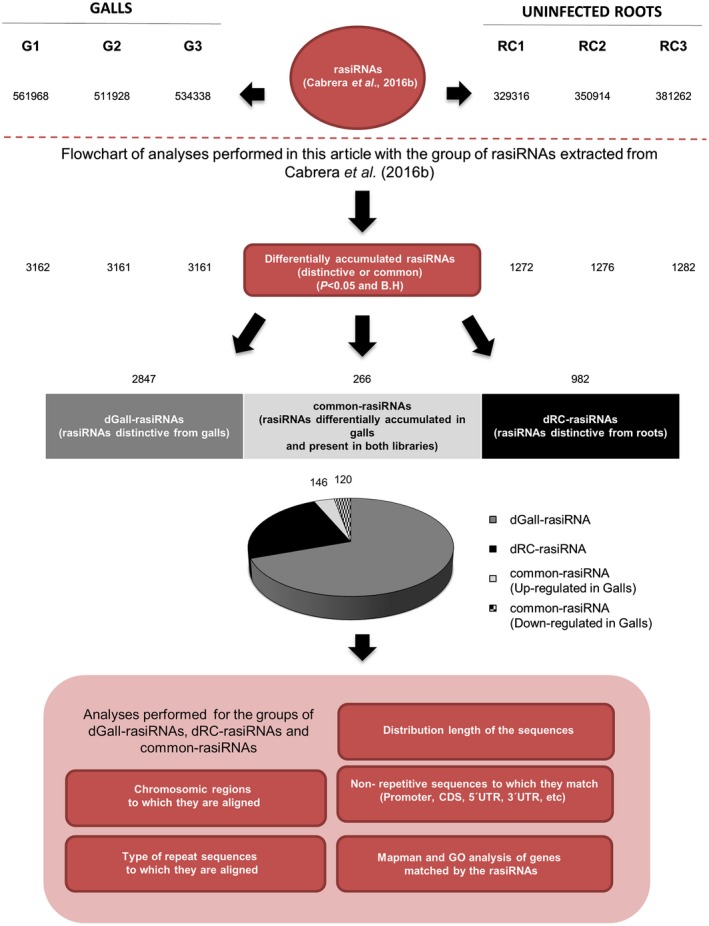
Flowchart of small RNA (sRNA) data processing and analysis. From top to bottom: number of repeat‐associated small interfering RNAs (rasiRNAs) from the six libraries published by Cabrera et al. (2016b); number of statistically significant rasiRNAs (P < 0.05) among the three independent replicates; number of distinctive rasiRNAs only present in gall (dGall‐rasiRNAs), distinctive rasiRNAs only present in root control (dRC‐rasiRNAs) and rasiRNAs present in gall and root control (common‐rasiRNAs). The study parameters analysed within the article for these sequences are indicated in the red boxes below. B.H, Benjamini–Hochberg; CDS, coding sequence; G1, G2 and G3, independent biological samples from galls; GO, gene ontology; RC1, RC2 and RC3, independent biological samples from control uninfected root; UTR, untranslated region. [Colour figure can be viewed at wileyonlinelibrary.com]
Surprisingly, a more stringent statistical analysis (t‐test, P < 0.01 and Benjamini–Hochberg correction with a false discovery rate of 1%; Table S2a, see Supporting Information) showed a number of unique reads for dGall‐rasiRNAs (2791; Table S2b), similar to that obtained for P < 0.05, whereas the number of dRC‐rasiRNAs and common‐rasiRNAs dropped drastically (9 and 40, respectively) (Tables S1a, S2c,d). These findings highlight the importance of the differential accumulation of rasiRNAs in galls at 3 dpi, suggesting that dGall‐rasiRNAs complementary to repetitive sequences may play a role in the regulation of gene expression in the early infection stages.
2.2. dGall‐rasiRNAs preferentially locate at pericentromeric regions with predominant sizes of 24 and 22 nucleotides
We explored whether dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs showed matching chromosome region preferences (Fig. 2a; Table S3a, see Supporting Information). Arabidopsis chromosomes can be categorized into five different regions: left arm (R1), left pericentromeric region (PC1), centromere (CEN), right pericentromeric region (PC2) and right arm (R2) (Arabidopsis Genome Initiative, 2000; Simon et al., 2015). The exact chromosome positions for each of the five regions for the five chromosomes are detailed in Table S3b. Our results show an important difference between dGall‐rasiRNAs and dRC‐rasiRNAs, i.e. dGall‐rasiRNAs clearly preferentially matched to PC regions in the five Arabidopsis chromosomes (Fig. 2b), whereas dRC‐rasiRNAs were distributed across the chromosome length, with the exception of CEN regions, and with preference to the R regions in Chr1, Chr2 and Chr5 (Fig. 2b; Table S3b). However, common‐rasiRNAs preferentially targeted CEN regions, except for those rasiRNAs that aligned to Chr1, which matched to PC1 (Fig. 2b; Table S3b).
Figure 2.
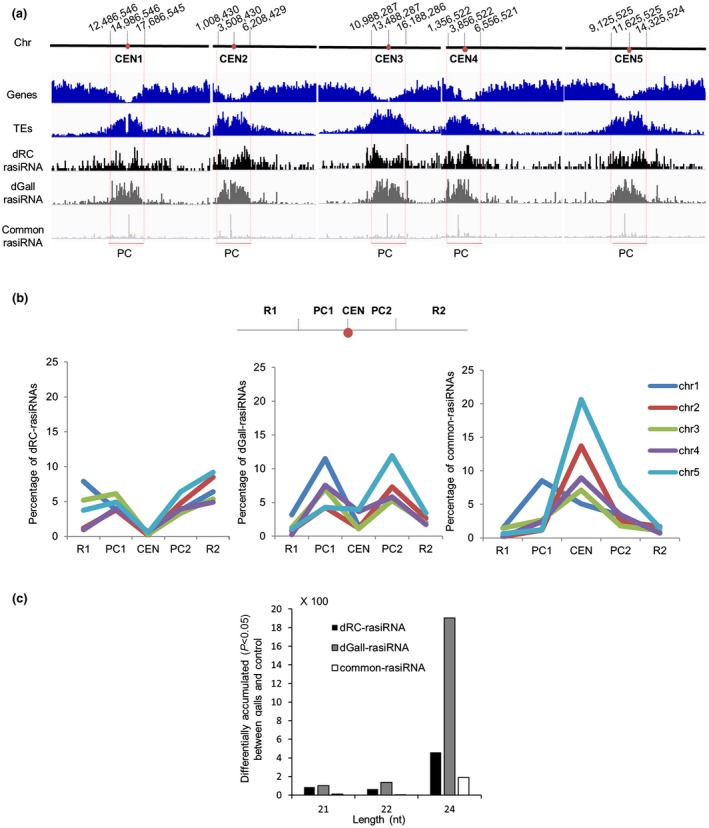
The distribution of repeat‐associated small interfering RNAs (rasiRNAs) in the Arabidopsis chromosomes shows distinctive (dGall‐rasiRNAs) preference for pericentromeric regions. (a) Arabidopsis TAIR10 genes and transposable elements (TEs) per chromosome (Chr) are coloured in blue. Classified groups of rasiRNA sequences: dGall‐rasiRNA (dark grey), dRC‐rasiRNA (black) and common‐rasiRNA (white) in the five Arabidopsis chromosomes are shown in the lower part of the figure. (b) Graph showing rasiRNA distribution for each of the classified groups: dGall‐rasiRNA (centre), dRC‐rasiRNA (left) and common‐rasiRNA (right). Arabidopsis chromosome regions on the x‐axis: left chromosome arm (R1), left pericentromeric region (PC1), centromere (CEN), right pericentromeric region (PC2) and right chromosome arm (R2). (c) Classification of rasiRNAs by nucleotide (nt) length: 21, 22 and 24 nt. [Colour figure can be viewed at wileyonlinelibrary.com]
We also grouped the differentially accumulated rasiRNAs (dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs ) into 21‐, 22‐ and 24‐nucleotide sequences, as products of DCL4, DCL2 and DCL3 activity, respectively, which could suggest the implication of PTGS (DCL2/DCL4) and/or canonical RdDM (DCL3) regulatory pathways (Fig. 2c). The majority were 24 nucleotides in length, corresponding mainly to dGall‐rasiRNAs (5764), followed by common‐rasiRNAs (2300) and dRC‐rasiRNAs (1207). The second most abundant size of dGall‐rasiRNAs was 22 nucleotides (8.4%; 915), whereas, in dRC‐rasiRNAs (9%; 269) and common‐rasiRNAs (6.9%; 281), 21‐nucleotide rasiRNAs were the second most abundant length (Fig. 2c; Table S3c). Our results also showed that 24‐nucleotide dGall‐rasiRNAs and DCL2‐dependent 22‐nucleotide rasiRNAs mainly matched to PC regions (Table S3d).
In summary, in galls, DCL3‐dependent 24‐nucleotide rasiRNAs are the major length category accumulated in PC regions, followed by DCL2‐dependent 22‐nucleotide rasiRNAs, in the five Arabidopsis chromosomes (Table S3d). Interestingly, this feature can be considered a signature of dGall‐rasiRNAs relative to dRC‐rasiRNAs, where DCL3‐dependent 24‐nucleotide rasiRNAs and DCL4‐dependent 21‐nucleotide rasiRNAs are mostly located in the R region (Table S3d).
2.3. Repression of retrotransposons correlates with the abundance of distinctive rasiRNAs from galls (24 and 22 nucleotides)
RdDM requires the biosynthesis of rasiRNAs to guide the methylation of TEs and repeats preventing their overexpression and proliferation (Zhang et al., 2007). Hence, we classified dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs according to their targets into five groups: (i) class I TEs, which are retrotransposons that function via intermediate RNAs and reverse transcriptase; (ii) class II TEs, which are DNA transposons that function via a DNA intermediate and transposase; (iii) ambiguous (ambi), which are sequences matching more than one TE; (iv) satellite DNA repeats, which are sequences localized within the centromeric regions; and (v) other repetitive regions. Interestingly, most dGall‐rasiRNAs (48.4%) were complementary to class I TEs, 27.4% to class II TEs, 14% matched more than one TE, 10% matched to satellite DNA sequences and only 0.13% matched to other repetitive sequences of the Arabidopsis genome (Fig. 3a). However, dRC‐rasiRNAs matched mainly to class II TEs (58.2%), followed by class I TEs (32.4%) and ambi (7.3%) (Fig. 3a).
Figure 3.
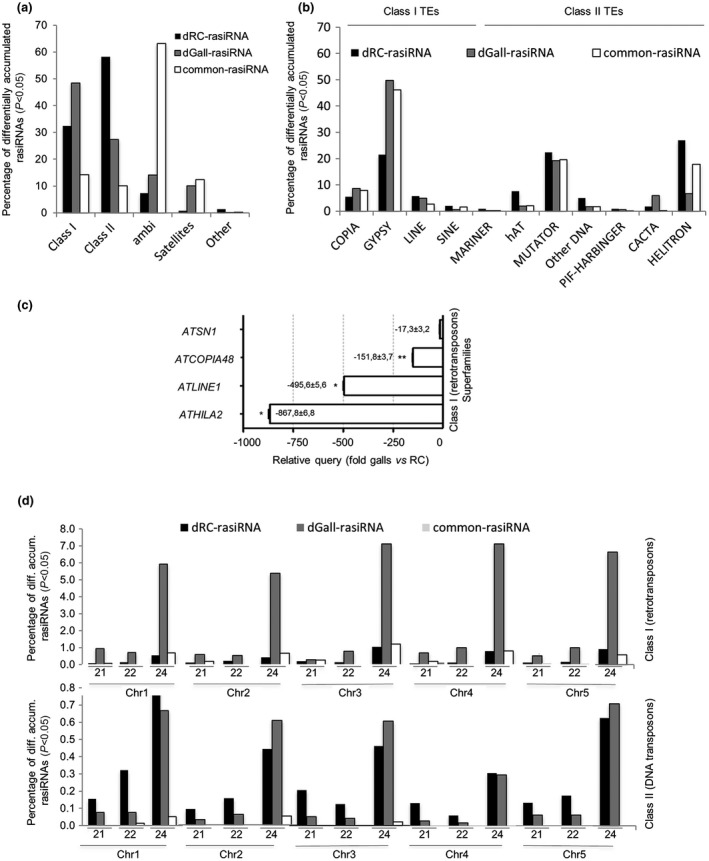
Repression of retrotransposons correlates with the abundance of distinctive gall repeat‐associated small interfering RNAs (rasiRNAs) (24 and 22 nucleotides). (a) Alignment of differentially accumulated rasiRNAs to genome repeat type: class I (retrotransposon), class II (DNA transposon), ambiguous (ambi; siRNAs complementary to more than one transposon element), satellites and other. (b) Percentage of differentially accumulated rasiRNAs that map to different transposon superfamilies also classified as class I and class II transposons. (c) Relative expression levels by quantitative polymerase chain reaction (qPCR) of specific candidates from retrotransposon superfamilies COPIA, GYPSY, LINE and SINE (i.e. ATCOPIA48, ATHILA2, ATLINE1 and ATSN1, respectively) in galls at 3 days post‐infection (dpi) vs. uninfected control roots. Values of two (ATSN1) and four (ATCOPIA48, ATLINE1 and ATHILA2) independent biological replicates, with three technical replicates each, were normalized to Glyceraldehyde‐3‐Phosphate Dehydrogenase (GADPH), used as internal control. Differences from control values were significant at *P < 0.05, **P < 0.01 (two‐tailed t‐test). (d) Mapping of reads normalized to the total amount of 21‐, 22‐ and 24‐nucleotide lengths in each of the five Arabidopsis chromosomes. dGall‐rasiRNA (dark grey), dRC‐rasiRNA (black) and common‐rasiRNA (white). Percentages in (a), (b) and (d) were calculated with respect to the total number of rasiRNAs in each group (dGall‐rasiRNAs, dRC‐rasiRNAs or common‐rasiRNAs).
Retrotransposons (class I TEs matched mostly by dGall‐rasiRNAs) can be further divided into those flanked by long terminal repeat (LTR) retrotransposons and non‐LTR retrotransposons (Slotkin and Martienssen, 2007), and can be classified into different superfamilies (COPIA, GYPSY, LINE and SINE). Our results showed that 49.7% (4071) of dGall‐rasiRNA sequences matched retrotransposons of the GYPSY superfamily, followed by 8.7% (711) of COPIA, 5.8% (400) of LINE and 0.5% (42) of SINE superfamilies (Fig. 3b; Table S1, Table S4). This trend was maintained for the common‐rasiRNAs and dRC‐rasiRNAs, in the latter case with a considerably fewer number of sequences.
However, dRC‐rasiRNAs matched mostly DNA transposons (class II TEs), divided into six classes: MuDR, EnSpm/CACTA, hAT, PIF/HARBINGER, the related POGO, Tc1 and MARINER elements, and rolling circle replicating HELITRON elements (Underwood et al., 2017). Our results showed that 58.2% of dRC‐rasiRNAs matched class II TEs, most from the HELITRON (27%; 751) and MUTATOR (22.3%; 620) families, followed by hAT (7.6%; 211) and POGO (5%; 139). In the case of common‐rasiRNAs, MUTATOR (194) and HELITRON (176) were the main families with similar percentages (19.6% and 17.8%, respectively; Fig. 3b; Table S1b–d).
Therefore, retrotransposons are the major rasiRNA targets in Arabidopsis galls induced by M. javanica at the early infection stages, matching predominantly RdDM targets, such as the retrotransposon GYPSY, whereas dRC‐rasiRNAs matched mainly DNA transposons (Fig. 3b). Hence, to assess the potential impact of M. javanica infection on TE silencing in galls through RdDM pathways, we monitored, by qPCR, the transcriptional status of several mRNA retrotransposons, which are well‐known targets of these pathways, i.e. ATLINE1, ATCOPIA48, ATSN1 and ATHILA2 (Zhang et al., 2006). All TE transcripts were drastically lower in 3‐dpi galls relative to control uninfected roots, and two, ATHILA2 and ATCOPIA48, with high significance (P < 0.05 and P < 0.01; Fig. 3c). These findings indicate that transcripts from retrotransposon families in pericentromeric regions (ATHILA2, ATCOPIA48 and ATLINE1) were strongly down‐regulated in galls. Interestingly, the drastic decrease in retrotransposon transcripts coincided with a strong accumulation of statistically significant dGall‐rasiRNAs (Table S3c) at centromeric and pericentromeric regions in Arabidopsis chromosomes (Fig. 2a,b).
To address the question of whether or not any specific rasiRNA population (21, 22 or 24 nucleotides in length) could be related to the regulation of the main retrotransposon and DNA transposons on M. javanica infection, we classified dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs with regard to their size (21, 22 and 24 nucleotides in length), type of TEs matched (class I and class II) and chromosome distribution (Chr1, Chr2, Chr3, Chr4, Chr5) (Fig. 3d; Table S1). Strikingly, a major signature of dGall‐rasiRNAs was the high abundance of DCL3‐dependent 24‐nucleotide rasiRNAs in retrotransposons in the five Arabidopsis chromosomes relative to that of dRC‐rasiRNAs and common‐rasiRNAs (Fig. 3d; Table S1). Consistent with the former data, DCL2‐dependent 22‐nucleotide rasiRNAs were also abundant amongst the dGall‐rasiRNAs in Chr3, Chr4 and Chr5 (Fig. 3d).
These differences were not found in DCL3‐dependent 24‐nucleotide rasiRNAs matching class II DNA TEs, as they were highly abundant in dGall‐rasiRNAs and dRC‐rasiRNAs (Fig. 3d; Table S1b).
2.4. RKN infection is compromised in some Arabidopsis mutants of key genes with altered expression in galls impaired in canonical and non‐canonical RDR6‐RdDM pathways
Therefore, our bioinformatic analyses suggest that, in Arabidopsis galls, transposons could be mainly regulated by DCL3‐dependent 24‐nucleotide rasiRNAs (Fig. 3d; Table S1). However, DCL2‐dependent 22‐nucleotide rasiRNAs, the second most abundant class in dGall‐rasiRNAs, may play an additional role in galls through the PTGS pathway. Moreover, both pathways (PTGS and canonical RdDM) are connected through the non‐canonical RdDM pathway (Fig. 4c; Cuerda‐Gil and Slotkin, 2016) for TE regulation, as mRNA cleavage by 22‐nucleotide rasiRNAs is a known trigger for additional RDR6 activity and the production of secondary rasiRNAs that may trans‐regulate other mRNAs with similar sequence (Chen et al., 2010; Cuperus et al., 2010).
Figure 4.
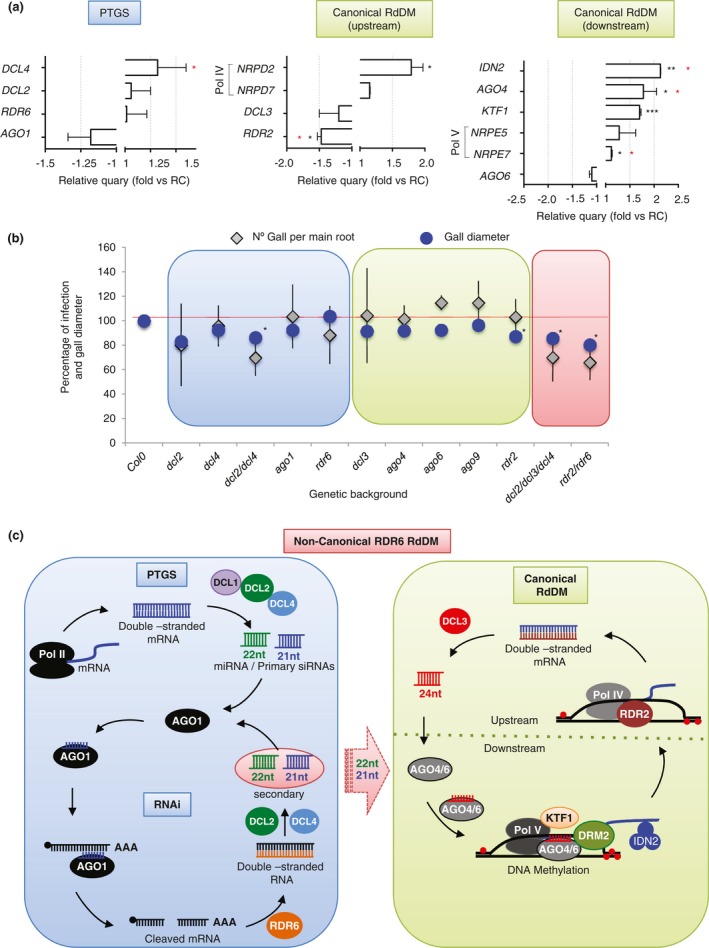
Root‐knot nematode (RKN) infection responses in Arabidopsis mutants of key genes with altered expression in galls impaired in canonical and non‐canonical RDR6‐RdDM pathways. (a) Relative expression levels of canonical, post‐transcriptional gene silencing (PTGS) and non‐canonical RNA‐directed DNA methylation (RdDM) representative genes obtained using quantitative polymerase chain reaction (qPCR) in galls at 3 days post‐infection (dpi) vs. uninfected control roots. The values of two to five independent biological replicates, with three technical replicates each, were normalized to the GADPH internal control. Differences from control values were significant at *P < 0.05, **P < 0.01, ***P < 0.001 (two‐tailed t‐test). A red asterisk indicates P < 0.05 from NEMATIC data (Cabrera et al., 2014). (b) Infection tests with Meloidogyne javanica showing the percentage of galls formed in mutant Arabidopsis lines impaired in repeat‐associated small interfering RNA (rasiRNA) biogenesis compared with the control Col‐0 (grey diamond), as well as the relative gall diameter (n ≥ 27 per line tested) at 14 dpi of the mutant lines relative to that of Col‐0 (blue dots). For infection tests, five to six independent experiments were performed per genetic background with at least 30 plants per independent experiment and transgenic Arabidopsis line. Blue background, mutant Arabidopsis lines from PTGS. Green background, mutant Arabidopsis lines from canonical RdDM pathway. Red background, mutant Arabidopsis lines from PTGS and canonical RdDM pathways. (c) Schematic diagram of non‐canonical RDR6‐RdDM pathway that connects PTGS and canonical RdDM through 21‐ and 22‐nucleotide rasiRNAs in Arabidopsis plants, based on Cuerda‐Gil and Slotkin (2016). PTGS: polymerase II (Pol II) transcription of transposable elements (TEs) or microRNA precursors generates primary small RNAs in an RNA‐dependent RNA polymerase (RDR)‐independent manner after cleavage by DICER‐LIKE (DCLs). Then, RDR6 generates double‐stranded RNAs (dsRNAs) that are triggered by DCL2 and DCL4 to produce secondary rasiRNAs. These can target additional copies of mRNA for the cleavage and production of more rasiRNAs through RNA interference (RNAi). Canonical RdDM: Pol IV and RDR2 produce dsRNA cleaved by DCL3. Then, 24‐nucleotide siRNAs are loaded into AGO4 and AGO6 to chromatin‐bound transcripts produced by Pol V (Upstream). After AGO4/6 interacts with Pol V transcripts, many other proteins will be recruited (IDN2, KTF1, etc.) to finally methylate the DNA through DNA methyltransferase DRM2 (Downstream). Non‐canonical RDR6‐RdDM: 21−22‐nucleotide rasiRNAs produced from Pol II–RDR6‐derived TE mRNAs are loaded into AGO6, which interacts with its target loci through a Pol V scaffolding transcript. [Colour figure can be viewed at wileyonlinelibrary.com]
To expand our knowledge of the implication of PTGS and RdDM pathways (Fig. 4c) during RKN infection, we performed qPCR and infection tests addressing the expression and role of key genes in both processes. Basic genes encoding members of the RdDM pathway biogenesis machinery, such as NRPD2 (encoding the second‐largest subunit of Pol IV), NRPE7, IDN2 and KTF1, were all induced in galls at 3 dpi (P < 0.05, P < 0.05, P < 0.01 and P < 0.001, respectively; Fig. 4a). AGO4 was also differentially up‐regulated in galls (P < 0.05), as were members of the non‐canonical RdDM pathway, such as DCL4, all of which showed a similar tendency in the qPCR analyses. Furthermore, the results of an independent technique, microarrays, compiled in the NEMATIC microarray database (Cabrera et al., 2014), showed that four of these genes (DCL4, IDN2, NRPE7 and AGO4) were also up‐regulated (P < 0.05). In contrast, RDR2 was down‐regulated in the qPCR analysis (P < 0.05; Fig. 4a), as well as in microarrays (P < 0.05; Fig. S1, see Supporting Information; Cabrera et al., 2014). All of these data confirm that M. javanica infection modifies the expression of key components of rasiRNA biogenesis participating in the PTGS and canonical RdDM pathways.
Next, we performed infection tests with mutant lines in which rasiRNA biogenesis was compromised (Fig. 4b). We challenged with M. javanica a set of loss‐of‐function single (ago1, ago4, ago6, dcl2, dcl3, dcl4, rdr2, rdr6), double (dcl2/dcl4, rdr2/rdr6) and triple (dcl2/dcl3/dcl4) mutants. A clear reduction in the infection rates of dcl2, dcl4, dcl2/4 and rdr6 mutant lines, compared with wild‐type Col‐0 plants, was observed, all lacking key functions for the non‐canonical RDR6‐RdDM pathway, although not statistically significant (Fig. 4c; Cuerda‐Gil and Slotkin, 2016). This is in agreement with previous infection tests performed with mutants for some of these genes in the context of plant–nematode interactions, in which a decrease in infection was observed, although not always significant (Hewezi et al., 2008; Medina et al., 2017). Moreover, double rdr2/rdr6 and triple dcl2/dcl3/dcl4 mutants impaired in both RdDM pathways (canonical and non‐canonical) showed an enhanced resistance (Fig. 4b). Interestingly, similar infection rates were found for Col‐0, ago1 and ago4, but not for ago6, which suggests that AGO4 and AGO6 respond divergently to M. javanica infection. To address whether the genes altered in mutants impaired in both RdDM pathways could be participating in gall development, we estimated the gall diameter on the same collection of mutant lines. Our results showed that, at 14 dpi, dcl2/dcl4, dcl2/dcl3/dcl4, rdr2 and rdr2/rdr6 galls were significantly smaller than Col‐0 galls (P < 0.05; Fig. 4b), i.e. mainly mutants for genes implicated in the PTGS pathway (DCL2, DCL4 and RDR6). These results are in accordance with our bioinformatic analysis, in which DCL3‐dependent 24‐nucleotide rasiRNAs and RDR6 secondary rasiRNAs (mainly 22 nucleotides) are highly abundant in Arabidopsis galls (Table S3c), and with the altered expression of key genes of the RdDM machinery (Fig. 4a), supporting the notion that both canonical RdDM and PTGS pathways are relevant for gall development in Col‐0 plants (Fig. 4c).
3. DISCUSSION
sRNA libraries from Arabidopsis galls induced by M. javanica (Cabrera et al., 2016b; 3 dpi) and M. incognita (Medina et al., 2017; 7 and 14 dpi), as well as syncytia induced by the cyst nematode H. schachtii (Hewezi et al., 2008; 4 and 7 dpi; Hewezi et al., 2017; 5 and 10 dpi), presented a significant enrichment in 24‐nucleotide sRNAs compared with control root libraries. In previous work by Cabrera et al. (2016b), an analysis of sRNA length distribution suggested that the abundance of 24‐nucleotide sRNAs constituted a gall hallmark, at least at early infection stages, whereas 21‐nucleotide sRNAs (the length of most plant miRNAs; Axtell, 2013; Xie et al., 2015) were more abundant in uninfected roots. We also reported that the group of sRNAs fully matching repetitive elements in the Arabidopsis genome (rasiRNAs; Bao et al., 2015) were more abundant in galls than in control roots (Cabrera et al., 2016b).
Therefore, in this work, we extended the analysis to rasiRNAs and found that they were significantly enriched in 3‐dpi galls relative to control roots, matching mainly TEs and satellite sequences (Fig. 3). In this respect, rasiRNAs perfectly matching TEs were also described in Arabidopsis roots infected with H. schachtii at 4 and 7 dpi, although they were not highly enriched in the total sRNA population (Hewezi et al., 2008).
The number of unique distinctive rasiRNAs was noticeably higher in galls than in uninfected roots after statistical analysis (P < 0.05) in the three library replicates. We classified them into three groups: gall‐distinctive (dGall‐rasiRNAs; 69.5%), control root‐distinctive (dRC‐rasiRNAs; 24%) and differentially accumulated rasiRNAs present in both galls and roots (common‐rasiRNAs; 6.5%; Fig. 1). A more restrictive analysis (P < 0.01) showed that the number of unique dGall‐rasiRNAs was similar, but dRC‐rasiRNAs and common‐rasiRNAs decreased drastically under the more stringent analysis (Table S1c,d). This indicates that distinct rasiRNAs from galls represent a robust group, suggesting that they may play a role in RKN feeding sites at the early stages of infection.
Interestingly, the unique rasiRNAs characteristic of galls (dGall‐rasiRNAs) matched mainly retrotransposons (class I TEs) from the GYPSY and COPIA superfamilies (Fig. 3). This is in agreement with the sense and antisense TE families previously identified as rasiRNA targets in Arabidopsis infected with H. schachtii at 4 and 7 dpi, suggesting a role in controlling the mobility and proliferation of TEs during cyst nematode infection (Hewezi et al., 2008). In contrast, dRC‐rasiRNAs matched mainly to DNA transposons (class II TEs) preferentially binding to HELITRON and MUTATOR superfamilies (Fig. 3). This finding also suggests a preferred location for dRC‐rasiRNAs, dGall‐rasiRNAs and common‐rasiRNAs at the early stages of M. javanica infection, because class I and class II TEs are concentrated at pericentromeric and chromosome arms, respectively (Arabidopsis Genome Initiative, 2000; Buisine et al., 2008) (Table S3d). This coincides with the distribution of dGall‐rasiRNAs and dRC‐rasiRNAs from our analyses within the Arabidopsis genome, in which a high and statistically significant accumulation of dGall‐rasiRNAs at centromeric and pericentromeric regions was encountered (Fig. 2; Table S1). In accordance with this bioinformatics analysis, transcript accumulation of retrotransposon families (ATHILA2, ATCOPIA48 and ATLINE1) was severely decreased in galls relative to uninfected control roots (Fig. 3). These results indicate that the preferential accumulation of dGall‐rasiRNAs could mediate a process not yet reported during plant–RKN interactions: the silencing of retrotransposon families, such as ATHILA2, ATCOPIA48 and ATLINE1, during the early stages of gall formation. Furthermore, this repression may be required for RKN infection. Similarly, Piya et al. (2017) found, by RNA‐sequencing (RNA‐seq), that several TEs (99 and 93 at 5 and 10 dpi, respectively) were differentially expressed in roots infected with H. schachtii vs. control roots, and one‐third were also down‐regulated. Interestingly, retrotransposons are also silenced in mammalian germ cells, in order to maintain genome integrity, whereas epigenetic control is relaxed to permit genome‐wide reprogramming (Yang and Wang, 2016). Little is yet known about the role of retrotransposon repression in similar situations in plants. For example, the tobacco Tnt1 retrotransposon is repressed in conditions that might favour germinal cell transposition (Grandbastien et al., 2005). Yet, in Arabidopsis, TE silencing in the developing embryo is assisted by endosperm‐derived 24‐nucleotide sRNAs (Bouyer et al., 2017). Similar to GCs within galls, columnella cells in the root meristem are rapidly differentiating cells (Kawakatsu et al., 2016). Interestingly, columnella cells show hypermethylated TEs and an increased abundance of transcripts encoding RdDM pathway components and 24‐nucleotide rasiRNAs (Kawakatsu et al., 2016). Thus, we can hypothesize that TE stability is required in early developing GCs and galls, which are newly formed cells and organs, for the maintenance of genome integrity during the rapid cell proliferation and differentiation. It is beyond question that GCs/galls experience an extensive reprogramming of gene expression in the initial stages of their differentiation from as yet poorly known vascular stem cells, as demonstrated by several transcriptomic analyses of galls and microdissected GCs (Jammes et al., 2005; Barcala et al., 2010; Damiani et al., 2012; Portillo et al., 2013). This reprogramming is probably achieved, at least in part, by silencing of genome‐unstable regions, such as retrotransposons, in the pericentromeric regions. Interestingly, CG‐rich heterochromatic states have recently been shown to be determinant features for the localization of DNA replication origins (ORIs) in heterochromatin. In particular, GYPSY retrotransposon elements co‐localized with ORIs at pericentromeric gene‐poor regions in Arabidopsis (Vergara et al., 2017). dGall‐rasiRNAs at 3 dpi are mainly accumulated at retrotransposon superfamilies, preferentially GYPSY superfamilies at pericentromeric regions, and expression analysis indicated that key members of several retrotransposon families, such as ATHILA2, were repressed (Fig. 3). Therefore, a putative scenario in GCs/galls, that suffer several rounds of mitosis and endoreduplication early in their development (de Almeida‐Engler et al., 2015), is the stabilization of transposons by RdDM pathway‐mediated methylation to ensure correct replication from the frequent ORIs. Further genome‐wide maps of ORIs are required to confirm their effect on gall development.
We examined the expression and function of key genes related to the canonical RdDM and PTGS pathways, the main pathways implicated in sRNA biogenesis (Ruiz‐Ferrer and Voinnet, 2009) in galls (Fig. 4). Several genes were differentially expressed in galls relative to controls at 3 dpi (P < 0.05; DCL4, NRPD2, RDR2, IDN2, AGO4, KTF1, NRPE7), the infection rate decreased and the gall size was significantly impaired mainly in double or triple mutants (Fig. 4). These results suggest the implication of canonical RdDM (through 24‐nucleotide rasiRNAs) and PTGS (mainly through 22‐nucleotide rasiRNAs; the second most abundant in dGall‐rasiRNAs) pathways in the regulation of TEs in galls (Fig. 4). Altogether, these findings suggest that DCL3‐ and DCL2‐dependent rasiRNAs play a role in the early stages of gall development. It is well documented that non‐canonical RdDM pathways are not completely independent of the canonical pathway and, as a consequence, many loci are simultaneously targeted by canonical and non‐canonical RdDM (Li et al., 2015; Nuthikattu et al., 2013; Panda et al., 2016). Our results agree with previous infection tests performed with cyst nematodes in Arabidopsis mutants impaired in RDR6 secondary siRNA biogenesis, which showed lower susceptibility (although not always statistically significant) to nematode infection (Hewezi et al., 2008). A restricted set of mutants was also tested for RKNs (M. incognita; Medina et al., 2017), showing results in agreement with the data presented here.
The functional role of the differentially accumulated rasiRNAs in galls and syncytia (Hewezi et al., 2008; this article) in TE regions, which are also differentially regulated in infected tissues vs. their corresponding uninfected control tissues, is still an interesting open field of study. In line with our data, the abundance of 24‐nucleotide rasiRNAs was associated with the hypermethylation of TEs and gene promoters during the interaction of Arabidopsis with cyst nematodes (Hewezi et al., 2017; Piya et al., 2017). Further experiments are required to correlate our results in galls with methylation at these sites and to understand their implications to gall development at the early stages of infection. In addition, we cannot discount that some dGall‐rasiRNAs could originate in M. javanica, as described for a Litomosoides sigmodontis (another nematode) miRNA in the body fluid of infected mice (Buck et al., 2014). Further experiments are also required to elucidate whether an inter‐kingdom sRNA transfer may occur between M. javanica and Arabidopsis plants on infection.
In conclusion, we have shown that rasiRNAs complementing repetitive sequences of the genome are enriched in Arabidopsis galls vs. uninfected roots, and those distinctive of galls are preferentially located in pericentromeric regions with predominant sizes of 24 and 22 nucleotides with retrotransposons as the major rasiRNA targets. qPCR analysis, as well as resistance testing with M. javanica mutants of key genes from the RdDM pathways, strongly suggests the implication of canonical RdDM (through 24‐nucleotide) and PTGS (mainly through 22‐nucleotide) pathways in the regulation of retrotransposons in galls, as they were severely repressed in galls. This is the first time that both RdDM pathways have been proposed to be involved in the plant–nematode interaction, probably in the repression of retrotransposons in early developing GCs. It is reasonable to consider that this may be crucial for the maintenance of the genomic integrity of galls/GCs during the dramatic reprogramming process from their vascular precursor cells.
4. EXPERIMENTAL PROCEDURES
4.1. Nematode population
Meloidogyne javanica Treub (1885) was maintained in vitro on etiolated cucumber (Cucumis sativus cv Hoffmanns Giganta) seedlings (Díaz‐Manzano et al., 2016b). Egg hatching was performed according to Díaz‐Manzano et al. (2016b).
4.2. Plant model and growth conditions
Arabidopsis (Arabidopsis thaliana) plants were grown vertically in controlled environment chambers under 16 h/8 h of light/dark at 22 ºC. Wild‐type and mutant homozygous lines used in the study were in the ecotype Col‐0 background and were PCR genotyped using the primers listed in Table S5. Arabidopsis mutant homozygous lines for ago4‐2, ago6‐3, ago9‐3, rdr2, rdr6, rdr2/6, dcl2, dcl3, dcl4, dcl2/4, dcl2/3, dlc3/4 and dcl2/3/4 mutant lines were donated by James Carrington (Donald Danforth Plant Science Center, St Louis, MO, USA).
4.3. Nematode infection tests and gall diameter estimation
For in vitro infection tests, five to six independent biological experiments were performed per genetic background. At least 30 plants per independent experiment and transgenic Arabidopsis line (ago4‐2, ago6‐3, ago9‐3, rdr2, rdr6, rdr2/6, dcl2, dcl3, dcl4, dcl2/4, dcl2/3, dlc3/4, dcl2/3/4 and ros1) were used with Col‐0 as control line. Seeds were sterilized, grown vertically and homogeneously inoculated following the protocol described by Olmo et al. (2017) in a long‐day (LD; 16 h : 8 h day : night) regime. The gall diameter was measured at 14 dpi (n ≥ 27 per line tested) per genetic background using the straight line and measurement tools in FIJI (Schindelin et al., 2012). For statistical analysis, Student’s t‐test was performed (P < 0.05).
4.4. Total RNA isolation and qPCR analysis
Control root segments from uninfected plants and galls from plants infected with M. javanica were hand dissected at 3 dpi according to our previous work (Barcala et al., 2010; Cabrera et al., 2016b). Total RNA extraction was performed using the AllPrep DNA/RNA/miRNA Universal reagent (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. One microgram of RNA from each sample was used for cDNA synthesis with a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) with selected primers (Table S5). The cDNA was diluted up to 1 : 50 for DCL2, DCL3, DCL4, RDR2, RDR6, AGO1, AGO4, AGO6, NRPD2, NRPD7, NRPE5, NRPR7, IDN2 and KTF1 genes and TEs (class I and class II). Relative gene expression was determined using the ΔΔCt method and a LightCycler® 480 II machine (Roche, Indianapolis, IN, USA). Undetectable transcripts (because of their very low abundance) were set to a value of 40 PCR cycles in order to perform the statistics (McCall et al., 2014). To obtain the expression values of galls vs. control roots, we used the relative quantity (RQ) as 2−ΔΔCt, where ΔΔCt for each gene is the difference between the average ΔCt in galls minus the average value of expression in control roots at 3 dpi. Values from two to five independent biological replicates were used as indicated in each figure legend, with three technical replicates each; all were normalized to the Glyceraldehyde‐3‐Phosphate Dehydrogenase (GADPH) internal control (Díaz‐Manzano et al., 2018). Differences from control values were significant at *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t‐test comparing ΔCt values, P < 0.05). A list of the sequence‐specific primers used in this study is provided in Table S5.
4.5. sRNA libraries and data analysis
Six independent sRNA libraries from three independent gall and control root samples (n = 300), generated in Cabrera et al. (2016b), were used in this study (GEO database accession number: GSE71563). For the present work, rasiRNAs from the six libraries that mapped to the Repbase database (https://www.girinst.org; Bao et al., 2015) were selected (Fig. 1). We used the R Stats package (Benjamini and Yekutieli, 2001; R Development Core Team, 2008) to perform Student’s t‐test analysis (rowttest) with P < 0.05 (Table S1) and P < 0.01 (Table S2), followed by a Benjamini–Hochberg correction, on Repbase rasiRNA sequences, identifying the unique rasiRNAs statistically significant among the three replicate libraries from galls and the three replicates from control roots. All statistically significant rasiRNAs were annotated using the TAIR10 Arabidopsis genome (chromosome, strand, start–end chromosome positions, length) and classified into three groups as described in the Results section (dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs). According to the nature of the repetitive elements, all statistically significant rasiRNAs were further classified as class I (retrotransposons), class II (DNA transposons), ambiguous (ambi; siRNAs able to match to more than one Arabidopsis TE), satellites and other repetitive sequences. Class I retrotransposons were subclassified into COPIA, GYPSY, LINE and SINE. In the same line, class II DNA transposons were divided into Tc‐MARINER, hAT, MUTATOR, PIF‐HARBINGER, CACTA, POGO and HELITRON. dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs were also classified according to the chromosome, length and regions. We classified each Arabidopsis chromosome into five regions: centromere (CEN), pericentromeric region 1 (PC1), pericentromeric region 2 (PC2) and chromosome arm regions 1 and 2 (R1 and R2). Pericentromeric regions are defined as regions in which the gene coverage in 1 Mb is equal to or lower than 40%. Chromosome positions for each of the five regions for the five chromosomes are detailed in Table S3b.
In order to find Arabidopsis chromosome regions matching (100% homology sequence) the unique statistically significant rasiRNAs, we used the following TAIR10 blast sets: 3_utr (3UTR), 5_utr (5UTR), cdna_20101214 (cDNA), cds_20111214 (cds), exon_20101028 (exon), intergenic_20101028 (intergenic), intron_20101028 (intron), pep_20101214 (pep), upstream_3000_translation_start_20101028 (promotor3kb), downstream_3000_translation_start_20101028 (downstream) and TEs (TE) (https://www.arabidopsis.org/download/index-auto.jsp?dir=%2Fdownload_files%2FSequences%2FTAIR10_blastsets). For genome annotation of the genes, we used TAIR10.
Supporting information
Fig. S1 Expression data of RNA‐directed DNA methylation (RdDM) genes at the NEMATIC database. Col‐0 Arabidopsis transposon elements and genes participating in canonical and non‐canonical RNA‐dependent RNA POLYMERASE 6 (RDR6)‐RdDM are included. Red, induced genes (P < 0.05; Cabrera et al., 2014); green, repressed genes (P < 0.05; Cabrera et al., 2014); grey, genes not differentially expressed relative to control uninfected roots.
Table S1 Statistically significant repetitive repeat‐associated small interfering RNAs (rasiRNAs) after t‐test (P < 0.05) and Benjamini–Hochberg correction. (a) Sequence data after statistical analysis. Order of the columns: rasiRNA sequence (column A), rasiRNA length (column B), biological independent replicates of gall (columns C–E), biological independent replicates of root control (columns F–H), repeat classification by Repbase database (column I), repeat type (column J), the difference in mean values (dm; column K), P value (column L) and P value after Benjamini–Hochberg correction (column M). (b) Location on the Arabidopsis genome of statistically significant rasiRNAs distinctive from galls (P < 0.05 and Benjamini–Hochberg correction). Order of the columns: rasiRNA sequence (column A), Arabidopsis chromosome (column B), start (column C) and end (column D) positions for the alignment, positive or negative strand for the alignment (column E), rasiRNA length (column F), number of normalized sequences for the three biological independent replicates of gall (columns G–I), number of normalized sequences for the three biological independent replicates of root control (columns J–L), repeat classification by Repbase database (column M), repeat type (column N), dm value (column O), P value (column P), P value after Benjamini–Hochberg correction (column Q) and gene annotation from TAIR10 datasets [CDS, gene, intergenic region, intron, promoter, transposable element (TE), untranslated region (UTR), 5′ UTR, cDNA, peptides and 3000 bp downstream]. (c) The same as (b) on distinctive rasiRNAs from root control. (d) The same as (b) on significant rasiRNAs commonly present in galls and root control libraries. (e) Classification of statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs that fully complement other regions of the Arabidopsis genome. All datasets were downloaded from TAIR10: annotated TE, intergenic regions, promoter (3000 bp upstream), 5′ UTR, 3′ UTR, exon, intron, 3000 bp downstream, cDNA, CDS and peptides. No. DE sequence, number of unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01). Unique No. of Hits, number of matches from unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01). Total No. of Hits, total number of matches from unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01).
Table S2 Statistically significant repetitive repeat‐associated small interfering RNAs (rasiRNAs) after t‐test (P < 0.01) and Benjamini–Hochberg correction. (a) Sequence data after statistical analysis. Order of the columns: rasiRNA sequence (column A), rasiRNA length (column B), number of normalized sequences for the three biological independent replicates of gall (columns C–E), number of normalized sequences for the three biological independent replicates of root control (columns F–H), repeat classification by Repbase database (column I), repeat type (column J), the difference in mean values (dm; column K), P value (column L) and P value after Benjamini–Hochberg correction (column M). (b) Location on the Arabidopsis genome of statistically significant rasiRNAs distinctive from galls (P < 0.01 and Benjamini–Hochberg correction). Order of the columns: rasiRNA sequence (column A), Arabidopsis chromosome (column B), start (column C) and end (column D) positions for the alignment, positive or negative strand for the alignment (column E), rasiRNA length (column F), number of normalized sequences for the three biological independent replicates of gall (columns G–I), number of normalized sequences for the three biological independent replicates of root control (columns J–L), repeat classification by Repbase database (column M), repeat type (column N), dm value (column O), P value (P), P value after Benjamini–Hochberg correction (column Q) and gene annotation from TAIR10 datasets [CDS, gene, intergenic region, intron, promoter, transposable element (TE), untranslated region (UTR), 5′ UTR, cDNA, peptides and 3000 bp downstream]. (c) The same as (b) on statistically distinctive rasiRNAs from root control. (d) The same as (b) on statistically significant rasiRNAs present in galls and root control.
Table S3 Bioinformatic analysis of the statistically significant distinctive repeat‐associated small interfering RNAs (rasiRNAs) in the libraries from galls infected with Meloidogyne javanica and from uninfected Arabidopsis roots. (a) Unique statistically significant rasiRNAs (P < 0.05 and Benjamini–Hochberg) distinctive from gall (dGall‐rasiRNA), root control (dRC‐rasiRNA) and commonly present in both (common‐rasiRNA) that fully matched to other regions of the Arabidopsis genome, classified by chromosomes (a) and strand (b). (b) The same groups of rasiRNAs described previously, but classified by chromosome regions. (c) The same groups of rasiRNAs described previously, but classified by length (18–26 nucleotides in length). (d) The same groups of rasiRNAs described previously, but classified by length (18–26 nucleotides in length) and also by Chromosome (Chr1–Chr5), strand (positive and negative) and chromosome regions. Arabidopsis chromosomes: Chr1, chromosome 1; Chr2, chromosome 2; Chr3, chromosome 3; Chr4, chromosome 4; Chr5, chromosome 5. Chromosome regions: left chromosome arm (R1), left pericentromeric region (PC1), centromere (CEN), right pericentromeric region (PC2) and right chromosome arm (R2).
Table S4 Classification of transposable elements (TEs) targeted by dGall‐rasiRNAs. (a) TEs targeted by dGall‐rasiRNAs (P < 0.01) were classified by TE name (column A), number of dGall‐rasiRNAs that matched to each TE (column B), TE family (column C) and TE superfamily (column D). (b) Members of TE families targeted by dGall‐rasiRNAs (P < 0.01). (c) Members of TE superfamilies targeted by dGall‐rasiRNAs (P < 0.01).
Table S5 List of primers used in this report.
ACKNOWLEDGEMENTS
This work was supported by the Spanish Government (grants AGL2013‐48787 and AGL2016‐75287‐R to C.E., and CSD2007‐057 and PCIN‐2013‐053 to C.F.) and by the Castilla‐La Mancha Government (PEII‐2014‐020‐P to C.F.). We wish to thank Dr. César Llave for kindly providing Arabidopsis mutant seeds and Dr. Ángel Arévalo for statistical R package support. We apologize to all colleagues whose work could not be cited because of the size limitations of the manuscript. The authors declare that there are no conflicts of interest with regard to the publication of this article.
REFERENCES
- Ali, M.A. , Abbas, A. , Azeem, F. , Javed, N. and Bohlmann, H. (2015) Plant–nematode interactions: from genomics to metabolomics. Int. J. Agric. Biol. 17, 1071–1082. [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana Nature 408, 796. [DOI] [PubMed] [Google Scholar]
- Axtell, M.J. (2013) Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 64, 137–159. [DOI] [PubMed] [Google Scholar]
- Bao, W. , Kojima, K.K. and Kohany, O. (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcala, M. , García, A. , Cabrera, J. , Casson, S. , Lindsey, K. , Favery, B. , García‐Casado, G. , Solano, R. , Fenoll, C. and Escobar, C. (2010) Early transcriptomic events in microdissected Arabidopsis nematode‐induced giant cells. Plant J. 61, 698–712. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. and Yekutieli, D. (2001) The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, No. 4 (Aug., 2001), pp. 1165–1188. [Google Scholar]
- Bird, A.F. (1962) The inducement of giant cells by Meloidogyne javanica. Nematologica. 8, 1–10. [Google Scholar]
- Bouyer, D. , Kramdi, A. , Kassam, M. , Heese, M. , Schnittger, A. , Roudier, F. and Colot, V. (2017) DNA methylation dynamics during early plant life. Genome Biol. 18, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, A.H. , Coakley, G. , Simbari, F. , McSorley, H.J. , Quintana, J.F. , Le Bihan, T. , Kumar, S. , Abreu‐Goodger, C. , Lear, M. and Harcus, Y. (2014) Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat. Commun. 5, 5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisine, N. , Quesneville, H. and Colot, V. (2008) Improved detection and annotation of transposable elements in sequenced genomes using multiple reference sequence sets. Genomics 91, 467–475. [DOI] [PubMed] [Google Scholar]
- Cabrera, J. , Bustos, R. , Favery, B. , Fenoll, C. and Escobar, C. (2014) NEMATIC: a simple and versatile tool for the in silico analysis of plant–nematode interactions. Mol. Plant Pathol. 15, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera, J. , Barcala, M. , Fenoll, C. and Escobar, C. (2016a) The power of omics to identify plant susceptibility factors and to study resistance to root‐knot nematodes. Curr. Issues Mol. Biol. 19, 53–72. [PubMed] [Google Scholar]
- Cabrera, J. , Barcala, M. , García, A. , Rio‐Machín, A. , Medina, C. , Jaubert‐Possamai, S. , Favery, B. , Maizel, A. , Ruiz‐Ferrer, V. and Fenoll, C. (2016b) Differentially expressed small RNAs in Arabidopsis galls formed by Meloidogyne javanica: a functional role for miR390 and its TAS3‐derived tasiRNAs. New Phytol. 209, 1625–1640. [DOI] [PubMed] [Google Scholar]
- Cavrak, V.V. , Lettner, N. , Jamge, S. , Kosarewicz, A. , Bayer, L.M. and Scheid, O.M. (2014) How a retrotransposon exploits the plant's heat stress response for its activation. PLoS Genet. 10, e1004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H.‐M. , Chen, L.‐T. , Patel, K. , Li, Y.‐H. , Baulcombe, D.C. and Wu, S.‐H. (2010) 22‐Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. 107, 15 269–15 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuerda‐Gil, D. and Slotkin, R.K. (2016) Non‐canonical RNA‐directed DNA methylation. Nat. Plants, 2, 16 163. [DOI] [PubMed] [Google Scholar]
- Cuperus, J.T. , Carbonell, A. , Fahlgren, N. , Garcia‐Ruiz, H. , Burke, R.T. , Takeda, A. , Sullivan, C.M. , Gilbert, S.D. , Montgomery, T.A. and Carrington, J.C. (2010) Unique functionality of 22‐nt miRNAs in triggering RDR6‐dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat. Struct. Mol. Biol. 17, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani, I. , Baldacci‐Cresp, F. , Hopkins, J. , Andrio, E. , Balzergue, S. , Lecomte, P. , Puppo, A. , Abad, P. , Favery, B. and Hérouart, D. (2012) Plant genes involved in harbouring symbiotic rhizobia or pathogenic nematodes. New Phytol. 194, 511–522. [DOI] [PubMed] [Google Scholar]
- de Almeida‐Engler, J. , Vieira, P. , Rodiuc, N. , de Sa, M.F.G. and Engler, G. (2015) The plant cell cycle machinery: usurped and modulated by plant‐parasitic nematodes In: Advances in Botanical Research, pp. 91–118. Amsterdam, the Netherlands: Elsevier Academic Press. [Google Scholar]
- Diao, X. , Freeling, M. and Lisch, D. (2005) Horizontal transfer of a plant transposon. PLoS Biol. 4, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz‐Manzano, F.E. , Barcala, M. , Engler, G. , Fenoll, C. , de Almeida‐Engler, J. and Escobar, C. (2016a) A reliable protocol for in situ microRNAs detection in feeding sites induced by root‐knot nematodes. Front. Plant Sci. 7, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz‐Manzano, F.E. , Olmo, R. , Cabrera, J. , Barcala, M. , Escobar, C. and Fenoll, C. (2016b) Long‐term in vitro system for maintenance and amplification of root‐knot nematodes in Cucumis sativus roots. Front. Plant Sci. 7, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz‐Manzano, F.E. , Cabrera, J. , Ripoll, J.J. , Olmo, I. , Andrés, M.F. , Silva, A.C. , Barcala, M. , Sánchez, M. , Ruíz‐Ferrer, V. and Almeida‐Engler, J. (2018) A role for the gene regulatory module microRNA172/TARGET OF EARLY ACTIVATION TAGGED 1/FLOWERING LOCUS T (miRNA172/TOE1/FT) in the feeding sites induced by Meloidogyne javanica in Arabidopsis thaliana. New Phytol. 217, 813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen, R.H. , Pelizzola, M. , Schmitz, R.J. , Lister, R. , Dowen, J.M. , Nery, J.R. , Dixon, J.E. and Ecker, J.R. (2012) Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. 109, E2183–E2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dropkin, V. (1972) Pathology of Meloidogyne—galling, giant cell formation, effects on host physiology. EPPO Bull. 2, 23–32. [Google Scholar]
- El‐Baidouri, M. , Carpentier, M.‐C. , Cooke, R. , Gao, D. , Lasserre, E. , Llauro, C. , Mirouze, M. , Picault, N. , Jackson, S.A. and Panaud, O. (2014) Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 24, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar, C. , Barcala, M. , Cabrera, J. and Fenoll, C. (2015) Overview of root‐knot nematodes and giant cells In: Advances in Botanical Research, pp. 1–32. Amsterdam, the Netherlands: Elsevier Academic Press. [Google Scholar]
- Grandbastien, M.‐A. , Audeon, C. , Bonnivard, E. , Casacuberta, J. , Chalhoub, B. , Costa, A.‐P. , Le, Q. , Melayah, D. , Petit, M. and Poncet, C. (2005) Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 110, 229–241. [DOI] [PubMed] [Google Scholar]
- Hewezi, T. , Howe, P. , Maier, T.R. and Baum, T.J. (2008) Arabidopsis small RNAs and their targets during cyst nematode parasitism. Mol. Plant–Microbe Interact. 21, 1622–1634. [DOI] [PubMed] [Google Scholar]
- Hewezi, T. , Lane, T. , Piya, S. , Rambani, A. , Rice, J.H. and Staton, M. (2017) Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiol. 174(1), 405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jammes, F. , Lecomte, P. , Almeida‐Engler, J. , Bitton, F. , Martin‐Magniette, M.L. , Renou, J.P. , Abad, P. and Favery, B. (2005) Genome‐wide expression profiling of the host response to root‐knot nematode infection in Arabidopsis. Plant J. 44, 447–458. [DOI] [PubMed] [Google Scholar]
- Kaur, P. , Shukla, N. , Joshi, G. , VijayaKumar, C. , Jagannath, A. , Agarwal, M. , Goel, S. and Kumar, A. (2017) Genome‐wide identification and characterization of miRNAome from tomato (Solanum lycopersicum) roots and root‐knot nematode (Meloidogyne incognita) during susceptible interaction. PloS One, 12, e0175178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakatsu, T. , Stuart, T. , Valdes, M. , Breakfield, N. , Schmitz, R.J. , Nery, J.R. , Urich, M.A. , Han, X. , Lister, R. and Benfey, P.N. (2016) Unique cell‐type‐specific patterns of DNA methylation in the root meristem. Nat. Plants, 2, 16 058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, J.A. , Du, J. , Hale, C.J. , Feng, S. , Krajewski, K. , Palanca, A.M.S. , Strahl, B.D. , Patel, D.J. and Jacobsen, S.E. (2013) Polymerase IV occupancy at RNA‐directed DNA methylation sites requires SHH1. Nature 498, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S. , Vandivier, L.E. , Tu, B. , Gao, L. , Won, S.Y. , Li, S. , Zheng, B. , Gregory, B.D. and Chen, X. (2015) Detection of Pol IV/RDR2‐dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res. 25, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López, A. , Ramírez, V. , García‐Andrade, J. , Flors, V. and Vera, P. (2011) The RNA silencing enzyme RNA polymerase V is required for plant immunity. PLoS Genet. 7, e1002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke, M.A. and Mosher, R.A. (2014) RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. [DOI] [PubMed] [Google Scholar]
- McCall, M.N. , McMurray, H.R. , Land, H. and Almudevar, A. (2014) On non‐detects in qPCR data. Bioinformatics 30(16), 2310–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue, A.D. , Nuthikattu, S. , Reeder, S.H. and Slotkin, R.K. (2012) Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8, e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, C. , Rocha, M. , Magliano, M. , Ratpopoulo, A. , Revel, B. , Marteu, N. , Magnone, V. , Lebrigand, K. , Cabrera, J. and Barcala, M. (2017) Characterization of microRNAs from Arabidopsis galls highlights a role for miR159 in the plant response to the root‐knot nematode Meloidogyne incognita. New Phytol. 216, 882–896. [DOI] [PubMed] [Google Scholar]
- Nuthikattu, S. , McCue, A.D. , Panda, K. , Fultz, D. , DeFraia, C. , Thomas, E.N. and Slotkin, R.K. (2013) The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21–22 nucleotide small interfering RNAs. Plant Physiol. 162, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmo, R. , Silva, A.C. , Díaz‐Manzano, F.E. , Cabrera, J. , Fenoll, C. and Escobar, C. (2017) A standardized method to assess infection rates of root‐knot and cyst nematodes in Arabidopsis thaliana mutants with alterations in root development related to auxin and cytokinin signaling. In: Dandekar T. and Naseem M. (eds.), Auxins and Cytokinins in Plant Biology, Methods in Molecular Biology (vol. 1569, pp 73‐‐81). New York, NY: Humana Press. [DOI] [PubMed] [Google Scholar]
- Panda, K. , Ji, L. , Neumann, D.A. , Daron, J. , Schmitz, R.J. and Slotkin, R.K. (2016) Full‐length autonomous transposable elements are preferentially targeted by expression‐dependent forms of RNA‐directed DNA methylation. Genome Biol. 17, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piya, S. , Bennett, M. , Rambani, A. and Hewezi, T. (2017). Transcriptional activity of transposable elements may contribute to gene expression changes in the syncytium formed by cyst nematode in arabidopsis roots. Plant Signal. Behav. 12(9), e1362521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo, M. , Cabrera, J. , Lindsey, K. , Topping, J. , Andrés, M.F. , Emiliozzi, M. , Oliveros, J.C. , García‐Casado, G. , Solano, R. and Koltai, H. (2013) Distinct and conserved transcriptomic changes during nematode‐induced giant cell development in tomato compared with Arabidopsis: a functional role for gene repression. New Phytol. 197, 1276–1290. [DOI] [PubMed] [Google Scholar]
- R Development Core Team (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3‐900051‐07‐0, URL: https://www.R-project.org. [Google Scholar]
- Ruiz‐Ferrer, V. and Voinnet, O. (2009) Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 60, 485–510. [DOI] [PubMed] [Google Scholar]
- Sarkies, P. , Selkirk, M.E. , Jones, J.T. , Blok, V. , Boothby, T. , Goldstein, B. , Hanelt, B. , Ardila‐Garcia, A. , Fast, N.M. and Schiffer, P.M. (2015) Ancient and novel small RNA pathways compensate for the loss of piRNAs in multiple independent nematode lineages. PLoS Biol. 13, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J. , Arganda‐Carreras, I. , Frise, E. , Kaynig, V. , Longair, M. , Pietzsch, T. , Preibisch, S. , Rueden, C. , Saalfeld, S. and Schmid, B. (2012) Fiji: an open‐source platform for biological‐image analysis. Nat. Methods, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, L. , Voisin, M. , Tatout, C. and Probst, A.V. (2015) Structure and function of centromeric and pericentromeric heterochromatin in Arabidopsis thaliana. Front. Plant Sci. 6, 1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter, A. , Daniel, X. , Flors, V. , Luna, E. , Hohn, B. and Mauch‐Mani, B. (2012) Descendants of primed Arabidopsis plants exhibit resistance to biotic stress. Plant Physiol. 158, 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin, R.K. and Martienssen, R. (2007) Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. [DOI] [PubMed] [Google Scholar]
- Subramanian, P. , Choi, I.‐C. , Mani, V. , Park, J. , Subramaniyam, S. , Choi, K.‐H. , Sim, J.‐S. , Lee, C.‐M. , Koo, J.C. and Hahn, B.‐S. (2016) Stage‐wise identification and analysis of miRNA from root‐knot nematode Meloidogyne incognita. Int. J. Mol. Sci. 17, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood, C.J. , Henderson, I.R. and Martienssen, R.A. (2017) Genetic and epigenetic variation of transposable elements in Arabidopsis. Curr. Opin. Plant Biol. 36, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara, Z. , Sequeira‐Mendes, J. , Morata, J. , Peiró, R. , Hénaff, E. , Costas, C. , Casacuberta, J.M. and Gutierrez, C. (2017) Retrotransposons are specified as DNA replication origins in the gene‐poor regions of Arabidopsis heterochromatin. Nucleic Acids Res. 45, 8358–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss, U. , Grundler, F.M. and Munch, A. (1992) The parasitic behaviour of second‐stage juveniles of Meloidogyne incognita in roots of Arabidopsis thaliana. Nematologica, 38, 98–111. [Google Scholar]
- Xie, M. and Yu, B. (2015) siRNA‐directed DNA methylation in plants. Curr. Genomics, 16, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, M. , Zhang, S. and Yu, B. (2015) microRNA biogenesis, degradation and activity in plants. Cell. Mol. Life Sci. 72, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. and Wang, P.J. (2016) Multiple LINEs of retrotransposon silencing mechanisms in the mammalian germline. Semin Cell Dev Biol. 59, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, A. , Lepère, G. , Jay, F. , Wang, J. , Bapaume, L. , Wang, Y. , Abraham, A.‐L. , Penterman, J. , Fischer, R.L. and Voinnet, O. (2013) Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. 110, 2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Henderson, I.R. , Lu, C. , Green, P.J. and Jacobsen, S.E. (2007) Role of RNA polymerase IV in plant small RNA metabolism. Proc. Natl. Acad. Sci. 104, 4536–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. , Kimatu, J.N. , Xu, K. and Liu, B. (2010) DNA cytosine methylation in plant development. J. Genet. Genomics, 37, 1–12. [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, Y. , Xie, F. , Li, C. , Zhang, B. , Nichols, R.L. and Pan, X. (2016) Identification and characterization of microRNAs in the plant parasitic root‐knot nematode Meloidogyne incognita using deep sequencing. Funct. Integr. Genomics, 16, 127–142. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Yazaki, J. , Sundaresan, A. , Cokus, S. , Chan, S.W.‐L. , Chen, H. , Henderson, I.R. , Shinn, P. , Pellegrini, M. and Jacobsen, S.E. (2006) Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhao, W. , Li, Z. , Fan, J. , Hu, C. , Yang, R. , Qi, X. , Chen, H. , Zhao, F. and Wang, S. (2015) Identification of jasmonic acid‐associated microRNAs and characterization of the regulatory roles of the miR319/TCP4 module under root‐knot nematode stress in tomato. J. Exp. Bot. 66, 4653–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Expression data of RNA‐directed DNA methylation (RdDM) genes at the NEMATIC database. Col‐0 Arabidopsis transposon elements and genes participating in canonical and non‐canonical RNA‐dependent RNA POLYMERASE 6 (RDR6)‐RdDM are included. Red, induced genes (P < 0.05; Cabrera et al., 2014); green, repressed genes (P < 0.05; Cabrera et al., 2014); grey, genes not differentially expressed relative to control uninfected roots.
Table S1 Statistically significant repetitive repeat‐associated small interfering RNAs (rasiRNAs) after t‐test (P < 0.05) and Benjamini–Hochberg correction. (a) Sequence data after statistical analysis. Order of the columns: rasiRNA sequence (column A), rasiRNA length (column B), biological independent replicates of gall (columns C–E), biological independent replicates of root control (columns F–H), repeat classification by Repbase database (column I), repeat type (column J), the difference in mean values (dm; column K), P value (column L) and P value after Benjamini–Hochberg correction (column M). (b) Location on the Arabidopsis genome of statistically significant rasiRNAs distinctive from galls (P < 0.05 and Benjamini–Hochberg correction). Order of the columns: rasiRNA sequence (column A), Arabidopsis chromosome (column B), start (column C) and end (column D) positions for the alignment, positive or negative strand for the alignment (column E), rasiRNA length (column F), number of normalized sequences for the three biological independent replicates of gall (columns G–I), number of normalized sequences for the three biological independent replicates of root control (columns J–L), repeat classification by Repbase database (column M), repeat type (column N), dm value (column O), P value (column P), P value after Benjamini–Hochberg correction (column Q) and gene annotation from TAIR10 datasets [CDS, gene, intergenic region, intron, promoter, transposable element (TE), untranslated region (UTR), 5′ UTR, cDNA, peptides and 3000 bp downstream]. (c) The same as (b) on distinctive rasiRNAs from root control. (d) The same as (b) on significant rasiRNAs commonly present in galls and root control libraries. (e) Classification of statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs that fully complement other regions of the Arabidopsis genome. All datasets were downloaded from TAIR10: annotated TE, intergenic regions, promoter (3000 bp upstream), 5′ UTR, 3′ UTR, exon, intron, 3000 bp downstream, cDNA, CDS and peptides. No. DE sequence, number of unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01). Unique No. of Hits, number of matches from unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01). Total No. of Hits, total number of matches from unique statistically significant dGall‐rasiRNAs, dRC‐rasiRNAs and common‐rasiRNAs (P < 0.05 and P < 0.01).
Table S2 Statistically significant repetitive repeat‐associated small interfering RNAs (rasiRNAs) after t‐test (P < 0.01) and Benjamini–Hochberg correction. (a) Sequence data after statistical analysis. Order of the columns: rasiRNA sequence (column A), rasiRNA length (column B), number of normalized sequences for the three biological independent replicates of gall (columns C–E), number of normalized sequences for the three biological independent replicates of root control (columns F–H), repeat classification by Repbase database (column I), repeat type (column J), the difference in mean values (dm; column K), P value (column L) and P value after Benjamini–Hochberg correction (column M). (b) Location on the Arabidopsis genome of statistically significant rasiRNAs distinctive from galls (P < 0.01 and Benjamini–Hochberg correction). Order of the columns: rasiRNA sequence (column A), Arabidopsis chromosome (column B), start (column C) and end (column D) positions for the alignment, positive or negative strand for the alignment (column E), rasiRNA length (column F), number of normalized sequences for the three biological independent replicates of gall (columns G–I), number of normalized sequences for the three biological independent replicates of root control (columns J–L), repeat classification by Repbase database (column M), repeat type (column N), dm value (column O), P value (P), P value after Benjamini–Hochberg correction (column Q) and gene annotation from TAIR10 datasets [CDS, gene, intergenic region, intron, promoter, transposable element (TE), untranslated region (UTR), 5′ UTR, cDNA, peptides and 3000 bp downstream]. (c) The same as (b) on statistically distinctive rasiRNAs from root control. (d) The same as (b) on statistically significant rasiRNAs present in galls and root control.
Table S3 Bioinformatic analysis of the statistically significant distinctive repeat‐associated small interfering RNAs (rasiRNAs) in the libraries from galls infected with Meloidogyne javanica and from uninfected Arabidopsis roots. (a) Unique statistically significant rasiRNAs (P < 0.05 and Benjamini–Hochberg) distinctive from gall (dGall‐rasiRNA), root control (dRC‐rasiRNA) and commonly present in both (common‐rasiRNA) that fully matched to other regions of the Arabidopsis genome, classified by chromosomes (a) and strand (b). (b) The same groups of rasiRNAs described previously, but classified by chromosome regions. (c) The same groups of rasiRNAs described previously, but classified by length (18–26 nucleotides in length). (d) The same groups of rasiRNAs described previously, but classified by length (18–26 nucleotides in length) and also by Chromosome (Chr1–Chr5), strand (positive and negative) and chromosome regions. Arabidopsis chromosomes: Chr1, chromosome 1; Chr2, chromosome 2; Chr3, chromosome 3; Chr4, chromosome 4; Chr5, chromosome 5. Chromosome regions: left chromosome arm (R1), left pericentromeric region (PC1), centromere (CEN), right pericentromeric region (PC2) and right chromosome arm (R2).
Table S4 Classification of transposable elements (TEs) targeted by dGall‐rasiRNAs. (a) TEs targeted by dGall‐rasiRNAs (P < 0.01) were classified by TE name (column A), number of dGall‐rasiRNAs that matched to each TE (column B), TE family (column C) and TE superfamily (column D). (b) Members of TE families targeted by dGall‐rasiRNAs (P < 0.01). (c) Members of TE superfamilies targeted by dGall‐rasiRNAs (P < 0.01).
Table S5 List of primers used in this report.