

Summary
Coffee leaf rust, caused by Hemileia vastatrix (Hv), represents the biggest threat to coffee production worldwide and ranks amongst the most serious fungal diseases in history. Despite a recent series of outbreaks and emergence of hypervirulent strains, the population evolutionary history and potential of this pathogen remain poorly understood. To address this issue, we used restriction site‐associated DNA sequencing (RADseq) to generate ∼19 000 single nucleotide polymorphisms (SNPs) across a worldwide collection of 37 Hv samples. Contrary to the long‐standing idea that Hv represents a genetically unstructured and cosmopolitan species, our results reveal the existence of a cryptic species complex with marked host tropism. Using phylogenetic and pathological data, we show that one of these lineages (C3) infects almost exclusively the most economically valuable coffee species (tetraploids that include Coffea arabica and interspecific hybrids), whereas the other lineages (C1 and C2) are severely maladapted to these hosts, but successfully infect diploid coffee species. Population dynamic analyses suggest that the C3 group may be a recent ‘domesticated’ lineage that emerged via host shift from diploid coffee hosts. We also found evidence of recombination occurring within this group, which could explain the high pace of pathotype emergence despite the low genetic variation. Moreover, genomic footprints of introgression between the C3 and C2 groups were discovered and raise the possibility that virulence factors may be quickly exchanged between groups with different pathogenic abilities. This work advances our understanding of the evolutionary strategies used by plant pathogens in agro‐ecosystems with direct and far‐reaching implications for disease control.
Keywords: fungi, Hemileia vastatrix, host shift, hybridization, plant pathogen
Introduction
In an era in which modern agro‐ecosystems create highly conducive environments for the appearance and dissemination of fungal pathogens, the assessment of the dynamics of these pathogens plays a crucial role in the generation of useful recommendations for disease management strategies (Drenth and Guest, 2016; Gandon et al., 2016; Stukenbrock and McDonald, 2008; Stukenbrock et al., 2011). However, if such strategies are to be effective and durable, they must include eco‐evolutionary principles that take into account the pathogen's evolutionary potential (Grünwald et al., 2016; Zhan et al., 2015). Pathogens with high evolutionary potential pose a greater risk of overcoming disease control strategies by usually having mixed reproductive systems, high gene flow, large effective population size and high mutation rates, features that may require special management practices (McDonald and Linde, 2002). In this regard, population genomics have provided an unprecedented amount of data to investigate the evolutionary processes that shape the structure of pathogen populations, pushing forward our understanding of pathogen biology and usually updating, or even inverting, a previous assessment of a pathogen's ability to respond to control measures in agro‐ecosystems (Menardo et al., 2016; Milgroom et al., 2014; Talas and McDonald, 2015).
Coffee leaf rust (CLR), caused by the obligate biotrophic fungus Hemileia vastatrix (Hv), is currently one of the biggest challenges to global coffee production and amongst the most serious crop diseases in history (Talhinhas et al., 2017). The disease causes premature defoliation on several species of the Coffea genus found at lower altitudes (<1000 m), but only C. canephora and C. arabica are considered to be economically relevant at a global scale (McCook and Vandermeer, 2015). Of these two hosts, C. arabica, which is a recent tetraploid hybrid between the diploid C. canephora and C. eugenioides species, represents the most economically valuable species and also the most susceptible to Hv attacks. From the first epidemic report in Sri Lanka in 1869, Hv has spread to virtually every coffee‐growing region in the world in about one and a half centuries (McCook and Vandermeer, 2015; Talhinhas et al., 2017). During this period, several major outbreaks have been registered in Asia and Africa and, more recently, since 2008, a cluster of outbreaks has been occurring across the Americas (Avelino et al., 2015; McCook and Vandermeer, 2015; Zambolim, 2016). Breeding for coffee resistance is considered to be the best long‐term solution to control CLR (McCook and Vandermeer, 2015), but the introduction of resistant varieties in the field has inevitably resulted in the loss of resistance as a result of adaptation of the pathogen. Coffee–rust interactions follow Flor's gene‐for‐gene model (Flor, 1942) within a race‐specific resistance system which imposes a coevolutionary ‘arms race’. The continuous exertion of selective pressure on the pathogen by the resistant varieties has led to the emergence of more than 50 races or pathotypes, which is remarkable for a supposedly asexual pathogen (Talhinhas et al., 2017). Hypervirulent CLR isolates able to infect coffee genotypes previously resistant to all known rust pathotypes have already been identified in India (Prakash et al., 2014).
The seriousness of CLR epidemics has triggered emergency actions across coffee‐producing nations and investigation of the pathogen's biology is gaining considerable momentum with attempts to gather genomic (Cristancho et al., 2014) and transcriptomic (Talhinhas et al., 2014) data. However, considerably less explored remains the evolutionary history of Hv populations, particularly at a global scale. Using random amplification of polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP) markers, population genetic studies of Hv populations have predominantly focused on restricted geographical areas, such as Brazil (Cabral et al., 2016; Maia et al., 2013; Nunes et al., 2009) and Colombia (Rozo et al., 2012), with the exception of Gouveia et al. (2005) which included isolates from Asia, Africa and America. These studies have consistently found no evidence of population structure with respect to pathotype, host or geographical origin, but have reported mixed results regarding the level of genetic variability. The sexuality of Hv is also a debatable subject in the literature, but Hv is generally considered to be an asexual pathogen because a sexual phase of its life cycle has not been identified so far and asexual urediniospores are the only known functional propagules (Silva et al., 2006; Talhinhas et al., 2017). However, meiosis has been discovered recently within the urediniospores in a supposedly hidden sexual reproductive cycle (Carvalho et al., 2011). Whether this means that recombination effectively occurs in natural populations of Hv remains an open question. Using RAPD and AFLP markers, some studies have been unable to detect recombination and support the asexual status of Hv (Gouveia et al., 2005; Rozo et al., 2012 ), whereas others have found evidence of recombination in some specific regions (Cabral et al., 2016; Maia et al., 2013).
In this study, we used restriction site‐associated DNA sequencing (RADseq) to generate thousands of molecular markers for Hv with the goal of investigating its genetic structure and population dynamics from a worldwide sample collection including a broad range of pathotypes and isolates infecting several coffee species. Specifically, our aims were as follows: (i) to produce a large single nucleotide polymorphism (SNP) dataset of high quality by controlling sequencing error using individual sample replicates; (ii) to investigate how genetic variation within Hv populations is distributed according to host, race pathotype and geographical origin; and (iii) to test for the presence of recombination within Hv.
Results
RADseq data assembly and quality control
Illumina RADseq of 38 Hv isolates from 11 geographical locations and different coffee hosts, and comprising 18 unique pathotypes, generated an average of 4.48 × 106 reads per sample, amounting to 4.09 × 108 base pairs per sample (Table S1, see Supporting Information). From the total sampling, nine isolates consisted of technical replicates used to test assembly parameters. Eleven de novo assemblies were performed and the results are summarized in Table S2 (see Supporting Information). The best assembly strategy resulted in a total locus error of 40.40%, partial locus error of 12.52%, allele error of 4.64% and SNP error of 3.92%, and yielded a final matrix of 19 505 SNPs across 14 556 loci and 29 isolates. The additional filter of excluding SNPs with a minor allele frequency (MAF) lower than 5% reduced the dataset to 8389 SNPs (6783 phylogenetically informative) across 6783 loci and 29 isolates.
Phylogenetic analysis
Phylogenetic reconstruction of the 29 Hv isolates with a concatenated dataset of 6783 variable loci using maximum likelihood (ML) and Bayesian methods yielded similar topologies with congruent branch support values (Fig. 1). The resulting best tree revealed the presence of three divergent and well‐supported groups (C1–C3), which appeared to be highly structured according to the host species. The eight isolates from groups C1 and C2 were sampled from diploid coffee species (C. canephora, C. racemosa, C. liberica and C. excelsa), whereas the remaining 21 isolates from group C3 were collected from C. arabica and interspecific hybrids, all of which were tetraploid. No other clear substructuring was observed concerning the geographical location or pathotype of the isolates. The only exception was a shallow, but well‐supported, five isolate group within C3 that included the most basic pathotypes (V5 and V1,5). In the basal position of the C3 group, there were three isolates of different geographical locations and pathotypes in a ladder‐like pattern (999, 2377 and 3624).
Figure 1.
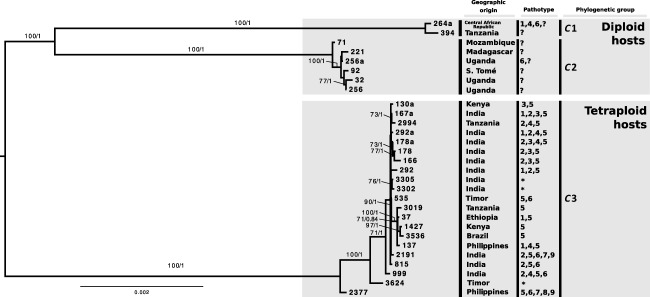
Phylogenetic relationships amongst 29 isolates of Hemileia vastatrix. Support values are provided above the branches with bootstrap values above 70 and posterior probability above 0.8. For each isolate, information about its geographical origin, pathotype and phylogenetic group is provided. *Hypervirulent isolates, i.e. isolates able to overcome all known resistance genes from tetraploid hosts.
Population structuring of Hv
fastSTRUCTURE analyses revealed that the most likely number of clusters (K) in our dataset was three, which corresponded to the same three clusters as identified in the phylogenetic analyses (Fig. 2). For the majority of the isolates, the membership coefficient was at the maximum value for the corresponding cluster, indicating a substantial degree of population differentiation. However, an admixture signal was detected for three isolates of the C3 group, which corresponded to the isolates found at the base of the C3 phylogenetic group (999, 2377 and 3624). This signal suggested that, for these isolates, a proportion of loci were more closely associated with the C2 group (infecting diploid hosts) than with their own group. Principal component analysis (PCA) also revealed the same pattern of three clusters as the previous analyses, with the first and second principal components explaining 35.37% and 20.34% of the variance, respectively (Fig. 3). It is noteworthy that, although most of the isolates from the C3 group were very closely clustered, five isolates (292, 999, 166, 3624 and 2377) appeared to stand apart from the group. Again, three of these isolates corresponded to the same isolates with admixture signal in the fastSTRUCTURE analysis and at the base of the C3 group phylogenetic tree. Estimates of F ST values for each population pair further supported a near‐complete genetic differentiation between each of the three groups, with weighted F ST values ranging from 0.92 (C1 × C2) to 0.95 (C1 × C3 and C2 × C3) (Fig. S1, see Supporting Information).
Figure 2.
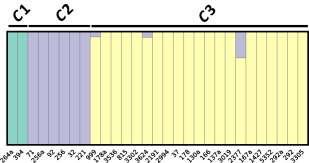
Structure plot of the 29 Hemileia vastatrix isolates with K = 3. Vertical bars represent an isolate and the colour proportion for each bar represents the posterior probability of assignment to one of the three clusters. The three groups identified in the phylogenetic tree are outlined above the plot.
Figure 3.
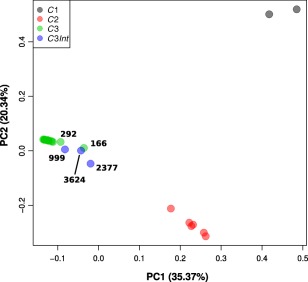
Principal component analysis of genomic diversity for 29 isolates of Hemileia vastatrix. Isolates are colour coded according to their assignment to the three phylogenetic groups. The three isolates of the C3 group that revealed a signal of allele sharing with the C2 group were further differentiated as a fourth C3Int group. PC, principal component.
Genetic diversity
For the estimation of genetic diversity indices, we focused only on the C2 and C3 groups because of the small number of samples of the C1 group (two isolates). From a total of 19 505 SNPs from the full dataset, only 2831 segregated within the C2 group and 7503 within the C3 group. When we applied a MAF filter that allowed only SNPs with more than one allele for each group (5% for the C3 group and 18% for the C2 group), the number of segregating SNPs was further reduced to 551 in C2 and 1563 in C3. This result was further corroborated by the allele frequency spectrum of each group, which revealed a distribution markedly skewed to lower frequencies of derived alleles (Fig. S2, see Supporting Information), and by the high proportion of singletons in C2 (91.81%) and C3 (90.03%) groups. Considering the inbreeding coefficient F IT for each isolate in the C2 and C3 groups, we found that the majority of the isolates had positive values ranging from 0.18 to 0.98, which indicates a slight to moderate excess of homozygotic SNPs compared with theoretical expectation (Fig. 4). Notable exceptions were the three isolates previously found at the base of the C3 phylogenetic group and showing the allele sharing signal, which consistently revealed negative F IT values, indicating an excess of heterozygotic SNPs.
Figure 4.
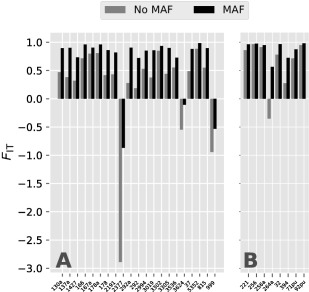
Bar plots of the inbreeding coefficient (F IT) for each isolate of Hemileia vastatrix from the C3 (A) and C2 (B) groups. For each isolate, F IT values were calculated for datasets with (MAF) and without (No MAF) minor allele frequency filtering.
Investigation of introgression between Hv groups
The allele sharing between the C3 and C2 groups was investigated for each isolate by scanning 4494 diagnostic SNPs. The results, summarized in Fig. 5, clearly reveal that most isolates of the C3 group had very few shared alleles with isolates of the C2 group, except for isolates 3624, 999 and 2377 (Fig. 5A). Indeed, from a total of 1938 SNPs that displayed at least one shared allele, 1619 (83.53%) were found exclusively in the three admixed isolates. Individually, these isolates revealed 5.83% (3624), 12.29% (999) and 30.65% (2377) of SNPs with shared alleles with the C2 group, with the vast majority of these SNPs being heterozygous. In contrast, these SNPs were mostly homozygous for the remaining isolates in the respective groups. We also verified whether there was a consistent signal of allele sharing across loci that contained two or more SNPs, which would be expected because of the linkage generated from their proximity. Indeed, between 99.02% and 99.58% of the SNPs in these loci agreed on the allele sharing signal. We also assessed the overlap of the SNPs with shared alleles between these three isolates, and found that only a small proportion were shared amongst the three isolates or paired combinations (Fig. S3, see Supporting Information). For instance, only seven SNPs with shared alleles were found in all three admixed isolates and, among the pair‐wise combinations, the overlap was never higher than 33%. The majority of the SNPs with shared alleles were exclusive to each of the three isolates (2377, 80.45%; 999, 58.80%; 3624, 50.22%). These analyses were also repeated with different F ST values used to generate the ‘signature’ SNPs (0.5 and 0.9) and the results were qualitatively equal and quantitatively similar. Finally, we assessed whether the introgression events occurred in both directions by performing these analyses considering the C2 group as the recipient. Although previous analyses did not hint at the presence of allele sharing in this direction, our SNP scan detected two C2 isolates with 9.96% (71) and 2.56% (256a) SNPs with shared alleles with the C3 group, the majority being heterozygous (Fig. 5B). The remaining C2 isolates displayed only vestigial amounts of shared SNPs.
Figure 5.
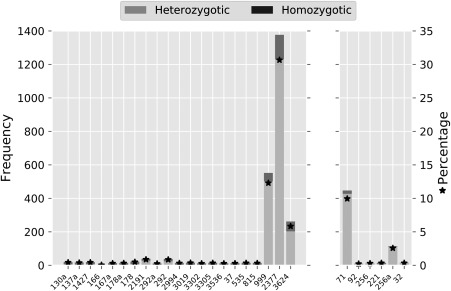
Summary of the diagnostic single nucleotide polymorphism (SNP) scanning for shared alleles between isolates of the C2 and C3 phylogenetic groups. The stacked bar plot represents the frequency, whereas the star point plot represents the percentage, of alleles that isolates shared with the other group.
Linkage disequilibrium (LD) and recombination
To estimate LD metrics and investigate the presence of recombination, we focused on the larger and epidemiologically more relevant C3 group. The mean value of D′ across SNP pairs was high (0.86 ± 0.27) and mean r 2 was low (0.13 ± 0.25) (Table S3, see Supporting Information). From the total pair‐wise SNP comparisons, only 17.8% SNP pairs could reject the null hypothesis of no association between genotypes, that is, of being in LD. Given the presence of isolates with a putative signal of introgression from the C2 group and the existence of a C3 subgroup containing isolates with basal pathotypes, we created one additional dataset in which the three introgressed isolates were removed (NoInt; n = 18) and another in which the isolates from the C3 subgroup were also removed (NoInt_NoV5; n = 13). These datasets were meant to remove potential biases that introgression and mild population structuring could introduce in genotype association, as these phenomena are known to inflate estimates of LD even in the presence of recombination (Agapow and Burt, 2001; Slatkin, 2008). The overall values of D′ and r 2 remained similar for the new datasets, but the percentage of SNP pairs rejecting the null hypothesis decreased to 7.73% in the NoInt dataset (D′ = 0.87 ± 0.29; r 2 = 0.08 ± 0.21) and 10.98% in the NoInt_NoV5 dataset (D′ = 0.89 ± 0.28; r 2 = 0.26) (Table S3).
We then estimated the standardized form of the index of association ( ) for the same datasets in order to assess whether the patterns of genetic variation in the C3 group were consistent with clonal or sexual reproduction. When using the complete C3 group (including putatively introgressed isolates and basal pathotypes), was low (0.05), but significantly higher than the expected null distribution of no linkage amongst markers (P = 0.001) (Fig. S4A, see Supporting Information). The same result was obtained for the NoInt dataset ( = 0.02; P = 0.001) (Fig. S4B), but, when both introgressed taxa and isolates from the C3 subgroup were removed, the index of association could not reject the null hypothesis of sexual reproduction ( = 0.01; P = 1) (Fig. 6).
Figure 6.
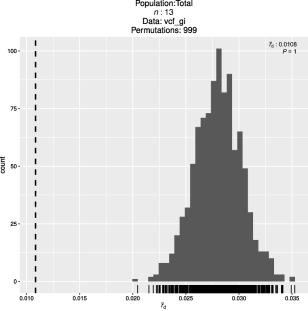
Results from the index of association (IA) analysis, using the standardized form ( ), for the isolates of the C3 group after removing putative introgressed isolates and an incipient but well‐supported subgroup. The histogram depicts the distribution of values expected from unlinked loci. The vertical broken line represents the observed value for the dataset.
Population dynamics of Hv in tetraploid hosts
The demographic history of Hv isolates from the C3 group was reconstructed using an extended Bayesian skyline analysis (Fig. 7). As there was no calibration available for our dataset, time estimates should be interpreted in relative terms. The historical demographic reconstruction revealed that the C3 group suffered a severe bottleneck during and shortly after the divergence from the remaining groups infecting diploid hosts (C1 and C2). After this bottleneck event, the effective population size seems to have remained low for most of the time until the onset of the diversification of the C3 group, at which point an increase in population size to values above those pre‐bottleneck is inferred (Fig. S5, see Supporting Information). We should note that the 95% high posterior density (HPD) intervals for these estimates can be quite large, particularly for the size of the expansion in recent times. However, the pattern of an ancient bottleneck with a recent population expansion still holds, even when different model specifications were experimented with during the skyline analyses.
Figure 7.
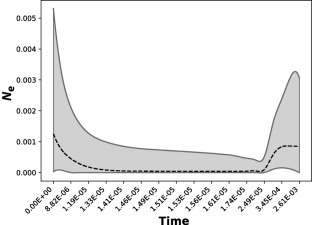
Extended Bayesian skyline plot depicting the population dynamics of the C3 group of Hemileia vastatrix through time. The x‐axis is in relative units of time, and the y‐axis corresponds to the effective population size (N e). The broken black line represents the median estimate of the effective population size, and the full grey lines delimit the 95% high posterior density.
Discussion
Revealing Hv as a potential cryptic species complex with host specialization
One of the first and most striking findings of this work was the discovery of three well‐diverged evolutionary lineages within our sampling of a supposedly single species, which is in disagreement with previous studies (Cabral et al., 2016; Maia et al., 2013; Rozo et al., 2012). Until now, disease management practices were heavily influenced by the idea that Hv represents a large unstructured population that is able to freely switch between coffee species (McCook and Vandermeer, 2015; Zambolim, 2016). By contrast, our results show a clear phylogenetic segregation between isolates infecting C. arabica and interspecific tetraploid hybrids (which will henceforth be collectively referred to as ‘tetraploids’) and isolates infecting C. canephora and other diploid coffee species with no commercial relevance. This structuring pattern was maintained even when an extended sampling of Hv (n = 119) was assessed in preliminary analyses of an ongoing project. The pathogenicity tests routinely performed at Centro de Investigação das Ferrugens do Cafeeiro (CIFC) (Oeiras, Portugal) also support this segregation, at least when considering the commercial coffee plants. Some coffee species of little or no commercial importance, such as C. racemosa and C. liberica, are universally susceptible to all Hv isolates, but their influence in the pathogen's dynamics is low because of their limited geographical distribution. However, this is clearly not the case for tetraploids and C. canephora. Isolates from the C3 group are consistently unable to infect several C. canephora differential genotypes, whereas isolates from the C1 and C2 groups are either unable to infect tetraploids, or trigger only mild symptoms with limited spore production in a few varieties. The immediate deduction from this is that adaptation to either tetraploids or C. canephora entails an adaptive trade‐off for Hv: the ability to successfully infect one host implies poor or no fitness in the other. This marked host tropism is quite intriguing when we consider that, at the cytological level, the infection process of compatible interactions is similar across all Hv isolates, regardless of the host (Silva et al., 2008). Indeed, without the knowledge of Hv's phylogenetic structure, there would be little reason to consider the output of cross‐inoculation tests anything other than the presence of different Hv races. Coincidentally, this would also explain the long‐standing perception that varieties of C. canephora are inherently more resistant to CLR (McCook and Vandermeer, 2015; Talhinhas et al., 2017). What seems to be the case, however, is that C. canephora is highly resistant to isolates adapted to tetraploids, which happen to represent the most widespread and epidemiological relevant group of Hv.
Given the implications of the discovery of multiple divergent and pathologically different lineages within Hv in understanding its evolutionary potential, we further investigated the genetic differentiation of these groups. Results from clustering analyses were unanimous in the confirmation of the three groups identified in the phylogenetic analysis, and further highlighted their high degree of genetic differentiation. With average weighted F ST values above 0.90 in all pair‐wise group comparisons, they seem to be almost completely isolated from each other at the genetic level. Notwithstanding, not only is the infection process identical across all isolates, but they are also morphologically similar (Silva et al., 2006). This raises the question of whether these groups should be considered as cryptic species. Cryptic species complexes are widespread in the fungal kingdom (Pringle et al., 2005; Silva et al., 2012b; Stukenbrock, 2013), including rust fungi (Bennett et al., 2011; Zhao et al., 2015). In fact, agro‐ecosystems have favoured the emergence of new and specialized closely related species by providing a new ecological niche (Silva et al., 2012a; Stukenbrock and Bataillon, 2012), and the correct identification of species boundaries can be difficult even with the aid of molecular data. However, in the case of Hv, there are implications that go beyond the eventual update of the taxonomic status for these groups. From an epidemiological standpoint, it is crucial to understand to what extent these groups are genetically isolated.
Investigation of the presence of introgression between groups of Hv infecting diploid and tetraploid hosts
Despite the high genetic differentiation, evidence of some allele sharing between the C2 and C3 groups was revealed early in our analyses. The challenge here was to assess whether this signal was the result of incomplete lineage sorting or introgression, as both can produce similar patterns of allele sharing (Twyford and Ennos, 2012). Similar to hybridization and introgression, incomplete lineage sorting can cause an excess of shared derived alleles, but this excess arises from purely non‐contemporary demographic phenomena, such as population fragmentation or non‐random mating in the ancient population (Eriksson and Manica, 2012). Nevertheless, these processes leave distinct signatures in the patterns of the genetic diversity of populations that can be unravelled by genome‐wide data (Staubach et al., 2012; Twyford and Ennos, 2012). For instance, considering the C3 group, our fine‐scale assessment of diagnostic SNPs revealed that as much as 84% of the SNPs with alleles shared with the C2 group were found exclusively in three isolates (3624, 999 and 2377). This markedly skewed distribution towards only a few isolates is coupled with the observation that virtually all SNPs in physical linkage have the same allele sharing signal. Contrary to the expectations of incomplete lineage sorting, where allele sharing is expected to be unlinked and reasonably distributed across all isolates, this is the expected result from introgression events. In this scenario, entire chromosomal segments of one species are inserted into the genetic background of the other species, creating blocks of linked SNPs found only on the introgressed offspring (Rheindt et al., 2014). The patterns of heterozygosity in SNPs with shared alleles also lend support to introgression over incomplete lineage sorting. The vast majority of the SNPs with shared alleles were homozygous for all non‐admixed isolates of the C2 and C3 groups, but heterozygous for the three admixed isolates. It is hard to conceive of a scenario in which purely demographic phenomena would lead to the fixation of alternative variants in two populations, with the consistent exception of a few isolates across hundreds of variable sites. However, hybridization and subsequent introgression are expected to produce this pattern of genetic variation (Harrison and Larson, 2014; Todesco et al., 2016). First (F1) or early generation hybrids will be heterozygous for nearly all sites segregating between the two parental species. Backcrossing and introgression of the hybrids into one of the parental species would result in heterozygous variants, as observed in the three admixed isolates. The introgression signal in the admixed isolates could also explain their basal position in the respective group, even though their pathotypes are highly derivative. It is widely known that hybridization and introgression can interfere with phylogenetic reconstructions, as introgressed individuals will appear at intermediate positions between the parental groups, depending on the extension of the introgression (Eaton and Ree, 2013).
Overall, although we did not detect any C2 × C3 F1 hybrids in our sampling, introgression between the C2 and C3 groups appears to be the most likely explanation for the shared polymorphism pattern in our data. Moreover, the presence of alleles in the C2 group that are shared with the C3 group suggests that introgression may be bidirectional. This has major implications for the epidemiology of Hv as it creates the possibility of genetic exchange between two divergent groups adapted to different sets of coffee hosts. In principle, this could quickly generate novel genetic recombinants and promote rapid adaptation to new resistant coffee hosts. Hybridization and introgression are being increasingly regarded as important for the rapid emergence of novel pathotypes or even species (Menardo et al., 2016; Stukenbrock, 2016; Stukenbrock and McDonald, 2008). In the case of Hv, this would certainly represent an opportunity for the pathogen to respond to the introgression of resistance genes from diploid coffee species into tetraploids. However, whether or not these introgression events are actually adaptive remains untested. In our sampling, we could only detect a clear signal of introgression in three isolates of the C3 group, but it is entirely possible that more events have already occurred. Our results are not consistent with this being a single and geographically restricted event, but are most likely the result of multiple independent events. The introgressed isolates were found in geographically distant locations and present a very small overlap in the introgressed SNPs. PCA of the C3 group alone also revealed that the introgressed isolates do not cluster together (Fig. S6, see Supporting Information). If we combine this information with the occurrence of recombination within the C3 group (see below), the dissemination of only a few adaptive key alleles that are harder to detect becomes a likely possibility, and one that may be more common than previously thought (Anderson et al., 2009; Rheindt et al., 2014).
The emergence and evolutionary history of the C3 group
With the clustering of isolates infecting the most economically relevant coffee hosts into a single group, our interest was shifted to how the C3 group emerged and subsequently evolved. Even with the application of thousands of SNPs, no significant genetic structuring was found according to geographical origin or pathotype within this group, apart from the incipient separation of isolates with basal pathotypes. To explain this apparent panmixia, in addition to the previously identified factors of worldwide expansion of coffee trade and high vagility of Hv spores (McCook and Vandermeer, 2015; Talhinhas et al., 2017), this work suggests two additional factors that may be tightly linked to the evolutionary potential of the C3 group: a recent origin and reticulate evolution, i.e. evolution shaped by hybridization/recombination between diverging lineages.
As the genetic structure of the C1–C3 groups is unknown to date, it has never been considered that the emergence of Hv in tetraploid hosts could have been the result of a recent introduction and adaptation event. Notwithstanding, this would be consistent with the recent origin of C. arabica from a hybridization event between C. eugenioides and C. canephora (Cenci et al., 2012). The reconstruction of the population dynamics of the C3 group lends support to this hypothesis by revealing a pattern of an early bottleneck followed by a relatively long stable period that culminated in a very recent population size explosion. The timing of the inferred bottleneck coincides with the divergence time between the C3 and C1–C2 groups and the population expansion closely matches the onset of the C3 group diversification. As only isolates of the C3 group seem to be adapted to tetraploid hosts, we suggest that this bottleneck could have been the result of a recent founder effect and/or a process of adaptation to C. arabica from a maladapted population. The most likely source of this introduction seems to be Hv isolates from diploid coffee hosts. Isolates infecting these hosts not only appear to be present for much longer, given their deep phylogenetic division, but they also share striking similarities in morphology and infection process with the C3 isolates. Indeed, the fact that some C2 and C1 isolates are able to infect some C. arabica genotypes to a very limited extent would provide an entry point to this new ecological niche. The opportunity to shift to the new host would be given by the regular presence of tetraploids and C. canephora plants, in either wild or commercial plantations, within cruising range of one another (McCook and Vandermeer, 2015). Given the high genetic homogeneity of cultivated varieties of tetraploids (Anthony et al., 2002), the eventual adaptation of this population could have resulted in the inferred pathogen population boom. As mentioned above, this adaptation entails a fitness trade‐off, as isolates of the C3 group are no longer able to infect C. canephora. Coincidentally, this would also produce an effective prezygotic reproductive barrier, known as immigrant inviability, where assortative mating arises as a byproduct of host specialization (Giraud, 2006; Giraud et al., 2010). Nevertheless, the opportunity to hybridize could still present itself in non‐commercial universally susceptible diploid coffee species. Interestingly, a similar scenario has been tied to the emergence of several recent pathogens in modern agro‐ecosystems (Geoghegan et al., 2016), including Colletotrichum kahawae causing coffee berry disease (Silva et al., 2012a).
What remains surprising in this scenario is how the substantial loss of genetic diversity after the emergence of this new ‘domesticated’ lineage is consistent with the rapid pace with which new rust pathotypes emerge. Despite the long‐standing assumption that Hv is an asexual pathogen (Silva et al., 2006), our work provides several lines of evidence that recombination is occurring at least in the C3 group. The presence of introgression between the C3 and C2 groups was the first hint pointing to the presence of recombination, as it is required for the backcrossing of genetic material from hybrids to the parental groups. Moreover, a very low proportion of SNP pairs was found to be under significant LD and the standardized index of association could not reject the null hypothesis of no linkage amongst markers. These metrics have been successfully used to uncover the existence of recombination in several fungal populations (Gladieux et al., 2011; Short et al., 2015), and their combination supports the existence of recombination of Hv isolates infecting tetraploids. Coincidentally, this would provide a viable mechanism for the quick generation of new allele combinations and, by extension, new pathotypes. The synergy of recombination, introgression and the ability of fungi to amplify favourable allele combinations through the massive production of asexual spores creates a plausible mechanism for pathogens to rapidly overcome resistant varieties, even from an initial pool of low genetic variation (Giraud et al., 2010). However, in the absence of functional sexual propagules, the mechanism by which recombination occurs still requires further investigation. Possible mechanisms include cryptosexuality within the urediniospores and parasexual nuclear recombination between two isolates after germ tube fusion or hyphal anastomosis, all of which can mimic the effects of sexual reproduction (Carvalho et al., 2011; Vittal et al., 2012; Wang and McCallum, 2009).
Conclusion and remarks on CLR disease management
Altogether, this work presents a striking example of how agro‐ecosystems can have a dramatic impact on the population biology and evolution of plant pathogens. The discovery of three divergent genetic lineages within Hv with specialized pathogenic behaviour towards commercial coffee species, and their ability to hybridize, represents a paradigm shift in our current understanding of this pathogen's evolutionary history. It is clear that the taxonomic status of the three genetic lineages reported here needs to be revised in future studies. Referring to Hv as a single cohesive genetic unit is not only of little practical use in the future, but may also be detrimental to quarantine practices. The introduction of isolates from the C1 or C2 groups into tetraploid coffee plantations does not carry the same risk as isolates from the C3 group, and the reverse is true for C. canephora plantations. Mixing different lineages with the ability to hybridize has the potential to increase the rate of pathotype emergence. In particular, greater care should be taken with nurseries of coffee germplasm and experimental stations, where multiple genotypes of different hybrids and species are stored nearby. Our results reveal that, if not adequately controlled and quarantined, isolates of Hv have strategies that make these locales ideal breeding grounds for the emergence of new hypervirulent pathotypes. Subsequent studies of the C3 group will be paramount to understand the frequency, direction and content that is transferred via introgression and recombination, which, ultimately, may be tightly linked to the capacity of the pathogen to overcome host resistance.
Experimental Procedures
Fungal material, sample preparation and RADseq
Twenty‐nine isolates of Hv from 11 geographical locations, collected from different diploid and tetraploid coffee hosts and comprising 18 unique pathotypes, were retrieved from a spore collection maintained at CIFC (Table S4, see Supporting Information). Pathotypes were determined based on inoculation assays on a set of coffee differentials bearing different resistance gene combinations under standard testing conditions (d'Oliveira, 1954). Coffee differentials include tetraploid coffee plants with resistance genotypes containing the nine major dominant genes (SH1–SH9), either individually or in combination, identified through classical genetics according to Flor's gene‐for‐gene model (Bettencourt and Rodrigues, 1988), and also six diploid coffee hosts (C. racemosa, C. excelsea, two C. canephora and two C. congensis). According to the virulence profile on the differential plants, isolates are classified into pathotypes (races) comprising virulence genes as inferred by Flor's theory, ranging from v1 to v9 in isolates derived from C. arabica and tetraploid interspecific hybrids, whereas those of the races that attack diploid coffee species are not known (v?). Individual sample replicates were added for nine isolates, chosen in order to encompass the largest possible range of geographical locations, pathotypes and hosts. The total sampling contained 38 isolates that were processed independently. DNA was extracted using a cetyltrimethylammonium bromide (CTAB)‐based protocol modified from Kolmer et al. (1995), and genomic DNA concentration and quality were checked by visual inspection on an agarose gel and with an ND‐1000 Nanodrop spectrophotometer. Three micrograms of high‐quality genomic DNA per individual were sent to Floragenex Inc. (Eugene, OR, USA) for RAD library preparation and sequencing, as described previously (Etter et al., 2011). Libraries with sample‐specific barcode sequences were produced from DNA digested with PstI enzyme, and single‐end (1 × 100‐bp) sequencing was performed in an Illumina (Portland, Oregon, USA) HiSeq 2000 machine.
RADseq assembly strategy and SNP calling
Sequence reads were de‐multiplexed and quality filtered with the process_radtags script (Catchen et al., 2013). Reads with uncalled bases or distance to barcodes higher than unity were removed. Base calls with a Phred score under 20 were converted to Ns and reads containing more than four Ns were discarded. RAD tags were de novo assembled and genotyped using PyRAD v3.0.63 (Eaton, 2014) because of its ability to handle gaps when clustering sequence reads. In order to optimize the assembly process, we used a similar strategy to that described in Mastretta‐Yanes et al. (2015), in which individual sample replicates were used to assess error rates across a range of values for three major assembly parameters. Four error rates were considered for each assembly: (i) total locus error; (ii) partial locus error; (iii) haplotype error; and (iv) SNP error. A complete description of the specified error rates is provided in Text S1 (see Supporting Information). Several values were tested for each of the following three assembly parameters separately, whilst fixing the remaining parameters: (i) minimum read depth (5–15); (ii) clustering threshold (0.80–097); and (iii) maximum shared heterozygosity (2–10). An overview of the parameter values for each assembly is provided in Table S2. Error rates and total numbers of SNPs were evaluated for each assembly, after the removal of SNPs with less than 50% of taxa representation, using the custom compare_pairs.py script. The assembly that yielded the lowest error rates whilst maximizing the total number of SNPs was then processed for further analyses. SNPs with a mismatch in at least one replicate were removed from the VCF file. Replicates were also removed, retaining whichever isolate from the pair that contained more data. Handling and exploration of alignment data matrices were performed using TriFusion v0.4.12 software (https://github.com/OdiogoSilva/TriFusion).
Phylogenetic analyses
To infer the phylogenetic relationships amongst our samples, we applied a supermatrix approach that included loci with SNPs represented in more than 50% of the isolates and a MAF above 5% into a single concatenated alignment. Concatenation and conversion of the alignment matrices to the appropriate formats were performed with TriFusion. ML reconstruction was performed using RAxML v8.2.6 (Stamatakis, 2014) with the GTRCAT model of sequence evolution and with bootstrap support estimated from 1000 replicates. The same data matrix was used for phylogeny estimation using a Bayesian framework as implemented in MrBayes v3.2.6 (Ronquist et al., 2012) with the GTR + Γ model of sequence evolution. Posterior probabilities were generated from 1 × 107 generations, sampling at every 1000th iteration, and the analysis was run three times with one cold and three incrementally heated Metropolis‐coupled Monte Carlo Markov chains, starting from random trees. The achievement of the stationary phase and mixing was checked for all parameters using Tracer V1.4, and 1 × 106 generations were discarded as burn‐in. Trees from different runs were combined using Logcombiner and summarized in a majority rule 50% consensus tree. Both RaxML and MrBayes runs were performed in the Cipres Science Gateway clusters (Miller et al., 2015).
Evaluation of Hv genetic structure
To detect potential admixture and population structure in our Hv sampling, the software fastSTRUCTURE v1.0 (Raj et al., 2014) wrapped in Structure_threader v0.4.3 (Pina‐Martins et al., 2017) was used with K values in the range 1–8. The optimal K value was found using the chooseK.py script bundled in Structure_threader. PCA was run using the SNPRELATE v1.8.0 R package (Zheng et al., 2012) after filtering non‐biallelic loci using the snpgdsPCA function. The discriminant analysis of principal components (DPAC) (Jombart et al., 2010) was also used to assess population clustering without the assumption of a population genetic model, and to provide a statistically sound method to detect the number of clusters that best fit the data. This analysis was conducted in the R environment with the package ADEGENET v2.0.1 (Jombart, 2008). The degree of population differentiation was also assessed by calculating the overall and distribution of SNP F ST values for each population pair using VCFTOOLS v0.1.14 (Danecek et al., 2011).
Introgression assessment
Early in this work, multiple genetically differentiated groups were detected within our sampling with some potential evidence of introgression found for specific isolates. To better assess this potential signal of introgression at the individual level, and to exclude the possibility of incomplete lineage sorting, we devised a simple SNP scanning strategy between the two groups of isolates. First, we filtered the SNP dataset so that only SNPs with an F ST value above 0.8 were retained. These were named ‘diagnostic’ SNPs, as they were able to almost completely differentiate both groups, whilst allowing the sharing of alleles for very few isolates (presumably the isolates with introgressed loci). Then, for each isolate in one of the groups, we scanned each ‘diagnostic’ SNP and kept score of how many alleles were shared with the genotype of the other group, and whether the shared alleles were homozygous or heterozygous. Our expectation for this assessment was that isolates with a signal of introgression would contain much higher numbers of shared alleles than would non‐introgressed taxa.
Recombination and LD
The LD metrics D′ and r 2 were used to estimate the pair‐wise LD between all SNPs that passed the MAF filter. Both metrics were calculated using the LD function of the GENETICS v1.3.8.1 R package, which is able to estimate the proportion of heterozygous genotypes using ML. In addition, a chi‐squared statistical test was calculated for each pair, providing a P value to test for marker independence. In addition, the fact that fungal populations may not be strictly clonal or sexual, but are often found in an intermediate position, was taken into account by estimating the standardized index of association ( ) (Agapow and Burt, 2001), as implemented in the POPPR v2.3.0 R package (Kamvar et al., 2014). The index of association tests to what extent individuals that are the same at one locus are more likely than random to be the same at other loci, and is thus a measure of LD based on the variance of the pair‐wise distance between individuals. The standardized form of the index of association provides a more unbiased test that is independent of the locus sample size. In order to obtain an expected null distribution and a P value, 999 permutations were performed on the data.
Population dynamics of Hv infecting tetraploid hosts
The demographic history of Hv populations was reconstructed using the extended Bayesian skyline model (Heled and Drummond, 2008) implemented in BEAST v1.8.3 (Drummond et al., 2012). The dataset for this analysis consisted only of RAD loci that were present in all analysed isolates (no missing data) and was assembled using TriFusion. A single molecular clock partition was established using the uncorrelated log‐normal prior. As there are no calibration points available within our dataset, the arbitrary starting value of unity was chosen for the ‘ucld.mean’ parameter of the clock model. The analysis was run twice, with default priors, for 1 × 108 generations, sampling at every 10 000th generation after an initial burn‐in of 10%. The performance of the Markov chain Monte Carlo (MCMC) procedure, namely the Effective Sample Size (ESS) values and mixing for each parameter, was assessed in Tracer v1.6 (Rambaut and Drummond, 2007).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Triangular matrix with pairwise F ST comparisons between the phylogenetic groups within Hemileia vastatrix. Scatter plots in the upper part of the matrix represent the F ST values for each individual single nucleotide polymorphism (SNP) segregating between the given group pair. Histograms in the lower part of the matrix represent the distribution of F ST values for the same segregating SNPs.
Fig. S2 Allele frequency spectrum for the single nucleotide polymorphisms (SNPs) of the C3 phylogenetic group (left) and the C2 group (right).The vertical broken line represents the mean of the dataset.
Fig. S3 Venn diagram of the overlap of the single nucleotide polymorphisms (SNPs) with shared alleles among the three Hemileia vastatrix isolates showing the admixture signal.
Fig. S4 Results from the index of association analysis, using the standardized form ( ), for the isolates of the complete C3 group (A) and after removing putative introgressed isolates (B). The histogram depicts the distribution of values expected from unlinked loci. The vertical broken line represents the observed value for the dataset.
Fig. S5 Extended Bayesian skyline plot depicting the population dynamics of the C3 group of Hemileia vastatrix through time with the inferred phylogeny overlapped. The x‐axis is in relative units of time, and the y‐axis corresponds to the effective population size. The broken black line represents the median estimate of the effective population size, and the full grey lines delimit the 95% high posterior density.
Fig. S6 Principal component analysis of genomic diversity amongst the 21 Hemileia vastatrix isolates from the C3 group. Isolates are colour coded to differentiate the three introgressed isolates from the remaining members of the C3 group.
Table S1 Summary statistics of read number and total base pairs for each Hemileia vastatrix isolate.
Table S2 Summary of the results from the 11 assemblies of restriction site‐associated DNA (RAD) data using PyRAD with information on the assembly parameters, error statistics and loci, and single nucleotide polymorphism (SNP) information.
Table S3 Summary of linkage disequilibrium statistics and results of the significance tests for the complete C3 dataset (Var1_MM50_maf), after removing putatively introgressed isolates (Var1_MM50_NoInt_maf) and after removing the isolates from the incipient C3 subgroup (Var1_MM50_NoInt_NoV5_maf)
Table S4 List of the Hemileia vastatrix isolates used in this study.
Text S1 Detailed overview of the restriction site‐associated DNA sequencing (RADseq) assembling strategy using technical replicates.
Acknowledgements
This project was funded by project grant PTDC/AGR‐GPL/119943/2010 from the Fundação para a Ciência e Tecnologia (FCT) (http://www.fct.pt/) and by FCT Unit funding UID/BIA/00329/2013 and UID/AGR/04129/2013. D.N.S. acknowledges the FCT grant SFRH/BD/86736/2012. D.B. acknowledges the FCT grant SFRH/BPD/104629/2014. We also acknowledge Ana Paula Pereira and the technical staff from CIFC/ISA for the support provided on isolate multiplication and pathotype testing.
References
- Agapow, P.‐M. and Burt, A. (2001) Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes, 1, 101–102. [Google Scholar]
- Anderson, T.M. , vonHoldt, B.M. , Candille, S.I. , Musiani, M. , Greco, C. , Stahler, D.R. , Smith, D.W. , Padhukasahasram, B. , Randi, E. , Leonard, J.A. , Bustamante, C.D. , Ostrander, E.A. , Tang, H. , Wayne, R.K. and Barsh, G.S. (2009) Molecular and evolutionary history of melanism in North American gray wolves. Science, 323, 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, F. , Combes, M.C. , Astorga, C. , Bertrand, B. , Graziosi, G. and Lashermes, P. (2002) The origin of cultivated Coffea arabica L. varieties revealed by AFLP and SSR markers. Theor. Appl. Genet. 104, 894–900. [DOI] [PubMed] [Google Scholar]
- Avelino, J. , Cristancho, M. , Georgiou, S. , Imbach, P. , Aguilar, L. , Bornemann, G. , Läderach, P. , Anzueto, F. , Hruska, A.J. and Morales, C. (2015) The coffee rust crises in Colombia and Central America (2008–2013): impacts, plausible causes and proposed solutions. Food Secur. 7, 303–321. [Google Scholar]
- Bennett, C. , Aime, M.C. and Newcombe, G. (2011) Molecular and pathogenic variation within Melampsora on Salix in western North America reveals numerous cryptic species. Mycologia, 103, 1004–1018. [DOI] [PubMed] [Google Scholar]
- Bettencourt, A.J. and Rodrigues, C.J Jr. (1988) Principles and practice of coffee breeding for resistance to rust and other diseases In: Coffee Agronomy, Vol. 4 (Clarke R.J. and Macrae R., eds), pp. 199–234. London and New York: Elsevier. [Google Scholar]
- Cabral, P.G.C. , Maciel‐Zambolim, E. , Oliveira, S.A.S. , Caixeta, E.T. and Zambolim, L. (2016) Genetic diversity and structure of Hemileia vastatrix populations on Coffea spp. Plant Pathol. 65, 196–204. [Google Scholar]
- Carvalho, C.R. , Fernandes, R.C. , Carvalho, G.M.A. , Barreto, R.W. and Evans, H.C. (2011) Cryptosexuality and the genetic diversity paradox in coffee rust, Hemileia vastatrix . PLoS One, 6, e26387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. and Cresko, W. A. (2013) Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci, A. , Combes, M.C. and Lashermes, P. (2012) Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant Mol. Biol. 78, 135–145. [DOI] [PubMed] [Google Scholar]
- Cristancho, M.A. , Botero‐Rozo, D.O. , Giraldo, W. , Tabima, J. , Riaño‐Pachón, D.M. , Escobar, C. , Rozo, Y. , Rivera, L.F. , Durán, A. , Restrepo, S. , Eilam, T. , Anikster, Y. and Gaitán, A.L. (2014) Annotation of a hybrid partial genome of the coffee rust (Hemileia vastatrix) contributes to the gene repertoire catalog of the Pucciniales. Front. Plant Sci. 5, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. , Lunter, G. , Marth, G.T. , Sherry, S.T. , McVean, G. and Durbin, R. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth, A. and Guest, D.I. (2016) Fungal and oomycete diseases of tropical tree fruit crops. Annu. Rev. Phytopathol. 54, 373–395. [DOI] [PubMed] [Google Scholar]
- Drummond, A.J. , Suchard, M. A. , Xie, D. and Rambaut, A. (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton, D.A.R. (2014) PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics, 30, 1844–1849. [DOI] [PubMed] [Google Scholar]
- Eaton, D.A.R. and Ree, R.H. (2013) Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). Syst. Biol. 62, 689–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson, A. and Manica, A. (2012) Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proc. Natl. Acad. Sci. USA, 109, 13 956–13 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter, P. , Bassham, S. , Hohenlohe, P. , Johnson, E. and Cresko, W. (2011) SNP discovery and genotyping for evolutionary genetics using RAD sequencing In: Molecular Methods for Evolutionary Genetics, Methods in Molecular Biology (Orgogozo V. and Rockman M., eds), pp. 157–178. Totowa, NJ: Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor, H.H. (1942) Inheritance of pathogenicity in Melampsora lini . Phytopathology, 32, 653–669. [Google Scholar]
- Gandon, S. , Day, T. , Metcalf, C.J.E. and Grenfell, B.T. (2016) Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends Ecol. Evol. 31, 776–788. [DOI] [PubMed] [Google Scholar]
- Geoghegan, J.L. , Senior, A.M. and Holmes, E.C. (2016) Pathogen population bottlenecks and adaptive landscapes: overcoming the barriers to disease emergence. Proc. R. Soc. London B: Biol. Sci. 283, 20160727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, T. (2006) Selection against migrant pathogens: the immigrant inviability barrier in pathogens. Heredity (Edinb), 97, 316–318. [DOI] [PubMed] [Google Scholar]
- Giraud, T. , Gladieux, P. and Gavrilets, S. (2010) Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol. Evol. 25, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladieux, P. , Guérin, F. , Giraud, T. , Caffier, V. , Lemaire, C. , Parisi, L. , Didelot, F. and Le Cam, B. (2011) Emergence of novel fungal pathogens by ecological speciation: importance of the reduced viability of immigrants. Mol. Ecol. 20, 4521–4532. [DOI] [PubMed] [Google Scholar]
- Gouveia, M.M.C. , Ribeiro, A. , Várzea, V.M.P. and Rodrigues, C.J. (2005) Genetic diversity in Hemileia vastatrix based on RAPD markers. Mycologia, 97, 396–404. [DOI] [PubMed] [Google Scholar]
- Grünwald, N. , McDonald, B.A. and Milgroom, M.G. (2016) Population genomics of fungal and oomycete pathogens. Annu. Rev. Phytopathol. 54, 323–346. [DOI] [PubMed] [Google Scholar]
- Harrison, R.G. and Larson, E.L. (2014) Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809. [DOI] [PubMed] [Google Scholar]
- Heled, J. and Drummond, A.J. (2008) Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics, 24, 1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , Devillard, S. and Balloux, F. (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamvar, Z.N. , Tabima, J.F. and Grünwald, N.J. (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ, 2, e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmer, J.A. , Liu, J.Q. and Sies, M. (1995) Virulence and molecular polymorphism in Puccinia recondita f. sp. tritici in Canada. Phytopathology, 85, 276–285. [Google Scholar]
- Maia, T. A. , Maciel‐Zambolim, E. , Caixeta, E.T. , Mizubuti, E.S.G. and Zambolim, L. (2013) The population structure of Hemileia vastatrix in Brazil inferred from AFLP. Australas. Plant Pathol. 42, 533–542. [Google Scholar]
- Mastretta‐Yanes, A. , Arrigo, N. , Alvarez, N. , Jorgensen, T.H. , Piñero, D. and Emerson, B.C. (2015) Restriction site‐associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Resour. 15, 28–41. [DOI] [PubMed] [Google Scholar]
- McCook, S. and Vandermeer, J. (2015) The big rust and the red queen: long‐term perspectives on coffee rust research. Phytopathology, 105, 1164–1173. [DOI] [PubMed] [Google Scholar]
- McDonald, B.A. and Linde, C. (2002) Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379. [DOI] [PubMed] [Google Scholar]
- Menardo, F. , Praz, C.R. , Wyder, S. , Ben‐David, R. , Bourras, S. , Matsumae, H. , McNally, K.E. , Parlange, F. , Riba, A. , Roffler, S. , Schaefer, L.K. , Shimizu, K.K. , Valenti, L. , Zbinden, H. , Wicker, T. and Keller, B. (2016) Hybridization of powdery mildew strains gives rise to pathogens on novel agricultural crop species. Nat. Genet. 48, 201–205. [DOI] [PubMed] [Google Scholar]
- Milgroom, M.G. , Jiménez‐Gasco, M. D M. , Olivares García, C. , Drott, M.T. and Jiménez‐Díaz, R.M. (2014) Recombination between clonal lineages of the asexual fungus Verticillium dahliae detected by genotyping by sequencing. PLoS One, 9, e106740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M.A. , Schwartz, T. , Pickett, B.E. , et al. (2015) A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evolutionary Bioinformatics 11, 43–48. doi: 10.4137/EBO.S21501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes, C.C. , Maffia, L.A. , Mizubuti, E.S.G. , Brommonschenkel, S.H. and Silva, J.C. (2009) Genetic diversity of populations of Hemileia vastatrix from organic and conventional coffee plantations in Brazil. Australas. Plant Pathol. 38, 445–452. [Google Scholar]
- d'Oliveira, B. (1954) As ferrugens do cafeeiro. Rev. Café Port, 1, 5–13. [Google Scholar]
- Pina‐Martins, F. , Silva, D.N. , Fino, J. and Paulo, O.S. (2017) Structure_threader: An improved method for automation and parallelization of programs structure, fastStructure and MavericK on multicore CPU systems. Molecular Ecology Resources, n/a-n/a. doi: 10.1111/1755-0998.12702. [DOI] [PubMed]
- Prakash, N. , Devasia, J. , Das Divya, K. , Manjunatha, B. , Seetharam, H. , Kumar, A. and Jayarama. (2014) Breeding for rust resistance in Arabica – where we are and what next? In: Proceedings of the 25th International Conference on Coffee Science (ASIC), Armenia, Colombia, p. B10.
- Pringle, A. , Baker, D.M. , Platt, J.L. , Wares, J.P. , Latgé, J.P. and Taylor, J.W. (2005) Cryptic speciation in the cosmopolitan and clonal human pathogenic fungus Aspergillus fumigatus . Evolution, 59, 1886–1899. [PubMed] [Google Scholar]
- Raj, A. , Stephens, M. and Pritchard, J.K. (2014) fastSTRUCTURE: variational inference of population structure in large SNP datasets. Genetics, 197, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. and Drummond, A.J. (2007) Tracer v1.4 Available at http://beast.bio.ed.ac.uk/Tracer [accessed on November 2017].
- Rheindt, F.E. , Fujita, M.K. , Wilton, P.R. and Edwards, S.V. (2014) Introgression and phenotypic assimilation in Zimmerius flycatchers (Tyrannidae): population genetic and phylogenetic inferences from genome‐wide SNPs. Syst. Biol. 63, 134–152. [DOI] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. , van der Mark, P. , Ayres, D.L. , Darling, A. , Höhna, S. , Larget, B. , Liu, L. , Suchard, M.A. and Huelsenbeck, J.P. (2012) Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozo, Y. , Escobar, C. , Gaitán, Á. and Cristancho, M. (2012) Aggressiveness and genetic diversity of Hemileia vastatrix during an epidemic in Colombia. J. Phytopathol. 160, 732–740. [Google Scholar]
- Short, D.P.G. , Gurung, S. , Gladieux, P. , Inderbitzin, P. , Atallah, Z.K. , Nigro, F. , Li, G. , Benlioglu, S. and Subbarao, K.V. (2015) Globally invading populations of the fungal plant pathogen Verticillium dahliae are dominated by multiple divergent lineages. Environ. Microbiol. 17, 2824–2840. [DOI] [PubMed] [Google Scholar]
- Silva, D.N. , Talhinhas, P. , Cai, L. , Manuel, L. , Gichuru, E.K. , Loureiro, A. , Várzea, V. , Paulo, O.S. and Batista, D. (2012a) Host‐jump drives rapid and recent ecological speciation of the emergent fungal pathogen Colletotrichum kahawae . Mol. Ecol. 21, 2655–2670. [DOI] [PubMed] [Google Scholar]
- Silva, D.N. , Talhinhas, P. , Várzea, V. , Cai, L. , Paulo, O.S. and Batista, D. (2012b) Application of the Apn2/MAT locus to improve the systematics of the Colletotrichum gloeosporioides complex: an example from coffee (Coffea spp.) hosts. Mycologia, 104, 396–409. [DOI] [PubMed] [Google Scholar]
- Silva, M.C. , Várzea, V. , Guerra‐Guimarães, L. , Azinheira, H. , Fernandez, D. , Petitot, A.‐S. , Bertrand, B. , Lashermes, P. and Nicole, M. (2006) Coffee resistance to the main diseases: leaf rust and coffee berry disease. Braz. J. Plant Physiol. 18, 119–147. [Google Scholar]
- Silva, M.C. , Guerra‐Guimarães, L. , Loureiro, A. and Nicole, M.R. (2008) Involvement of peroxidases in the coffee resistance to orange rust (Hemileia vastatrix). Physiol. Mol. Plant Pathol. 72, 29–38. [Google Scholar]
- Slatkin, M. (2008) Linkage disequilibrium – understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubach, F. , Lorenc, A. , Messer, P.W. , Tang, K. , Petrov, D.A. and Tautz, D. (2012) Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus). PLoS Genet. 8, e1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock, E.H. (2013) Evolution, selection and isolation: a genomic view of speciation in fungal plant pathogens. New Phytol. 199, 895–907. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. (2016) Hybridization speeds up the emergence and evolution of a new pathogen species. Nat. Genet. 48, 113–115. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. and Bataillon, T. (2012) A population genomics perspective on the emergence and adaptation of new plant pathogens in agro‐ecosystems. PLoS Pathog. 8, e1002893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock, E.H. and McDonald, B.A. (2008) The origins of plant pathogens in agro‐ecosystems. Annu. Rev. Phytopathol. 46, 75–100. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. , Bataillon, T. , Dutheil, J.Y. , Hansen, T.T. , Li, R. , Zala, M. , McDonald, B. A. , Wang, J. and Schierup, M.H. (2011) The making of a new pathogen: insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res. 21, 2157–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talas, F. and McDonald, B.A. (2015) Genome‐wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genomics, 16, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhinhas, P. , Azinheira, H.G. , Vieira, B. , Loureiro, A. , Tavares, SÃ‐L. , Batista, D. , Morin, E. , Petitot, A.‐S. , Paulo, O.S. , Poulain, J. , Da Silva, C. , Duplessis, S. , Silva, Mdo C. and Fernandez, D. (2014) Overview of the functional virulent genome of the coffee leaf rust pathogen Hemileia vastatrix with an emphasis on early stages of infection. Front. Plant Sci. 5, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhinhas, P. , Batista, D. , Diniz, I. , Vieira, A. , Silva, D.N. , Loureiro, L. , Tavares, S. , Pereira, A.P. , Azinheira, H.G. , Guerra‐Guimarães, L. , Várzea, V. and Silva, M.D.C. (2017) Pathogen profile: the coffee leaf rust pathogen Hemileia vastatrix: one and a half centuries around the tropics. Mol. Plant Pathol. 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todesco, M. , Pascual, M.A. , Owens, G.L. , Ostevik, K.L. , Moyers, B.T. , Hübner, S. , Heredia, S.M. , Hahn, M.A. , Caseys, C. , Bock, D.G. and Rieseberg, L.H. (2016) Hybridization and extinction. Evol. Appl. 9, 892–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford, A. D. and Ennos, R. A. (2012) Next‐generation hybridization and introgression. Heredity (Edinb), 108, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittal, R. , Yang, H.C. and Hartman, G.L. (2012) Anastomosis of germ tubes and migration of nuclei in germ tube networks of the soybean rust pathogen, Phakopsora pachyrhizi . Eur. J. Plant Pathol. 132, 163–167. [Google Scholar]
- Wang, X. and McCallum, B. (2009) Fusion body formation, germ tube anastomosis, and nuclear migration during the germination of urediniospores of the wheat leaf rust fungus, Puccinia triticina . Phytopathology, 99, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Zambolim, L. (2016) Current status and management of coffee leaf rust in Brazil. Trop. Plant Pathol. 41, 1–8. [Google Scholar]
- Zhan, J. , Thrall, P.H. , Papaïx, J. , Xie, L. and Burdon, J.J. (2015) Playing on a pathogen's weakness: using evolution to guide sustainable plant disease control strategies. Annu. Rev. Phytopathol. 53, 19–43. [DOI] [PubMed] [Google Scholar]
- Zhao, P. , Wang, Q.‐H. , Tian, C.‐M. , Kakishima, M. and Papp, T. (2015) Integrating a numerical taxonomic method and molecular phylogeny for species delimitation of Melampsora species (Melampsoraceae, Pucciniales) on willows in China. PLoS One, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S.M. , Laurie, C. and Weir, B.S. (2012) A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28, 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Triangular matrix with pairwise F ST comparisons between the phylogenetic groups within Hemileia vastatrix. Scatter plots in the upper part of the matrix represent the F ST values for each individual single nucleotide polymorphism (SNP) segregating between the given group pair. Histograms in the lower part of the matrix represent the distribution of F ST values for the same segregating SNPs.
Fig. S2 Allele frequency spectrum for the single nucleotide polymorphisms (SNPs) of the C3 phylogenetic group (left) and the C2 group (right).The vertical broken line represents the mean of the dataset.
Fig. S3 Venn diagram of the overlap of the single nucleotide polymorphisms (SNPs) with shared alleles among the three Hemileia vastatrix isolates showing the admixture signal.
Fig. S4 Results from the index of association analysis, using the standardized form ( ), for the isolates of the complete C3 group (A) and after removing putative introgressed isolates (B). The histogram depicts the distribution of values expected from unlinked loci. The vertical broken line represents the observed value for the dataset.
Fig. S5 Extended Bayesian skyline plot depicting the population dynamics of the C3 group of Hemileia vastatrix through time with the inferred phylogeny overlapped. The x‐axis is in relative units of time, and the y‐axis corresponds to the effective population size. The broken black line represents the median estimate of the effective population size, and the full grey lines delimit the 95% high posterior density.
Fig. S6 Principal component analysis of genomic diversity amongst the 21 Hemileia vastatrix isolates from the C3 group. Isolates are colour coded to differentiate the three introgressed isolates from the remaining members of the C3 group.
Table S1 Summary statistics of read number and total base pairs for each Hemileia vastatrix isolate.
Table S2 Summary of the results from the 11 assemblies of restriction site‐associated DNA (RAD) data using PyRAD with information on the assembly parameters, error statistics and loci, and single nucleotide polymorphism (SNP) information.
Table S3 Summary of linkage disequilibrium statistics and results of the significance tests for the complete C3 dataset (Var1_MM50_maf), after removing putatively introgressed isolates (Var1_MM50_NoInt_maf) and after removing the isolates from the incipient C3 subgroup (Var1_MM50_NoInt_NoV5_maf)
Table S4 List of the Hemileia vastatrix isolates used in this study.
Text S1 Detailed overview of the restriction site‐associated DNA sequencing (RADseq) assembling strategy using technical replicates.