

Summary
Quantitative disease resistance (QDR) is the predominant form of resistance against necrotrophic pathogens. The genes and mechanisms underlying QDR are not well known. In the current study, the Arabidopsis–Alternaria brassicae pathosystem was used to uncover the genetic architecture underlying resistance to A. brassicae in a set of geographically diverse Arabidopsis accessions. Arabidopsis accessions revealed a rich variation in the host responses to the pathogen, varying from complete resistance to high susceptibility. Genome‐wide association (GWA) mapping revealed multiple regions to be associated with disease resistance. A subset of genes prioritized on the basis of gene annotations and evidence of transcriptional regulation in other biotic stresses was analysed using a reverse genetics approach employing T‐DNA insertion mutants. The mutants of three genes, namely At1g06990 (GDSL‐motif lipase), At3g25180 (CYP82G1) and At5g37500 (GORK), displayed an enhanced susceptibility relative to the wild‐type. These genes are involved in the development of morphological phenotypes (stomatal aperture) and secondary metabolite synthesis, thus defining some of the diverse facets of quantitative resistance against A. brassicae.
Keywords: Arabidopsis, CYP82G1, GORK, GWA mapping, necrotrophs, quantitative resistance
Introduction
Fungal pathogens are one of the highly evolved groups of microorganisms affecting various plant species and strongly differ in important life history traits, such as dispersal mechanisms, type of reproduction and modes of parasitism. Pathogenic fungi obtain resources from their hosts principally in two different ways: as biotrophs or necrotrophs. Necrotrophic pathogens extract nutrients from dead cells of the host killed during invasion. In crops, economic damage caused by fungal diseases is estimated to be above US$200 billion annually (Birren et al., 2002). A recent survey has indicated that losses caused by necrotrophic pathogens far exceed those resulting from biotrophic pathogens (Murray and Brennan, 2009). Broad host‐range necrotrophs (BHNs), such as Sclerotinia sclerotiorum and Botrytis cinerea, can infect more than 300 different plant species. BHNs typically deploy a diverse arsenal of effectors, including cell‐wall degrading enzymes (CWDEs), phytotoxic compounds and reactive oxygen species (ROS), to induce necrosis. The diversity of virulence strategies thus warrants a multifaceted defence by the host to successfully ward off the attack (Roux et al., 2014). In contrast, narrow host‐range necrotrophs (NHNs), such as Cochliobolus victoriae, Pyrenophora tritici‐repentis and Stagonosporum nodorum, tend to rely on host‐specific toxins (HSTs) that are directed at specific targets present only in some species or subtypes of a particular species (Condon et al., 2013; Lorang et al., 2012). The recognition of these HSTs by the host machinery thus leads to susceptibility. Plant disease resistance can be either qualitative, which is conferred by single resistance (R) genes, or quantitative, which is mostly mediated by multiple genes. Host resistance against BHNs is known to be usually quantitative (St Clair, 2010). Some of the quantitative resistance loci (QRLs) for the paradigmatic BHNs, such as B. cinerea, S. sclerotiorum and Plectosphaerella cucumerina, have been identified, but the underlying mechanisms of most of these QRLs are unknown (Denby et al., 2004; Llorente et al., 2005; Micic et al., 2004; Rowe and Kliebenstein, 2008). Few genes which recognize the HSTs of NHNs have been identified (Faris et al., 2010; Friesen et al., 2007; Liu et al., 2012; Lorang et al., 2007). Unlike NHNs, necrotrophs such as Alternaria brassicae and Alternaria brassicicola infect only the members of the Brassicaceae family, including the wild and cultivated species (Sharma et al., 2002). These necrotrophs thus represent an intermediate class between BHNs and NHNs. The genetic architecture of resistance to these necrotrophs is relatively unexplored when compared with that of BHNs and NHNs. There are currently no known resistance loci identified in any of the natural hosts (Brassica crops) for resistance to A. brassicae.
Arabidopsis has been used as a model host to study the host–pathogen interactions of many plant pathogens. Arabidopsis has a very extensive global distribution, and local adaptation to various environments has led to substantial phenotypic variation in its natural populations (Koornneef et al., 2004). Also, an extensive catalogue of genetic variation [approximately 250 000 single nucleotide polymorphisms (SNPs)] in approximately 1300 accessions has been developed (Horton et al., 2012). Most of the Arabidopsis accessions tested with A. brassicicola have shown complete resistance to the pathogen (Kagan and Hammerschmidt, 2002; Mukherjee et al., 2009). In comparison, in an earlier work on a few accessions of Arabidopsis, a number of highly susceptible and resistant accessions were found (Rajarammohan et al., 2017). In the crosses between highly susceptible and resistant accessions, both major and minor loci conferring resistance were mapped. Further, it was hypothesized that different loci/mechanisms conferring resistance might exist in different accessions of Arabidopsis.
The major objectives of this study were as follows: (i) to investigate whether there is variation for resistance against A. brassicae amongst a set of diverse Arabidopsis accessions; (ii) to study the genetic architecture underlying this variation (if any); and (iii) to obtain insights into the genes and mechanisms involved in the mediation of resistance against A. brassicae.
Results
Natural variation in response to A. brassicae in Arabidopsis accessions
A collection of 176 accessions was selected for this study. These accessions were chosen as they were genetically diverse and were collected from throughout the native range of the species (Cao et al., 2011; Nordborg et al., 2005). Some of the accessions for which very few seeds were available, had poor germination efficiency and a very late flowering time were not included in the analysis. A total number of 123 accessions was used for genome‐wide association (GWA) mapping. The geographical location and disease indices of these accessions are listed in Table S1 (see Supporting Information). The number of infection foci developing into lesions was considered as a measure of susceptibility (disease index, DI). DI was normalized to a susceptible control accession (normalized disease index, NDI; described in Experimental procedures) in each experiment to account for experimental variation. The accessions exhibited wide variation in their response to the pathogen (Fig. 1A). Disease susceptibility varied substantially with the NDI varying from 0 to 1.40. The continuous variation observed indicated that the trait is quantitative in nature. Significant differences in the NDI were observed amongst the accessions [analysis of variance (ANOVA): F = 37.6265, P < 2.0 × 10−16], suggesting that genetic variation could explain a major part of the phenotypic variation observed. Subsequently, the broad‐sense heritability (H 2) was found to be 0.863 (confidence interval, 0.826–0.895). The NDI was not correlated with latitude or longitude of the origin of the collection of accessions (–0.07 < r < 0.13, P > 0.16). In order to ascertain whether resistance or susceptibility was predominant in the population tested, the accessions were broadly classified as resistant (NDI = 0–0.20), intermediate (NDI = 0.21–0.75), susceptible (NDI = 0.76–0.95) and highly susceptible (NDI = 0.96–1.40) based on their disease reactions. The majority of the tested accessions were found to be intermediate (48%, or 59 accessions), 23% (28 accessions) were resistant and 29% (36 accessions) were categorized as susceptible and highly susceptible (Fig. 1B).
Figure 1.
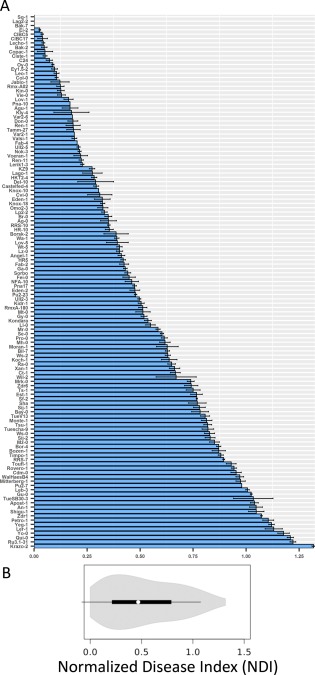
Natural variation in disease resistance to Alternaria brassicae in 123 accessions of Arabidopsis thaliana. (A) Normalized disease index (NDI) scores of the 123 accessions used in the study. The mean and standard error are shown (n ≥⩾ 12). (B) Violin plot (box‐and‐whisker plot overlaid with a kernel density plot) of the phenotypic variation in the collection of 123 accessions.
GWA mapping of resistance to A. brassicae in Arabidopsis accessions
To study the genetic basis of resistance to A. brassicae, GWA mapping was performed using the NDI scores and the 214 000 SNP dataset that is commonly employed for GWA studies in Arabidopsis (Kim et al., 2007). The significance of association was evaluated using three different models, namely a linear regression model (LM), a non‐parametric test [Kruskal–Wallis test (KW)] and an accelerated mixed model (AMM). Each of the methods has their advantages and drawbacks (Korte and Farlow, 2013; Vilhjalmsson and Nordborg, 2013). The rationale behind the use of three different models for GWA mapping was to achieve a balance between the false‐positive (LM and KW) and false‐negative (AMM) rates. GWA mapping using all three models revealed multiple peaks of moderate significance rather than a single strong association, as seen in some GWA studies (Fig. 2A–C) (Baxter et al., 2010; Chao et al., 2012; Meijon et al., 2013). This result suggests that the variation mapped is polygenic and is controlled by multiple genes. The most significant SNP association that was consistent across all the methods was at chromosome 3: 9168769 (P = 8.95 × 10−7). However, as the trait is known to be quantitative, a set of heuristic parameters was used to determine additional candidate loci associated with resistance to A. brassicae (Fig. 2D). Many studies have developed different criteria to prioritize and validate the candidate genes (Chan et al., 2010; Verslues et al., 2014).
Figure 2.
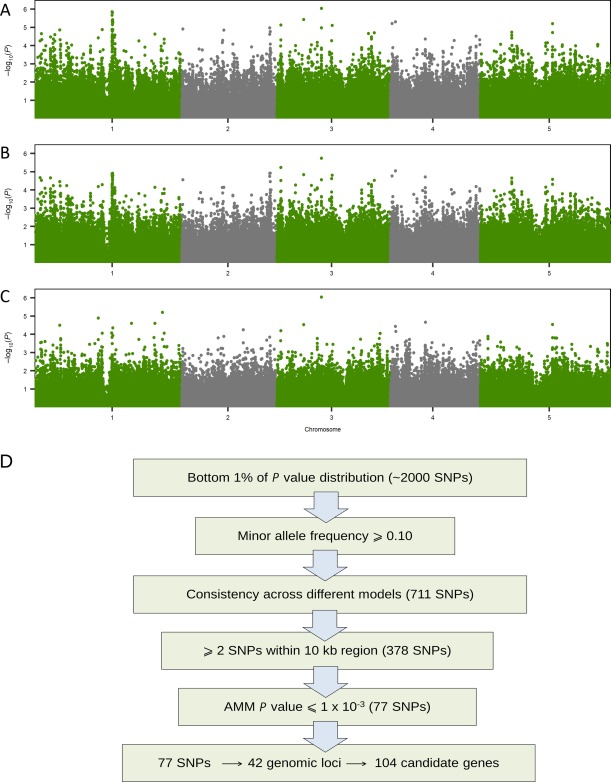
Manhattan plots of single nucleotide polymorphism (SNP) association with disease resistance phenotype (normalized disease index, NDI) using a linear regression model (LM) (A), non‐parametric test [Kruskal–Wallis test (KW)] (B) and an accelerated mixed model (AMM) (C). SNPs along each chromosome are represented along the x‐axis, and the –log10(P) value is shown along the y‐axis. Arabidopsis chromosomes 1–5 are shown in contrasting colours on the x‐axis. (D) Schematic diagram of the pipeline to narrow down the list of SNPs to candidate genes (details given in the text).
Arbitrarily, SNPs from the bottom 1% of the P value distribution of each method (approximately 2000 SNPs) and a minor allele frequency (MAF) > 10% were considered for further analysis. Although the ranks of the top SNPs varied between the methods, there was an overall concordance between the SNPs from the three methods (Fig. S6, see Supporting Information). The SNPs that were consistently significant across all three methods were considered for further analysis. A total of 711 SNP associations (P values ranging from 8.94 × 10−7 to 4.44 × 10−3) were found to be common between the three methods. Previous reports have shown that, for true associations, multiple SNPs per region are found to be associated with the trait (Chan et al., 2010; Zhao et al., 2007). Therefore, we prioritized the list based on the presence of two or more significant SNPs from a particular genomic region (Chan et al., 2010), which led to a comprehensive list of 378 SNPs. A final P value cut‐off (AMM) of 1 × 10−3 was used to demarcate a set of candidate SNPs. Subsequently, 77 candidate SNPs were mapped to the genome, leading to the identification of 42 loci associated with disease resistance. The list of candidate genes was determined after taking into account other SNPs that were in linkage disequilibrium (LD) with the candidate SNPs. An LD value of r 2 > 0.6 was used to demarcate the SNPs that could be directly or indirectly associated with the trait. Consequently, 106 candidate genes were identified (Table S4, see Supporting Information). As there was no a priori information about the genetics of host response to A. brassicae, it was challenging to narrow down the list of candidates that could be functionally validated. Therefore, gene annotations and the expression profiles of these genes under various biotic stresses from public databases (Botany Array Resource, GEO and AtGenExpress) were used to select probable candidate genes. Subsequently, a subset of 16 candidate genes was prioritized based on the evidence from functional annotations (relating to defence responses) or gene expression profiles (as described above), or both, for functional validation using a reverse genetics approach.
Functional validation of candidate genes
To determine whether the identified genes influence disease progression, the knockout effect of the candidate genes was tested using T‐DNA insertion mutants. T‐DNA insertion mutants in six of the candidate genes were not available from the stock centre as indicated in Table 1. One of the candidate genes (At1g58160) was annotated to be a pseudogene in the Col‐0 accession, and hence the T‐DNA insertion mutant in this gene was not studied. T‐DNA insertions for three candidates were present in the promoter regions. As it is unclear how insertions in the promoter would affect the activity of the gene, and hence its role in disease resistance, these mutants were also not considered. Finally, homozygous insertion mutants in the locus of six candidate genes were obtained from the stock centre (Table 1). Seeds of the mutant line SALK_104813C (At3g23280) did not germinate and hence could not be used for further analysis. The presence of homozygous T‐DNA insertions in the genes was confirmed using polymerase chain reaction (PCR) and the null mutation was confirmed by reverse transcription‐polymerase chain reaction (RT‐PCR) (Figs S1–S3, see Supporting Information). Disease resistance, as measured by the normalized cumulative disease index (NCDI), was determined for the mutants and compared with that of wild‐type Col‐0 (Table 2). The mutants showed varying degrees of disease establishment, as evident from the macroscopic lesions formed. F‐tests derived from linear models for NCDI revealed that the mutants of genes At3g25180, At5g37500 and At1g06990 were significantly different from the wild‐type, while accounting for variation between experiments and individual replicates. The effect of experiments and individual replicates was significantly higher than the effect of the genotype for mutants of the genes At2g44240 and At2g44270 (Tables 2, S5, see Supporting Information).
Table 1.
The subset of 16 candidate genes prioritized from the 106 genes identified in the genome‐wide association (GWA) mapping.
| Gene | Description | P value of most significant SNP * | Annotated role in defence † | Expression levels in biotic stress ‡ | SALK line | Location of insertion |
|---|---|---|---|---|---|---|
| AT1G06990 | Encodes a GDSL‐like lipase superfamily protein | 1.79E‐04 | Yes | No | SALK_097961C | Exon |
| AT1G58160 | Encodes a mannose‐binding lectin superfamily protein (JAX1) | 5.44E‐05 | Yes | No | Pseudogene in Col‐0 | — |
| AT1G79310 | Encodes a putative metacaspase (MC7) | 2.49E‐04 | Yes | No | SALK_148148C | Promoter |
| AT1G79320 | Encodes a putative metacaspase (MC6) | 2.49E‐04 | Yes | No | Not available § | — |
| AT2G43860 | Encodes a pectin lyase‐like superfamily protein | 2.346E‐05 | Yes | No | SALK_098125C | Promoter |
| AT2G44240 | Encodes a plant protein of unknown function (DUF239) | 1.71E‐05 | No | Yes | SALK_075708 | Exon |
| AT2G44270 | Encodes ROL5 (repressor of lxr1) | 1.71E‐05 | No | Yes | SALK_078566C | Exon |
| AT3G10195 | Encodes a defensin‐like (DEFL) family protein | 6.29E‐04 | Yes | No | SALK_017693C | Promoter |
| AT3G23280 | Encodes a ubiquitin ligase (XBAT35) | 6.57E‐05 | No | Yes | SALK_104813C | Exon |
| AT3G25180 | Encodes a cytochrome P450 monooxygenase (CYP82G1) | 8.94E‐07 | Yes | Yes | CS436097 | Intron |
| AT3G52600 | Encodes a cell wall invertase 2 (CWINV2) | 3.34E‐05 | Yes | Yes | Not available § | — |
| AT3G57380 | Encodes a glycosyltransferase family 61 protein | 2.49E‐04 | Yes | Yes | Not available | — |
| AT4G02330 | Encodes a pectin methylesterase (PME41) | 4.88E‐06 | Yes | Yes | Not available § | — |
| AT5G13190 | Encodes a plasma membrane localized LITAF domain protein (GILP) | 8.40E‐05 | Yes | Yes | Not available | — |
| AT5G37500 | Encodes a guard cell outward potassium channel GORK | 1.73E‐04 | No | Yes | SALK_144737C | Exon |
| AT5G37610 | Encodes a eukaryotic porin family protein | 1.90E‐05 | No | Yes | Not available § | — |
*P value of most significant single nucleotide polymorphism (SNP) from the linear model.
†Gene annotations from TAIR 10 and gene ontology (GO) terms.
‡mRNA expression levels in various biotic stresses from public databases.
§Not available at the time of the experiment.
Table 2.
Normalized cumulative disease index (NCDI) values and significance of the genotype terms for the T‐DNA insertion mutants.
| Line | NCDI (mean ± SE) | % increase in susceptibility | F value | P value: genotype |
|---|---|---|---|---|
| At3g25180 | 0.280 ± 0.03 | 110.5263158 | 40.7498 | 2.41E‐05 |
| At5g37500 | 0.300 ± 0.03 | 125.5639098 | 29.3237 | 7.16E‐05 |
| At1g06990 | 0.201 ± 0.02 | 51.12781955 | 6.8566 | 0.019371 |
| At2g44240 | 0.082 ± 0.02 | −38.34586466 | 5.0509 | 0.04125 |
| At2g44270 | 0.161 ± 0.08 | 21.05263158 | 0.7882 | 0.39774 |
| Col‐0 | 0.133 ± 0.02 | – | – | – |
Summary of disease indices (NCDI) and P values of an F‐test from a linear model comparing each mutant separately with wild‐type Col‐0. The model is described in Experimental procedures and the expanded table can be found as Table S5 (see Supporting Information).
Natural variation at the CYP82G1 locus mediates resistance to A. brassicae
The gene At3g25180 encodes a cytochrome P450 monooxygenase (CYP82G1) that catalyses the production of two volatile homoterpenes, (E,E)‐4,8,12‐trimethyl‐1,3,7,11‐tridecatetraene (TMTT) and 4,8‐dimethylnona‐1,3,7‐triene (DMNT), although it produces TMTT only in planta (Lee et al., 2010). Multiple significant SNPs were present within the exonic region of At3g25180 (Fig. 3A). For the highly significant SNP positioned at Chr3–9168769 (P = 8.94 × 10−7), most of the resistant accessions had the G allele, whereas the susceptible accessions had the alternative T allele (Fig. 3B). CYP82G1 contained a total of eight non‐synonymous changes, which clearly segregated between 10 highly resistant and 10 highly susceptible accessions (Fig. S4, see Supporting Information). Four of these non‐synonymous changes were found to have a potentially damaging effect on the protein structure based on in silico studies (Table S6, see Supporting Information). Furthermore, the expression levels of the gene in a highly resistant and highly susceptible accession on infection were studied. The gene was significantly up‐regulated in the resistant accession (CIBC‐5), whereas it was down‐regulated in the susceptible accession (Zdr1) (Fig. 3C). Therefore, these preliminary analyses show that the natural variation in defence against A. brassicae contributed by CYP82G1 seems to be driven by both expression‐level and protein structure‐level changes. To determine whether CYP82G1 mediates resistance to A. brassicae, a T‐DNA insertion mutant (CS436097) was tested for its response to the pathogen. This was the only insertion mutant available for this gene from the stock centre and had already been characterized to be a null mutant in a previous study (Lee et al., 2010). The mutant of the gene At3g25180 (CS436097) caused >110% increase in susceptibility relative to the wild type (Col‐0) (Fig. 3D,E, Table 2).
Figure 3.
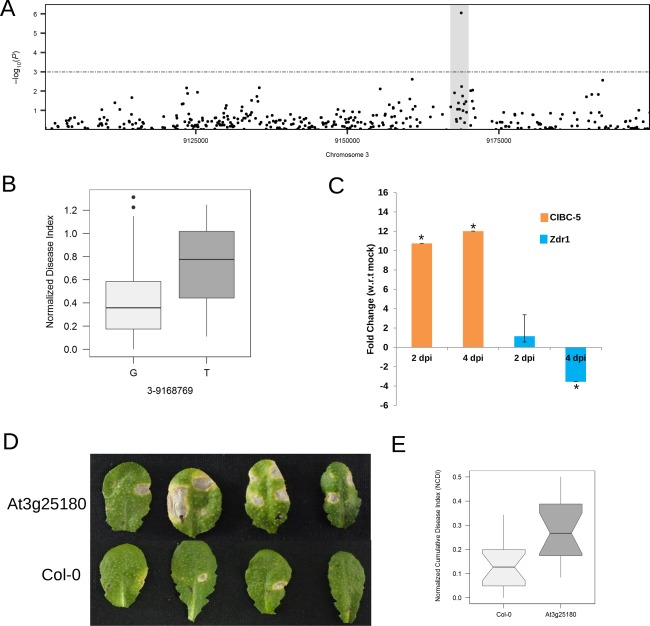
Functional analysis of the gene At3g25180. (A) Local association plot (accelerated mixed model, AMM) zoomed in on region 21 (chromosome 3) containing the gene At3g25180. (B) Boxplot of normalized disease index (NDI) sorted by the alleles (G and T) at the single nucleotide polymorphism (SNP) position Chr3‐9168769. (C) Expression pattern of At3g25180 in CIBC‐5 and Zdr1, at 2 and 4 days post‐infection (dpi), with reference to (w.r.t.) mock infected (distilled water). The mean values (± standard deviation) of three biological replicates are shown. *P < 0.05 by Mann–Whitney U‐test. (D) Representative images of infected leaves of the mutant (At3g25180) and wild‐type (Col‐0) at 7 dpi. (E) Boxplot showing normalized cumulative disease index (NCDI) of wild‐type (Col‐0) and mutant.
Stomatal closure regulated by a Guard cell outward rectifying K+ channel (GORK) aids in resistance to A. brassicae
The gene At5g37500 contains multiple nominally significant SNPs (Fig. 4A). It encodes a Guard cell outward rectifying K+ channel (GORK) of the Shaker family of K+ channels, which is involved in stomatal opening and closure. Sorting the accessions based on the alleles (G and A) of the most significant SNP in this region positioned at Chr5‐14892180 (P = 1.73 × 10−4) showed a clear demarcation between the resistant and susceptible accessions (Fig. 4B). A mutant containing the T‐DNA insertion in At5g37500 showed a highly significant increase in susceptibility relative to the wild‐type (>125% increase in susceptibility) (Fig. 4C,D, Table 2).
Figure 4.
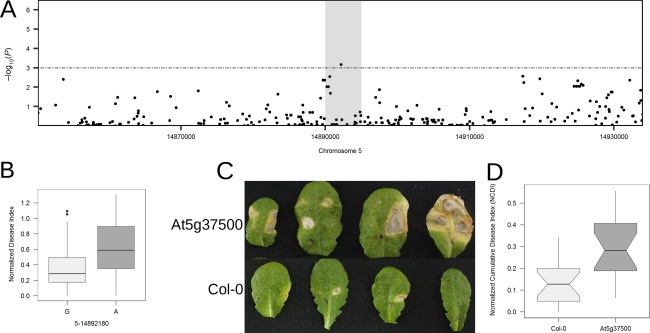
Functional analysis of the gene At5g37500. (A) Local association plot (accelerated mixed model, AMM) zoomed in on region 38 (chromosome 5) containing the gene At5g37500. (B) Boxplot of normalized disease index (NDI) sorted by the alleles (G and A) at the single nucleotide polymorphism (SNP) position Chr5‐14892180. (C) Representative images of infected leaves of the mutant (At5g37500) and wild‐type (Col‐0) at 7 days post‐infection (dpi). (D) Boxplot showing normalized cumulative disease index (NCDI) of wild‐type (Col‐0) and mutant.
Possible role of the GDSL‐motif containing lipase (At1g06990) in resistance to A. brassicae
The intergenic region between At1g06980 and At1g06990 contains two nominally significant SNPs (Fig. 5A). The gene At1g06990 encodes a GDSL‐motif esterase/acyltransferase/lipase. GDSL‐motifs containing lipases, such as GLIP1 and GLIP2, have been implicated in resistance to A. brassicicola (Kwon et al., 2009; Lee et al., 2009). Sorting the accessions based on the alleles (G and T) of the most significant SNP in this region positioned at Chr1‐2145568 (P = 1.79 × 10−4) showed a clear demarcation between the resistant and susceptible accessions (Fig. 5B). The mutant in the exonic region of this gene showed a nominal increase (>54%) in susceptibility (Fig. 5C,D, Table 2).
Figure 5.
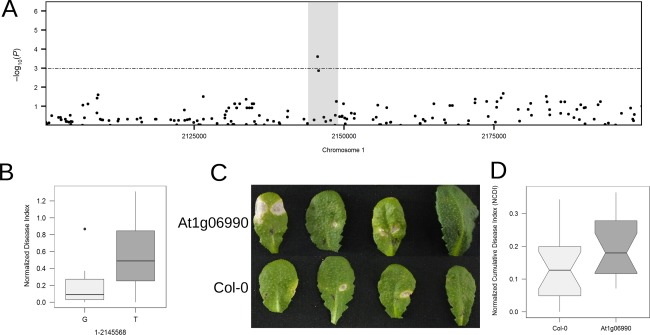
Functional analysis of the gene At1g06990. (A) Local association plot (accelerated mixed model, AMM) zoomed in on region 2 (chromosome 1) containing the gene At1g06990. (B) Boxplot of normalized disease index (NDI) sorted by the alleles (G and T) at the single nucleotide polymorphism (SNP) position Chr1‐2145568. (C) Representative images of infected leaves of the mutant (At1g06990) and wild‐type (Col‐0) at 7 days post‐infection (dpi). (D) Boxplot showing normalized cumulative disease index (NCDI) of wild‐type (Col‐0) and mutant.
Discussion
Multiple genes govern variation in resistance to A. brassicae in Arabidopsis
In the current study, we studied the extent of variation in a set of diverse accessions of Arabidopsis and analysed the genetic architecture of Arabidopsis responses to A. brassicae, a necrotrophic pathogen affecting Brassicaceae crops. We observed a continuous variation in the disease responses of the accessions, which is indicative of the quantitative nature of resistance (Fig. 1). This variation encompasses the whole range of resistance responses from completely resistant to highly susceptible accessions, which is unlike the reports for the other BHNs, such as B. cinerea and S. sclerotiorum, where complete resistance is not known (Denby et al., 2004; Perchepied et al., 2010). The availability of a whole gamut of variation for resistance in diverse Arabidopsis accessions thus presents an excellent genetic model to study the mechanisms of resistance against A. brassicae. Very few population‐wide studies for disease resistance have been carried out in Arabidopsis. Except for a recent study which aimed to unravel the quantitative basis of resistance to B. cinerea (Corwin et al., 2016), other studies have focused on resistance to bacterial and biotrophic pathogens (Aoun et al., 2017; Aranzana et al., 2005; Huard‐Chauveau et al., 2013; Nemri et al., 2010). This study is one of the first instances in which GWA mapping has been utilized to dissect the underlying genetic basis of disease resistance to necrotrophic fungi in Arabidopsis.
GWA studies have been used extensively in Arabidopsis, and various statistical methods have been developed to correct for population structure‐dependent confounding. In this study, three different statistical models were used for GWA mapping. The combinatorial use of three different models allowed for a balance between false‐positive and false‐negative rates. A total of 106 candidate genes mapping to 42 loci were identified to be associated with resistance to A. brassicae. The presence of multiple peaks of nominal significance, rather than a single strong peak, indicated that the variation mapped is polygenic and additive. It is possible that low‐frequency alleles with a large effect may be present in some accessions, but did not appear in the GWA mapping as minor alleles (MAF < 10%) were excluded from the study. This is supported by the observation that there are very few highly resistant accessions, which may contain these low‐frequency alleles of major effect. Furthermore, quantitative trait locus (QTL) mapping in biparental populations for resistance to A. brassicae revealed major loci at different genomic positions which were not identified in GWA mapping (Rajarammohan et al., 2017). These loci might therefore represent the minor alleles of large effect. The genes identified in GWA mapping, such as CYP82G1, may represent loci that are likely to be broadly important in the natural populations. Therefore, the variation defined in this study may serve to uncover both common loci of minor effects and rare loci of large effects.
Novel genes influencing disease resistance to A. brassicae
The gene At3g25180 encodes a cytochrome P450 monooxygenase (CYP82G1) that catalyses the production of two volatile homoterpenes, TMTT and DMNT, although it produces TMTT only in planta (Lee et al., 2010). DMNT and TMTT are constituents of the volatile blend that is induced on herbivore or pathogen attack in many angiosperms (Mumm et al., 2008; Tholl et al., 2011). Although CYP82G1 catalyses the production of DMNT and TMTT in vitro, it is known to act as a TMTT synthase in vivo because Arabidopsis leaves lack E‐nerolidol, the precursor of DMNT (Herde et al., 2008). Cytochrome P450s in Arabidopsis have been studied extensively, and many have been implicated in the biosynthesis of various secondary metabolites (Bak et al., 2011). A recent study has also suggested a possible role for CYP82G1 in long‐chain alkyl glucosinolate biosynthesis in response to allyl glucosinolate treatment (Francisco et al., 2016). CYP82G1 is the only member of the G subfamily of the CYP82 family of proteins in Arabidopsis. CYP82C2, a member of the same family, has been shown to be involved in resistance to a BHN, B. cinerea (Liu et al., 2010). Homoterpenes have been shown to play a role in defence against microbial pathogens. DMNT is formed in the roots by an independent pathway, where it is induced by the root‐rot necrotrophic pathogen Pythium irregulare and thus contributes to root defence (Sohrabi et al., 2015). Further, it was shown that the germination of zoospores of P. irregulare was inhibited in medium supplemented with DMNT. However, medium supplemented with TMTT did not affect either mycelial growth or spore germination of A. brassicae (Rajarammohan et al., unpublished data). TMTT is induced by virulent Pseudomonas syringae and methyl jasmonate application, but the absence of TMTT did not alter the defence response of the plants against the virulent pathogen (Attaran et al., 2008). Also, TMTT has been shown to induce defence gene expression in lima bean leaves (Arimura et al., 2000). Therefore, CYP82G1, which catalyses the production of TMTT, might modify the defence response to A. brassicae by priming the plant against pathogen invasion. In addition, CYP82G1 may also be involved in as yet unknown defence response pathways.
The gene At5g37500 encodes a GORK of the Shaker family of K+ channels involved in stomatal closure (Ache et al., 2000). The gene is also known to be up‐regulated in the case of water deprivation and cold stress (Becker et al., 2003; Hosy et al., 2003). Plants actively close their stomata in response to pathogen invasion through the activation of anion channels, membrane depolarization and subsequent activation of potassium channels (Sawinski et al., 2013). Studies have shown that pathogens modulate stomatal function to gain access to the host. A well‐studied example is that of a toxin (coronatine) that is secreted by Pseudomonas syringae, which relaxes guard cells to aid host penetration (Melotto et al., 2006; Zeng and He, 2010). Fusicoccin, a toxin produced by Fusicoccum amygdali, also targets H+‐ATPases to perturb stomatal closure (de Boer and de Vries‐van Leeuwen, 2012). The GORK mutant has been reported to show limited stomatal closure in response to abscisic acid (ABA) and jasmonic acid (JA) (Becker et al., 2003). A previous study in Brassica juncea (Giri et al., 2013) and studies from our laboratory (Mandal et al., 2017) have shown that A. brassicae preferentially penetrates through stomatal openings. The culture filtrate of A. brassicae caused infection‐like symptoms on both resistant and susceptible accessions and mimicked the spore inoculation symptoms very closely. Application of this culture filtrate to wild‐type (Col‐0) plants resulted in stomatal opening (Fig. S5A,B, see Supporting Information). Therefore, A. brassicae may employ as yet unknown effectors, which may target stomatal opening/closure through direct or indirect interaction with GORK to establish itself in the host. The enhanced susceptibility of the mutant may therefore result from the limited stomatal closure, which might increase the accessibility of the host to the invading pathogen.
GDSL‐type esterases/lipases are a class of hydrolytic enzymes that can bind to a broad range of substrates and are known to have regiospecificity and stereoselectivity (Lai et al., 2017). They have been implicated in plant development, morphogenesis and defence responses (Brick et al., 1995; Ling et al., 2006; Oh et al., 2005). A secretome analysis in Arabidopsis identified GDSL lipase‐like 1 (GLIP1) as a secreted lipase, which is a component of disease resistance against A. brassicicola. The GLIP1 protein was found to directly affect the spore germination of A. brassicicola by disrupting the structure of the fungal cell wall (Oh et al., 2005). Secondarily, GLIP1 has also been shown to regulate local and systemic resistance through the ethylene signalling pathway (Kwon et al., 2009). GLIP2, another GDSL‐motif containing protein, is known to play a role in resistance to Erwinia carotovora via the negative regulation of auxin signalling (Lee et al., 2009). CaGLIP1 in hot pepper, which also harbours a GDSL‐motif, has been shown to be involved in the pathogen and wounding stress response (Hong et al., 2008; Kim et al., 2008). In addition, ESM1, which contains a GDSL‐motif, has been shown to be a myrosinase‐associated protein that alters the hydrolysis of glucosinolates and favours the production of isothiocyanates (Zhang et al., 2006). As multiple members of the GDSL‐motif containing family of proteins have been implicated in biotic stress responses, At1g06990, which also contains a GDSL‐motif, may mediate resistance against A. brassicae via certain unknown pathways, and thus warrants further investigation.
Poland et al. (2009) described several possible hypotheses related to the mechanisms underlying quantitative disease resistance (QDR) genes or QRLs. One of the possible hypotheses was the conditioning of resistance by genes regulating morphological and developmental phenotypes. One of the potential candidates, At5g37500 (GORK), is involved in stomatal opening, but also seems to participate in defence against A. brassicae, as evident from the enhanced susceptibility of the mutant. The gene At3g25180 (CYP82G1) is involved in the biosynthesis of secondary metabolites which might participate in the chemical defence against pathogens. The initial list of candidates contained genes from various biological processes, which are directly or indirectly involved in defence‐related functions. Various disease resistance genes or leucine‐rich repeat (LRR)‐domain‐containing genes were also present in the initial list of candidates. This shows that there might be a repertoire of receptors involved in the recognition of effectors from necrotrophic pathogens. Therefore, we cannot rule out the possibility of the role of the other candidates in mediating QDR against A. brassicae. The list of likely candidates identified as contributing to QDR provides the raw genetic material to unravel the multiple facets of QDR against A. brassicae. The present study, whilst revealing certain novel genes and mechanisms involved in defence against A. brassicae, also highlights the complexity of QDR against necrotrophs.
The genes identified in this study need to be confirmed for their role in disease resistance against A. brassicae by the analysis of additional mutants in the genes and the performance of global transcriptomic studies to unravel the underlying mechanisms. Some of the mutations of the candidate genes did not result in any significant difference in CDI relative to wild‐type plants. This could be attributed to the genetic background of the mutation (Col‐0), or to the small effect contributed by the gene towards the phenotype (as is the case in many quantitative traits). Alternatively, the natural variation in these genes might have resulted in a change‐of‐function variation rather than a complete loss‐of‐function (T‐DNA insertion), leading to failure by this validation method. The creation of site‐specific mutations in different accessions using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)‐Cas9 system might be able to help validate the role of such variants in disease resistance. Finally, there is a possibility that the candidate might be a false positive picked up in the association mapping. Also, the other nearby genes that did not have any functional annotations, but were in LD with the genes tested in this study, may contribute to disease resistance.
To the best of our knowledge, the current study is the first report of GWA mapping of disease resistance to the necrotrophic fungus A. brassicae in Arabidopsis. The A. brassicae–Arabidopsis pathosystem is well adapted to study the genetic bases of QDR as it displays a huge variation in resistance, ranging from complete resistance to extreme susceptibility. Analysis of the natural variation in response to A. brassicae in Arabidopsis accessions has revealed multiple genes which might be involved in disease resistance. In addition, A. brassicae is a pathogen that specifically affects the Brassicaceae family to which Arabidopsis also belongs. The further dissection of the mechanisms underlying resistance conferred by the genes identified in this study, such as CYP82G1, may help to develop resistance in the cultivated brassicas.
Experimental Procedures
Plant materials and fungal culture conditions
The natural accessions of Arabidopsis thaliana used in this study were obtained from the Arabidopsis Biological Resource Center (ABRC), Columbus, OH, USA (Table S1 contains the list of accessions, their geographical locations and DIs). The plants were grown at 22 °C and 60% humidity under a 10‐h/14‐h light/dark cycle for 5 weeks before challenging them with the pathogen.
Single‐spore cultures were established from an isolate (J3) collected from the experimental field station in the village of Jaunty, New Delhi, India. The A. brassicae isolate was maintained on radish root sucrose agar (RRSA) medium (Thakur and Kolte, 1985). The fungal cultures were regularly subcultured and kept at 22 °C under a 12‐h/12‐h light/dark cycle. The pathogen was periodically passaged through B. juncea var. Varuna (its natural host) to preserve the virulence of the strain. The fungus re‐isolated from infected tissue was used for the pathogenicity assays.
Pathogen bioassay
Bioassays were carried out using spores from 15‐day‐old cultures. Cultures were flooded with distilled water to collect the spores. The spore suspension was filtered, and the spore concentration was adjusted to (3–5) × 103 spores/mL before inoculation. Five‐week‐old Arabidopsis plants were infected by the drop inoculation method. Four drops of a 5‐µL spore suspension were applied onto the adaxial surface of each of the 8–10 similarly aged leaves. Inoculated plants were kept at 100% humidity at 20 ± 1 °C under a 10‐h/14‐h light/dark cycle. The plants were evaluated for symptoms at 7 days post‐infection (dpi).
Disease evaluation
The disease was discerned by the formation of necrotic lesions encircled by chlorotic halos at the spots of inoculation. The number of necrotic lesions was used to calculate a DI for each accession.
A total of 123 accessions of A. thaliana were screened for resistance to A. brassicae. Two independent experiments were performed, with each experiment containing six to eight plants of every accession. Therefore, at least 12 independent replicates were available for each accession. The block design was a completely random unbalanced design owing to the unequal number of replicates. The unequal number of replicates resulted from the retarded growth of some plants of the accessions, which were then not taken forward for infection assays. In addition, Gre‐0, a previously characterized susceptible accession, was included in all the experiments to serve as a control. Inoculations were performed on 8–10 leaves of each plant of each accession. The accessions were quantitatively evaluated using a DI score based on the number of necrotic spots developing post‐inoculation (McKinney, 1923). The DI score was calculated as follows:
where DI is the disease index, K is the total number of leaves (usually eight), ns is the number of necrotic spots/leaf and nsmax is the maximum number of necrotic spots/leaf (usually four).
nsmax, denoting the maximum number of necrotic spots per leaf, is usually a constant value (four spots), as each leaf is inoculated at four sites with 5 µL of spore suspension. The DI scores were calculated individually for each plant of each accession. The DI scores thus calculated were normalized to the DI score of the control accession (Gre‐0) in each experiment to minimize the effect of experimental variation on the phenotype. This was denoted as NDI. The median value of the NDI scores of each accession was used for association mapping. The effect of the experiment (replicates) on the phenotype was determined using a generalized linear model (GLM):
The accession genotype (A) and replicates (P) were both denoted as fixed effects and the residual error was assumed to be normal. An ANOVA using the F‐test was conducted on the GLM to determine the effect of experimental replicates on the phenotype. The ‘heritability’ package in R was used to calculate the broad‐sense heritability (H 2). Assuming that all differences between the accessions are genetic, H 2 was estimated as described in Kruijer et al. (2015).
GWA analysis of disease resistance
GWA mapping was performed using the GWAPP web interface (http://gwas.gmi.oeaw.ac.at) (Seren et al., 2012). All three models, namely LM, KW and AMM, were used with the default settings to identify associations between the phenotype of 123 accessions and the 206 000 SNPs available on the web interface.
Validation of putative candidate genes
T‐DNA knockout lines in the Col‐0 background were identified in the SALK and GABI‐Kat library and obtained from ABRC (Alonso et al., 2003; Kleinboelting et al., 2012). The homozygosity of the mutants was confirmed using standard PCR‐based indel markers developed by the SALK Institute Genomic Analysis Laboratory (http://signal.salk.edu/tdnaprimers.2.html) (the list of primers used for confirmation is given in Table S2, see Supporting Information). The plants were grown and infected as described above for the natural accessions.
The differences in the disease resistance phenotype between the T‐DNA insertion mutants and the wild‐type plants may be small. Therefore, to determine the differences in the disease phenotype, the mutants were scored using a cumulative disease index (CDI) calculated from the number of necrotic spots and the size of each spot using the formula:
where CDI is the cumulative disease index, K is the total number of leaves, ns is the total number of necrotic spots/leaf, ss is the size of the necrotic spot, nsmax is the maximum number of necrotic spots/leaf (usually four) and ssmax is the maximum size of the necrotic spot.
Three independent experiments were carried out, with each experiment containing six plants of each mutant. Therefore, 18 independent replicates were available for each mutant as well as the wild‐type. A randomized block design was adopted for this experiment, with each experiment indicative of a block. The CDI was scored for each individual plant in each experiment. The accession Zdr‐1 was used as a susceptible control in these experiments, as we found the disease phenotype of Zdr‐1 to be robust and even more susceptible than Gre‐0 during the screening of 123 accessions. Therefore, for the validation experiments, the CDI scores were normalized to the CDI score of the susceptible accession Zdr‐1 in each experiment to minimize the effect of experimental variation on the phenotype. This was denoted as the normalized CDI (NCDI) and was used for comparisons with the wild‐type. In order to estimate the effect of the experimental variation on the phenotype, a GLM was used:
The fixed effects were denoted by L, E and P, which represent the mutant genotype, experimental block and individual plant, respectively. The individual plant was also included as a fixed factor in the model to account for the variation observed between plants of the same genotype. The residual error was assumed to be normal. An ANOVA using the F‐test was conducted on the GLM to determine the effects of the experiments on the phenotype.
The percentage increase in susceptibility for each mutant was calculated with respect to the wild‐type (Col‐0) as:
Quantitative RT‐PCR assays
Total RNA was extracted from the leaves collected from six plants in each experiment using an RNeasy plant mini kit according to the manufacturer's recommendations (Qiagen, Gaithersburg, MD, USA). On‐column DNase digestion was carried out to remove gDNA contamination. First‐strand cDNA was synthesized from 1 µg of total RNA using an MMLV Reverse Transcriptase First‐Strand cDNA Synthesis Kit (Epicentre Biotechnologies, Madison, WI, USA) according to the manufacturer's protocol.
Each reaction contained 10 ng of cDNA, 1 pmol of each primer (Table S1) and 2 × SYBR Select Master Mix (ThermoFisher Scientific, Waltham, MA, USA). PCRs were carried out using the standard setting in a QuantStudio 6 Flex Real‐Time PCR System (ThermoFisher Scientific, Waltham, MA, USA). The mean cycle threshold (Ct) values of the genes were calculated from replicate wells. The mean Ct values of the target genes were normalized to the Ct values of the endogenous control (TIP41‐like gene, At4g34270) for each sample (denoted by dCt). Further, to calculate the relative expression levels, the dCt values of the infected samples were normalized to the mock (distilled water)‐inoculated samples of each time point. These values from the three individual biological experiments were subjected to data standardization and compared using a described method (Willems et al., 2008). Subsequently, a Mann–Whitney U‐test (non‐parametric test) was used to test the statistical significance of the fold changes. The sequences of the primers used in quantitative PCR are listed in Table S3 (see Supporting Information).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Confirmation of T‐DNA insertion in At3g25180. (A) Position of the T‐DNA insertion in the gene At3g25180. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory. (C) Confirmation of null allele by checking the expression levels of At3g25180 in cDNA of wild‐type and T‐DNA mutant.
Fig. S2 Confirmation of T‐DNA insertion in At5g37500. (A) Position of the T‐DNA insertion in the gene At5g37500. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory. (C) Confirmation of null allele by checking the expression levels of At5g37500 in cDNA of wild‐type and T‐DNA mutant.
Fig. S3 Confirmation of T‐DNA insertion in At1g06990. (A) Position of the T‐DNA insertion in the gene At1g06990. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory.
Fig. S4 Non‐synonymous changes in the coding region of At3g25180. (A) The gene model of At3g25180 with the approximate positions of the non‐synonymous variants in the different exons of the gene. (B) Segregation of the alleles corresponding to the non‐synonymous variants in At3g25180 (CYP82G1) in 10 highly resistant and 10 highly susceptible accessions.
Fig. S5 Response of wild‐type (Col‐0) to culture filtrate of Alternaria brassicae. (A) Panel representing stomatal opening in response to treatment (3 h) with culture filtrate (CF) of A. brassicae and potato dextrose broth (PDB) in 4‐week‐old wild‐type Col‐0 leaves, Scale bar, 500 µm. Leaves were stained with 20 µm propidium iodide (PI) prior to treatment (CF and PDB). (B) Boxplot showing the stomatal aperture index (SAI) for the CF‐treated and mock (PDB)‐treated leaves of Col‐0. Data from two independent biological experiments, with the number of stomata measured for SAI indicated in the figure.
Fig. S6 Venn diagram depicting the overlap between the top 2000 single nucleotide polymorphisms (SNPs) from each method, namely the linear regression model (LM), Kruskal–Wallis test (KW) and accelerated mixed model (AMM).
Table S1 Accessions used in the current study together with their geographical location and normalized disease index (NDI).
Table S2 List of primers used to confirm the homozygosity of the T‐DNA insertion mutants.
Table S3 List of primers used in quantitative polymerase chain reaction (qPCR).
Table S4 Candidate genes (106) identified after prioritization of the single nucleotide polymorphisms (SNPs) arising from genome‐wide association (GWA) mapping.
Table S5 P values of F‐tests of T‐DNA insertion mutants. A summary table of P values of each term in the linear model for each mutant against wild‐type Col‐0.
Table S6 In silico prediction of the effect of non‐synonymous changes in the coding region of At3g25180. Predictions shown with two different commonly used algorithms: SIFT (Kumar et al., 2009) and POLYPHEN 2.0 (Adzhubei et al., 2010).
Acknowledgements
We thank Professor Wilhelm Boland and Dr Stefan Bartram (Max Planck Institute for Chemical Ecology, Jena, Germany) for kindly providing TMTT ((E,E)‐4,8,12‐trimethyl‐1,3,7,11‐tridecatetraene). This work was supported by grants from Department of Biotechnology (DBT) projects BT/IN/Indo‐UK/CGAT/12/DP/2014‐15 and BT/01/COE/08/06‐II and Science and Engineering Research Board (SERB) project SB/FT/LS‐327/2012. S.R. acknowledges the receipt of a research fellowship from UGC and DBT. The authors also acknowledge the Department of Science and Technology ‐ Fund for Improvement of S&T infrastructure (DST‐FIST)‐funded Central Instrumentation Facility of the Department of Genetics, University of Delhi South Campus.
References
- Ache, P. , Becker, D. , Ivashikina, N. , Dietrich, P. , Roelfsema, M.R. and Hedrich, R. (2000) GORK, a delayed outward rectifier expressed in guard cells of Arabidopsis thaliana, is a K(+)‐selective, K(+)‐sensing ion channel. FEBS Lett. 486, 93–98. [DOI] [PubMed] [Google Scholar]
- Alonso, J.M. , Stepanova, A.N. , Leisse, T.J. , Kim, C.J. , Chen, H. , Shinn, P. , Stevenson, D.K. , Zimmerman, J. , Barajas, P. , Cheuk, R. , Gadrinab, C. , Heller, C. , Jeske, A. , Koesema, E. , Meyers, C.C. , Parker, H. , Prednis, L. , Ansari, Y. , Choy, N. , Deen, H. , Geralt, M. , Hazari, N. , Hom, E. , Karnes, M. , Mulholland, C. , Ndubaku, R. , Schmidt, I. , Guzman, P. , Aguilar‐Henonin, L. , Schmid, M. , Weigel, D. , Carter, D.E. , Marchand, T. , Risseeuw, E. , Brogden, D. , Zeko, A. , Crosby, W.L. , Berry, C.C. and Ecker, J.R. (2003) Genome‐wide insertional mutagenesis of Arabidopsis thaliana . Science, 301, 653–657. [DOI] [PubMed] [Google Scholar]
- Aoun, N. , Tauleigne, L. , Lonjon, F. , Deslandes, L. , Vailleau, F. , Roux, F. and Berthomé, R. (2017) Quantitative disease resistance under elevated temperature: genetic basis of new resistance mechanisms to Ralstonia solanacearum . Front. Plant Sci. 8, 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranzana, M.J. , Kim, S. , Zhao, K. , Bakker, E. , Horton, M. , Jakob, K. , Lister, C. , Molitor, J. , Shindo, C. , Tang, C. , Toomajian, C. , Traw, B. , Zheng, H. , Bergelson, J. , Dean, C. , Marjoram, P. and Nordborg, M. (2005) Genome‐wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura, G‐I. , Ozawa, R. , Shimoda, T. , Nishioka, T. , Boland, W. and Takabayashi, J. (2000) Herbivory‐induced volatiles elicit defence genes in lima bean leaves. Nature, 406, 512–515. [DOI] [PubMed] [Google Scholar]
- Attaran, E. , Rostas, M. and Zeier, J. (2008) Pseudomonas syringae elicits emission of the terpenoid (E,E)‐4,8,12‐trimethyl‐1,3,7,11‐tridecatetraene in Arabidopsis leaves via jasmonate signaling and expression of the terpene synthase TPS4. Mol. Plant–Microbe Interact. 21, 1482–1497. [DOI] [PubMed] [Google Scholar]
- Bak, S. , Beisson, F. , Bishop, G. , Hamberger, B. , Höfer, R. , Paquette, S. and Werck‐Reichhart, D. (2011) Cytochromes p450. Arabidopsis Book, 9, e0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter, I. , Brazelton, J.N. , Yu, D. , Huang, Y.S. , Lahner, B. , Yakubova, E. , Li, Y. , Bergelson, J. , Borevitz, J.O. , Nordborg, M. , Vitek, O. , Salt, D.E. and Copenhaver, G.P. (2010) A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1;1. PLoS Genet. 6, e1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D. , Hoth, S. , Ache, P. , Wenkel, S. , Roelfsema, M.R.G. , Meyerhoff, O. , Hartung, W. and Hedrich, R. (2003) Regulation of the ABA‐sensitive Arabidopsis potassium channel gene GORK in response to water stress. FEBS Lett. 554, 119–126. [DOI] [PubMed] [Google Scholar]
- Birren, B. , Fink, G. and Lander, E. (2002) Fungal genome initiative: white paper developed by the Fungal Research Community. Available at https://www.genome.gov/pages/research/sequencing/seqproposals/fungalinitiative_genome.pdf[accessed 27 December 2017].
- de Boer, A.H. and de Vries‐van Leeuwen, I.J. (2012) Fusicoccanes: diterpenes with surprising biological functions. Trends Plant Sci. 17, 360–368. [DOI] [PubMed] [Google Scholar]
- Brick, D.J. , Brumlik, M.J. , Buckley, J.T. , Cao, J.X. , Davies, P.C. , Misra, S. , Tranbarger, T.J. and Upton, C. (1995) A new family of lipolytic plant enzymes with members in rice, Arabidopsis and maize. FEBS Lett. 377, 475–480. [DOI] [PubMed] [Google Scholar]
- Cao, J. , Schneeberger, K. , Ossowski, S. , Günther, T. , Bender, S. , Fitz, J. , Koenig, D. , Lanz, C. , Stegle, O. , Lippert, C. , Wang, X. , Ott, F. , Müller, J. , Alonso‐Blanco, C. , Borgwardt, K. , Schmid, K.J. and Weigel, D. (2011) Whole‐genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. [DOI] [PubMed] [Google Scholar]
- Chan, E.K. , Rowe, H.C. and Kliebenstein, D.J. (2010) Understanding the evolution of defense metabolites in Arabidopsis thaliana using genome‐wide association mapping. Genetics, 185, 991–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, D.‐Y. , Silva, A. , Baxter, I. , Huang, Y.S. , Nordborg, M. , Danku, J. , Lahner, B. , Yakubova, E. , Salt, D.E. and Bomblies, K. (2012) Genome‐wide association studies identify heavy metal ATPase3 as the primary determinant of natural variation in leaf cadmium in Arabidopsis thaliana . PLoS Genet. 8, e1002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon, B.J. , Leng, Y. , Wu, D. , Bushley, K.E. , Ohm, R.A. , Otillar, R. , Martin, J. , Schackwitz, W. , Grimwood, J. , MohdZainudin, NurAinIzzati , Xue, C. , Wang, R. , Manning, V.A. , Dhillon, B. , Tu, Z.J. , Steffenson, B.J. , Salamov, A. , Sun, H. , Lowry, S. , LaButti, K. , Han, J. , Copeland, A. , Lindquist, E. , Barry, K. , Schmutz, J. , Baker, S.E. , Ciuffetti, L.M. , Grigoriev, I.V. , Zhong, S. , Turgeon, B.G. and Madhani, H.D. (2013) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 9, e1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corwin, J.A. , Copeland, D. , Feusier, J. , Subedy, A. , Eshbaugh, R. , Palmer, C. , Maloof, J. , Kliebenstein, D.J. and Koenig, D. (2016) The quantitative basis of the Arabidopsis innate immune system to endemic pathogens depends on pathogen genetics. PLoS Genet. 12, e1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denby, K.J. , Kumar, P. and Kliebenstein, D.J. (2004) Identification of Botrytis cinerea susceptibility loci in Arabidopsis thaliana . Plant J. 38, 473–486. [DOI] [PubMed] [Google Scholar]
- Faris, J.D. , Zhang, Z. , Lu, H. , Lu, S. , Reddy, L. , Cloutier, S. , Fellers, J.P. , Meinhardt, S.W. , Rasmussen, J.B. , Xu, S.S. , Oliver, R.P. , Simons, K.J. and Friesen, T.L. (2010) A unique wheat disease resistance‐like gene governs effector‐triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. USA, 107, 13 544–13 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco, M. , Joseph, B. , Caligagan, H. , Li, B. , Corwin, J.A. , Lin, C. , Kerwin, R.E. , Burow, M. and Kliebenstein, D.J. (2016) Genome wide association mapping in Arabidopsis thaliana identifies novel genes involved in linking allyl glucosinolate to altered biomass and defense. Front. Plant Sci. 7, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen, T.L. , Meinhardt, S.W. and Faris, J.D. (2007) The Stagonospora nodorum–wheat pathosystem involves multiple proteinaceous host‐selective toxins and corresponding host sensitivity genes that interact in an inverse gene‐for‐gene manner. Plant J. 51, 681–692. [DOI] [PubMed] [Google Scholar]
- Giri, P. , Taj, G. , Meena, P.D. and Kumar, A. (2013) Microscopic study of Alternaria brassicae infection processes in Brassica juncea cultivars by drop plus agarose method. Afr. J. Microbiol. Res. 7, 4284–4290. [Google Scholar]
- Herde, M. , Gartner, K. , Kollner, T.G. , Fode, B. , Boland, W. , Gershenzon, J. , Gatz, C. and Tholl, D. (2008) Identification and regulation of TPS04/GES, an Arabidopsis geranyllinalool synthase catalyzing the first step in the formation of the insect‐induced volatile C16‐homoterpene TMTT. Plant Cell, 20, 1152–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, J.K. , Choi, H.W. , Hwang, I.S. , Kim, D.S. , Kim, N.H. , Choi, D.S. , Kim, Y.J. and Hwang, B.K. (2008) Function of a novel GDSL‐type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta, 227, 539–558. [DOI] [PubMed] [Google Scholar]
- Horton, M.W. , Hancock, A.M. , Huang, Y.S. , Toomajian, C. , Atwell, S. , Auton, A. , Muliyati, N.W. , Platt, A. , Sperone, F.G. , Vilhjálmsson, B.J. , Nordborg, M. , Borevitz, J.O. and Bergelson, J. (2012) Genome‐wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 44, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosy, E. , Vavasseur, A. , Mouline, K. , Dreyer, I. , Gaymard, F. , Poree, F. , Boucherez, J. , Lebaudy, A. , Bouchez, D. , Very, A.‐A. , Simonneau, T. , Thibaud, J.‐B. and Sentenac, H. (2003) The Arabidopsis outward K+ channel GORK is involved in regulation of stomatal movements and plant transpiration. Proc. Natl. Acad. Sci. USA, 100, 5549–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard‐Chauveau, C. , Perchepied, L. , Debieu, M. , Rivas, S. , Kroj, T. , Kars, I. , Bergelson, J. , Roux, F. and Roby, D. (2013) An atypical kinase under balancing selection confers broad‐spectrum disease resistance in Arabidopsis . PLOS Genet. 9, e1003766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan, I.A. and Hammerschmidt, R. (2002) Arabidopsis ecotype variability in camalexin production and reaction to infection by Alternaria brassicicola . J. Chem. Ecol. 28, 2121–2140. [DOI] [PubMed] [Google Scholar]
- Kim, K.J. , Lim, J.H. , Kim, M.J. , Kim, T. , Chung, H.M. and Paek, K.H. (2008) GDSL‐lipase1 (CaGL1) contributes to wound stress resistance by modulation of CaPR‐4 expression in hot pepper. Biochem. Biophys. Res. Commun. 374, 693–698. [DOI] [PubMed] [Google Scholar]
- Kim, S. , Plagnol, V. , Hu, T.T. , Toomajian, C. , Clark, R.M. , Ossowski, S. , Ecker, J.R. , Weigel, D. and Nordborg, M. (2007) Recombination and linkage disequilibrium in Arabidopsis thaliana . Nat. Genet. 39, 1151–1155. [DOI] [PubMed] [Google Scholar]
- Kleinboelting, N. , Huep, G. , Kloetgen, A. , Viehoever, P. and Weisshaar, B. (2012) GABI‐Kat SimpleSearch: new features of the Arabidopsis thaliana T‐DNA mutant database. Nucleic Acids Res. 40, D1211–D1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koornneef, M. , Alonso‐Blanco, C. and Vreugdenhil, D. (2004) Naturally occurring genetic variation in Arabidopsis thaliana . Annu. Rev. Plant Biol. 55, 141–172. [DOI] [PubMed] [Google Scholar]
- Korte, A. and Farlow, A. (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods, 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruijer, W. , Boer, M.P. , Malosetti, M. , Flood, P.J. , Engel, B. , Kooke, R. , Keurentjes, J.J.B. and van Eeuwijk, F.A. (2015) Marker‐based estimation of heritability in immortal populations. Genetics, 199, 379–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon, S.J. , Jin, H.C. , Lee, S. , Nam, M.H. , Chung, J.H. , Kwon, S.I. , Ryu, C.‐M. and Park, O.K. (2009) GDSL lipase‐like 1 regulates systemic resistance associated with ethylene signaling in Arabidopsis . Plant J. 58, 235–245. [DOI] [PubMed] [Google Scholar]
- Lai, C.‐P. , Huang, L.‐M. , Chen, L.‐F.O. , Chan, M.‐T. and Shaw, J.‐F. (2017) Genome‐wide analysis of GDSL‐type esterases/lipases in Arabidopsis . Plant Mol. Biol. 95, 181–197. [DOI] [PubMed] [Google Scholar]
- Lee, D.S. , Kim, B.K. , Kwon, S.J. , Jin, H.C. and Park, O.K. (2009) Arabidopsis GDSL lipase 2 plays a role in pathogen defense via negative regulation of auxin signaling. Biochem. Biophys. Res. Commun. 379, 1038–1042. [DOI] [PubMed] [Google Scholar]
- Lee, S. , Badieyan, S. , Bevan, D.R. , Herde, M. , Gatz, C. and Tholl, D. (2010) Herbivore‐induced and floral homoterpene volatiles are biosynthesized by a single P450 enzyme (CYP82G1) in Arabidopsis . Proc. Natl. Acad. Sci. USA, 107, 21205–21210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, H. , Zhao, J. , Zuo, K. , Qiu, C. , Yao, H. , Qin, J. , Sun, X. and Tang, K. (2006) Isolation and expression analysis of a GDSL‐like lipase gene from Brassica napus L. J. Biochem. Mol. Biol. 39, 297–303. [DOI] [PubMed] [Google Scholar]
- Liu, F. , Jiang, H. , Ye, S. , Chen, W.‐P. , Liang, W. , Xu, Y. , Sun, B. , Sun, J. , Wang, Q. , Cohen, J.D. and Li, C. (2010) The Arabidopsis P450 protein CYP82C2 modulates jasmonate‐induced root growth inhibition, defense gene expression and indole glucosinolate biosynthesis. Cell Res. 20, 539–552. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Zhang, Z. , Faris, J.D. , Oliver, R.P. , Syme, R. , McDonald, M.C. , McDonald, B.A. , Solomon, P.S. , Lu, S. , Shelver, W.L. , Xu, S. , Friesen, T.L. and Tyler, B. (2012) The cysteine rich necrotrophic effector SnTox1 produced by Stagonospora nodorum triggers susceptibility of wheat lines harboring Snn1. PLOS Pathogens, 8, e1002467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente, F. , Alonso‐Blanco, C. , Sanchez‐Rodriguez, C. , Jorda, L. and Molina, A. (2005) ERECTA receptor‐like kinase and heterotrimeric G protein from Arabidopsis are required for resistance to the necrotrophic fungus Plectosphaerella cucumerina . Plant J. 43, 165–180. [DOI] [PubMed] [Google Scholar]
- Lorang, J. , Kidarsa, T. , Bradford, C.S. , Gilbert, B. , Curtis, M. , Tzeng, S.‐C. , Maier, C.S. and Wolpert, T.J. (2012) Tricking the guard: exploiting plant defense for disease susceptibility. Science, 338, 659–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorang, J.M. , Sweat, T.A. and Wolpert, T.J. (2007) Plant disease susceptibility conferred by a “resistance” gene. Proc. Natl. Acad. Sci. USA, 104, 14 861–14 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal, S. , Rajarammohan, S. and Kaur, J. (2017) Alternaria brassicae interactions with the model Brassicaceae member Arabidopsis thaliana closely resembles those with Mustard (Brassica juncea). Physiology and Molecular Biology of Plants. 10.1007/s12298-017-0486-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, H.H. (1923) Influence of soil temperature and moisture on infection of wheat seedlings by Helminthosporium sativum . J. Agric. Res. XXVI, 195–217. [Google Scholar]
- Meijon, M. , Satbhai, S.B. , Tsuchimatsu, T. and Busch, W. (2013) Genome‐wide association study using cellular traits identifies a new regulator of root development in Arabidopsis . Nat. Genet. 46, 77–81. [DOI] [PubMed] [Google Scholar]
- Melotto, M. , Underwood, W. , Koczan, J. , Nomura, K. and He, S.Y. (2006) Plant stomata function in innate immunity against bacterial invasion. Cell, 126, 969–980. [DOI] [PubMed] [Google Scholar]
- Micic, Z. , Hahn, V. , Bauer, E. , Schön, C.C. , Knapp, S.J. , Tang, S. and Melchinger, A.E. (2004) QTL mapping of Sclerotinia midstalk‐rot resistance in sunflower. Theor. Appl. Genet. 109, 1474–1484. [DOI] [PubMed] [Google Scholar]
- Mukherjee, A.K. , Lev, S. , Gepstein, S. and Horwitz, B.A. (2009) A compatible interaction of Alternaria brassicicola with Arabidopsis thaliana ecotype DiG: evidence for a specific transcriptional signature. BMC Plant Biol. 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm, R. , Posthumus, M.A. and Dicke, M. (2008) Significance of terpenoids in induced indirect plant defence against herbivorous arthropods. Plant Cell Environ. 31, 575–585. [DOI] [PubMed] [Google Scholar]
- Murray, G.M. and Brennan, J.P. (2009) Estimating disease losses to the Australian wheat industry. Australas. Plant Pathol. 38, 558–570. [Google Scholar]
- Nemri, A. , Atwell, S. , Tarone, A.M. , Huang, Y.S. , Zhao, K. , Studholme, D.J. , Nordborg, M. and Jones, J.D.G. (2010) Genome‐wide survey of Arabidopsis natural variation in downy mildew resistance using combined association and linkage mapping. Proc. Natl. Acad. Sci. USA, 107, 10 302–10 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M. , Hu, T.T. , Ishino, Y. , Jhaveri, J. , Toomajian, C. , Zheng, H. , Bakker, E. , Calabrese, P. , Gladstone, J. , Goyal, R. , Jakobsson, M. , Kim, S. , Morozov, Y. , Padhukasahasram, B. , Plagnol, V. , Rosenberg, N.A. , Shah, C. , Wall, J.D. , Wang, J. , Zhao, K. , Kalbfleisch, T. , Schulz, V. , Kreitman, M. and Bergelson, J. (2005) The pattern of polymorphism in Arabidopsis thaliana . PLoS Biol. 3, e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, I.S. , Park, A.R. , Bae, M.S. , Kwon, S.J. , Kim, Y.S. , Lee, J.E. , Kang, N.Y. , Lee, S. , Cheong, H. and Park, O.K. (2005) Secretome analysis reveals an Arabidopsis lipase involved in defense against Alternaria brassicicola . Plant Cell, 17, 2832–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perchepied, L. , Balagué, C. , Riou, C. , Claudel‐Renard, C. , Rivière, N. , Grezes‐Besset, B. and Roby, D. (2010) Nitric oxide participates in the complex interplay of defense‐related signaling pathways controlling disease resistance to Sclerotinia sclerotiorum in Arabidopsis thaliana . Mol. Plant–Microbe Interact. 23, 846–860. [DOI] [PubMed] [Google Scholar]
- Poland, J.A. , Balint‐Kurti, P.J. , Wisser, R.J. , Pratt, R.C. and Nelson, R.J. (2009) Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14, 21–29. [DOI] [PubMed] [Google Scholar]
- Rajarammohan, S. , Kumar, A. , Gupta, V. , Pental, D. , Pradhan, A.K. and Kaur, J. (2017) Genetic architecture of resistance to Alternaria brassicae in Arabidopsis thaliana: QTL mapping reveals two major resistance‐conferring loci. Front. Plant Sci. 8, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, F. , Voisin, D. , Badet, T. , Balagué, C. , Barlet, X. , Huard‐Chauveau, C. , Roby, D. and Raffaele, S. (2014) Resistance to phytopathogens e tutti quanti: placing plant quantitative disease resistance on the map. Mol. Plant Pathol. 15, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe, H.C. and Kliebenstein, D.J. (2008) Complex genetics control natural variation in Arabidopsis thaliana resistance to Botrytis cinerea . Genetics, 180, 2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawinski, K. , Mersmann, S. , Robatzek, S. and Bohmer, M. (2013) Guarding the green: pathways to stomatal immunity. Mol. Plant–Microbe Interact. 26, 626–632. [DOI] [PubMed] [Google Scholar]
- Seren, U. , Vilhjalmsson, B.J. , Horton, M.W. , Meng, D. , Forai, P. , Huang, Y.S. , Long, Q. , Segura, V. and Nordborg, M. (2012) GWAPP: a web application for genome‐wide association mapping in Arabidopsis . Plant Cell, 24, 4793–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, G. , Dinesh Kumar, V. , Haque, A. , Bhat, S.R. , Prakash, S. and Chopra, V.L. (2002) Brassica coenospecies: a rich reservoir for genetic resistance to leaf spot caused by Alternaria brassicae . Euphytica, 125, 411–417. [Google Scholar]
- Sohrabi, R. , Huh, J.‐H. , Badieyan, S. , Rakotondraibe, L.H. , Kliebenstein, D.J. , Sobrado, P. and Tholl, D. (2015) In planta variation of volatile biosynthesis: an alternative biosynthetic route to the formation of the pathogen‐induced volatile homoterpene DMNT via triterpene degradation in Arabidopsis roots. Plant Cell, 27, 874–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair, D.A. (2010) Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 48, 247–268. [DOI] [PubMed] [Google Scholar]
- Thakur, R. and Kolte, S.J. (1985) Radish root extract agar, a suitable medium for the growth and sporulation of Alternaria brassicae . Cruciferae Newsl. 10, 117–118. [Google Scholar]
- Tholl, D. , Sohrabi, R. , Huh, J.H. and Lee, S. (2011) The biochemistry of homoterpenes–common constituents of floral and herbivore‐induced plant volatile bouquets. Phytochemistry, 72, 1635–1646. [DOI] [PubMed] [Google Scholar]
- Verslues, P.E. , Lasky, J.R. , Juenger, T.E. , Liu, T.W. and Kumar, M.N. (2014) Genome‐wide association mapping combined with reverse genetics identifies new effectors of low water potential‐induced proline accumulation in Arabidopsis . Plant Physiol. 164, 144–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhjalmsson, B.J. and Nordborg, M. (2013) The nature of confounding in genome‐wide association studies. Nat. Rev. Genet. 14, 1–2. [DOI] [PubMed] [Google Scholar]
- Willems, E. , Leyns, L. and Vandesompele, J. (2008) Standardization of real‐time PCR gene expression data from independent biological replicates. Anal. Biochem. 379, 127–129. [DOI] [PubMed] [Google Scholar]
- Zeng, W. and He, S.Y. (2010) A prominent role of the flagellin receptor FLAGELLIN‐SENSING2 in mediating stomatal response to Pseudomonas syringae pv tomato DC3000 in Arabidopsis . Plant Physiol. 153, 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Ober, J.A. and Kliebenstein, D.J. (2006) The gene controlling the quantitative trait locus EPITHIOSPECIFIER MODIFIER1 alters glucosinolate hydrolysis and insect resistance in Arabidopsis . Plant Cell, 18, 1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K. , Aranzana, M.J. , Kim, S. , Lister, C. , Shindo, C. , Tang, C. , Toomajian, C. , Zheng, H. , Dean, C. , Marjoram, P. and Nordborg, M. (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Confirmation of T‐DNA insertion in At3g25180. (A) Position of the T‐DNA insertion in the gene At3g25180. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory. (C) Confirmation of null allele by checking the expression levels of At3g25180 in cDNA of wild‐type and T‐DNA mutant.
Fig. S2 Confirmation of T‐DNA insertion in At5g37500. (A) Position of the T‐DNA insertion in the gene At5g37500. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory. (C) Confirmation of null allele by checking the expression levels of At5g37500 in cDNA of wild‐type and T‐DNA mutant.
Fig. S3 Confirmation of T‐DNA insertion in At1g06990. (A) Position of the T‐DNA insertion in the gene At1g06990. (B) Confirmation of T‐DNA insertion using polymerase chain reaction (PCR)‐based indel markers developed from the SALK Institute Genomic Analysis Laboratory.
Fig. S4 Non‐synonymous changes in the coding region of At3g25180. (A) The gene model of At3g25180 with the approximate positions of the non‐synonymous variants in the different exons of the gene. (B) Segregation of the alleles corresponding to the non‐synonymous variants in At3g25180 (CYP82G1) in 10 highly resistant and 10 highly susceptible accessions.
Fig. S5 Response of wild‐type (Col‐0) to culture filtrate of Alternaria brassicae. (A) Panel representing stomatal opening in response to treatment (3 h) with culture filtrate (CF) of A. brassicae and potato dextrose broth (PDB) in 4‐week‐old wild‐type Col‐0 leaves, Scale bar, 500 µm. Leaves were stained with 20 µm propidium iodide (PI) prior to treatment (CF and PDB). (B) Boxplot showing the stomatal aperture index (SAI) for the CF‐treated and mock (PDB)‐treated leaves of Col‐0. Data from two independent biological experiments, with the number of stomata measured for SAI indicated in the figure.
Fig. S6 Venn diagram depicting the overlap between the top 2000 single nucleotide polymorphisms (SNPs) from each method, namely the linear regression model (LM), Kruskal–Wallis test (KW) and accelerated mixed model (AMM).
Table S1 Accessions used in the current study together with their geographical location and normalized disease index (NDI).
Table S2 List of primers used to confirm the homozygosity of the T‐DNA insertion mutants.
Table S3 List of primers used in quantitative polymerase chain reaction (qPCR).
Table S4 Candidate genes (106) identified after prioritization of the single nucleotide polymorphisms (SNPs) arising from genome‐wide association (GWA) mapping.
Table S5 P values of F‐tests of T‐DNA insertion mutants. A summary table of P values of each term in the linear model for each mutant against wild‐type Col‐0.
Table S6 In silico prediction of the effect of non‐synonymous changes in the coding region of At3g25180. Predictions shown with two different commonly used algorithms: SIFT (Kumar et al., 2009) and POLYPHEN 2.0 (Adzhubei et al., 2010).