

Summary
Erwinia amylovora is the causal agent of fire blight, one of the most devastating diseases of apple and pear. Erwinia amylovora is thought to have originated in North America and has now spread to at least 50 countries worldwide. An understanding of the diversity of the pathogen population and the transmission to different geographical regions is important for the future mitigation of this disease. In this research, we performed an expanded comparative genomic study of the Spiraeoideae‐infecting (SI) E. amylovora population in North America and Europe. We discovered that, although still highly homogeneous, the genetic diversity of 30 E. amylovora genomes examined was about 30 times higher than previously determined. These isolates belong to four distinct clades, three of which display geographical clustering and one of which contains strains from various geographical locations (‘Widely Prevalent’ clade). Furthermore, we revealed that strains from the Widely Prevalent clade displayed a higher level of recombination with strains from a clade strictly from the eastern USA, which suggests that the Widely Prevalent clade probably originated from the eastern USA before it spread to other locations. Finally, we detected variations in virulence in the SI E. amylovora strains on immature pear, and identified the genetic basis of one of the low‐virulence strains as being caused by a single nucleotide polymorphism in hfq, a gene encoding an important virulence regulator. Our results provide insights into the population structure, distribution and evolution of SI E. amylovora in North America and Europe.
Keywords: Erwinia amylovora, fire blight, genomics, pathogen distribution, virulence variations
Introduction
Fire blight, caused by Erwinia amylovora, is one of the most devastating diseases of Rosaceae plants, such as apple and pear (van der Zwet et al., 2012). E. amylovora can infect various organs of host plants, including flowers, shoots, leaves, fruits and rootstocks. Following initial infection, pathogen cells can migrate systemically in host plants through xylem vessels, where biofilms are formed through the production of exopolysaccharides (Koczan et al., 2009), eventually causing plant death (Malnoy et al., 2012; van der Zwet et al., 2012). As a result of often severe damage and wide prevalence, annual losses to fire blight and costs of control are estimated at over $100 million in the USA alone (Norelli et al., 2003).
It is well accepted that E. amylovora was indigenous on wild hosts, such as crab apple (Malus sp.), service berry (Amelanchier sp.) and mountain ash (Sorbus sp.), in North America (van der Zwet et al., 2012). The introduction of apple (M. × domestica) and pear (Pyrus communis) to North America by European settlers in the 1600s–1700s created the opportunity for E. amylovora to shift from its wild hosts to these two agriculturally important crops for the first time (van der Zwet et al., 2012). Denning (1794) provided the first written documentation of fire blight in the Hudson Valley of New York. Fire blight was subsequently observed in northeastern, midwestern and western USA from the early 1800s to the early 1900s (Fletcher, 1931–1933; Hotson, 1916; Isaac, 1902; Kennicot, 1850; Lowell, 1826). By the mid‐1900s, fire blight was observed in all major apple‐producing regions in the USA (van der Zwet et al., 2012). This disease also spread globally to New Zealand (1919), Europe (1950s) and the Middle East (1988), most probably through the transport of fire blight‐infected plant materials (van der Zwet et al., 2012). Currently, fire blight has been reported in at least 50 countries worldwide (Myung et al., 2016; Rhouma et al., 2014; Soukainen et al., 2015; van der Zwet et al., 2012).
Based on differences in host specificity, E. amylovora strains can be divided into Spiraeoideae‐infecting (SI) and Rubus‐infecting (RI) strains: SI strains can cause disease on hosts in the subfamily Spiraeoideae, such as apple, pear, hawthorn (Crataegus sp.), quince (Cydonia sp.) and loquat (Eriobotrya japonica); RI strains can only infect Rubus plants, such as raspberry (R. strigosus) and blackberry (R. allegheniensis) (Braun and Hildebrand, 2005; Reies and Otterbacher, 1977). Despite the differences in host preference, the overall genetic diversity of E. amylovora, for both SI and RI strains, is very low (Mann et al., 2013; Zhao and Qi, 2011). A previous genomic analysis of 12 E. amylovora strains (nine SI strains and three RI strains) determined the genome‐wide amino acid identity (AAI) of these strains as 99.72% (Mann et al., 2013). The genetic diversity in the SI isolates was even lower than that of the RI isolates (AAI values for SI isolates and RI isolates are 99.98% and 99.42%, respectively). As a result of the high genetic homogeneity, it was hypothesized that the SI E. amylovora population has undergone a recent genetic bottleneck associated with the large‐scale cultivation of apples and pears starting in the 1700s (Malnoy et al., 2012; Mann et al., 2013; McGhee and Sundin, 2012). Because of the high level of genome homogeneity, limited efforts have been spent in the genome sequencing of additional SI E. amylovora strains in recent years. Only four genomes of E. amylovora have been deposited in the National Center for Biotechnology Information (NCBI) in the past 8 years.
Although the previous genomic study suggested that SI E. amylovora strains were highly homogenous (Mann et al., 2013), there is evidence indicating that the population may be more diverse than previously thought. For example, SI isolates have been shown to exhibit different levels of virulence in causing infections on different apple cultivars and in immature pear assays (McGhee et al., 2011; Norelli et al., 1984; Wang et al., 2010). In addition, most SI E. amylovora strains characterized in the previous genomic study (Mann et al., 2013) were not isolated from the geographic origin of the pathogen (seven of nine strains were from Europe, only two strains were from North America). When analysed by strain tracking and genotyping methods, such as clustered regularly interspaced short palindromic repeats (CRISPRs) and minisatellites, the SI population seemed to be more diverse, and could be grouped into different genotype groups (Bühlmann et al., 2014; McGhee and Sundin, 2012; Rezzonico et al., 2011). However, the clustering and grouping generated by these methods does not reflect the true phylogeny and could not be used to determine genome‐wide genetic diversity (McGhee and Sundin, 2012).
Underlying this work, we hypothesized that a more representative genetic diversity and phylogeny of SI E. amylovora could be determined by the examination of more strains from the origin of the disease (North America). Through an effort to survey the E. amylovora population for streptomycin resistance, we collected a large number of E. amylovora strains from the northeastern USA. In this study, genomes of 19 representative strains from northeastern, midwestern and western USA were sequenced and analysed for their genetic diversity, phylogeny and level of recombination. Our study revealed that the population of SI E. amylovora in North America and Europe is more diverse than previously thought, with strains belonging to four distinct clades. The high level of recombination between strains from the ‘Widely Prevalent’ clade and strains from a clade of eastern US origin suggested that eastern USA is probably the origin of the strains causing the current global fire blight epidemics. Finally, we identified variations in bacterial virulence, and attributed the low‐virulence phenotype of one strain to a single nucleotide polymorphism (SNP) in hfq, a global virulence regulatory gene in E. amylovora.
Results and Discussion
Collection of fire blight samples and isolation of E. amylovora in northeastern USA
In the summer of 2015 and 2016, we collected 89 fire blight samples from a total of 23 commercial orchards in seven states of the northeastern USA [Rhode Island (RI), Connecticut (CT), Massachusetts (MA), New Hampshire (NH), Vermont (VT), Maine (ME) and New York (NY)] (Fig. 1). These samples included shoots, blossoms, immature fruits as well as ooze droplets. The majority of the samples were collected from apples, with ‘Macoun’, ‘Macintosh’ and ‘Gala’ being the predominant varieties, and the rest were collected from pears (‘Bartlett’ and ‘Bosc’). Seventy‐eight E. amylovora strains were isolated from these samples (42 in 2015 and 36 in 2016). The identities of the strains were confirmed using an E. amylovora diagnostic primer set targeting chromosomal DNA (Bereswill et al., 1995). The virulence of most isolates was confirmed by pathogenicity assay on immature pear fruits, with at least two isolates from each sampling location tested.
Figure 1.
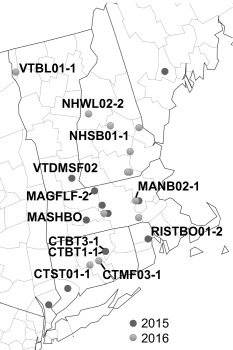
Sample collection sites. Map of six New England states (CT, RI, MA, NH, VT, ME) and New York is displayed. Dots represent orchards in which fire blight samples were collected. Colours indicate year of sampling.
The SI E. amylovora population in North America and Europe displays much higher genetic diversity than previously determined and belongs to four monophyletic clades
The acquisition of the large collection of E. amylovora strains from the northeastern USA allowed us to use genomic approaches to further characterize their phylogeny and diversity. We performed genome sequencing on 19 SI E. amylovora strains: 12 were isolated from New England in our 2015–2016 survey and seven were isolated from other parts of North America in previous studies (Table 1). Five complete genomes and 14 high‐quality draft genomes were generated (Table 1). In addition to the 19 genomes sequenced from this study, 11 genomes of SI E. amylovora strains from the NCBI genome database (https://www.ncbi.nlm.nih.gov/genome/) were also included in the analysis. The 30 SI E. amylovora strains analysed in this study represent a wide geographical distribution, including eastern and western USA, Canada, Mexico and Europe (Table 1).
Table 1.
Genomes generated and analysed in this study
| Strain ID | Geographical location | Host | Year of isolation | Sequencing technology_status | Genome size (bp) | Plasmids | Reference | NCBI WGS accession |
|---|---|---|---|---|---|---|---|---|
| CTBT1‐1 | Glastonbury, CT, USA | Pyrus communis (pear) | 2015 | PacB‐complete | 3830072 | pEA29; pEA72 | This study | NQJP00000000 |
| CTBT3‐1 | Glastonbury, CT, USA | Pyrus communis (pear) | 2015 | PacB‐complete | 3826717 | pEA29; pEA72 | This study | NQJO00000000 |
| MAGFLF 2 | Greenfield, MA, USA | Malus domestica (apple) | 2015 | PacB‐complete | 3830233 | pEA29 | This study | NQJN00000000 |
| MASHBO | Southampton, MA, USA | Pyrus communis (pear) | 2015 | PacB‐draft | 3859870 | pEA29 | This study | NQJK00000000 |
| RISTBO01–2 | Scituate, RI, USA | Malus domestica (apple) | 2015 | PacB‐complete | 3831709 | pEA72 | This study | NQJM00000000 |
| VTDMSF02 | Dummerston, VT, USA | Malus domestica (apple) | 2015 | PacB‐complete | 3833606 | pEA29; pEA72 | This study | NQJL00000000 |
| CTMF03‐1 | Middlefield, CT, USA | Pyrus communis (pear) | 2016 | Miseq‐draft | 3814230 | pEA29 | This study | NQKB00000000 |
| CTST01‐1 | Southington, CT, USA | Malus domestica (apple) | 2016 | Miseq‐draft | 3886446 | pEA29; pEA72 | This study | NQKA00000000 |
| MANB02‐1 | Nothboro, MA, USA | Malus domestica (apple) | 2016 | Miseq‐draft | 3807992 | pEA29 | This study | NQJX00000000 |
| NHSB01‐1 | Sanbornton, NH, USA | Malus domestica (apple) | 2016 | Miseq‐draft | 3802373 | pEA29 | This study | NQJW00000000 |
| NHWL02‐2 | West Lebanon, NH, USA | Malus domestica (apple) | 2016 | Miseq‐draft | 3858243 | pEA29; pEA72 | This study | NQJV00000000 |
| VTBL01‐1 | Burlington, VT, USA | Malus domestica (apple) | 2016 | Miseq‐draft | 3803426 | pEA29 | This study | NQJR00000000 |
| CA3R | CA, USA | Malus domestica (apple) | 1995 | Miseq‐draft | 4042114 | pEA29; pEA72 | This study; McManus and Jones ( 1995) | NQKC00000000 |
| Ea110 | Ingham, MI, USA | Malus domestica (apple) | 1975 | Miseq‐draft | 3805200 | pEA29 | This study; Ritchie and Klos (1977) | NQJZ00000000 |
| LA092 | WA, USA | Pyrus communis (pear) | 1988 | Miseq‐draft | 3870152 | pEA29; pEA72 | This study; McManus and Jones ( 1995) | NQJY00000000 |
| OR1 | OR, USA | Pyrus communis (pear) | N/A | Miseq‐draft | 3834797 | pEA29 | This study; McManus and Jones ( 1995) | NQJU00000000 |
| OR6 | OR, USA | Pyrus communis (pear) | N/A | Miseq‐draft | 3910597 | pEA29; pEA72 | This study; McManus and Jones ( 1995) | NQJT00000000 |
| UT5P4 | UT, USA | Malus domestica (apple) | 2000 | Miseq‐draft | 3812516 | pEA29 | This study; McGhee and Sundin ( 2012) | NQJS00000000 |
| WSDA87‐73 | WA, USA | Malus domestica (apple) | N/A | Miseq‐draft | 3811504 | pEA29 | This study; McManus and Jones ( 1995) | NQJQ00000000 |
| 01SF‐BO | Ravenna, Italy | Sorbus sp. | 1991 | From NCBI‐draft | 3795813 | pEA29 | K. Geider (Julius Kühn‐Institut, Quedlinburg, Germany); Mann et al. ( 2013) | CAPA01 |
| ATCC49946 | NY, USA | Malus domestica (apple) | 1973 | From NCBI‐complete | 3905604 | pEA29; pEA72 | K. Geider (Julius Kühn‐Institut, Quedlinburg, Germany); Sebaihia et al. ( 2010) | GCA_000027205.1 |
| ACW56400 | Fribourg, Switzerland | Pyrus communis (pear) | 2007 | From NCBI‐draft | 3860835 | N/A | ACW; Mann et al. ( 2013) | AFHN01 |
| CFBP1232 | UK | Pyrus communis (pear) | 1959 | From NCBI‐draft | 3795525 | N/A | CFBP; Mann et al. ( 2013) | CAPB01 |
| CFBP1430 | Lille, France | Crataegus sp. (hawthorn) | 1972 | From NCBI‐complete | 3833832 | pEA29 | CFBP; Smits et al. (2010) | GCA_000091565.1 |
| CFBP2585 | Ireland | Sorbus sp. | 1986 | From NCBI‐draft | 3821701 | N/A | CFBP; Mann et al. ( 2013) | CAOZ01 |
| Ea266 | Ontario, Canada | Malus sp. | 1981 | From NCBI‐draft | 3786928 | N/A | CFBP; Mann et al. ( 2013) | CAOY01 |
| Ea356 | Germany | Cotoneaster sp. | 1979 | From NCBI‐draft | 3792204 | N/A | Mann et al. ( 2013) | CAOX01 |
| LA635 | Cuauhtémoc, Mexico | Malus domestica (apple) | N/A | From NCBI‐draft | 3791024 | N/A | Smits et al. (2014) | CBVS01 |
| LA636 | Cuauhtémoc, Mexico | Malus domestica (apple) | N/A | From NCBI‐draft | 3789866 | N/A | Smits et al. (2014) | CBVT01 |
| LA637 | Guerrero, Mexico | N/A | N/A | From NCBI‐draft | 3869546 | N/A | Smits et al. (2014) | CBVU01 |
| Ea644 | Massachusetts, USA | Rubus idaeus | 2003 | From NCBI‐draft | 3832325 | pEA29 | Mann et al. ( 2013) | CAPD01 |
| MR1 | Alcona, MI, USA | Rubus sp. (raspberry) | 1995 | From NCBI‐draft | 3813203 | pEA29 | Mann et al. ( 2013) | CAPE01 |
ACW, Forschungsanstalt Agroscope Changins‐Wadenswil; ATCC, American Type Culture Collection; CFBP, French collection of plant pathogenic bacteria; N/A, Information unavailable.
We performed a whole‐genome alignment of the 30 SI E. amylovora genomes. A total of 11 857 SNPs was identified within a 3 658 056‐bp aligned genome sequence, with the pairwise nucleotide identity determined as 99.90%. The overall diversity of the 30 SI E. amylovora genomes, as determined by Watterson's theta (Watterson, 1975), was 8.18 ×10−4 per site, which is at least 100‐fold lower than that of other bacterial plant pathogens, such as the kiwifruit pathogen Pseudomonas syringae pv. actinidiae and the turfgrass pathogen Acidovorax avenae (Watterson's theta values determined as 0.008 and 0.0279, respectively) (McCann et al., 2013; Zeng et al., 2017). However, when compared with a previous comparative genomics study of E. amylovora, the genetic diversity of E. amylovora determined by our study was approximately 33 times higher (nucleotide pairwise identity was 99.90% in this study, compared with the amino acid pairwise identity of 99.98% of coding regions, or nucleotide pairwise identity of 99.997% on the whole genome, in the previous study). In addition, we observed a higher diversity in North American isolates (Watterson's theta value of 8.56 ×10−4) than in European isolates (Watterson's theta value of 2.14 ×10−5).
Furthermore, we determined the phylogeny of the 30 SI E. amylovora strains based on the SNPs identified at the genome level (Bertels et al., 2014). Our results showed that the SI E. amylovora strains in North America and Europe fell into four monophyletic clades, three of which exhibited strict phylogeographical structure (Fig. 2A). Three strains isolated from the western USA formed a single clade (named Western U.S. 1), with a total number of 160 SNPs identified amongst all strains (pairwise identity, 99.997%). Nine strains isolated from the eastern USA formed one clade (Eastern U.S.), which differed by 416 SNPs (pairwise identity, 99.996%). One single strain (CA3R) from the western USA formed its own clade (Western U.S. 2). Compared with the three clades with strict phylogeographical structure, 17 E. amylovora strains from the eastern USA, western USA, Canada, Mexico and Europe formed a single clade. As this clade contained strains from multiple locations, we named this clade the ‘Widely Prevalent’ clade. A total of 802 SNPs was identified in these strains (pairwise identity, 99.995%). The structure of the phylogenetic tree was highly robust, as all nodes were well supported by high bootstrap values (Fig. 2). We also observed that all SI E. amylovora clades were clustered on the phylogenetic tree, apart from the RI E. amylovora strains (Fig. 2B).
Figure 2.

Phylogeny of Spiraeoideae‐infecting (SI) (A), and SI and Rubus‐infecting (RI) (B), Erwinia amylovora strains, inferred from a whole‐genome alignment. The phylogenetic tree was generated based on a merged whole‐genome nucleotide alignment generated by REALPHY. The tree was constructed using the maximum likelihood method with the general time reversible model, and was midpoint rooted. Bootstrap support values are 100% unless indicated. Colours indicate the regions in which the strains were isolated (green, eastern USA; red, western USA and Mexico; blue, Europe).
Pairwise analysis of the number of SNPs revealed that any two single strains from different clades of Widely Prevalent, Eastern U.S. or Western U.S. 1 differed from one another by approximately 2500 SNPs, whereas two strains from a single clade mentioned above differed from each other by 4–259 SNPs (Fig. S1, see Supporting Information). Western U.S. 2 strain CA3R differed from strains from other clades by approximately 8700 SNPs (Fig. S1). No significant genome rearrangement was observed (Fig. S2, see Supporting Information).
Two plasmids, pEA29 and pEA72, were the most prevalent in the 19 strains sequenced. Nearly all strains contained the ubiquitous, non‐conjugative plasmid pEA29, with the exception of RISTBO01–2 (Table 1). This echoes previous findings that pEA29 may not be universal in E. amylovora (Llop et al., 2006, 2011). pEA72, a plasmid first reported in ATCC 49946 (Sebaihia et al., 2010), was found in approximately 42% of the strains sequenced, including RISTBO01–2, the strain lacking pEA29 (Table 1). Other conjugative plasmids, such as pEA34, as found in streptomycin‐resistant strains in Michigan (Chiou and Jones, 1991), and pEI70 (Llop et al., 2011), were not found in the strains of this study.
Prior to this study, the New England region had never been surveyed for fire blight, and E. amylovora strains from this region were not available and thus were not included in any previous population genetics studies (McGhee and Sundin, 2012; McGhee et al., 2002; McManus and Jones, 1995; Rezzonico et al., 2011). Because fire blight was first reported in the northeastern USA, we hypothesized that strains from this region would be critical for the reflection of the true diversity and phylogeny of E. amylovora. This point was confirmed by our diversity and phylogenetic analyses, and is further discussed in the subsequent two paragraphs.
We observed that the genetic diversity of E. amylovora was much higher than previously thought (McCann et al., 2013). This could be because our study included an enlarged collection of strains from North America (24 strains from western and eastern North America, six strains from Europe) than the previous study (only two strains from North America, seven strains from Europe). It is well accepted that E. amylovora originated from North America and later spread to Europe and other parts of the world. As the centre of origin maintains the highest pathogen population diversity (Stukenbrock and McDonald, 2008), we observed a much higher genetic diversity amongst North American isolates than amongst European isolates. We believe that the genetic diversity calculated from this study is a better representation of the true scenario of the SI E. amylovora strains. Despite this, the overall diversity of SI E. amylovora strains was still very low in comparison with other pathogens that have emerged in recent years (McCann et al., 2013; Potnis et al., 2011, 2015; Zeng et al., 2017).
We discovered that the SI E. amylovora strains in North America and Europe belonged to four distinct clades. Many of the strains used in this study were also used in previous genotyping studies (McGhee and Sundin, 2012; McGhee et al., 2002; McManus and Jones, 1995; Rezzonico et al., 2011). From a comparison of which groups these strains belong to in our study vs. previous studies, we can determine how the four clades identified in this study are similar to or different from the genotype groups identified in previous studies. Both ribotyping (McManus and Jones, 1995) and groEL sequencing (McGhee et al., 2002) grouped the SI E. amylovora strains into two genotypes (McGhee and Sundin, 2012). The type 1 strains determined by both of these methods were also determined as monophyletic in our study (as the Widely Prevalent clade), with the exception of CA3R, a strain determined as type 1 by ribotyping and groEL sequencing, but separately grouped as a single clade in our study (Western U.S. 2 clade). The second genotype strains (type 2 for groEL sequence typing or type 3 strains for ribotyping) were also identified as monophyletic in our study, as the Western U.S. 1 clade. In recent studies of the clustering and tracking of E. amylovora using CRISPR analysis (McGhee and Sundin, 2012; Rezzonico et al., 2011), strains of the Widely Prevalent clade from this study were also grouped together in one cluster (as Group 1 in Rezzonico et al., 2011). Furthermore, the CRISPR analysis also clustered these strains with strains from New Zealand and the Middle East, which further suggests that strains from the Widely Prevalent clade are widely distributed at a global level, causing disease epidemics worldwide. In addition, consistent with our study, strains of the Western U.S. 1 clade were clustered together in the CRISPR analysis (as Group 3 in Rezzonico et al., 2011), and CA3R was separately delineated by CRISPR (McGhee and Sundin, 2012). However, as CRISPR groups strains on the basis of the difference in CRISPR arrays and may not reflect true phylogeny, there are situations in which the phylogenetic relationships of strains cannot be clearly resolved by CRISPR. For example, it is unclear whether strain UT5P4 belongs to the Widely Prevalent clade or the Western U.S. 1 group in the CRISPR analysis (McGhee and Sundin, 2012). According to Rezzonico et al. (2011), UT5P4 would be grouped with strains of Group 2. In contrast, the phylogeny inferred by the genome SNP analysis of this study clearly suggests that UT5P4 belongs to the Widely Prevalent clade (or Group 1 of Rezzonico et al., 2011) with a high bootstrap support (Fig. 2A). Finally, none of the previous genotyping studies identified any SI E. amylovora strains that are of eastern US origin, but do not belong to the Widely Prevalent clade. Here, we identified a new clade (Eastern U.S.) of SI E. amylovora strains in the New England region that are phylogenetically distinct from those of the Widely Prevalent clade. There could be a couple of explanations why this clade was not seen in previous studies. It is possible that, although this clade existed in eastern USA, previous sampling was not sufficiently intensive to include these isolates, or the research methods employed by previous studies were not sufficiently sensitive to differentiate this clade from other clades. It is also possible that this clade recently emerged as a result of recombination (illustrated later in this article), as all the isolates were only isolated in recent years.
With improved resolution, the phylogeny showed that five of the six European strains analysed in this study (01SFR‐BO, CFBP1430, ACW56400, CFBP1232, CFBP2585) were highly clonal (Fig. 2), forming a monophyletic phylogroup with only 89 SNPs distinguishing these strains. This supports the hypothesis that the spread of E. amylovora to European countries (UK, Ireland, Italy, France and Switzerland) may have been through one single transmission event rather than multiple events. As the other European strain (Ea356) belongs to a different phylogroup, with 179 SNPs different from the other European strains, it is possible that the spread of Ea356 was through a separate transmission event. Our data also suggest that, under natural conditions, the spontaneous mutation rate in E. amylovora is very low, considering that a strain isolated in 2007 from Switzerland (ACW56400) differed from a potentially ancestral strain isolated in 1959 from the UK (CFBP1430) by only 46 SNPs.
Spatially structured populations are observed in the Eastern and Western U.S. clades, but not in the Widely Prevalent clade
To test whether the phylogenetic relationships were shaped by geography, we also determined the distances between isolates and correlated them with genetic distances. No significant correlations between genetic distance and geographical distance of the SI E. amylovora strains were observed (Fig. 3A), suggesting that the dispersal rates of E. amylovora were too high to maintain spatially structured populations. However, when we only included strains from the Eastern U.S., Western U.S. 1 and Western U.S. 2 clades, a significant correlation between genetic distance and geographical distance was detected (Fig. 3B). These results suggest that human activities have played a critical role in the dispersal of strains from the Widely Prevalent clade, but have played less of a role in the dispersal of E. amylovora strains of clades that are locally present in North America.
Figure 3.

Mantel's test of genetic distance by geographical distance. (A) Mantel's test using all strains except those of the ‘Widely Prevalent’ clade. (B) Mantel's test using all 29 Spiraeoideae‐infecting (SI) Erwinia amylovora strains. OR6 is excluded because of a lack of GPS coordinates.
Strains of the Widely Prevalent clade display evidence of recombination with strains of the Eastern U.S. clade
Homologous recombination, defined as a segment of DNA from one microorganism being transferred and recombined with the genome of another microorganism in prokaryotes, has been commonly observed in plant‐pathogenic bacteria, and is thought to play a critical role in pathogen evolution and host adaptation (Awadalla, 2003; Baltrus et al., 2017; Mhedbi‐Hajri et al., 2013; Yan et al., 2008; Zeng et al., 2017). We determined the level of recombination that occurred between the four clades of SI E. amylovora (Fig. 4). Our results revealed that an average of 3.39% of the genome showed evidence of recombination, marked by gene conversion, between any two strains of different phylogroups. More specifically, a high level of recombination occurred between the Widely Prevalent clade and the Eastern U.S. clade (Fig. 4), with an average of 4.54% of genomes showing evidence of recombination; limited recombination occurred between the Western U.S. clades (1 and 2) and other clades (Eastern U.S. clade and Widely Prevalent clade, Fig. 4), with an average of only 1.75% of the genome showing signs of recombination. Detailed examination of the number of recombination events, total size of the recombination events, the number of SNPs on the recombined sections and the total number of SNPs identified from the genome (Table S1, see Supporting Information) revealed that, although strains from the Western U.S. clades (OR1 and LA092) displayed a much lower level of recombination with the mixed group strain (VTBL01‐1 and Ea110) than with strains of the Eastern U.S. clade (CTBT3‐1 and VTDMSF02), these strains differed by a similar number of SNPs (about 2500 SNPs pairwise).
Figure 4.

Recombination between any two Spiraeoideae‐infecting (SI) Erwinia amylovora strains at the genome level. Gene conversion fragments were identified by GENECONV using the nucleotide alignment of the whole‐genome sequences. The percentage was calculated by dividing the size of cumulative gene conversion fragments by the size of the aligned sequences.
In addition to the variations in the level of recombination between different clades mentioned above, we also observed that strains of the Eastern U.S. clade displayed different levels of recombination with strains of the Widely Prevalent clade. Four strains, CTBT1‐1, CTBT3‐1, NHWL02‐2 and MANB02‐1, which formed a subgroup within the Eastern U.S. clade, displayed high levels of recombination with all strains in the Widely Prevalent clade, even though these Widely Prevalent strains were from different origins of isolation (eastern USA, western USA, Canada, Mexico and Europe, Fig. 4). This suggests that recombination may have occurred between the ancestor of CTBT1‐1, CTBT3‐1, NHWL02‐2 and MANB02‐1, and the ancestor of Widely Prevalent strains. Interestingly, other Eastern U.S. strains (RISTBO01–2, CTST1‐1, MAGFLF‐2, VTDMSF02 and CTMF03‐1) displayed different levels of recombination with strains of the Widely Prevalent clade, and the level of recombination was in accordance with the origin of isolation of the Widely Prevalent strains: this subgroup of Eastern U.S. strains displayed high levels of recombination with Widely Prevalent strains that were isolated from eastern US locations and low levels of recombination with Widely Prevalent strains that were isolated from other locations, such as Europe, western USA and Mexico, with the exception of Ea356 (Fig. 4).
As the SI E. amylovora strains were highly clonal (pairwise identity, 99.9%) and infected the same hosts, recombination would occur at a similar rate between these strains. One critical condition for recombination to occur is that the two strains (donor and recipient of the DNA exchange) must be in close proximity at the same time (Wicker et al., 2012). Thus, two strains that are physically present in the same region for a long period of time may have a higher chance to share DNA, recombine and thus display a higher level of recombination, compared with two strains that are geographically or chronologically separated. Because of this, we can use the level of recombination to infer the geographical and chronological coexistence of different lineages of strains. Amongst the four clades of SI strains identified in this study, Western U.S. (1, 2) and Eastern U.S. clades caused infection only at regional levels, but the Widely Prevalent clade was widespread throughout eastern and western USA, Canada, Mexico, Europe and, possibly, New Zealand and the Middle East. There have been debates about the original location in which strains of this clade resided prior to their global spread. In a previous study of E. amylovora involving strain typing by CRISPR, the authors hypothesized that the Widely Prevalent strains may have originated from the eastern USA because the number of Eastern strains in this clade is overwhelmingly higher than the number of strains of western US origin (McGhee and Sundin, 2012). However, there is no further evidence to support this hypothesis. Our results showed that the Widely Prevalent strains displayed a high level of recombination with a clade of strains that were strictly present in the eastern USA (Eastern U.S. clade), whilst displaying a low level of recombination with clades of strains that were only present in the western USA (Western U.S. 1 and 2 clades). This observation suggests that the Widely Prevalent strains may have originated from the eastern USA, where these strains may have had a higher chance to recombine with the eastern U.S. strains (Eastern U.S. clade) than with strains from the western USA (Western U.S. 1 and 2 clades).
Furthermore, although it is well accepted that North America is the origin of fire blight, it is not clear in which part of North America E. amylovora originally arose. Fire blight was first observed in eastern USA in 1790 (Hudson River Valley in New York) (Denning, 1794; van der Zwet et al., 2012) and was later reported in western USA in the late 1800s (Hotson, 1916; Hutt, 1903; Isaac, 1902; Jackson, 1910). There are debates over whether E. amylovora causing fire blight on apples in western USA was originally present in western USA or spread from eastern to western USA through the transportation of infected plant materials and/or human activities. Here, we have provided evidence that the Western U.S. clades (Western U.S. 1 and 2) and Eastern U.S. clade may have been present in western and eastern USA, respectively, for a long time (Eastern and Western clades are phylogenetically distinct with a low level of recombination between the two groups). This suggests that both eastern and western USA were colonized by E. amylovora before the discovery of the New World. Despite this, we observed that some Eastern‐originating strains have spread to the western USA (strains in the Widely Prevalent clades with a western US origin) as a result of human activities.
Although we observed convincing patterns of recombination variations over a fairly large number of strains, there are potential limitations in this study. In addition to geographical coexistence, bacterial defence systems may also potentially affect the level of recombination. We determined the presence and absence of two major defence systems in the sequenced E. amylovora genomes: restriction modification and CRISPR/Cas systems. Both systems were present in all 30 genomes, although the number of CRISPR repeats varied in strains of different phylogroups (Table 2). In general, our data suggested that variations in defence systems were not correlated with variations in the level of recombination. Furthermore, as the strains used in this study were isolated from different years, this may also affect the level of recombination. Future studies with a larger collection of strains isolated in recent years from a larger geographical area, especially from Europe and western USA, may help to overcome these limitations.
Table 2.
Presence of restriction modification and clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein (CRISPR/Cas) systems in Erwinia amylovora strains
| Strain ID | Phylogenetic group* | CRISRP group† | Presence of restriction modification systems‡ | Presence of cas1–cas5 | Number of CRISPR1 repeats§ | Number of CRISPR2 repeats§ |
|---|---|---|---|---|---|---|
| La637 | Epidemic | I | Yes | Yes | 38 | 23 |
| La635 | Epidemic | I | Yes | Yes | 28 | 23 |
| La636 | Epidemic | I | Yes | Yes | 36 | 23 |
| Ea110 | Epidemic | I | Yes | Yes | 37 | 23 |
| MASHBO | Epidemic | I | Yes | Yes | 37 | 26 |
| NHSB01‐1 | Epidemic | I | Yes | Yes | 28 | 26 |
| Ea266 | Epidemic | I | Yes | Yes | 10 | 22 |
| UT5P4 | Epidemic | II | Yes | Yes | 36 | 35 |
| ATCC49946 | Epidemic | I | Yes | Yes | 28 | 32 |
| 01SFR‐BO | Epidemic | I | Yes | Yes | 37 | 34 |
| CFBP1430 | Epidemic | I | Yes | Yes | 28 | 34 |
| ACW56400 | Epidemic | I | Yes | Yes | 37 | 34 |
| CFBP 1232 | Epidemic | I | Yes | Yes | 36 | 32 |
| CFBP2585 | Epidemic | I | Yes | Yes | 37 | 34 |
| Ea356 | Epidemic | I | Yes | Yes | 36 | 36 |
| WSDA87‐73 | Epidemic | I | Yes | Yes | 36 | 34 |
| VTBL01‐1 | Epidemic | I | Yes | Yes | 30 | 35 |
| VTDMSF02 | Eastern | ND | Yes | Yes | 81 | 55 |
| CTMF03‐1 | Eastern | ND | Yes | Yes | 84 | 55 |
| MAGFLF‐2 | Eastern | ND | Yes | Yes | 55 | 40 |
| CTST01‐1 | Eastern | ND | Yes | Yes | 82 | 55 |
| CTBT3‐1 | Eastern | ND | Yes | Yes | 94 | 25 |
| CTBT1‐1 | Eastern | ND | Yes | Yes | 94 | 25 |
| NHWL02‐2 | Eastern | ND | Yes | Yes | 90 | 25 |
| MANBO2‐1 | Eastern | ND | Yes | Yes | 91 | 29 |
| RISTBO01‐2 | Eastern | ND | Yes | Yes | 94 | 58 |
| OR6 | Western 1 | III | Yes | Yes | 97 | 49 |
| LA092 | Western 1 | III | Yes | Yes | 56 | 49 |
| OR1 | Western 1 | III | Yes | Yes | 98 | 49 |
| CA3R | Western 2 | ND | Yes | Yes | 23 | 49 |
*Epidemic, Widely Prevalent clade; Eastern, Eastern U.S. clade; Western 1, Western U.S. 1 clade; Western 2, Western U.S. 2 clade.
†The CRISPR groups were determined according to the groups named by Rezzonico et al. (2011). ND indicates that the strain was not included in the previous CRISPR study and the CRISPR pattern did not match any of the groups determined in that study.
‡Presence of 13 restriction modification system genes, EAM_0060, EAM_0061, EAM_0062, EAM_0280, EAM_0862, EAM_0863, EAM_0864, EAM_2079, EAM_2080, EAM_3033, EAM_3237, EAM_3405, EAM_3406, identified by the REBASE database (Roberts et al., 2015).
§Number of CRISPR repeats was calculated by CRT1.1 (Bland et al., 2007).
Erwinia amylovora strains display variation in virulence amongst different phylogroups
As we identified genetic variation amongst the E. amylovora strains, we further tested whether these strains displayed any difference in virulence. In addition to the strains in Fig. 2, we also included 10 additional SI E. amylovora strains isolated from our fire blight survey in northeastern USA, and RBA4, an RI E. amylovora strain which is not expected to be virulent on immature pear (Braun and Hildebrand, 2005). When inoculated onto immature pear, all SI E. amylovora strains tested caused symptoms of necrosis and ooze, whereas the RI strain RBA4 was non‐pathogenic (Fig. 5A,B). However, variations in the size of maceration area were observed amongst the different strains (Fig. 5A). The average size of the maceration area caused by all strains tested, indicated by the dark lesion surrounding the inoculation site, was 210.4 mm2. Compared with the average size of the maceration area, a dramatic reduction in size was observed for four strains: CTBT1‐1, a strain of the Eastern U.S. clade, and OR1, OR6 and LA092, three strains that belong to the Western U.S. 1 clade. Interestingly, all strains in the Western U.S. 1 clade uniformly displayed reduced virulence.
Figure 5.
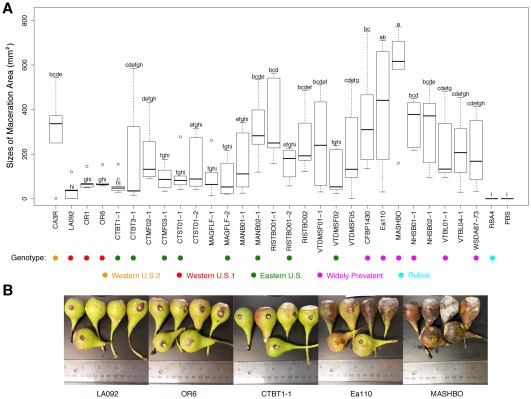
Virulence variations of Erwinia amylovora strains on immature pear. (A) Quantification of the sizes of the maceration area on immature pear fruits. (B) Virulence phenotypes of representative high‐virulence and low‐virulence strains. Photographs of the pears with maceration symptoms were taken at 8 days post‐inoculation and the sizes of maceration were quantified using ImageJ.
The low‐virulence phenotype of CTBT1‐1 is attributed to an SNP in hfq
To determine whether the variations in virulence could be attributed to any genetic variations, we performed a comparative genomic analysis of one of the low‐virulence strains, CTBT1‐1, with the phylogenetically closely related high‐virulence strain CTBT3‐1. CTBT1‐1 and CTBT3‐1 were isolated from two individual pear trees with fire blight symptoms in the same orchard. Genome alignment of CTBT1‐1 and CTBT3‐1 revealed very minimum genome rearrangement between the two strains (Fig. S3A, see Supporting Information). Calling of SNPs and small INDELs identified three SNPs between CTBT1‐1 and CTBT3‐1 (Fig. S3B). One of the three SNPs was located at nucleotide 47 in hfq (G in CTBT3‐1 and A in CTBT1‐1), a gene encoding a small RNA chaperone. This SNP results in a polymorphism at amino acid 16 (‘CGT’ encoding for ‘arginine’ in CTBT3‐1 and ‘CAT’ encoding for ‘histidine’ in CTBT1‐1). The other two SNPs were located in a gene with uncharacterized function (EAM_2564) and in an intergenic region (between EAM_0327 and EAM_0328).
To determine whether the polymorphism located in hfq may explain the low‐virulence phenotype in CTBT1‐1, we generated deletion mutants of hfq (Δhfq) in CTBT1‐1 and CTBT3‐1 and complemented both mutants with either pCLhfq16R or pCLhfq16H (pCL1920 vector carrying hfq, with R or H at amino acid 16). The virulence of the wild‐type and Δhfq of CTBT1‐1 and CTBT3‐1, as well as the complementation strains, was compared in an immature pear assay (Fig. 6). High‐ and low‐virulence phenotypes were observed in CTBT3‐1 and CTBT1‐1, respectively. Mutation of hfq caused a reduction in virulence in CTBT3‐1, but not in CTBT1‐1. The low‐virulence phenotype in Δhfq of both CTBT1‐1 and CTBT3‐1 can be complemented to the high‐virulence phenotype by introduction of a copy of hfq16R (CTBT3‐1 version), but not by pCLhfq16H (CTBT1‐1 version). Finally, the phenotypes of amylovoran production and motility of CTBT1‐1 were the same as Δhfq of both CTBT3‐1, and could be complemented by pCLhfq16R (Fig. S4, see Supporting Information). These results confirmed that the SNP at nucleotide 47 of hfq is the genetic cause of the low‐virulence phenotype in CTBT1‐1.
Figure 6.

Virulence variations of Erwinia amylovora strains on immature pear. (A) Virulence of CTBT3‐1, CTBT1‐1, Δhfq of the two strains and Δhfq strains complemented by pCLhfq16R and pCLhfq16H. Photographs were taken at 9 days post‐inoculation. (B) Alignment of the first 54 nucleotides of hfq in CTBT3‐1 and CTBT1‐1, illustrating the single nucleotide polymorphism at nucleotide 47 (amino acid 16).
Variations in virulence are commonly observed in many bacterial pathogens. In many cases, the variation is caused either by the acquisition/deletion of a virulence gene (Adhikari et al., 2013) or by sequence variations of a virulence gene (Ma et al., 2006). Here, we presented evidence that the variation in virulence could also be caused by sequence variations of virulence regulators. Hfq and Hfq‐dependent regulatory small RNAs are essential for full virulence in E. amylovora (Zeng and Sundin, 2014; Zeng et al., 2013). In Hfq, amino acid 16, at which polymorphism occurs, is part of a conserved α‐helix structure immediately upstream of the Sm‐1 motif of Hfq. The Sm‐1 motif is important for binding to the uridine‐rich region of sRNAs and forms a ring‐like heteroheptamer (Sun et al., 2002). Thus, the reduction in virulence in CTBT1‐1 could be explained by a reduced activity of Hfq‐16H. This point mutation is not prevalent in other E. amylovora strains. CTBT1‐1 is the only strain of all 30 E. amylovora strains sequenced in this study that contains a histidine at amino acid 16 of Hfq instead of an arginine. We are currently elucidating the genetic causes and evolutionary forces of other low‐virulence strains.
Conclusions
Our comparative genomic analysis of E. amylovora, including 12 new strains from the eastern USA, revealed increased diversity in this pathogen and enabled us to distinguish four clades. Founder events, and subsequent genetic bottlenecks, together with a relatively short number of years (c. 250–300 years) since the establishment on apple and pear, are thought to have limited the overall genetic diversity observed within E. amylovora. However, there has been a lack of inclusion of strains from eastern USA, the presumed geographical origin of E. amylovora, in previous genomic studies. By including these strains, we have increased the breadth of knowledge of the evolution of E. amylovora. SNP analysis of two very closely related strains with significant differences in virulence enabled us to identify a mutation in the global regulator hfq. Although genetic differences between strains of different levels of virulence can be detected by comparative genomics, the confirmation of the identified genes as the true cause of the difference in virulence can be difficult as, frequently, many genetic differences are detected between genomes of high‐ and low‐virulence strains. The fact that E. amylovora genomes are highly homogeneous (99.9% identical), whereas we are still able to observe variations in the virulence of these strains, provides us with a unique opportunity to understand how genetic variations can cause phenotypic variations, and how pathogens evolve and adapt to a plant host.
Experimental Procedures
Bacterial strains and growth conditions
Erwinia amylovora strains were cultured in Luria–Bertani (LB) medium at 28 °C. Strains were stored at −80 °C in 15% glycerol. When required, antibiotics were added to the medium at the following concentrations: chloramphenicol, 30 μg/mL; kanamycin, 50 μg/mL; ampicillin, 100 μg/mL.
Collection of fire blight samples and isolation of E. amylovora strains
Samples were collected through a collaborative effort of the Connecticut Agricultural Experiment Station, University of Massachusetts, extension associates and researchers from New England states, as well as the Connecticut and Massachusetts Apple Grower Associations. Three samples were collected from different trees in each orchard when possible. Samples were placed in enclosed plastic bags with a damp paper towel and brought or shipped to the CAES Jenkins‐Waggoner laboratory for pathogen isolation. Tissues were surface sterilized by 70% ethanol for 3 min, rinsed in sterile distilled water, dissected into 5‐mm pieces using a razor blade and placed in microcentrifuge tubes with sterile water. Sterile water containing the pathogens was streaked onto a CCT plate (Ishimaru and Klos, 1984). Single, dome‐shaped colonies of E. amylovora were picked and restreaked onto an LB agar plate to produce pure cultures. The identity of E. amylovora was confirmed by polymerase chain reaction (PCR) using diagnostic primers that detect the ams genes from the chromosome (Bereswill et al., 1995).
Genome sequencing and assembly
Total DNA was isolated using a Qiagen DNeasy Kit (Qiagen, Valencia, CA, USA). The quality and quantity of the purified DNA were checked with a Nanodrop 2000C (Nanodrop Products, Wilmington, DE, USA). Genomic DNA was sequenced by either an Illumina MiSeq instrument (Illumina Inc, San Diego, CA, USA) or a Pacific Bioscience RS II instrument (Pacific Biosciences, Menlo Park, CA, USA) at the Yale Center for Genome Analysis using the same procedure as described in our previous work (Zeng et al., 2017).
Genomes were assembled using Canu version 1.3 (Koren et al., 2017) with default parameters, except that the estimated genome size was set to 4.0 Mbp. Consensus sequence polishing was performed using the Quiver consensus model in Genomic Consensus version 0.92 (Chin et al., 2013). Base modification was performed by mapping raw reads against a draft genome assembly with the BLASR aligner (Chaisson and Tesler, 2012) in SMRT analysis Suite version 2.3 (Pacific Biosciences) using default settings. Illumina sequencing reads were de novo assembled using SPAdes version 3.9.0 (Bankevich et al., 2012) with automatic Kmer selection and default settings. Genome assembly evaluation quality was evaluated using QUAST version 4.3 (Gurevich et al., 2013).
Genome alignment, SNP identification, phylogenetic analysis and identification of recombination
Genomes of E. amylovora were aligned using a reference sequence alignment‐based phylogeny (REALPHY) analysis (Bertels et al., 2014). REALPHY produced a merged alignment from two individual reference sequence alignments (ATCC49946 and CTBT1‐1) using default parameters. The number of SNPs as well as the pairwise identity between different genomes or genome groups were calculated using Geneious (9.1.6) (Kearse et al., 2012). Watterson's theta value (Watterson, 1975) was calculated using the Egglib package (De Mita and Siol, 2012). The phylogeny of these genomes was inferred from the genome alignment generated by REALPHY, using a maximum likelihood (ML) method with a general time reversible (GTR) substitution model and with 1000 bootstrap replicates. The phylogenetic tree was rooted using a midpoint method.
The genome alignment of 30 E. amylovora strains generated by REALPHY was used as input in GENCONV (http://www.math.wustl.edu/~sawyer/geneconv/) with default settings (Sawyer, 1989) to predict the amount of gene conversion fragments. The multiple comparison‐corrected P values representing the possibility of gene conversion events were calculated with 10 000 random permutations. Fragments with P < 0.05 were considered as significant gene conversion fragments. The percentage of genome recombination was calculated using the cumulative base pair of gene conversion fragments to divide the size of the genome alignment (3 658 056 bp). The percentage of recombination between any two genomes was displayed in a heat map generated by Plotly (https://plot.ly).
Mantel's test
The genome nucleotide alignment from REALPHY was used as the input file to calculate the pairwise genetic distance, employing the ‘ape’ package (Paradis et al., 2004) in R with the TN93 evolutionary model (Tamura and Nei, 1993). The pairwise geographical distance was calculated based on the GPS coordinates of the isolation origin using the ‘geosphere’ package in R (https://cran.rproject.org/package=geosphere). Mantel's test was performed using the ‘ade4’ package (Dray and Dufour, 2007) in R with 10 000 permutations.
Detection of restriction modification and CRISPR/Cas systems in E. amylovora genomes
Genes encoding the restriction modification system in E. amylovora (EAM_0060, EAM_0061, EAM_0062, EAM_0280, EAM_0862, EAM_0863, EAM_0864, EAM_2079, EAM_2080, EAM_3033, EAM_3237, EAM_3405, EAM_3406) were acquired from the REBASE database (Roberts et al., 2015) and their nucleotide sequences were used to search the 30 E. amylovora genomes for their presence and absence using Geneious (Kearse et al., 2012). The nucleotide sequences of Cas systems (cas1, cas2, cas3, cas4, cas5) in the E. amylovora ATCC49946 genome were also used to determine their presence in the genomes analysed in this research. The number of repeats in CRR1 and CRR2 were determined by first identifying their genome location by searching for their upstream and downstream limits using CR1 and CR2 sequences (Table S2, see Supporting Information), followed by determination of the number of CRISPR repeats by CRT1.1 (Bland et al., 2007).
Virulence test
The virulence of the E. amylovora strains was tested using an immature pear fruit assay as described previously (Zhao et al., 2005). Briefly, bacteria were cultured in LB broth overnight, and adjusted to 2 × 105 colony‐forming units (CFU)/mL in sterile 0.5 × phosphate‐buffered saline (PBS). Immature pears (cultivar ‘Bosc’) were surface sterilized with 10% bleach, rinsed with sterile distilled water, wounded by a 0.2‐mm syringe needle and placed in an enclosed glass tray with sterile wet paper towels on the bottom to maintain humidity. Five microlitres of the bacterial suspension were added to the wounded area on immature pears, and the inoculated pears were incubated at 28 °C under high relative humidity conditions. Photographs of the lesion symptoms were taken at 8 days post‐inoculation. All assays were repeated at least twice, with five biological replicates in each experiment. The lesion sizes on immature pears were quantified using ImageJ software (Schneider et al., 2012). Statistical analysis was performed using the one‐way analysis of variance (ANOVA) model in the ‘stats’ package in R.
Identification of SNPs between CTBT1‐1 and CTBT3‐1
Corrected Pacbio reads from CTBT3‐1 were aligned to the CTBT1‐1 complete genome assembly using BWA MEM version 0.7.15 (Li, 2013) with the –x parameter sets to ‘pacbio’. Aligned reads were sorted and indexed using SAMtools version 1.3.1 (Li et al., 2009) and duplicated reads were marked using PicardTools version 1.140 (Picard Tools ‐ By Broad Institute; http://broadinstitute.github.io/picard/). Mutations were identified from aligned reads using FreeBayes version 1.1.0 (Garrison and Marth, 2012) and the options set as follows: ‘‐p 1 –C 30’. The impacts of the SNPs were manually evaluated.
Deletion mutagenesis and complementation
Chromosomal deletion mutants of E. amylovora were constructed using a previously reported λ red recombinase method (Datsenko and Wanner, 2000; Zhao et al., 2009). Briefly, recombination fragments consisting of 50‐nucleotide homology arms of regions of hfq flanking a chloramphenicol resistance cassette were amplified from plasmid pKD4 using hfqmut_F and hfqmut_R (Table S2). PCR products were purified and electroporated into E. amylovora strains expressing recombinase genes from the helper plasmid pKD46. Mutants were selected on LB medium amended with chloramphenicol and ampicillin. Successful deletion of the target gene was confirmed by PCR and sequencing.
The hfq gene with the polymorphism at nucleotide 47 was PCR amplified using the genomic DNA isolated from CTBT3‐1 and CTBT1‐1, and primers Chfq F_Xbal and Chfq R_SacI (Table S2). The amplified PCR fragment was confirmed by electrophoresis, purified by gel purification and digested by the restriction enzymes XbaI and SacI. The digested fragments were cloned into the complementation vector pCL1920 (Lerner and Inouye, 1990) between the XbaI and SacI sites. The sequence and polymorphism at nucleotide 47 in hfq in pCL1920 were confirmed by sequencing. The resulting plasmids pCLhfq16R and pCLhfq16H were introduced into the E. amylovora strains CTBT3‐1 and CTBT1‐1 and the hfq mutants by electroporation.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Pairwise comparison of the number of single nucleotide polymorphisms (SNPs) of the 30 genomes. SNPs were identified from the whole‐genome alignment by the Egglib package. Heat map was generated by Plotly.
Fig. S2 MAUVE alignment of six complete genomes of Erwinia amylovora. Conserved regions of the genome are represented by coloured boxes. Coloured lines suggest translocations. Same coloured blocks on opposite sides of the line suggest inversions.
Fig. S3 (A) Dotplot of CTBT3‐1 and CTBT1‐1 produced by MUMmer. (B) Single nucleotide polymorphisms (SNPs) identified between CTBT3‐1 and CTBT1‐1 by SNP calling. The upper parts of the figure show the sequence variations in CTBT3‐1, whereas the lower parts show the mapped reads of CTBT1‐1. The image was generated using IGV version 2.3.81.
Fig. S4 Amylovoran production (A) and swarming motility (B) of CTBT3‐1, CTBT1‐1, CTBT3‐1Δhfq and CTBT1‐1Δhfq C‐hfq‐16R. Amylovoran production and swarming motility were tested using the same protocols as described previously (Zeng et al., 2013). Amylovoran production was measured at 72 h post‐inoculation in MBMA medium (3 g KH2PO4, 7 g K2HPO4, 1 g [NH4]2SO4, 2 ml glycerol, 0.5 g citric acid, 0.03 g MgSO4; amended with 1% sorbitol). The amount of amylovoran was quantified by measurement of the turbidity [optical density at 600 nm (OD600)] after the addition of cetylpyrimidinium chloride (CPC) to the supernatant of MBMA medium. Statistical analysis was performed using the one‐way analysis of variance (ANOVA) model in the ‘stats’ package in R. For motility assay, bacterial cells were inoculated at the centre of swarming agar plates (0.3% agar). The inoculated plates were incubated at 28 °C at high humidity. Photographs were taken at 20 h post‐inoculation.
Table S1 Recombination events between strains of different clades, detected by Geneconv.
Table S2 DNA oligonucleotide sequences used in this study.
Acknowledgements
This study was supported by the Northeastern IPM Center partnership grant, United States Department of Agriculture, National Institute of Food and Agriculture‐Organic Transitions (2017–51106‐27001), USDA‐NIFA‐Agriculture and Food Research Initiative‐Exploratory Research (2016–67030‐24856), USDA‐Hatch (CONH00650) to Q.Z., and Michigan State University AgBioResearch to G.W.S. We thank Y. Li, M. Concklin, J. Clements and H. Faubert for assistance in sample collection, G. Wang and the Yale Center for Genome Analysis for the genome, and S. Lefrancois and A. Swett for technical support. Finally, we thank the New England apple growers, and the Connecticut and Massachusetts fruit grower associations for their collaboration.
References
- Adhikari, B.N. , Hamilton, J.P. , Zerillo, M.M. , Tisserat, N. , Lévesque, C.A. and Buell, C.R. (2013) Comparative genomics reveals insight into virulence strategies of plant pathogenic oomycetes. PLoS One, 8, e75072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla, P. (2003) The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4, 50–60. [DOI] [PubMed] [Google Scholar]
- Baltrus, D.A. , McCann, H.C. and Guttman, D.S. (2017) Evolution, genomics and epidemiology of Pseudomonas syringae: challenges in bacterial molecular plant pathology. Mol. Plant Pathol. 18, 152–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. , Lesin, V.M. , Nikolenko, S.I. , Pham, S. , Prjibelski, A.D. , Pyshkin, A.V. , Sirotkin, A.V. , Vyahhi, N. , Tesler, G. , Alekseyev, M.A. and Pevzner, P.A. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereswill, S. , Bugert, P. , Bruchmuller, I. and Geider, K. (1995) Identification of the fire blight pathogen, Erwinia amylovora, by PCR assays with chromosomal DNA. Appl. Environ. Microbiol. 61, 2636–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertels, F. , Silander, O.K. , Pachkov, M. , Rainey, P.B. and van Nimwegen, E. (2014) Automated reconstruction of whole‐genome phylogenies from short‐sequence reads. Mol. Biol. Evol. 31, 1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland, C. , Ramsey, T.L. , Sabree, F. , Lowe, M. , Brown, K. , Kyrpides, N.C. and Hugenholtz, P. (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics, 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, P.G. and Hildebrand, P.D. (2005) Infection, carbohydrate utilization, and protein profiles of apple, pear, and raspberry isolates of Erwinia amylovora . Can. J. Plant Pathol. 27, 338–348. [Google Scholar]
- Bühlmann, A. , Dreo, T. , Rezzonico, F. , Pothier, J.F. , Smits, T.H.M. , Ravnikar, M. , Frey, J.E. and Duffy, B. (2014) Phylogeography and population structure of the biologically invasive phytopathogen Erwinia amylovora inferred using minisatellites. Environ. Microbiol. 16, 2112–2125. [DOI] [PubMed] [Google Scholar]
- Chaisson, M.J. and Tesler, G. (2012) Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics, 13, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, C.‐S. , Alexander, D.H. , Marks, P. , Klammer, A.A. , Drake, J. , Heiner, C. , Clum, A. , Copeland, A. , Huddleston, J. , Eichler, E.E. , Turner, S.W. and Korlach, J. (2013) Nonhybrid, finished microbial genome assemblies from long‐read SMRT sequencing data. Nat Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Chiou, C.S. and Jones, A.L. (1991) The analysis of plasmid‐mediated streptomycin resistance in Erwinia amylovora . Phytopathology, 81, 710–714. [Google Scholar]
- Datsenko, K.A. and Wanner, B.L. (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc. Natl. Acad. Sci. USA, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mita, S. and Siol, M. (2012) EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet. 13, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning, W. (1794) On the decay of apple trees. N.Y. Soc. Promotion. Agric. Arts Manuf. Trans. 2, 219–222. [Google Scholar]
- Dray, S. and Dufour, A.B. (2007) The ade4 package: implementing the duality diagram for ecologists. J. Stat. Software, 22, 1–20. [Google Scholar]
- Fletcher, S.W. (1931. –1933) History of fruit growing in Pennsylvania: part I, part II, and part III. Pa. State. Hortic. Assoc. Proc. 72, 1–26. [Google Scholar]
- Garrison, E. and Marth, G. (2012) Haplotype‐based variant detection from short‐read sequencing. arXiv, 1207.3907.
- Gurevich, A. , Saveliev, V. , Vyahhi, N. and Tesler, G. (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotson, J.W. (1916) Observations on fire blight in the Yakima Valley, Washington. Phytopathology, 6, 288–292. [Google Scholar]
- Hutt, W.N. (1903) Pear‐blight. Utah Agric. Exp. Stn. Bull. 85, 45–52. [Google Scholar]
- Isaac, J. (1902) The pear‐blight in California. Calif. State Hortic. Assoc. Bienn. Rep. 135, 3–14. [Google Scholar]
- Ishimaru, C. and Klos, E.J. (1984) New medium for detecting Erwinia amylovora and its use in epidemiological studies. Phytopathology, 74, 1342–1345. [Google Scholar]
- Jackson, H.S. (1910) Fire blight of pear and apple. Oreg. Agric. Exp. Stn. Circ. Bull. 7. [Google Scholar]
- Kearse, M. , Moir, R. , Wilson, A. , Stones‐Havas, S. , Cheung, M. , Sturrock, S. , Buxton, S. , Cooper, A. , Markowitz, S. , Duran, C. , Thierer, T. , Ashton, B. , Meintjes, P. and Drummond, A. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennicot, J.A. (1850) Observations on pomology. Ohio Agric. Rep. 1850, 464. [Google Scholar]
- Koczan, J.M. , McGrath, M.J. , Zhao, Y. and Sundin, G.W. (2009) Contribution of Erwinia amylovora exopolysaccharides amylovoran and levan to biofilm formation: implications in pathogenicity. Phytopathology, 99, 1237–1244. [DOI] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner, C.G. and Inouye, M. (1990) Low copy number plasmids for regulated low‐level expression of cloned genes in Escherichia coli with blue/white insert screening capability. Nucleic Acids Res. 18, 4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv, 1303.3997v1.
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. and Durbin, R. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llop, P. , Donat, V. , Rodríguez, M. , Cabrefiga, J. , Ruz, L. , Palomo, J.L. , Montesinos, E. and López, M.M. (2006) An indigenous virulent strain of Erwinia amylovora lacking the ubiquitous plasmid pEA29. Phytopathology, 96, 900–907. [DOI] [PubMed] [Google Scholar]
- Llop, P. , Cabrefiga, J. , Smits, T.H.M. , Dreo, T. , Barbé, S. , Pulawska, J. , Bultreys, A. , Blom, J. , Duffy, B. , Montesinos, E. , López, M.M. and Yang, C.‐H. (2011) Erwinia amylovora novel plasmid pEI70: complete sequence, biogeography, and role in aggressiveness in the fire blight phytopathogen. PLoS One, 6, e28651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell, J. (1826) Pear blight. N. Engl. Farmer. 5, 1–2. [Google Scholar]
- Ma, W. , Dong, F.F. , Stavrinides, J. and Guttman, D.S. (2006) Type III effector diversification via both pathoadaptation and horizontal transfer in response to a coevolutionary arms race. PLoS Genet. 2, e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnoy, M. , Martens, S. , Norelli, J.L. , Barny, M.‐A. , Sundin, G.W. , Smits, T.H.M. and Duffy, B. (2012) Fire blight: applied genomic insights of the pathogen and host. Annu. Rev. Phytopathol. 50, 475–494. [DOI] [PubMed] [Google Scholar]
- Mann, R.A. , Smits, T.H.M. , Bühlmann, A. , Blom, J. , Goesmann, A. , Frey, J.E. , Plummer, K.M. , Beer, S.V. , Luck, J. , Duffy, B. and Rodoni, B. (2013) Comparative genomics of 12 strains of Erwinia amylovora identifies a pan‐genome with a large conserved core. PLoS One, 8, e55644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann, H.C. , Rikkerink, E.H.A. , Bertels, F. , Fiers, M. , Lu, A. , Rees‐George, J. , Andersen, M.T. , Gleave, A.P. , Haubold, B. , Wohlers, M.W. , Guttman, D.S. , Wang, P.W. , Straub, C. , Vanneste, J. , Rainey, P.B. , Templeton, M.D. and Dangl, J.L. (2013) Genomic analysis of the Kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog. 9, e1003503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee, G.C. and Sundin, G.W. (2012) Erwinia amylovora CRISPR elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS One, 7, e41706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee, G.C. , Guasco, J. , Bellomo, L.M. , Blumer‐Schuette, S.E. , Shane, W.W. , Irish‐Brown, A. and Sundin, G.W. (2011) Genetic analysis of streptomycin‐resistant (Sm(R)) strains of Erwinia amylovora suggests that dissemination of two genotypes is responsible for the current distribution of Sm(R) E. amylovora in Michigan. Phytopathology, 101, 182–191. [DOI] [PubMed] [Google Scholar]
- McGhee, G.C. , Schnabel, E.L. , Maxson‐Stein, K. , Jones, B. , Stromberg, V.K. , Lacy, G.H. and Jones, A.L. (2002) Relatedness of chromosomal and plasmid DNAs of Erwinia pyrifoliae and Erwinia amylovora . Appl. Environ. Microbiol. 68, 6182–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus, P.S. and Jones, A.L. (1995) Genetic fingerprinting of Erwinia amylovora strains isolated from tree‐fruit crops and Rubus spp. Phytopathology, 85, 1547–1553. [Google Scholar]
- Mhedbi‐Hajri, N. , Hajri, A. , Boureau, T. , Darrasse, A. , Durand, K. , Brin, C. , Saux, M.F.‐L. , Manceau, C. , Poussier, S. , Pruvost, O. , Lemaire, C. , Jacques, M.‐A. and Crandall, K.A. (2013) Evolutionary history of the plant pathogenic bacterium Xanthomonas axonopodis . PLoS One, 8, e58474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung, I.‐S. , Lee, J.‐Y. , Yun, M.‐J. , Lee, Y.‐H. , Lee, Y.‐K. , Park, D.‐H. and Oh, C.‐S. (2016) Fire blight of apple, caused by Erwinia amylovora, a new disease in Korea. Plant Dis. 100, 1774. [Google Scholar]
- Norelli, J.L. , Aldwinckle, H.S. and Beer, S.V. (1984) Differential host × pathogen interactions among cultivars of apple and strains of Erwinia amylovora . Phytopathology, 74, 136–139. [Google Scholar]
- Norelli, J.L. , Jones, A.L. and Aldwinckle, H.S. (2003) Fire blight management in the twenty‐first century: using new technologies that enhance host resistance in apple. Plant Dis. 87, 756–765. [DOI] [PubMed] [Google Scholar]
- Paradis, E. , Claude, J. and Strimmer, K. (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics, 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Potnis, N. , Krasileva, K. , Chow, V. , Almeida, N.F. , Patil, P.B. , Ryan, R.P. , Sharlach, M. , Behlau, F. , Dow, J.M. , Momol, M.T. , White, F.F. , Preston, J.F. , Vinatzer, B.A. , Koebnik, R. , Setubal, J.C. , Norman, D.J. , Staskawicz, B.J. and Jones, J.B. (2011) Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics, 12, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potnis, N. , Timilsina, S. , Strayer, A. , Shantharaj, D. , Barak, J.D. , Paret, M.L. , Vallad, G.E. and Jones, J.B. (2015) Bacterial spot of tomato and pepper: diverse Xanthomonas species with a wide variety of virulence factors posing a worldwide challenge. Mol. Plant Pathol. 16, 907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reies, S.M. and Otterbacher, A.G. (1977) Occurrence of fire blight on thornless blackberry in Illinois. Plant Dis. Rep. 61, 232–235. [Google Scholar]
- Rezzonico, F. , Smits, T.H. and Duffy, B. (2011) Diversity, evolution, and functionality of clustered regularly interspaced short palindromic repeat (CRISPR) regions in the fire blight pathogen Erwinia amylovora . Appl. Environ. Microbiol. 77, 3819–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhouma, A. , Helali, F. , Chettaoui, M. , Hajjouji, M. and Hajlaoui, M.R. (2014) First report of fire blight caused by Erwinia amylovora on pear in Tunisia. Plant Dis. 98, 158. [DOI] [PubMed] [Google Scholar]
- Ritchie, D.R. and Klos, E.J. (1977) Isolation of Erwinia amylovora bacteriophage from the aerial parts of apple trees. Phytopathology, 67, 101–104. [Google Scholar]
- Roberts, R.J. , Vincze, T. , Posfai, J. and Macelis, D. (2015) REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S. (1989) Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6, 526–538. [DOI] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. and Eliceiri, K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebaihia, M. , Bocsanczy, A.M. , Biehl, B.S. , Quail, M.A. , Perna, N.T. , Glasner, J.D. , DeClerck, G.A. , Cartinhour, S. , Schneider, D.J. , Bentley, S.D. , Parkhill, J. and Beer, S.V. (2010) Complete genome sequence of the plant pathogen Erwinia amylovora strain ATCC 49946. J. Bacteriol. 192, 2020–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits, T.H. , Guerrero‐Prieto, V.M. , Hernandez‐Escarcega, G. , Blom, J. , Goesmann, A. , Rezzonico, F. , Duffy, B. , and Stockwell, V.O. (2014) Whole‐genome sequencing of Erwinia amylovora strains from Mexico detects single nucleotide polymorphisms in rpsL conferring streptomycin resistance and in the avrRpt2 effector altering host interactions. Genome Announc. 2, e01229–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits, T.H. , Rezzonico, F. , Kamber, T. , Blom, J. , Goesmann, A. , Frey, J.E. , and Duffy, B. (2010) Complete genome sequence of the fire blight pathogen Erwinia amylovora CFBP 1430 and comparison to other Erwinia spp. Mol. Plant Microbe Interact. 23, 384–393. [DOI] [PubMed] [Google Scholar]
- Soukainen, M. , Santala, J. and Tegel, J. (2015) First report of Erwinia amylovora, the causal agent of fire blight, on pear in Finland. Plant Dis. 99, 1033. [Google Scholar]
- Stukenbrock, E.H. and McDonald, B.A. (2008) The origins of plant pathogens in agro‐ecosystems. Annu. Rev. Phytopathol. 46, 75–100. [DOI] [PubMed] [Google Scholar]
- Sun, X. , Zhulin, I. and Wartell, R.M. (2002) Predicted structure and phyletic distribution of the RNA‐binding protein Hfq. Nucleic Acids Res. 30, 3662–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. and Nei, M. (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. [DOI] [PubMed] [Google Scholar]
- Wang, D. , Korban, S.S. and Zhao, Y. (2010) Molecular signature of differential virulence in natural isolates of Erwinia amylovora . Phytopathology, 100, 192–198. [DOI] [PubMed] [Google Scholar]
- Watterson, G.A. (1975) On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276. [DOI] [PubMed] [Google Scholar]
- Wicker, E. , Lefeuvre, P. , de Cambiaire, J.C. , Lemaire, C. , Poussier, S. and Prior, P. (2012) Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 6, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, S. , Liu, H. , Mohr, T.J. , Jenrette, J. , Chiodini, R. , Zaccardelli, M. , Setubal, J.C. and Vinatzer, B.A. (2008) Role of recombination in the evolution of the model plant pathogen Pseudomonas syringae pv. tomato DC3000, a very atypical tomato strain. Appl. Environ. Microbiol. 74, 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Q. and Sundin, G.W. (2014) Genome‐wide identification of Hfq‐regulated small RNAs in the fire blight pathogen Erwinia amylovora discovered small RNAs with virulence regulatory function. BMC Genomics, 15, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Q. , McNally, R.R. and Sundin, G.W. (2013) Global small RNA chaperone Hfq and regulatory small RNAs are important virulence regulators in Erwinia amylovora . J. Bacteriol. 195, 1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Q. , Wang, J. , Bertels, F. , Giordano, P. , Chilvers, M.I. , Huntley, R.B , Vargas, J. M. , Sundin, G. W. , Jacobs, J. L. and Yang, C. H. (2017) Recombination of virulence genes in divergent Acidovorax avenae strains that infect a common host. Mol. Plant–Microbe Interact. 30, 813–828. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. and Qi, M. (2011) Comparative genomics of Erwinia amylovora and related Erwinia species—what do we learn? Genes (Basel), 2, 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Blumer, S.E. and Sundin, G.W. (2005) Identification of Erwinia amylovora genes induced during infection of immature pear tissue. J. Bacteriol. 187, 8088–8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Sundin, G.W. and Wang, D. (2009) Construction and analysis of pathogenicity island deletion mutants of Erwinia amylovora . Can. J. Microbiol. 55, 457–464. [DOI] [PubMed] [Google Scholar]
- van der Zwet, T. , Orolaza‐Halbrendt, N. and Zeller, W. (2012) Fire Blight History, Biology, and Management. St Paul, MN: American Phytopathological Society Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Pairwise comparison of the number of single nucleotide polymorphisms (SNPs) of the 30 genomes. SNPs were identified from the whole‐genome alignment by the Egglib package. Heat map was generated by Plotly.
Fig. S2 MAUVE alignment of six complete genomes of Erwinia amylovora. Conserved regions of the genome are represented by coloured boxes. Coloured lines suggest translocations. Same coloured blocks on opposite sides of the line suggest inversions.
Fig. S3 (A) Dotplot of CTBT3‐1 and CTBT1‐1 produced by MUMmer. (B) Single nucleotide polymorphisms (SNPs) identified between CTBT3‐1 and CTBT1‐1 by SNP calling. The upper parts of the figure show the sequence variations in CTBT3‐1, whereas the lower parts show the mapped reads of CTBT1‐1. The image was generated using IGV version 2.3.81.
Fig. S4 Amylovoran production (A) and swarming motility (B) of CTBT3‐1, CTBT1‐1, CTBT3‐1Δhfq and CTBT1‐1Δhfq C‐hfq‐16R. Amylovoran production and swarming motility were tested using the same protocols as described previously (Zeng et al., 2013). Amylovoran production was measured at 72 h post‐inoculation in MBMA medium (3 g KH2PO4, 7 g K2HPO4, 1 g [NH4]2SO4, 2 ml glycerol, 0.5 g citric acid, 0.03 g MgSO4; amended with 1% sorbitol). The amount of amylovoran was quantified by measurement of the turbidity [optical density at 600 nm (OD600)] after the addition of cetylpyrimidinium chloride (CPC) to the supernatant of MBMA medium. Statistical analysis was performed using the one‐way analysis of variance (ANOVA) model in the ‘stats’ package in R. For motility assay, bacterial cells were inoculated at the centre of swarming agar plates (0.3% agar). The inoculated plates were incubated at 28 °C at high humidity. Photographs were taken at 20 h post‐inoculation.
Table S1 Recombination events between strains of different clades, detected by Geneconv.
Table S2 DNA oligonucleotide sequences used in this study.