

Summary
PecS is one of the major global regulators controlling the virulence of Dickeya dadantii, a broad‐host‐range phytopathogenic bacterium causing soft rot on several plant families. To define the PecS regulon during plant colonization, we analysed the global transcriptome profiles in wild‐type and pecS mutant strains during the early colonization of the leaf surfaces and in leaf tissue just before the onset of symptoms, and found that the PecS regulon consists of more than 600 genes. About one‐half of these genes are down‐regulated in the pecS mutant; therefore, PecS has both positive and negative regulatory roles that may be direct or indirect. Indeed, PecS also controls the regulation of a few dozen regulatory genes, demonstrating that this global regulator is at or near the top of a major regulatory cascade governing adaptation to growth in planta. Notably, PecS acts mainly at the very beginning of infection, not only to prevent virulence gene induction, but also playing an active role in the adaptation of the bacterium to the epiphytic habitat. Comparison of the patterns of gene expression inside leaf tissues and during early colonization of leaf surfaces in the wild‐type bacterium revealed 637 genes modulated between these two environments. More than 40% of these modulated genes are part of the PecS regulon, emphasizing the prominent role of PecS during plant colonization.
Keywords: Arabidopsis, Dickeya dadantii, epiphytic colonization, in planta transcriptome, regulatory networks, T6SS, virulence
Introduction
Gene regulation plays a prominent role in enabling plant‐pathogenic bacteria to respond to environmental changes during different stages of their life cycle, both inside and outside their hosts. Sophisticated regulatory networks coordinate pathogen gene expression to optimize energy conservation, disease development, host defence evasion and pathogen dispersal. These regulatory networks rely on global regulators and cell‐to‐cell communication systems (Mole et al., 2007).
Dickeya dadantii is a broad‐host‐range phytopathogenic bacterium responsible for soft rot disease on many crops and ornamentals, including Brassicaceae (Golkhandan et al., 2016; Parkinson et al., 2009). Pathogen spread is mainly a result of wounds caused by field or planting operations, insects, splashing rain or infested water pooled in plant leaf whorls (Pérombelon, 2002). Often, after penetration into its host plant, D. dadantii resides latently in the plant intercellular spaces without provoking any symptoms, and disease occurs only when environmental conditions are favourable for massive bacterial multiplication and the production of cell wall‐degrading enzymes (Pérombelon, 2002). Soft rot symptoms are mainly caused by the production and secretion of plant cell wall‐degrading enzymes. These include several pectinases and a cellulase secreted by the Out type II secretion system (T2SS), together with four proteases secreted by the Prt type I secretion system (T1SS) (Barras et al., 1994; Charkowski et al., 2012). Like many other Gram‐negative pathogenic bacteria, D. dadantii also possesses an Hrp type III secretion system (T3SS) that has been shown to play a role in pathogenesis at both low infection inoculum (Bauer et al., 1994, 1995) and in semi‐tolerant Saintpaulia plants (Yang et al., 2002). Numerous other factors, such as surface polysaccharides [lipopolysaccharides (LPS) and extracellular polysaccharides (EPS)], adhesins and efficient iron uptake systems are, however, essential in the first stages of plant–bacterium interactions leading to efficient plant colonization and, ultimately, disease expression (Barras et al., 1994; Charkowski et al., 2012). Dickeya dadantii is also able to cope with the chemical and environmental stresses encountered in planta, and to escape from infection‐induced plant defence responses by the accumulation of osmoprotectants (Gouesbet et al., 1996) or antioxidants (Reverchon et al., 2002), and by the production of several factors involved in the detoxification of antibacterial compounds or cellular repair (El Hassouni et al., 1999; Lopez‐Solanilla et al., 1998).
Complex regulatory networks, including several global regulators, tightly control this shift from latent infection to disease expression (Charkowski et al., 2012; Reverchon and Nasser, 2013). The response to three of these regulators—GacA, MfbR and PecS—has been analysed in planta by quantitative polymerase chain reaction (qPCR) analysis (Lebeau et al., 2008; Mhedbi‐Hajri et al., 2011; Reverchon et al., 2010). The PecS major regulator is striking as it belongs to a horizontally acquired genomic island present exclusively in the Dickeya genus and some members of the related soft rot genus Pectobacterium (Deochand et al., 2016). Yet, PecS plays a prominent role in the regulation of virulence, specifically in Dickeya, even though many of the genes it controls are widespread in all soft rot enterobacteria. Indeed, as revealed by phenotypic and gene expression analyses, PecS negatively controls the production of pectinases and cellulase, the antioxidant molecule indigoidine and the harpin HrpN, as well as motility, both in culture and in planta (Mhedbi‐Hajri et al., 2011; Reverchon et al., 1994). Genome‐wide characterization of the in‐culture PecS regulon broadens the number of PecS target genes to 134 under these artificial conditions (Hommais et al., 2008). On various host plants, pecS mutants induce symptoms more rapidly and with more efficiency than the wild‐type strain (Hommais et al., 2008; Mhedbi‐Hajri et al., 2011).
Taken together, these results suggest that PecS has very broad effects on D. dadantii transcription, and that it plays a particularly important role during plant infection and soft rot disease development. Transcriptome analysis of bacterial pathogens provides a powerful approach for the identification and analysis of the patterns of genes that are differentially expressed during infection, with whole‐genome analyses giving a broad picture of adaptation to the host. Recent reports have presented such in planta analyses for various phytopathogenic bacteria (Chapelle et al., 2015; Jacobs et al., 2012; Soto‐Suarez et al., 2010; Yu et al., 2013), and whole‐genome analyses have deciphered the involvement of global regulators known to be important in plant–bacteria interactions in Pectobacterium atrosepticum and Pseudomonas syringae (Liu et al., 2008; Yu et al., 2014).
To define the PecS regulon in the natural context of an infected host plant, we compared the whole‐genome expression profiles of D. dadantii and a pecS mutant strain at both an early stage of leaf colonization and at the onset of soft rot symptoms. This analysis extended the PecS regulon to about 13% of the D. dadantii gene content. This new PecS regulon comprises genes encoding the previously identified virulence factors as well as new targets, such as genes involved in primary metabolism and other regulators. It includes both positively and negatively regulated genes. Definition of the genes modulated during the switch from epiphytic to mesophyll colonizing state in the wild‐type strain confirmed the major role of PecS in the control of infection.
Results and Discussion
Comparison of D. dadantii gene expression profiles in the wild‐type strain and pecS mutant during plant infection
The expression of D. dadantii virulence genes during plant infection has been mainly analysed after inoculation of the model plant Arabidopsis thaliana (Lebeau et al., 2008; Mhedbi‐Hajri et al., 2011; Reverchon et al., 2010). For our infection kinetics, we used a non‐invasive inoculation method by briefly immersing 6‐week‐old Arabidopsis plants into a D. dadantii bacterial suspension in the presence of a surfactant, as described previously (Chapelle et al., 2015; Lebeau et al., 2008). Under these conditions, the first maceration spotty symptoms appeared at about 30 h post‐inoculation (hpi). Microscopic analysis using green fluorescent protein (GFP)‐tagged D. dadantii revealed that, at 8 hpi, D. dadantii bacteria are in deep association with plant tissue, either deeply nestled at the junctions of epidermal cells or in the apoplast between parenchyma cells (Chapelle et al, 2015). We chose to compare the transcriptomic profiles of D. dadantii wild‐type strain and the A4476 pecS mutant at two early stages of the infection process: epiphytic colonization of the leaf surface (6 hpi) and leaf invasion just before the onset of visible symptoms (24 hpi, hereafter referred to as leaf‐infecting bacteria). The comparison of the in planta growth rate of the bacteria during these first stages of infection confirmed that the pecS mutant multiplies as well as the wild‐type strain on leaves (Fig. S1, see Supporting Information). RNA purification was achieved by taking advantage of the recent development of a protocol allowing the isolation of bacteria from infected plant tissues (Chapelle et al., 2015). We measured gene transcription by cDNA hybridization on separate coding sequence (CDS) pangenomic microarrays, and identified genes that were differentially expressed (i) in the wild‐type versus the pecS mutant strain and (ii) at 6 hpi versus 24 hpi, using the ANAIS INRA program (Simon and Biot, 2010). When comparing the expression profiles of wild‐type and pecS bacteria collected from leaf surfaces at 6 hpi, 997 genes were found to be differentially expressed with a false discovery rate (FDR) of 0.05, 575 of which (306 up‐regulated and 269 down‐regulated) exhibited a fold change (FC) difference of two or more. In contrast, only 137 genes (56 up‐regulated and 81 down‐regulated) were differentially expressed (FDR limit = 0.05, FC cut‐off ≥ 2) in pecS versus wild‐type bacteria collected from inside the leaves at 24 hpi. Our transcriptomic analysis identified all genes previously shown to be under PecS regulation in planta by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR)‐based expression analysis, providing a reassuring internal control (Mhedbi‐Hajri et al., 2011). To validate the microarray data further, the activation of eight genes covering the range of activation levels was checked by qRT‐PCR on total RNA of infected plants (Fig. S2, see Supporting Information).
Characterization of the D. dadantii PecS regulon during epiphytic colonization of leaves
We manually classified into functional categories the 575 genes differentially expressed at 6 hpi between the pecS mutant and the wild‐type parent (Table S1, see Supporting Information), and the relative distribution of these categories is presented in Fig. 1. For more than one‐third of the differentially expressed genes (209 genes, 36%), no precise function could be assigned, and two‐thirds of these were down‐regulated. The other highly represented categories comprised genes involved in protein secretion and secreted proteins, metabolism, transport and regulation. Genes whose modulation of expression might be relevant for interactions with plants are presented in Table 1 and are discussed below.
Figure 1.
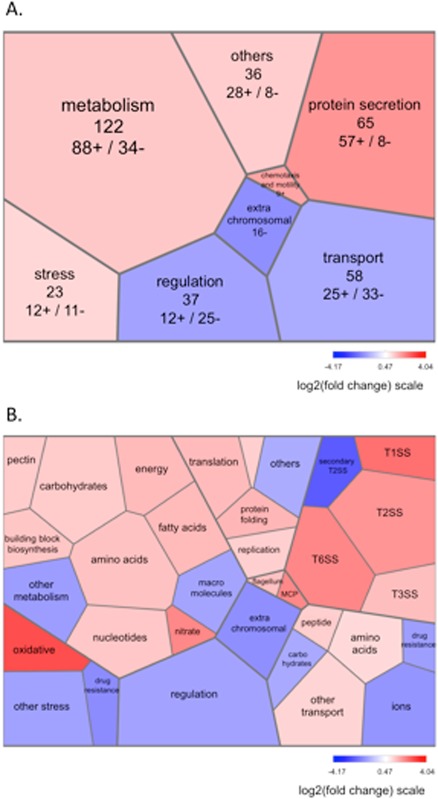
Tree maps of differentially expressed functional gene clusters in the pecS mutant during epiphytic leaf colonization. Gene expression changes in the pecS mutant relative to the wild‐type parent at 6 h post‐inoculation (hpi) on the leaf surface are visualized by Voronoi tree maps. The mean ratios of the expression data in the different gene clusters (on a log2 scale) are colour coded using a divergent colour gradient. The blue and red tiles represent down‐ and up‐regulated gene clusters, respectively. Two levels of classification are displayed (A, B) according to our custom functional classification (corresponding to category A/subcategory B in Table S1, see Supporting Information). In (A), for each category, the total number of modulated genes is indicated, and the numbers of up (+) and down‐regulated (–) genes are provided. MCP, methyl‐accepting chemotaxis protein; T1SS, type I secretion system; T2SS, type II secretion system; T3SS, type III secretion system; T6SS, type VI secretion system.
Table 1.
A subset of genes for which expression is dependent on PecS during epiphytic leaf colonization on leaf surfaces at 6 h post‐inoculation (hpi).
| Gene ID | Gene product description | FC pecS/WT at 6 hpi | P | FC WT 24 hpi/6 hpi | P | |||
|---|---|---|---|---|---|---|---|---|
| Secreted protein and secretion systems | ||||||||
| T1SS | ||||||||
| inh | ABF‐0017125 | Protease inhibitor | Protein secretion | T1SS | 6.31 | 1.84E‐05 | 6.58 | 2.40E‐07 |
| prtA | ABF‐0020373 | Secreted protease | Protein secretion | T1SS | 11.1 | 2.07E‐02 | 13.61 | 3.27E‐06 |
| prtB | ABF‐0047107 | Secreted protease | Protein secretion | T1SS | 15.92 | 4.86E‐04 | 10.32 | 1.33E‐04 |
| prtC | ABF‐0020371 | Secreted protease | Protein secretion | T1SS | 2.85 | 1.20E‐03 | 3.17 | 4.54E‐04 |
| prtD | ABF‐0017126 | Alkaline protease secretion ATP‐binding protein | Protein secretion | T1SS | 5.27 | 7.75E‐03 | 5.91 | 9.73E‐07 |
| prtE | ABF‐0017127 | Protease secretion protein | Protein secretion | T1SS | 5.58 | 3.17E‐05 | 7.39 | 4.15E‐05 |
| prtF | ABF‐0017128 | Protease secretion protein | Protein secretion | T1SS | 4.51 | 5.63E‐03 | 5.75 | 2.17E‐05 |
| prtG | ABF‐0017123 | Secreted protease | Protein secretion | T1SS | 4.96 | 3.92E‐04 | 6.37 | 9.48E‐05 |
| T2SS | ||||||||
| avrL | ABF‐0019143 | Avirulence protein | Protein secretion | T2SS | 7.46 | 1.71E‐02 | 6.87 | 3.22E‐04 |
| avrM | ABF‐0015381 | Avirulence protein | Protein secretion | T2SS | 11.75 | 1.20E‐03 | 7.80 | 9.48E‐05 |
| celZ | ABF‐0018772 | Secreted cellulase | Protein secretion | T2SS | 2.55 | 5.55E‐03 | 4.65 | 1.53E‐08 |
| nip | ABF‐0015387 | Necrosis‐inducing protein | Protein secretion | T2SS | 3.23 | 1.28E‐02 | 3.03 | 2.78E‐04 |
| outD | ABF‐0018230 | T2SS component | Protein secretion | T2SS | 3.89 | 1.07E‐04 | 4.33 | 4.15E‐05 |
| outE | ABF‐0018231 | T2SS component | Protein secretion | T2SS | 3.08 | 3.07E‐04 | 3.55 | 7.86E‐05 |
| outG | ABF‐0018233 | T2SS component | Protein secretion | T2SS | 3.62 | 1.01E‐04 | 3.86 | 1.82E‐04 |
| outH | ABF‐0018234 | T2SS component | Protein secretion | T2SS | 3.01 | 2.00E‐03 | 3.16 | 3.09E‐03 |
| outI | ABF‐0018235 | T2SS component | Protein secretion | T2SS | 3.32 | 6.50E‐04 | 2.79 | 1.93E‐03 |
| outJ | ABF‐0018236 | T2SS component | Protein secretion | T2SS | 2.55 | 1.92E‐04 | 2.75 | 1.34E‐03 |
| outK | ABF‐0018237 | T2SS component | Protein secretion | T2SS | 2.29 | 1.22E‐03 | 2.33 | 6.06E‐03 |
| outL | ABF‐0018238 | T2SS component | Protein secretion | T2SS | 2.71 | 9.14E‐06 | 3.53 | 1.96E‐05 |
| outM | ABF‐0018239 | T2SS component | Protein secretion | T2SS | 3.83 | 1.95E‐05 | 3.80 | 4.15E‐05 |
| paeY | ABF‐0019649 | Pectin acetylesterase | Protein secretion | T2SS | 5.44 | 2.92E‐03 | 11.79 | 6.99E‐04 |
| pelA | ABF‐0019643 | Pectate lyase | Protein secretion | T2SS | 9.51 | 6.83E‐03 | ||
| pelB | ABF‐0020840 | Pectate lyase | Protein secretion | T2SS | 4.62 | 3.59E‐04 | 6.56 | 1.39E‐04 |
| pelC | ABF‐0020837 | Pectate lyase | Protein secretion | T2SS | 2.32 | 8.70E‐03 | 4.01 | 1.90E‐03 |
| pelD | ABF‐0019648 | Pectate lyase | Protein secretion | T2SS | 4.94 | 1.02E‐02 | 8.03 | 7.32E‐04 |
| pelI | ABF‐0014586 | Pectate lyase | Protein secretion | T2SS | 5.28 | 1.38E‐03 | 22.71 | 1.16E‐05 |
| pelL | ABF‐0018773 | Pectate lyase | Protein secretion | T2SS | 3.4 | 4.44E‐02 | 3.25 | 3.06E‐02 |
| pelN | ABF‐0019391 | Pectate lyase | Protein secretion | T2SS | 4.29 | 1.42E‐05 | 4.70 | 7.74E‐05 |
| pelZ | ABF‐0020835 | Pectate lyase | Protein secretion | T2SS | 11.18 | 4.45E‐06 | 9.88 | 1.09E‐04 |
| pem | ABF‐0019650 | Pectin esterase | Protein secretion | T2SS | 4.49 | 5.57E‐06 | 9.65 | 1.31E‐06 |
| Secondary T2SS Stt | ||||||||
| pnlH | ABF‐0020720 | Pectin lyase | Protein secretion | Secondary T2SS | −4.91 | 5.99E‐03 | −2.90 | 1.85E‐02 |
| sttD | ABF‐0020342 | Secondary T2SS component | Protein secretion | Secondary T2SS | −5.47 | 5.23E‐05 | −3.01 | 5.25E‐04 |
| sttE | ABF‐0047167 | Secondary T2SS component | Protein secretion | Secondary T2SS | −4.77 | 1.27E‐05 | −3.35 | 8.78E‐03 |
| sttF | ABF‐0020728 | Secondary T2SS component | Protein secretion | Secondary T2SS | −4.95 | 2.68E‐03 | −3.53 | 1.13E‐03 |
| sttG | ABF‐0020727 | Secondary T2SS component | Protein secretion | Secondary T2SS | −5.84 | 1.58E‐03 | −4.54 | 1.01E‐02 |
| sttI | ABF‐0020726 | Secondary T2SS component | Protein secretion | Secondary T2SS | −11.42 | 2.06E‐03 | −8.23 | 1.61E‐02 |
| sttL | ABF‐0020723 | Secondary T2SS component | Protein secretion | Secondary T2SS | −8.26 | 1.34E‐03 | −6.71 | 3.24E‐02 |
| T3SS | ||||||||
| dspE | ABF‐0019012 | T3SS secreted effector | Protein secretion | T3SS | 4.09 | 4.67E‐03 | 2.34 | 1.52E‐02 |
| dspF | ABF‐0019013 | DspE chaperone | Protein secretion | T3SS | 2.30 | 4.60E‐03 | ||
| hrcQ | ABF‐0015585 | T3SS component | Protein secretion | T3SS | 2.28 | 7.64E‐03 | ||
| hrpG | ABF‐0020865 | T3SS component | Protein secretion | T3SS | 2.21 | 7.23E‐08 | 2.30 | 2.89E‐06 |
| hrpN | ABF‐0020784 | Harpin, T3SS secreted | Protein secretion | T3SS | 5.12 | 4.19E‐04 | 3.24 | 3.39E‐03 |
| hrpV | ABF‐0020783 | T3SS component | Protein secretion | T3SS | 2.63 | 2.60E‐04 | ||
| hrpW | ABF‐0019009 | T3SS secreted protein | Protein secretion | T3SS | 3.59 | 2.11E‐03 | ||
| ABF‐0019006 | Potential HrpW‐specific chaperone | Protein secretion | T3SS | 3.11 | 4.64E‐03 | |||
| T4SS | ||||||||
| virB1 | ABF‐0018723 | T4SS component | Protein secretion | T4SS | −2.83 | 5.92E‐03 | −2.06 | 4.79E‐02 |
| T6SS | ||||||||
| clpB | ABF‐0015858 | ClpB protein, T6SS | Protein secretion | T6SS | 4.38 | 3.25E‐03 | ||
| hcp | ABF‐0015593 | Hcp protein, external part of T6SS apparatus | Protein secretion | T6SS | 3.29 | 3.92E‐02 | 2.35 | 4.43E‐02 |
| hcpA | ABF‐0017270 | Hcp protein, external part of T6SS apparatus | Protein secretion | T6SS | 4.85 | 5.19E‐04 | ||
| hcpB | ABF‐0018743 | Hcp protein, external part of T6SS apparatus | Protein secretion | T6SS | 7.42 | 3.58E‐06 | ||
| hcpC | ABF‐0015893 | Hcp protein, external part of T6SS apparatus | Protein secretion | T6SS | 4.65 | 3.29E‐04 | ||
| icmF | ABF‐0015854 | T6SS component | Protein secretion | T6SS | 3.52 | 4.29E‐06 | ||
| impA | ABF‐0015853 | T6SS component | Protein secretion | T6SS | 3.82 | 1.20E‐03 | ||
| impB | ABF‐0015869 | T6SS component | Protein secretion | T6SS | 4.46 | 2.53E‐02 | 2.33 | 4.21E‐02 |
| impC | ABF‐0015868 | T6SS component | Protein secretion | T6SS | 5.35 | 5.48E‐05 | 2.77 | 1.22E‐03 |
| impG | ABF‐0015865 | T6SS component | Protein secretion | T6SS | 5.05 | 2.35E‐06 | ||
| impH | ABF‐0015864 | T6SS component | Protein secretion | T6SS | 2.92 | 9.06E‐06 | ||
| impI | ABF‐0015862 | T6SS component | Protein secretion | T6SS | 3.67 | 1.79E‐03 | 2.40 | 2.03E‐04 |
| impJ | ABF‐0015860 | T6SS component | Protein secretion | T6SS | 4.1 | 6.03E‐06 | 2.58 | 1.46E‐04 |
| impK | ABF‐0015859 | T6SS component | Protein secretion | T6SS | 4.76 | 5.64E‐06 | ||
| sciN | ABF‐0015861 | T6SS component | Protein secretion | T6SS | 4.14 | 2.90E‐04 | 2.71 | 4.93E‐03 |
| vgrGA | ABF‐0017269 | T6SS component | Protein secretion | T6SS | 2.96 | 2.27E‐02 | ||
| ABF‐0015866 | T6SS component | Protein secretion | T6SS | 5.16 | 1.06E‐04 | 2.85 | 4.09E‐03 | |
| Secreted proteins | ||||||||
| virK | ABF‐0020481 | VirK protein | Others | 3.71 | 6.48E‐06 | 2.47 | 1.06E‐04 | |
| yoaJ | ABF‐0014642 | Endoglucanase | 2.92 | 3.15E‐04 | 8.36 | 7.74E‐05 | ||
| Metabolism and transport | ||||||||
| Genes involved in pectin metabolism and transport | ||||||||
| kdgA | ABF‐0019960 | 2‐Dehydro‐3‐deoxyphosphogluconate aldolase | Metabolism | Pectin | 3.52 | 1.31E‐04 | 3.49 | 1.09E‐04 |
| kdgF | ABF‐0014524 | Pectin degradation protein | Metabolism | Pectin | 3.2 | 3.49E‐02 | ||
| kduD | ABF‐0047127 | 2‐Deoxy‐d‐gluconate 3‐dehydrogenase | Metabolism | Carbohydrates | 3.67 | 4.22E‐02 | ||
| kduI | ABF‐0020174 | 4‐Deoxy‐l‐threo‐5‐hexosulose‐uronate ketol‐isomerase | Metabolism | Carbohydrates | 3.89 | 3.31E‐02 | ||
| ogl | ABF‐0020118 | Oligogalacturonate lyase | Metabolism | Pectin | 3.9 | 2.72E‐03 | 2.67 | 2.55E‐02 |
| pehK | ABF‐0014876 | Polygalacturonase | Metabolism | Pectin | −2.01 | 3.25E‐02 | ||
| pehN | ABF‐0020789 | Exo‐poly‐alpha‐d‐galacturonosidase | Metabolism | Pectin | −2.84 | 1.16E‐04 | ||
| pel | ABF‐0019932 | Pectate lyase | Metabolism | Pectin | −2.38 | 3.47E‐03 | ||
| pelW | ABF‐0019626 | Exopolygalacturonate lyase | Metabolism | Pectin | 2.34 | 1.57E‐04 | 3.38 | 2.36E‐03 |
| togT | ABF‐0016394 | Oligogalacturonide transporter | Transport | Carbohydrates | 2.24 | 3.56E‐04 | ||
| Glycolysis and acetate dissimilation | ||||||||
| aceE | ABF‐0019243 | Pyruvate dehydrogenase, decarboxylase component E1 | Metabolism | Energy | 2.11 | 3.06E‐04 | ||
| aceF | ABF‐0019241 | Pyruvate dehydrogenase, dihydrolipoyltransacetylase component E2 | Metabolism | Energy | 4.2 | 1.28E‐03 | ||
| ackA | ABF‐0020586 | Acetate kinase A and propionate kinase 2 | Metabolism | Carbohydrates | 2.08 | 2.31E‐03 | ||
| actP | ABF‐0018003 | Acetate permease | Transport | −2.74 | 3.34E‐03 | 2.77 | 3.82E‐02 | |
| fumA | ABF‐0016800 | Fumarate hydratase class I | Metabolism | Energy | 2.67 | 6.01E‐03 | ||
| mqo | ABF‐0015393 | Malate:quinone oxidoreductase | Metabolism | Energy | 3.63 | 1.50E‐03 | ||
| pgi | ABF‐0017827 | Glucose‐6‐phosphate isomerase | Metabolism | Carbohydrates | 2.88 | 2.42E‐05 | ||
| pps | ABF‐0019893 | Phosphoenolpyruvate synthase | Metabolism | −4.57 | 3.01E‐03 | |||
| pta | ABF‐0020585 | Phosphate acetyltransferase | 2.09 | 7.11E‐03 | ||||
| ptsG | ABF‐0017988 | PTS system glucose‐specific IICB component | Transport | Carbohydrates | 8.86 | 3.28E‐02 | ||
| pykA | ABF‐0019963 | Pyruvate kinase II | Metabolism | Carbohydrates | 4.57 | 2.59E‐04 | 3.76 | 1.04E‐03 |
| Fatty acid metabolism | ||||||||
| accA | ABF‐0017473 | Acetyl‐CoA carboxylase, carboxytransferase, α subunit | Metabolism | Fatty acids | 5.48 | 4.51E‐05 | 2.98 | 8.04E‐03 |
| accB | ABF‐0020596 | Acetyl CoA carboxylase, BCCP subunit | Metabolism | Fatty acids | 4.21 | 8.15E‐06 | 2.39 | 2.82E‐02 |
| accC | ABF‐0017046 | Acetyl‐CoA carboxylase, biotin carboxylase subunit | Metabolism | Fatty acids | 5.2 | 1.76E‐02 | 2.82 | 2.27E‐02 |
| accD | ABF‐0017783 | accD|acetyl‐CoA carboxylase, β (carboxyltranferase) subunit | Metabolism | Fatty acids | 2.01 | 2.22E‐04 | 2.06 | 4.44E‐04 |
| fabF | ABF‐0017981 | 3‐Oxoacyl‐[acyl‐carrier‐protein] synthase, KASII | Metabolism | Fatty acids | 2.55 | 1.53E‐02 | 2.01 | 1.57E‐02 |
| fabG | ABF‐0047181 | 3‐Oxoacyl‐[acyl‐carrier‐protein] reductase | Metabolism | Fatty acids | 3.36 | 7.46E‐03 | ||
| fabH | ABF‐0020855 | 3‐Oxoacyl‐[acyl‐carrier‐protein] synthase III | Metabolism | Fatty acids | 2.21 | 1.66E‐03 | ||
| fabI | ABF‐0019259 | Enoyl‐[acyl‐carrier‐protein] reductase [NADH] | Metabolism | Building block biosynthesis | 2.83 | 1.25E‐03 | ||
| Nitrogen metabolism | ||||||||
| nasA | ABF‐0017402 | Assimilatory nitrate reductase large subunit (EC:1.7.99.4) | Metabolism | Nitrate | 4.29 | 3.94E‐02 | ||
| nasD | ABF‐0017399 | Nitrate ABC transporter, ATP‐binding protein | Transport | Nitrate | 3.12 | 4.71E‐02 | ||
| nasF | ABF‐0017397 | Nitrate ABC transporter, nitrate‐binding protein | Transport | Nitrate | 2.74 | 2.14E‐02 | ||
| nirB | ABF‐0017401 | Nitrite reductase [NAD(P)H] large subunit | Metabolism | Nitrate | 7.34 | 1.78E‐02 | ||
| nirE | ABF‐0017403 | Uroporphyrinogen‐III methyltransferase | Metabolism | Building block biosynthesis | 2.31 | 3.97E‐03 | ||
| Energy production | ||||||||
| atpA | ABF‐0014787 | F1 sector of membrane‐bound ATP synthase, α subunit | Metabolism | Energy | 3.26 | 9.24E‐03 | 4.59 | 3.95E‐04 |
| atpC | ABF‐0014784 | F1 sector of membrane‐bound ATP synthase, ε subunit | Metabolism | Energy | 3.07 | 8.75E‐03 | 2.54 | 6.43E‐03 |
| cyoB | ABF‐0017593 | Cytochrome o ubiquinol oxidase subunit I | Metabolism | Energy | 2.28 | 1.86E‐04 | ||
| hydN | ABF‐0016264 | Electron transport protein hydN | Metabolism | Energy | 2.78 | 1.84E‐05 | ||
| nuoG | ABF‐0017326 | NADH:ubiquinone oxidoreductase, chain G | Metabolism | Energy | 2.62 | 2.24E‐02 | ||
| nuoN | ABF‐0017335 | NADH:ubiquinone oxidoreductase, membrane subunit N | Metabolism | Energy | 3.46 | 4.78E‐02 | ||
| Other metabolic genes | ||||||||
| bglA | ABF‐0016574 | 6‐Phospho‐β‐glucosidase | Metabolism | Carbohydrates | 4.36 | 1.81E‐03 | 3.62 | 4.67E‐03 |
| bglX | ABF‐0018933 | Periplasmic β‐glucosidase | Metabolism | Carbohydrates | 3.14 | 2.70E‐06 | 2.23 | 1.06E‐02 |
| nrdD | ABF‐0016556 | Anaerobic ribonucleoside‐triphosphate reductase | Metabolism | Nucleotides | −6.15 | 6.60E‐04 | −2.17 | 4.59E‐02 |
| nrdG | ABF‐0016554 | Anaerobic ribonucleotide reductase activating protein | Metabolism | −3.31 | 3.18E‐02 | |||
| iaaH | ABF‐0016563 | Indoleacetamide hydrolase | Metabolism | −2.86 | 5.77E‐03 | −2.67 | 1.87E‐02 | |
| Chemotaxis and motility | ||||||||
| cheB | ABF‐0018751 | Chemotaxis response regulator protein‐glutamate methylesterase | Chemotaxis and motility | 2.26 | 8.13E‐03 | |||
| flgF | ABF‐0017936 | Flagellar basal‐body rod protein flgF | Chemotaxis and motility | Flagellum | 2.34 | 3.94E‐03 | 3.21 | 1.78E‐02 |
| flhE | ABF‐0018581 | Flagellar protein flhE | Chemotaxis and motility | Flagellum | 4.64 | 1.96E‐02 | 2.83 | 4.43E‐02 |
| fliH | ABF‐0017954 | Flagellar assembly protein | Chemotaxis and motility | Flagellum | 2.44 | 2.04E‐04 | 2.25 | 9.21E‐03 |
| fliI | ABF‐0017953 | Flagellum‐specific ATP synthase | Chemotaxis and motility | Flagellum | 2.54 | 5.60E‐03 | 2.19 | 2.53E‐02 |
| rhlA | ABF‐0019839 | (R)‐3‐hydroxydecanoyl‐ACP:CoA transacylase | Chemotaxis and motility | 10.16 | 3.29E‐04 | 6.75 | 8.81E‐04 | |
| ABF‐0014536 | Methyl‐accepting chemotaxis protein | Chemotaxis and motility | Methyl‐accepting chemotaxis protein | 4.36 | 1.94E‐02 | 5.01 | 7.50E‐03 | |
| ABF‐0015600 | Methyl‐accepting chemotaxis protein | Chemotaxis and motility | Methyl‐accepting chemotaxis protein | 2.54 | 1.24E‐02 | |||
| ABF‐0017674 | Methyl‐accepting chemotaxis protein | Chemotaxis and motility | Methyl‐accepting chemotaxis protein | 5.15 | 3.36E‐02 | 8.18 | 2.40E‐07 | |
| Genes involved in regulation | ||||||||
| asnC | ABF‐0014800 | DNA‐binding transcriptional dual regulator | Regulation | −4.84 | 1.27E‐04 | −4.02 | 2.57E‐03 | |
| birA | ABF‐0020875 | Bifunctional biotin‐[acetylCoA carboxylase] holoenzyme synthetase/DNA‐binding transcriptional repressor | Regulation | −2.33 | 3.78E‐04 | |||
| fruR | ABF‐0018397 | Fructose repressor FruR, LacI family | −2.21 | 1.19E‐02 | −2.63 | 7.03E‐03 | ||
| glnG | ABF‐0016307 | Nitrogen regulation protein NR(I), two‐component regulatory system | Regulation | 2.19 | 2.10E‐05 | |||
| glnL | ABF‐0016308 | Sensory histidine kinase in two‐component regulatory system with GlnG | Regulation | 2.36 | 9.58E‐05 | |||
| lexA | ABF‐0018677 | DNA‐binding transcriptional repressor of SOS regulon | Regulation | 2.35 | 5.68E‐05 | |||
| pecM | ABF‐0016087 | Signal receptor | Regulation | 2.68 | 2.48E‐02 | |||
| pecS | ABF‐0016089 | DNA‐binding transcriptional regulatory protein | Regulation | 9.05 | 7.53E‐07 | |||
| rpoS | ABF‐0020446 | RNA polymerase, sigma S (sigma 38) factor | Regulation | −2.32 | 2.73E‐02 | |||
| vfmA | ABF‐0016079 | 3‐Oxoacyl‐[acyl‐carrier‐protein] synthase, KASIII | Regulation | 8.56 | 1.91E‐03 | |||
| vfmB | ABF‐0016077 | VfmB protein | Regulation | 8.67 | 9.17E‐05 | 2.10 | 4.75E‐02 | |
| vfmC | ABF‐0016076 | Probable na+‐driven multidrug efflux pump transmembrane protein | Regulation | 4.41 | 2.07E‐03 | |||
| vfmD | ABF‐0016075 | Probable hydrolase | Regulation | 5.39 | 7.54E‐05 | |||
| Stress | ||||||||
| indA | ABF‐0016084 | Indigoidine biosynthesis protein | Stress | Oxidative | 11.98 | 4.97E‐04 | ||
| indB | ABF‐0016083 | Similar to phosphoglycolate phosphatase | Stress | Oxidative | 12.93 | 1.25E‐03 | ||
| indC | ABF‐0016081 | Indigoidine synthase | Stress | Oxidative | 16.45 | 2.54E‐06 | 2.18 | 3.24E‐03 |
| proQ | ABF‐0020257 | ProP effector | Stress | Osmotic | 2.11 | 2.31E‐03 | ||
| rsxA | ABF‐0015559 | Inner membrane subunit of SoxR‐reducing complex | Stress | Oxidative | 2.26 | 6.67E‐04 | ||
| tehB | ABF‐0016795 | Tellurite resistance protein | Stress | Drug resistance | −2.85 | 1.31E‐03 | −3.04 | 2.70E‐04 |
| ABF‐0019342 | Catalase | Stress | Oxidative | 4.6 | 2.52E‐04 | |||
FC, fold change; T1SS, type I secretion system; T2SS, type II secretion system; T3SS, type III secretion system; T4SS, type IV secretion system; T6SS, type VI secretion system; WT, wild‐type parent.
Cell wall degradation and pectin metabolism
As already reported both in vitro (Hommais et al., 2008) and during infection (Mhedbi‐Hajri et al., 2011), genes encoding cell wall‐degrading enzymes were overexpressed in the pecS mutant. These included genes encoding the four proteases secreted by the Prt T1SS and most pectinases and cellulase secreted by the Out T2SS (Table 1, Fig. 2). In contrast, the pehKN genes, encoding polyglacturonases, were slightly down‐regulated in accordance with the reported control by PecS of these genes in in vitro conditions (Hugouvieux‐Cotte‐Pattat et al., 2002). However, the expression of the periplasmic PelX pectate lyase‐ and periplasmic PehVWX polygalacturonase‐encoding genes did not appear to be modulated in the pecS mutant. Several genes involved in the transport and catabolism of pectin degradation products were also overexpressed in the pecS mutant, such as genes encoding the TogT oligogalacturonide transporter, the cytoplasmic PelW pectate lyase and Ogl oligogalacturonide lyase, and KduI, KduD and KdgA, which are enzymes that further catabolize oligogalacturonide catabolism products (Fig. 2). Most D. dadantii cell wall degradation capacities are thus negatively controlled by PecS during the epiphytic colonization of leaves.
Figure 2.
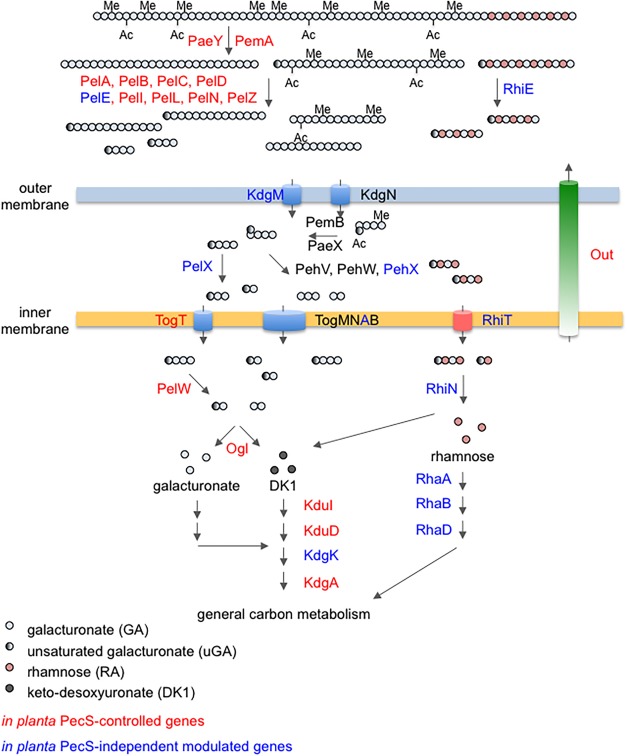
Role of PecS in the regulation of the pectin metabolic pathway during interactions with Arabidopsis. PecS‐controlled genes are indicated in red, PecS‐independent in planta modulated genes are indicated in blue. Pectinases (Pae, pectin acetylesterase; Pem, pectin methyl esterase; Pel, pectate lyase; RhiE, rhamnogalacturonate lyase) are secreted by the Out type II secretion system in the plant apoplast, where they degrade pectin. The pectin degradation products are transported into the bacterial periplasm by the KdgMN porins and further degraded by PelX and PehVWX pectinases (Peh, polygalacturonases). The resulting small oligomers are transported into the cytoplasm by Tog and RhiT transporters, and further degraded to 2‐keto‐3‐deoxygluconate (Kdg), which is metabolized to enter the general carbon metabolism (adapted from Hugouvieux‐Cotte‐Pattat et al., 2014).
Protein secretion systems
Dickeya dadantii 3937 possesses all six protein secretion systems commonly found in Gram‐negative bacteria (T1SS–T6SS) (Charkowski et al., 2012). The transcriptomic data indicated that five of the six secretion systems were differentially expressed in the pecS mutant relative to the wild‐type strain at 6 hpi (Table 1, Fig. 1B). Genes encoding the constituents of both Prt T1SS and Out T2SS were overexpressed by 2.3–5.6‐fold in the pecS mutant. As reported previously both in vitro and during infection (Hommais et al., 2008; Lebeau et al., 2008), the hrpN gene encoding a T3SS effector was also highly overexpressed in the mutant. Our data also showed the overexpression of genes encoding two other T3SS effectors—DspE and HrpW—and their cognate or putative chaperones, as well as the slight induction of three genes encoding components of the T3SS apparatus (HrcQ, HrpG, HrpV) (Table 1). Finally, all genes involved in the biosynthesis of T6SS were up‐regulated in the pecS mutant, as well as four hcp genes and one vgrG gene that encode the external constituents of the T6SS ‘needle’. However, no differences in expression were observed for the various rhs genes proposed to be T6SS effectors in D. dadantii (Koskiniemi et al., 2013). Genes encoding additional proteins shown to be part of the D. dadantii secretome or predicted by the pSORT program to be exported were also up‐regulated in the pecS mutant. These included genes encoding the two Avr‐like AvrL and AvrM proteins (Kazemi‐Pour et al., 2004) or proteins shown to be involved in virulence in other plant‐pathogenic bacteria, such as NipE, which was characterized as a necrosis‐inducing protein in Pectobacterium (Mattinen et al., 2004), the putative endoglucanase YoaJ or the Agrobacterium tumefaciens VirK protein of unknown function.
In contrast, genes encoding other protein secretion systems were down‐regulated in the pecS mutant, such as the virB1 gene encoding a component of T4SS, and the stt and pnlH genes encoding, respectively, a secondary T2SS and the pectin lyase that is exposed on the outer side of the outer membrane by the Stt system (Ferrandez and Condemine, 2008). As already largely revealed by the characterization of the in vitro PecS regulon (Hommais et al., 2008), PecS is thus a major regulator of the D. dadantii secretome at both the level of the secretion systems and for the production of the secreted proteins.
Metabolism and transport
Interestingly, in addition to genes involved in the catabolism of pectin degradation products, several genes involved in primary metabolism were also differentially expressed in the pecS mutant. Most genes involved in the conversion of glucose‐6‐phosphate to acetate were up‐regulated, whereas the ActP transporter encoding gene was down‐regulated (Table 1, Fig. 3), pointing to the control of the acetate dissimilation pathway by PecS. Similarly, genes related to energy production, such as those encoding electron transporters or some of the membrane‐anchored ATPase subunits, were differentially expressed in the mutant relative to the wild‐type strain. The nasADEFBnirE operon, encoding genes involved in NO3 transport and assimilation, was overexpressed in the mutant, as were genes involved in the fatty acid biosynthetic pathway. These included accA, accC and accD, which are involved in the initial steps of fatty acid biosynthesis, and fabB, fabI, fabH, fabD and fabG, which are involved in fatty acid elongation. Several transport systems encoding genes were also controlled by PecS, including systems predicted to transport carbohydrates, amino acids or peptides, as well as systems that might be involved in drug resistance. These data indicate that the PecS regulon extends to primary functions far beyond the genes dedicated to plant cell wall degradation and pectin metabolism.
Figure 3.
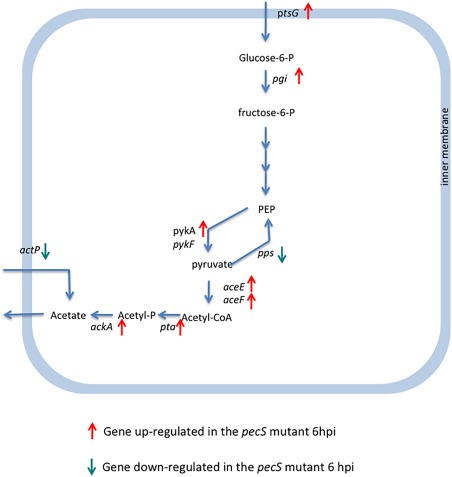
PecS‐controlled genes in glucose catabolism and acetate dissimilation pathways on leaf surfaces at 6 h post‐inoculation (hpi). Glucose is metabolized to pyruvate and acetyl‐coenzyme A (acetyl‐CoA) by the glycolytic pathway. Acetyl‐CoA is metabolized in acetate by the Pta‐AckA dissimilation pathway and is excreted. The ActP acetate permease is an active transporter that pumps acetate to the bacterial cytoplasm. Genes up‐ and down‐regulated in the pecS mutant are indicated by ascending and descending arrows, respectively.
Oxidative stress responses
The pecS mutant has been shown previously in vitro to harbour a high resistance to oxidative stress, mainly mediated by the overproduction of the reactive oxygen species (ROS) scavenger indigoidine (Reverchon et al., 2002). Consistent with this observation, the corresponding biosynthetic indABC genes were highly expressed in the pecS mutant on leaf surfaces. Apart from this biosynthetic pathway, however, only a few genes involved in oxidative stress were differentially expressed in planta between the mutant and the wild‐type strain, including genes encoding a putative catalase (ABF‐0019342), the GroL chaperone, which has been shown to be important for resistance to oxidative stress (Farr and Kogoma, 1991), and RsxA, which modulates the activity of the oxidative stress‐related SoxR regulator in Escherichia coli (Koo et al., 2003). Interestingly, these genes are different from the oxidative stress‐related genes controlled by PecS in vitro (Hommais et al., 2008). Such a situation was also observed in P. syringae, where antioxidant enzymes encoding genes expressed in vitro during oxidative stress were different from those expressed in planta (Yu et al., 2013). Intriguingly, in addition to the genes involved in the biosynthesis of the ROS scavenger indigoidine, very few oxidative stress‐related genes appear to be controlled by PecS on leaf surfaces. This may indicate either that indigoidine overproduction is sufficient to scavenge plant‐produced ROS during infection, or that D. dadantii does not recognize the plant leaf surface as an oxidatively hostile environment.
Motility and chemotaxis
PecS has been shown to repress flagellar genes and the pecS mutant is hypermotile in vitro (Hommais et al., 2008; Rouanet et al., 2004). On leaf surfaces, four genes involved in flagellum synthesis were slightly up‐regulated in the mutant at 6 hpi. As observed in vitro, on both leaf surfaces and inside the plant, the pecS mutant also highly overexpressed the rhlA gene, which encodes an enzyme which catalyses the production of a surfactant that promotes swarming motility (Hommais et al., 2008). In addition, both the chemotaxis‐regulating CheB protein‐ and three methyl‐accepting chemotaxis protein (MCP)‐encoding genes were also up‐regulated in the mutant. Together, these results indicate that PecS controls D. dadantii chemotaxis and motility during pathogen epiphytic colonization of plant leaves.
Regulators
It has been reported previously that in vitro PecS represses its own synthesis (Battesti et al., 2011) and, consistent with this finding, the truncated pecS gene present in the insertion‐derived pecS mutant is itself highly expressed at 6 hpi. The pecS mutant also overexpresses the vfmABCD operon, responsible for the biosynthesis of the signal produced by the newly identified D. dadantii quorum‐sensing system (Nasser et al., 2013). In addition, certain metabolism regulatory genes were slightly differentially expressed in the pecS mutant, such as the genes encoding the GlnGL two‐component system and the AsnC regulator, both involved in nitrogen assimilation, and the fructose metabolism‐controlling FruR repressor. PecS also slightly activates the expression of the rpoS gene. This gene encodes the s38 sigma factor, believed to prepare bacterial cells to respond to subsequent stresses (Battesti et al., 2011), such as those that D. dadantii may encounter during plant infection.
Most of the additional 35 predicted regulatory genes that were differentially expressed in the pecS mutant at 6 hpi are of unknown function, and the corresponding regulated genes and functions are unknown. It should be noted that about two‐thirds of these were down‐regulated (Fig. 1, Table S1). This indicates that PecS also positively controls, directly or indirectly, a substantial number of regulatory genes and their targets that are important for D. dadantii adaptation to the epiphytic environment encountered on leaf surfaces. This demonstrates that PecS is at or near the top of a major regulatory cascade, with many additional constituents still to be identified. Such a regulatory cascade is reminiscent of that described during potato tuber infection by the Pe. atrosepticum soft rot pathogen. Indeed, Liu et al. (2008) showed that, during tuber infection, the ExpI/R‐mediated quorum‐sensing system controlled more than 1100 genes, including genes encoding over 70 regulators. Notably, and contrary to PecS, the ExpI/R system appears to control most other global regulators known to be associated with plant cell wall‐degrading enzyme production in Pectobacterium. In contrast, in P. syringae, although the GacA/gacS regulatory system modulates about 360 genes in epiphytic bacteria, it controls only a few known global regulatory genes (Yu et al., 2014).
Comparison of D. dadantii in planta‐modulated genes with those expressed during the epiphytic colonization of leaves
To better understand the importance of PecS regulation of the D. dadantii transcriptome during plant infection, we compared, in the wild‐type strain, the expression profiles on leaf surfaces at 6 hpi and inside the plant tissues just before the onset of maceration symptoms at 24 hpi. A total of 637 genes were differentially expressed between these two conditions (FDR < 0.05, FC > 2 or FC < −2; Table S2, see Supporting Information). Again, a significant proportion of these genes were down‐regulated (53%).
A known or predicted function could be assigned to 358 genes (Table S2), which were manually classified into the functional categories (Table S2, Fig. 4) discussed below.
Figure 4.
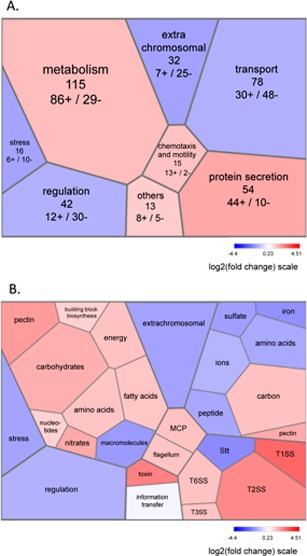
Tree maps of differentially expressed functional gene clusters in the Dickeya dadantii wild‐type strain during the epiphytic/leaf‐infecting growth transition. Gene expression changes in bacteria isolated from infected leaves at 24 h post‐inoculation (hpi) relative to those in bacteria isolated from leaf surfaces at 6 hpi are visualized by Voronoi tree maps. The mean ratios of expression data in the different gene clusters (on a log2 scale) were colour coded with a divergent colour gradient. The blue and red tiles represent down‐ and up‐regulated pathways, respectively. Two levels of classification are displayed (A, B) according to our custom functional classification (corresponding to category A/subcategory B in Table S2, see Supporting Information). In (A), for each category, the total number of modulated genes is indicated and the numbers of up (+) and down‐regulated (–) genes are provided. MCP, methyl‐accepting chemotaxis protein; T1SS, type I secretion system; T2SS, type II secretion system; T3SS, type III secretion system; T6SS, type VI secretion system.
Cell wall degradation, pectin metabolism and protein secretion
As expected from previous reports (see review of Charkowski et al., 2012), most genes involved in plant cell wall degradation and pectin metabolism were more strongly expressed at the onset of maceration symptoms (24 hpi) than during the epiphytic colonization of leaves (6 hpi) in the wild‐type strain (Fig. 2). They include genes encoding extracellular cell wall‐degrading enzymes and their respective secretion systems, several of which have been identified previously as in planta‐induced genes by qRT‐PCR (Lebeau et al., 2008; Mhedbi‐Hajri et al., 2011; Reverchon et al., 2010). In addition, the pehX and pelX periplasmic pectinase‐encoding genes were up‐regulated at 24 hpi, as were the genes encoding the entire degradation pathway of the rhamnogalacturonan I backbone, a side chain of pectin. These genes encode the rhamnogalacturonan lyase RhiE, the RhiT transporter, RhiN and RhaDAB catabolic enzymes, and the RhaSR regulators. Their role in virulence has been demonstrated previously, as has their high expression in planta (Hugouvieux‐Cotte‐Pattat, 2004).
Our analysis also showed, for the first time in Dickeya, the in planta induction of T6SS‐encoding genes, an activation already reported for T6SS in P. atrosepticum (Liu et al., 2008). The expression of three other protein secretion system‐related genes was also modulated in leaf‐infecting relative to epiphytic bacteria. The hrpN and DspA/E T3SS effector‐encoding genes were up‐regulated, and some T4SS genes and the entire stt operon encoding the secondary T2SS were down‐regulated at 24 hpi. All of these data further stress the importance of the different components of the secretome in the interactions with plants.
Metabolism and transport
The expression of about 200 genes involved in metabolism and transport was modulated in leaf‐infecting relative to epiphytic bacteria. In addition to the enzymes and transporters involved in pectin and rhamnogalacturonan catabolic pathways, several genes involved in β‐glucoside transport and catabolism were up‐regulated. The related substrates include aromatic β‐glucosides, such as arbutin and salicin, which are very common in plants. Leaf infection also promoted the activation of genes encoding the narGHIJ and frdABCD operons encoding NO3‐reductase and fumarate reductase complexes. These complexes function as terminal electron acceptors during anaerobic respiration in E. coli (Jones et al., 2011) and, together with the F0F1‐ATPase, their activation points to the modulation of energy conversion during leaf infection. We also observed the activation of several genes involved in fatty acid metabolism, such as acc, fad and fab genes, which might indicate a remodelling of membrane composition. Interestingly, several transport systems were down‐regulated in leaf‐infecting versus epiphytic bacteria, highlighting the different environment encountered on leaf surfaces relative to the leaf interior. A similar transcriptomic study performed using the P. syringae foliar pathogen has already indicated that these two habitats offer distinct environments for bacteria (Yu et al., 2013).
Chemotaxis and motility
Eight genes involved in motility were activated in the plant interior (Table S2, Fig. 4). Indeed, seven flagellar genes were slightly modulated and the rhlA gene involved in swarming was strongly up‐regulated in leaf‐infecting bacteria. Although a non‐motile FliC mutant was impaired in disease progression, the inactivation of rhlA did not show an effect on virulence (Hommais et al., 2008). Seven genes encoding MCP were also modulated in leaf‐infecting versus epiphytic bacteria, pointing again to the recognition of specific metabolites inside plants.
Regulation
Forty‐two regulatory genes were also differentially expressed in planta at 24 hpi relative to leaf surfaces at 6 hpi, 30 of which were down‐regulated. For most of these regulators, the regulated genes are unknown. Only two of these genes encode known global regulators: the slightly up‐regulated expR gene, which encodes an N‐acyl homoserine lactone (AHL)‐dependent negative regulator, and the down‐regulated pecT gene, which encodes a global negative regulator of pectinase production, motility and EPS synthesis (Condemine et al, 1999, Charkowski et al., 2012, Reverchon and Nasser, 2013). The analysis of exp mutations only revealed a minor role of the AHL‐mediated ExpIR quorum‐sensing system on virulence or on the expression of virulence genes in planta during infection (Mhedbi‐Hajri et al., 2011). In contrast, a pecT mutant is hypervirulent and, interestingly, down‐regulation of pecT during infection has been demonstrated previously by qRT‐PCR (Lebeau et al., 2008).
The large number of genes whose expression was modulated between leaf‐infecting and epiphytic bacteria confirms the expected activation of most well‐characterized virulence genes at the onset of disease, but also reveals the modulation of hundreds of genes activated either on leaf surfaces or in the plant interior. It thus highlights an important and distinct adaptation to these two niches.
The PecS‐regulated genes constitute a large part of the D. dadantii in planta‐modulated genes
Of the 637 genes modulated in planta at 24 hpi versus on leaves at 6 hpi in the wild‐type strain, 41% were also differentially expressed in the pecS mutant relative to the wild‐type strain on leaf surfaces at 6 hpi (Fig. 5). As expected from previous reports (for reviews, see Charkowski et al., 2012; Reverchon and Nasser, 2013), these genes included most genes encoding protein secretion systems and secreted proteins, as 89% of the in planta‐modulated genes in the wild‐type strain belonging to this category are part of the PecS regulon (Table S2, Fig. 5). Notably, however, the PehX and PelX periplasmic pectinase‐ and the RhiE rhamnogalacturonan lyase‐encoding genes were up‐regulated only in the wild‐type strain at 24 hpi. This confirms the previously assigned important role of PecS in the repression of virulence genes in plant colonization before the onset of disease (Mhedbi‐Hajri et al., 2011). About one‐half of the in planta‐modulated genes in the wild‐type strain related to chemotaxis and motility are part of the PecS regulon. These include four flagellar genes and the gene encoding the RhlA surfactant involved in swarming motility. Among the seven genes encoding MCP that were differentially expressed 24 hpi versus 6 hpi in the wild‐type strain, however, five did not appear to be part of the PecS regulon. As three other MCP‐encoding genes were up‐regulated on leaves in the pecS mutant relative to the wild‐type strain, this suggests that PecS may have complex interactions with chemotactic functions, depending on the infection stage. One‐half of the in planta‐modulated genes involved in response to stress were also part of the PecS regulon, as well as around one‐third of the genes involved in transport, regulation and metabolism (Table S2). This again highlights a wide PecS control that stretches far beyond the already characterized virulence factors.
Figure 5.

PecS dependence of regulated genes during the epiphytic leaf colonization/leaf infection transition visualized by Voronoi tree maps. The genes of each category represented were sorted following their modulation in the pecS mutant after epiphytic leaf colonization [6 h post‐inoculation (hpi)] and/or leaf infection (24 hpi) (Table S2, see Supporting Information). The colour gradient indicates gene expression changes in the wild‐type bacteria at 6 relative to 24 hpi. The areas marked with dots correspond to the PecS‐controlled genes.
The role of PecS regulation is mainly restricted to the early stages of infection
To assess the role of the PecS regulator during the later stages of infection, we compared the transcriptional profiles of pecS and wild‐type bacteria present inside plant tissues at 24 hpi. In these conditions, the transcriptomes of both strains were much more similar, as only 137 genes harbouring differential expression were identified, 58% of which were only slightly modulated (–2.5 < FC < 2.5; Table S3, see Supporting Information). Remarkably, among the most highly up‐regulated genes in the pecS mutant, nine were clustered in the same region of the chromosome around the pecS gene itself, and are thus probably part of the horizontally acquired genomic island including the pecS gene. They are involved in the production of indigoidine or in the biosynthesis of the Vfm quorum‐sensing signal (Table S3). The expression of these genes appears to be directly dependent on the presence of the PecS protein, as they were also highly overexpressed in the pecS mutant during epiphytic colonization of leaf surfaces (Table S1), as well as in in vitro conditions (Hommais et al., 2008). In addition to the pilNMC genes which encode components of type IV pili, other modulated genes encoding proteins are scattered in different metabolic pathways or transport systems. Notably, in accordance with previous data (Mhedbi‐Hajri et al., 2011), only a few genes encoding known virulence factors were differentially expressed in the pecS mutant inside plant tissues at 24 hpi, and the differences in expression were low (FC ∼ 2). Our whole‐genome transcriptomic data thus reinforce the assumption that an important role of PecS is to prevent the premature expression of virulence genes in the first stages of colonization, as proposed previously (Mhedbi‐Hajri et al., 2011).
Conclusions
Several studies have already reported the prominent role of the PecS master regulator in the complex regulatory cascade controlling the production of D. dadantii virulence factors (Charkowski et al., 2012; Reverchon and Nasser, 2013), and an in vitro transcriptomic analysis has defined 134 genes that constitute the PecS regulon during growth in culture (Hommais et al., 2008). Our in planta analysis enlarged the PecS regulon to about 600 genes that were more than two‐fold differentially expressed in the mutant relative to the wild‐type 3937 strain. This corresponds to about 13% of the total pathogen genome. The differentially expressed functions include most of those previously described as being part of the PecS regulon, such as plant cell wall‐degrading enzymes, multiple secretion systems and flagellar components, and indigoidine biosynthesis (Hommais et al., 2008). The present analysis added several type II and type III secreted proteins, and constituents of T6SS, to the PecS regulon, as well as various T6SS secreted Hcp and VgrG proteins. It also extended the role of PecS to the control of genes involved in metabolism, transport and chemotaxis, as well as more than 40 regulatory genes including the Vfm and RpoS master regulators. It remains to be determined which PecS target genes are directly controlled at the DNA level by this regulator and which might be controlled via the regulation of other regulatory genes.
This substantial number of regulatory genes whose expression is affected in the pecS mutant demonstrates that PecS is at or near the top of a major regulatory cascade, with many additional constituents still to be identified.
Although most of the previously described members of the PecS regulon consist of repressed genes, our study revealed that about one‐half of the genes modulated during interaction with the plant host are positively regulated by PecS during epiphytic leaf colonization. This confirms that PecS acts mainly in the first stages of infection to prevent virulence gene induction, but also points to an active role in the adaptation of the bacterium to the epiphytic habitat. Such a dual role of PecS has been suggested previously by the in vitro transcriptomic analysis of the PecS regulon (Hommais et al., 2008).
PecS controls the expression of 41% of the more than 600 D. dadantii genes modulated during the switch from epiphytic colonization to plant invasion, extending from well‐known virulence genes to numerous genes involved in metabolism, transport or chemotaxis. It is fascinating that such a recently acquired regulator, mostly found in Dickeya species, recruited under its control so many genes, including virulence‐related genes that are shared by other soft rot bacteria, such as the closely related Pectobacterium.
The work presented here, as well as previous studies (Jacobs et al., 2012; Yu et al., 2013, 2014), also shows that plant‐pathogenic bacteria not only carefully regulate the expression of genes encoding virulence factors to maximize success at each phase of disease, but also respond to triggers encountered in different niches during plant colonization. Culture studies cannot generate a complete understanding of this critical aspect of bacterial pathogenesis, emphasizing the importance of in planta analyses.
Experimental Procedures
Bacteria, plant material and strains, and infection procedure
Dickeya dadantii 3937 strain (our collection) and the pecS mutant (A4476 pecS::uidA‐KmR, Reverchon et al., 1994) were grown at 30 °C overnight on Luria–Bertani (LB) plates. Bacteria were resuspended in 50 mm KPO4, pH 6.8, buffer containing 0.01% (v/v) of Silwet L‐77 surfactant (van Meeuwen Chemicals BV, Weesp, the Netherlands). Col‐0 Arabidopsis plants were grown for 6 weeks under short‐day conditions at 24/19 °C (8 h light/16 h dark). Six‐week‐old plants were inoculated by rapid immersion in a bacterial suspension at 108 colony‐forming units (cfu)/mL and incubated at 24 °C/19 °C (day/night) in long‐day conditions (16 h of light) in small transparent enclosed containers (BHR, Bouillard Frères SA, Saint‐Germain du Plain, France: ref B*MX; length, 51 cm; width, 38.5 cm; height, 36 cm) with abundant watering to give 100% humidity.
Isolation of bacteria from infected plants
Bacteria were isolated from plant tissues following the protocol described by Chapelle et al. (2015). Bacteria present on leaf surfaces were recovered by rinsing the inoculated leaves in RNA freezing buffer [50 mm KPO4, pH 6.8, buffer supplemented with 20% of Genelock™/AssayAssure™ RNA‐stabilizing reagent (Sierra Molecular Inc., Incline Village, NV, USA, Cat#33300)], filtering the bacterial suspension through 10‐µm cheesecloth to remove contaminating soil and recovering the bacterial pellet by centrifugation at 10 000 g for 15 min.
For the isolation of bacteria present inside plant leaves at 24 hpi, plant aerial parts were collected in freezing buffer, plant tissues were disrupted with a blender and the homogenate was filtered through 25‐µm and then 10‐µm cheesecloth to remove large plant tissue debris. The flow‐throughs were further cleaned by a 500 g centrifugation for 10 min, and the bacterial cells were collected by centrifuging the resulting supernatant at 10 000 g for 15 min. Bacteria isolated either from leaf surfaces or from inside the plants were further purified on a Gentodenz density gradient, as described previously (Chapelle et al., 2015), resuspended in 200 µL of ethanol/phenol stop solution (5% water‐saturated phenol in ethanol), recovered by quick centrifugation, frozen in liquid nitrogen and stored at −80 °C.
RNA extraction, microarray design and hybridizations
RNA from purified bacteria was isolated by a hot phenol procedure. The bacterial pellet was resuspended in 600 µL of ice‐cold A buffer [20 mm sodium acetate, pH 5.2, 1 mm ethylenediaminetetraacetic acid (EDTA)]. Cells were immediately lysed by the addition of 40 µL of 10% sodium dodecylsulfate (SDS) and 600 µL of hot acidic phenol (65 °C), vortexed and incubated for 6–8 min at 65 °C. RNA was further cleaned by two additional phenol–chloroform extractions and ethanol precipitated.
The microarrays used in this study were custom designed and produced by NimbleGen Systems Inc. (Madison, WI, USA) based on the annotated sequence (version number 6) of D. dadantii (Glasner et al., 2011; available at https://asap.ahabs.wisc.edu/asap/logon.php). These arrays comprised 4753 CDSs. The microarrays consisted of five probes of 60 nucleotides per CDS (4597 CDSs present) and three copies of each probe per array separated into three blocks.
For microarray analyses, double‐strand cDNAs were synthesized according to the NimbleGen protocol using random hexamer primers for first‐strand cDNA synthesis. cDNAs were labelled and hybridized by NimbleGen Systems, Inc.
Microarray data analysis
One‐colour NimbleGen expression data were processed using the ANAIS web interface (Simon and Biot, 2010; http://anais.versailles.inra.fr/). Normalization was performed with the robust multi‐array analysis (RMA) background correction process. To determine differentially expressed genes, FC was calculated for each experimental condition, normalized to the referring experimental condition and one‐way analysis of variance (ANOVA) was performed on all log10‐transformed normalized data. The raw P values were adjusted by the false discovery rate Bonferroni (FDR(BH)) method (type I error, 5%) to control for the family‐wise error rate and to limit false positives drastically in a multiple comparison context (Benjamini and Hochberg, 1995). Log2 fold changes for the whole genome genes for the three studied comparisons are given in Table S4. The microarray data were deposited in the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under the accession number GSE94713 (NCBI tracking system #18259698).
Voronoi tree map visualization of the functional classification was performed by Voronto mapper software (Santamaria and Pierre, 2012).
Expression analysis by quantitative reverse transcription‐polymerase chain reaction
Total RNA was extracted from 6‐week‐old Arabidopsis plants infected by rapid immersion in a bacterial suspension (108 cfu/mL) in 50 mm KPO4, pH 6.8, buffer containing 0.01% (v/v) Silwet L‐77 surfactant (van Meeuwen Chemicals BV). Aerial plant tissues were frozen in liquid N2 and ground to a fine powder. One to three milligrams of powder were mixed with 3.2 mL of extraction buffer (4 m guanidium isothiocyanate, 27 mm sodium acetate, 143 mm β‐mercaptoethanol) and centrifuged at 9700 g for 35 min at 4 °C. RNAs were pelleted by centrifugation (185 000 g for 5 h at 20 °C) on a caesium chloride cushion (5.7 m CsCl, 25 mm sodium acetate, pH 6). Pelleted RNAs were washed twice with 70% ethanol and resuspended in 50 µL of H2O.
RNA samples were treated with RNAse‐free DNAse I (Invitrogen Carlsbad, CA, USA) to remove any DNA contamination. First‐strand cDNAs were then synthesized from 3 µg of total RNA using Superscript II RNAse H‐reverse transcriptase (Invitrogen Waltham, Massachusetts, USA) and random hexamer primers (Invitrogen) following the manufacturer's instructions.
For quantitative PCR analysis, cDNAs were amplified using Maxima® SYBR Green/ROX qPCR Master Mix (Fermentas) according to the manufacturer's license in an Applied Biosystems 7300 Real Time PCR System using the following conditions: 10 min at 95 °C, followed by 40 amplification cycles, each consisting of 15 s at 95 °C and 60 s at 60 °C. The results were analysed using Applied Biosystems Sequence Detection Software v1.3.1. The constitutive lpxC gene encoding a UDP‐N‐acetylglucosamine deacetylase involved in lipid A biosynthesis was used as internal control to normalize the expression data (Hommais et al., 2011).
Competing Interests
The authors declare that they have no competing interests.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Growth kinetics of Dickeya dadantii wild‐type (WT) and pecS mutant on Arabidopsis infection. hpi, hours post‐inoculation.
Fig. S2 Validation of microarray results by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR). hpi, hours post‐inoculation; WT, wild‐type.
Table S1 Differentially expressed genes in the pecS mutant relative to the wild‐type strain during epiphytic leaf colonization at 6 h post‐inoculation (hpi).
Table S2 Differentially expressed genes during leaf infection at 24 h post‐inoculation (hpi) relative to epiphytic leaf colonization at 6 hpi in the wild‐type strain.
Table S3 Differentially expressed genes in the pecS mutant relative to the wild‐type strain at 24 h post‐inoculation (hpi).
Table S4 Differential expression whole‐genome data for the three studied comparisons log2 fold changes are given).
Acknowledgements
We gratefully acknowledge Caitilyn Allen for fruitful discussions and critical reading of the manuscript, Emma Rochelle‐Newall for English corrections and Lydie Blottière for her help with the statistical treatment of numeration data. This work was supported by the REGUPATH grant from the French ‘ANR blanc 2007’ Programme (ANR‐07‐BLAN‐0212).
References
- Barras, F. , Van Gijsegem, F. and Chatterjee, A.K. (1994) Extracellular enzymes and pathogenesis of soft‐rot Erwinia . Annu. Rev. Phytopathol. 32, 201–234. [Google Scholar]
- Battesti, A. , Majdalani, N. and Gottesman, S. (2011) The RpoS‐mediated general stress response in Escherichia coli . Annu. Rev. Microbiol. 65, 189–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, D.W. , Bogdanove, A.J. , Beer, S.V. and Collmer, A. (1994) Erwinia chrysanthemi hrp genes and their involvement in soft rot pathogenesis and elicitation of the hypersensitive response. Mol. Plant–Microbe Interact. 7, 573–581. [DOI] [PubMed] [Google Scholar]
- Bauer, D.W. , Wei, Z.M. , Beer, S.V. and Collmer, A. (1995) Erwinia chrysanthemi harpinEch: an elicitor of the hypersensitive response that contributes to soft‐rot pathogenesis. Mol. Plant–Microbe Interact. 8, 484–491. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B, 57, 289–300. [Google Scholar]
- Chapelle, E. , Alunni, B. , Malfatti, P. , Solier, L. , Pédron, J. , Kraepiel, Y. and Van Gijsegem, F. (2015) A straightforward and reliable method for bacterial in planta transcriptomics: application to the Dickeya dadantii/Arabidopsis thaliana pathosystem. Plant J. 82, 352–362. [DOI] [PubMed] [Google Scholar]
- Charkowski, A. , Blanco, C. , Condemine, G. , Expert, D. , Franza, T. , Hayes, C. , Hugouvieux‐Cotte‐Pattat, N. , López Solanilla, E. , Low, D. , Moleleki, L. , Pirhonen, M. , Pitman, A. , Perna, N. , Reverchon, S. , Rodríguez Palenzuela, P. , San Francisco, M. , Toth, I. , Tsuyumu, S. , van der Waals, J. , van der Wolf, J. , Van Gijsegem, F. , Yang, C.H. and Yedidia, I. (2012) The role of small molecules in soft rot Enterobacteriaceae pathogenicity. Annu. Rev. Phytopathol. 50, 425–449. [DOI] [PubMed] [Google Scholar]
- Condemine, G. , Castillo, A. , Passeri, F. and Enard, C. (1999) The PecT repressor coregulates synthesis of exopolysaccharides and virulence factors in Erwinia chrysanthemi . Mol. Plant–Microbe Interact. 12, 45–52. [DOI] [PubMed] [Google Scholar]
- Deochand, D.K. , Meariman, J.K. and Grove, A. (2016) pH‐dependent DNA distortion and repression of gene expression by Pectobacterium atrosepticum PecS. ACS Chem. Biol. 11, 2049–2056. [DOI] [PubMed] [Google Scholar]
- El Hassouni, M.E. , Chambost, J.P. , Expert, D. , Van Gijsegem, F. and Barras, F. (1999) The minimal gene set member msrA, encoding peptide methionine sulfoxide reductase, is a virulence determinant of the plant pathogen Erwinia chrysanthemi . Proc. Natl. Acad. Sci. USA, 96, 887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, S.B. and Kogoma, T. (1991) Oxidative stress responses in Escherichia coli and Salmonella typhimurium . Microbiol. Rev. 55, 561–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandez, Y. and Condemine, G. (2008) Novel mechanism of outer membrane targeting of proteins in Gram‐negative bacteria. Mol. Microbiol. 69, 1349–1357. [DOI] [PubMed] [Google Scholar]
- Glasner, J.D. , Yang, C.‐H. , Reverchon, S. , Hugouvieux‐Cotte‐Pattat, N. , Condemine, G. , Bohin, J.‐P. , Van Gijsegem, F. , Yang, S. , Franza, T. , Expert, D. , Plunkett, G., 3rd , San Francisco, M.J. , Charkowski, A.O. , Py, B. , Bell, K. , Rauscher, L. , Rodriguez‐Palenzuela, P. , Toussaint, A. , Holeva, M.C. , He, S.Y. , Douet, V. , Boccara, M. , Blanco, C. , Toth, I. , Anderson, B.D. , Biehl, B.S. , Mau, B. , Flynn, S.M. , Barras, F. , Lindeberg, M. , Birch, P.R. , Tsuyumu, S. , Shi, X. , Hibbing, M. , Yap, M.N. , Carpentier, M. , Dassa, E. , Umehara, M. , Kim, J.F. , Rusch, M. , Soni, P. , Mayhew, G.F. , Fouts, D.E. , Gill, S.R. , Blattner, F.R. , Keen, N.T. and Perna, N.T. (2011) Genome sequence of the plant‐pathogenic bacterium Dickeya dadantii 3937. J. Bacteriol. 193, 2076–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkhandan, E. , Kamaruzaman, S. , Zainalabidin, M.A. and Nasehi, A. (2016) First report of soft rot disease on pak choi (Brassica chinensis) caused by Dickeya dadantii subsp dieffenbachiae in Malaysia. Plant Dis. 100, 209. [Google Scholar]
- Gouesbet, G. , Trautwetter, A. , Bonnassie, S. , Wu, L.F. and Blanco, C. (1996) Characterization of the Erwinia chrysanthemi osmoprotectant transporter gene ousA . J. Bacteriol. 178, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommais, F. , Oger‐Desfeux, C. , Van Gijsegem, F. , Castang, S. , Ligori, S. , Expert, D. , Nasser, W. and Reverchon, S. (2008) PecS is a global regulator of the symptomatic phase in the phytopathogenic bacterium Erwinia chrysanthemi 3937. J. Bacteriol. 190, 7508–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommais, F. , Zghidi‐Abouzid, O. , Oger‐Desfeux, C. , Pineau‐Chapelle, E. , Van Gijsegem, F. , Nasser, W. and Reverchon, S. (2011) lpxC and yafS are the most suitable internal controls to normalize Real Time RT‐qPCR expression in the phytopathogenic bacterium Dickeya dadantii . PLoS One, 6, e20269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugouvieux‐Cotte‐Pattat, N. (2004) The RhaS activator controls the Erwinia chrysanthemi 3937 genes rhiN, rhiT and rhiE involved in rhamnogalacturonan catabolism. Mol. Microbiol. 51, 1361–1374. [DOI] [PubMed] [Google Scholar]
- Hugouvieux‐Cotte‐Pattat, N. , Shevchik, V.E. and Nasser, W. (2002) PehN, a polygalacturonase hornologue with a low hydrolase activity, is coregulated with the other Erwinia chrysanthemi polygalacturonases. J. Bacteriol. 184, 2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugouvieux‐Cotte‐Pattat, N. , Condemine, G. and Shevchik, V.E. (2014) Bacterial pectate lyases, structural and functional diversity. Environ. Microbiol. Rep. 6, 427–440. [DOI] [PubMed] [Google Scholar]
- Jacobs, J.M. , Babujee, L. , Meng, F. , Milling, A. and Allen, C. (2012) The in planta transcriptome of Ralstonia solanacearum: conserved physiological and virulence strategies during bacterial wilt of Tomato. mBio, 3, e00114–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S.A. , Gibson, T. , Maltby, R.C. , Chowdhury, F.Z. , Stewart, V. , Cohen, P.S. and Conway, T. (2011) Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun. 79, 4218–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi‐Pour, N. , Condemine, G. and Hugouvieux‐Cotte‐Pattat, N. (2004) The secretome of the plant pathogenic bacterium Erwinia chrysanthemi . Proteomics, 4, 3177–3186. [DOI] [PubMed] [Google Scholar]
- Koo, M.S. , Lee, J.H. , Rah, S.Y. , Yeo, W.S. , Lee, J.W. , Lee, K.L. , Koh, Y.S. , Kang, S.O. and Roe, J.H. (2003) A reducing system of the superoxide sensor SoxR in Escherichia coli . EMBO J. 22, 2614–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskiniemi, S. , Lamoureux, J.G. , Nikolakakis, K.C. , T'kint de Roodenbeke, C. , Kaplan, M.D. , Low, D.A. and Hayes, C.S. (2013) Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. USA, 110, 7032–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau, A. , Reverchon, S. , Gaubert, S. , Kraepiel, Y. , Simond‐Cote, E. , Nasser, W. and Van Gijsegem, F. (2008) The GacA global regulator is required for the appropriate expression of Erwinia chrysanthemi 3937 pathogenicity genes during plant infection. Environ. Microbiol. 10, 545–559. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Coulthurst, S.J. , Pritchard, L. , Hedley, P.E. , Ravensdale, M. , Humphris, S. , Burr, T. , Takle, G. , Brurberg, M.B. , Birch, P.R. , Salmond, G.P. and Toth, I.K. (2008) Quorum sensing coordinates brute force and stealth modes of infection in the plant pathogen Pectobacterium atrosepticum . PLoS Pathog. 4, e1000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Solanilla, E. , Garcia‐Olmedo, F. and Rodriguez‐Palenzuela, P. (1998) Inactivation of the sapA to sapF locus of Erwinia chrysanthemi reveals common features in plant and bacterial pathogenesis. Plant Cell, 10, 917–924. [PMC free article] [PubMed] [Google Scholar]
- Mattinen, L. , Tshuikina, M. , Mae, A. and Pirhonen, M. (2004) Identification and characterization of Nip, necrosis‐inducing virulence protein of Erwinia carotovora subsp. carotovora . Mol. Plant–Microbe Interact. 17, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Mhedbi‐Hajri, N. , Malfatti, P. , Pédron, J. , Gaubert, S. , Reverchon, S. and Van Gijsegem, F. (2011) PecS is an important player in the regulatory network governing the coordinated expression of virulence genes during the interaction between Dickeya dadantii 3937 and plants. Environ. Microbiol. 13, 2901–2914. [DOI] [PubMed] [Google Scholar]
- Mole, B.M. , Baltrus, D.A. , Dangl, J.L. and Grant, S.R. (2007) Global virulence regulation networks in phytopathogenic bacteria. Trends Microbiol. 15, 363–371. [DOI] [PubMed] [Google Scholar]
- Nasser, W. , Dorel, C. , Wawrzyniak, J. , Van Gijsegem, F. , Groleau, M.‐C. , Déziel, E. and Reverchon, S. (2013) Vfm a new quorum sensing system controls the virulence of Dickeya dadantii . Environ. Microbiol. 15, 865–880. [DOI] [PubMed] [Google Scholar]
- Parkinson, N. , Stead, D. , Bew, J. , Heeney, J. , Tsror, L. and Elphinstone, J. (2009) Dickeya species relatedness and clade structure determined by comparison of recA sequences. Int. J. Syst. Evol. Microbiol. 59, 2388–2393. [DOI] [PubMed] [Google Scholar]
- Pérombelon, M.C.M. (2002) Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. [Google Scholar]
- Reverchon, S. and Nasser, W. (2013) Dickeya ecology, environment sensing and regulation of virulence program. Environ. Microbiol. Rep. 5, 622–636. [DOI] [PubMed] [Google Scholar]
- Reverchon, S. , Nasser, W. and Robert‐Baudouy, J. (1994) pecS: a locus controlling pectinase, cellulase and blue pigment production in Erwinia chrysanthemi . Mol. Microbiol. 11, 1127–1139. [DOI] [PubMed] [Google Scholar]
- Reverchon, S. , Rouanet, C. , Expert, D. and Nasser, W. (2002) Characterization of indigoidine biosynthetic genes in Erwinia chrysanthemi and role of this blue pigment in pathogenicity. J. Bacteriol. 184, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverchon, S. , Van Gijsegem, F. , Effantin, G. , Zghidi‐Abouzid, O. and Nasser, W. (2010) Systematic targeted mutagenesis of the MarR/SlyA family members of Dickeya dadantii 3937 reveals a role for MfbR in the modulation of virulence gene expression in response to acidic pH. Mol. Microbiol. 78, 1018–1037. [DOI] [PubMed] [Google Scholar]
- Rouanet, C. , Reverchon, S. , Rodionov, D.A. and Nasser, W. (2004) Definition of a consensus DNA‐binding site for PecS, a global regulator of virulence gene expression in Erwinia chrysanthemi and identification of new members of the PecS regulon. J. Biol. Chem. 279, 30 158–30 167. [DOI] [PubMed] [Google Scholar]
- Santamaria, R. and Pierre, P. (2012) Voronto: mapper for expression data to ontologies. Bioinformatics, 28, 2281–2282. [DOI] [PubMed] [Google Scholar]
- Simon, A. and Biot, E. (2010) ANAIS: analysis of NimbleGen arrays interface. Bioinformatics, 26, 2468–2469. [DOI] [PubMed] [Google Scholar]
- Soto‐Suarez, M. , Bernal, D. , Gonzalez, C. , Szurek, B. , Guyot, R. , Tohme, J. and Verdier, V. (2010) In planta gene expression analysis of Xanthomonas oryzae pathovar oryzae, African strain MAI1. BMC Microbiol. 10, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C.‐H. , Gavilanes‐Ruiz, M. , Okinaka, Y. , Vedel, R. , Berthuy, I. , Boccara, M. , Chen, J.W. , Perna, N.T. and Keen, N.T. (2002) hrp genes of Erwinia chrysanthemi 3937 are important virulence factors. Mol. Plant–Microbe Interact. 15, 472–480. [DOI] [PubMed] [Google Scholar]
- Yu, X. , Lund, S.P. , Scott, R.A. , Greenwald, J.W. , Records, A.H. , Nettleton, D. , Lindow, S.E. , Gross, D.C. and Beattie, G.A. (2013) Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. Proc. Natl. Acad. Sci. USA, 110, E425–E434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Lund, S.P. , Greenwald, J.W. , Records, A.H. , Scott, R.A. , Nettleton, D. , Lindow, S.E. , Gross, D.C. and Beattie, G.A. (2014) Transcriptional analysis of the global regulatory networks active in Pseudomonas syringae during leaf colonization. mBio, 5, e01683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Growth kinetics of Dickeya dadantii wild‐type (WT) and pecS mutant on Arabidopsis infection. hpi, hours post‐inoculation.
Fig. S2 Validation of microarray results by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR). hpi, hours post‐inoculation; WT, wild‐type.
Table S1 Differentially expressed genes in the pecS mutant relative to the wild‐type strain during epiphytic leaf colonization at 6 h post‐inoculation (hpi).
Table S2 Differentially expressed genes during leaf infection at 24 h post‐inoculation (hpi) relative to epiphytic leaf colonization at 6 hpi in the wild‐type strain.
Table S3 Differentially expressed genes in the pecS mutant relative to the wild‐type strain at 24 h post‐inoculation (hpi).
Table S4 Differential expression whole‐genome data for the three studied comparisons log2 fold changes are given).