

Summary
The ascomycete fungal pathogen Fusarium graminearum causes the globally important Fusarium head blight (FHB) disease on cereal hosts, such as wheat and barley. In addition to reducing grain yield, infection by this pathogen causes major quality losses. In particular, the contamination of food and feed with the F. graminearum trichothecene toxin deoxynivalenol (DON) can have many adverse short‐ and long‐term effects on human and animal health. During the last decade, the interaction between F. graminearum and both cereal and model hosts has been extensively studied through transcriptomic analyses. In this review, we present an overview of how such analyses have advanced our understanding of this economically important plant–microbe interaction. From a host point of view, the transcriptomes of FHB‐resistant and FHB‐susceptible cereal genotypes, including near‐isogenic lines (NILs) that differ by the presence or absence of quantitative trait loci (QTLs), have been studied to understand the mechanisms of disease resistance afforded by such QTLs. Transcriptomic analyses employed to dissect host responses to DON have facilitated the identification of the genes involved in toxin detoxification and disease resistance. From the pathogen point of view, the transcriptome of F. graminearum during pathogenic vs. saprophytic growth, or when infecting different cereal hosts or different tissues of the same host, have been studied. In addition, comparative transcriptomic analyses of F. graminearum knock‐out mutants with altered virulence have provided new insights into pathogenicity‐related processes. The F. graminearum transcriptomic data generated over the years are now being exploited to build a systems level understanding of the biology of this pathogen, with an ultimate aim of developing effective and sustainable disease prevention strategies.
Keywords: barley, Fusarium graminearum, Fusarium head blight, maize, stalk rot, transcriptome, wheat
Introduction
The fungal pathogen Fusarium graminearum, the causative agent of Fusarium head blight (FHB, also known as scab or ear blight) disease in wheat and barley, and ear and stalk rot disease in maize, is responsible for substantial economic losses every year. The mycotoxins, such as the trichothecene toxin deoxynivalenol (DON) and the oestrogenic mycotoxin zearalenone (ZEA), produced by this pathogen during disease development, significantly reduce grain quality (Wegulo, 2012). The consumption of food and feed produced from toxin‐contaminated plants can have serious adverse effects on human and animal health (Sobrova et al., 2010). Therefore, various countries or jurisdictions (e.g. the European Union) have imposed strict regulations on permissible DON levels (e.g. EFSA CONTAM Panel, 2013). For these reasons, F. graminearum, which is considered to be one of the most important fungal plant pathogens in the world (Dean et al., 2012), has the potential to cause serious economic, social, environmental and health issues.
The sexual spores (e.g. ascospores) of the fungus are the primary cause of infection of cereal flowers by F. graminearum (Ma et al., 2013; Trail, 2009). After initial infection, the pathogen moves within the spike through the rachis tissue, colonizing the developing seeds. Warm and moist weather during anthesis creates conditions conducive for disease development with potentially disastrous outcomes (Goswami and Kistler, 2004). Fusarium graminearum is also capable of infecting the roots and young seedlings of wheat and barley, causing crown rot, root rot and seedling blight (Stephens et al., 2008; Wang et al., 2015). During the infection of wheat roots, F. graminearum employs strategies (e.g. the formation of penetration pegs to invade the root epidermis) typically used by root‐infecting pathogens. No lesion development is observed at the early stages of root infection by F. graminearum. However, subsequently, lesions develop on the infected roots and stems, and the infection process from this point on resembles that used during head infection (Wang et al., 2015). The infection of maize stems by F. graminearum causes stalk rot disease. The disintegration of pith tissue during stalk rot weakens the maize plant, making it susceptible to lodging (Sutton, 1982). Fusarium graminearum overwinters in the soil and/or plant debris in the form of asexual macroconidia and starts new infection cycles in the spring (Bai and Shaner, 2004).
Fungicides can be used to control FHB. However, the cost, difficulties associated with the identification of the correct application timing, potential environmental issues and the development of fungicide resistance in pathogen populations limit chemical control options (Yuen and Schoneweis, 2007). Therefore, the improvement of cereal cultivars with relatively high levels of FHB resistance is an essential breeding objective. However, despite major efforts, most cereal cultivars available today show only moderate levels of FHB resistance. This is at least partly because resistance against this pathogen acts quantitatively, implying that many genes in the host, each with relatively small effects, are involved in disease resistance (Bai and Shaner, 2004).
A better understanding of the mechanisms involved in pathogen infection and host defence can reveal essential new knowledge that can be utilized to develop FHB‐resistant cereals. For instance, this knowledge can identify potential weaknesses in the pathogen that can be targeted by genetic [e.g. host‐produced RNA interference (RNAi)] or chemical (e.g. fungicides) means (Cheng et al., 2015; Koch et al., 2013). Similarly, the identification and modification of candidate genes encoding potential susceptibility factors (i.e. host components exploited by pathogens to cause disease) in the host can be an option to increase disease resistance (Garvin et al., 2015). In the face of constantly changing pathogen populations and environmental factors, innovative research and development are needed to avoid potential FHB epidemics and to ensure that food and feed supplies are free from mycotoxin contamination. To better understand the host factors that provide resistance to FHB, transcriptomes of the cereal–F. graminearum interactions have been extensively studied during the last decade under a variety of conditions. The availability of a complete genome sequence for F. graminearum and the construction of gene chips for both cereal hosts and the pathogen (Close et al., 2004; Cuomo et al., 2007; Guangsheng et al., 2012; Güldener et al., 2006) greatly facilitated earlier transcriptomic studies. The increasing availability of sequence information from cereal host species is also contributing to current transcriptomic studies using RNA‐sequencing (RNA‐seq) technologies (Hofstad et al., 2016; Nussbaumer et al., 2015; Powell et al., 2017a, 2017b).
The aim of this review is to present an overview of what we have learned so far from these efforts about this economically important plant–pathogen interaction, and how this knowledge can be exploited to develop effective and environmentally friendly disease protection strategies. To date, most transcriptomic studies have been conducted on the wheat–F. graminearum interaction and this will be reflected in the coverage of this review.
How the Host Plant Defends Itself Against F. graminearum: Transcriptomic Analysis of Host Responses to Pathogen Infection
To identify the host genes and defence‐associated plant processes activated by pathogen infection, initial transcriptomic studies have either analysed the responses of single cereal genotypes or compared the transcriptome of susceptible and resistant cereal genotypes to F. graminearum infection. One of the earlier transcriptomic studies identified differentially expressed genes (induced or repressed relative to mock‐treated plants) in the Chinese wheat cultivar Ning 7840 with known FHB resistance. Genes involved in primary metabolism and photosynthesis, plant defence, transcriptional regulation and protein synthesis were amongst the functional categories induced in this cultivar by F. graminearum (Kong et al., 2007).
Using microarray analysis, coupled with suppressive subtractive hybridization, another study comparing the transcriptomes of the FHB‐resistant and FHB‐susceptible wheat cultivars Ning 7840 and Clark, respectively, found that defence‐related genes were induced earlier in the resistant cultivar Ning 7840 than in the susceptible cultivar Clark after F. graminearum inoculations. Therefore, the ability to mount an earlier and stronger defence response seems to be a factor contributing to FHB resistance (Bernardo et al., 2007). One of the genes differentially expressed in this study encodes a CYP71C subfamily P450 enzyme potentially involved in the biosynthesis of the wheat defence compound DIMBOA (2,4‐dihydroxy‐7‐methoxy‐1,4‐benzoxazin‐3‐one), which is known to be toxic to F. graminearum (Glenn et al., 2001). However, recent studies have also shown that F. graminearum is able to degrade this defence compound (Kettle et al., 2015).
The defence gene expression profiles triggered by F. graminearum during the infection of the crown tissue of a moderately susceptible and a susceptible wheat cultivar have been comparatively analysed in another transcriptomic study. The genes encoding membrane‐associated proteins, transcription factors, in particular bZIPs and WRKYs, and carbohydrate metabolism‐related transcripts were significantly enriched amongst the differentially expressed genes in the moderately susceptible wheat genotype (Erayman et al., 2015). Comparative transcriptomic analyses of moderately resistant and susceptible wheat genotypes have also indicated that the suppression of pathogen virulence mechanisms by the host might be a factor contributing to disease resistance. This suggestion was supported by the finding that the expression of genes encoding ABC transporters, uridine diphosphate (UDP)‐glucosyltransferases and protease inhibitors, potentially involved in the detoxification of pathogen effectors (e.g. toxins and enzymes), was higher in the FHB‐resistant wheat genotypes Dream and Sumai 3 than in the FHB‐susceptible cultivar Lynx (Gottwald et al., 2012). Similar categories of genes differentially and/or temporally regulated between partially FHB‐resistant and FHB‐susceptible wheat genotypes have also been reported by other transcriptomic studies (Kosaka et al., 2015; Muhovski et al., 2012).
Overall, these transcriptomic studies have shown that F. graminearum activates host defences that are largely similar to those induced after pathogen infection in many other plant–microbe interactions. For instance, the genes encoding pathogenesis‐related (PR) proteins are activated in F. graminearum‐infected plants (Fig. 1) (Golkari et al., 2009). Confirming the roles of these genes in plant defence, transgenic expression of alpha‐1‐purothionin, thaumatin‐like protein 1 and β‐1‐3‐glucanase in wheat reduces FHB severity, DON accumulation and the percentage of grains with discoloured or bleached appearance under both glasshouse and field conditions (Mackintosh et al., 2007).
Figure 1.
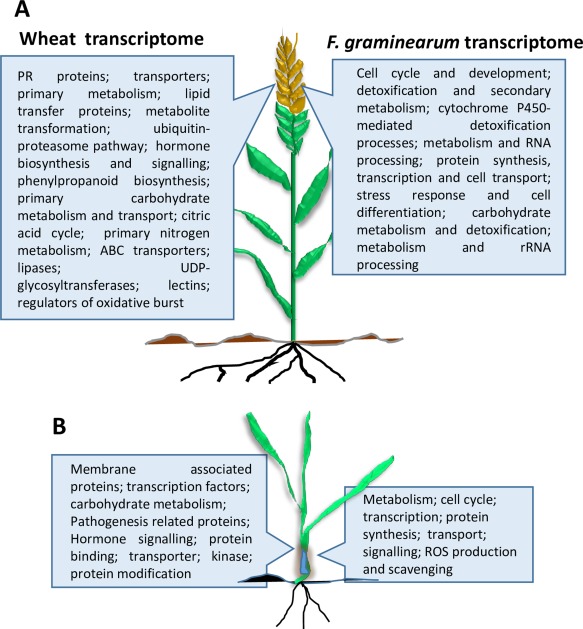
General classes of host and pathogen genes altered in heads (A) and seedlings (B) during the wheat–Fusarium graminearum interaction. Major processes proposed to be involved in F. graminearum during the infection of heads were taken from the network analysis conducted by Guo et al. (2016), which used publically available transcriptomic data from multiple studies. Please refer to the text for additional information. PR, pathogenesis‐related; ROS, reactive oxygen species; UDP, uridine diphosphate.
Transcriptomic Analyses of Host Phytohormone Signalling Pathways during FHB
Pathogen infection activates phytohormone signalling pathways which, in turn, control many aspects of plant immunity (Schenk et al., 2000). However, pathogens have developed innovative ways to interfere with plant hormone signalling pathways (Kazan and Lyons, 2014). These concepts are also applicable to cereal–F. graminearum interactions. For instance, in comparative transcriptomic analyses conducted between the resistant wheat Wangshuibai and its susceptible mutant Nauh117, the signalling pathway of the plant hormone ethylene was found to be associated with FHB susceptibility (Xiao et al., 2013). The involvement of ethylene signalling in FHB susceptibility has been demonstrated previously in both dicotyledonous and monocotyledonous plants based on the characterization of Arabidopsis and wheat ein2 mutants (Chen et al., 2009). EIN2 (ETHYLENE INSENSITIVE 2) encodes a protein involved in ethylene signalling, and the increased FHB resistance observed in ein2 mutants suggests that F. graminearum exploits the host ethylene signalling pathway to cause disease. However, other transcriptomic studies have implicated ethylene in FHB resistance. For instance, an earlier transcriptomic study comparing the transcriptomes of the FHB‐resistant wheat cultivar Sumai 3 and the FHB‐susceptible wheat landrace Y‐1193‐6 concluded that jasmonate (JA) and ethylene signalling pathways positively contribute to FHB resistance in wheat, whereas the contribution of the salicylic acid (SA) pathway to FHB resistance is negligible (Li and Yen, 2008).
Another study comparing the transcriptomes of Wangshuibai and Meh0106, a susceptible mutant derived from Wangshuibai, showed that SA and Ca2+ signalling, followed by the JA pathway, was rapidly activated after infection, suggesting that the coordinated induction of phytohormone signalling pathways contributes to FHB resistance. In contrast, the activation of the SA pathway was delayed in the susceptible genotype (Ding et al., 2011). In addition, the genes involved in JA, ethylene and gibberellin (GA) signalling were differentially expressed during priming‐mediated resistance induced in wheat by F. graminearum mutants defective in virulence, such as tri6 (unable to accumulate DON) and noxAB (NADH oxidase mutant) (Ravensdale et al., 2014). Another study comparing the transcriptomes of the moderately resistant winter wheat variety Dream and the susceptible variety Lynx suggested that a non‐specific resistance mechanism, possibly induced by JA and ethylene, may be contributing to disease resistance by regulating the expression of defence‐associated genes encoding lipid transfer proteins, thionins, defensins and GDS‐like lipases (Fig. 1) (Gottwald et al., 2012). In addition, GA and abscisic acid (ABA) seem to antagonistically modulate resistance to F. graminearum (Buhrow et al., 2016). As suggested by Kazan and Lyons (2014), the involvement of plant hormones and hormonal crosstalk in any plant–microbe interaction is complex, and future studies are required to fully understand the effect of plant hormones on cereal–F. graminearum interactions.
What Makes a Host Resistant to F. graminearum: Comparative Transcriptomics of FHB‐Resistant and FHB‐Susceptible Near‐Isogenic Lines (NILs)
Over the past 15 years, large numbers of quantitative trait loci (QTLs) providing quantitative resistance to F. graminearum have been identified in diverse cereal germplasm (Buerstmayr et al., 2009). Although the genes underlying most of these QTLs are yet to be identified, as reviewed briefly below, comparative transcriptomic analyses of NILs that differ by the presence/absence of resistance‐related loci have so far revealed many new insights into the potential mechanisms conferred by these QTLs (Zhuang et al., 2013) (Fig. 2).
Figure 2.
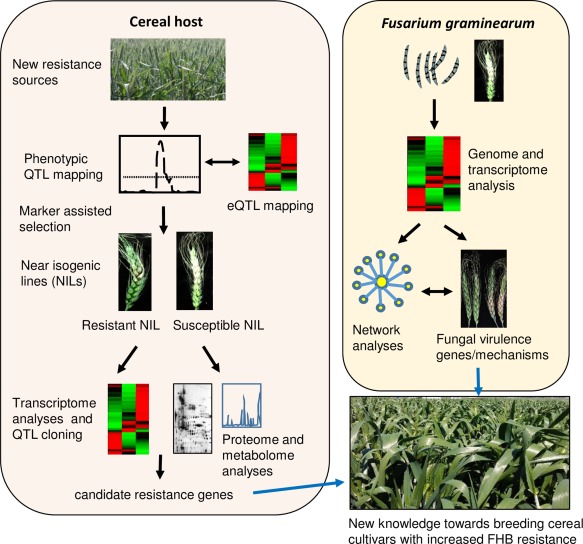
Combined use of genetic and genomic (e.g. transcriptomic and metabolomic analyses) approaches to understand the cereal host–Fusarium graminearum interaction and improve disease resistance in cereals. Fusarium head blight (FHB)‐resistant cereal germplasm and genetically characterized near‐isogenic lines (NILs) are subjected to transcriptomic, proteomic and metabolomic analysis to identify and validate candidate genes and processes that are associated with disease resistance in the host. Similarly, genetic and genomic approaches, including network analyses, in the pathogen identify genes and processes associated with pathogen virulence. The combination of the knowledge obtained from both the host and the pathogen is expected to contribute to the development of new cultivars with increased FHB resistance. eQTL, expressed QTL; QTL, quantitative trait locus. Please see the text for additional information.
2DL
The 2DL QTL, derived from the wheat cultivar Wuhan‐1, provides FHB resistance by reducing the spread of the pathogen within the head, which is termed type 2 resistance (Mesterhazy, 1995). The mode of action of 2DL has been investigated using NILs that differ by this locus (Long et al., 2015). Strong down‐regulation of phytohormone‐regulated genes, including those involved in auxin signalling, was observed in the susceptible NIL (Long et al., 2015). Therefore, the 2DL‐mediated resistance appears to act by antagonizing the interference of phytohormone pathways by the pathogen. Interestingly, significant differences observed between resistant and susceptible NILs in basal expression levels of a subset of defence genes were not affected by F. graminearum, suggesting that the 2DL‐mediated resistance may also be mediated by increased levels of basal defences (Long et al., 2015). Indeed, metabolomic analyses have shown that basal levels of various resistance‐related metabolites are higher in resistant than susceptible NILs (Hamzehzarghani et al., 2008). This initial finding has recently led to the identification of the gene underlying the 2DL QTL (see below).
Fhb1
Fhb1 and Qfhs.ifa‐5A provide the most effective partial resistance to FHB in wheat. Both QTLs are derived from the Chinese Spring wheat cultivar Sumai 3. Fhb1 is located on the short arm of chromosome 3B and is known to provide type 2 resistance (Cuthbert et al., 2006).
To understand the molecular mechanisms of FHB resistance provided by Fhb1, different wheat genotypes or NILs with and without this locus have been subjected to transcriptomic analyses (Schweiger et al., 2016) (Fig. 2). For instance, the transcriptomic analysis of Sumai 3 and two susceptible NILs revealed that the genes encoding β‐1,3‐glucanase (PR‐2), wheatwins (PR‐4) and thaumatin‐like proteins (PR‐5) showed higher levels of expression in Sumai 3 than in the susceptible NIL following inoculation with F. graminearum (Fig. 1) (Golkari et al., 2009). In addition, phenylalanine ammonia‐lyase (PAL) was induced only in Sumai 3, suggesting that defence‐related metabolites produced by the PAL pathway contribute to FHB resistance in this wheat cultivar (Golkari et al., 2009). In another study, comparative analyses of the defence responses of the FHB‐resistant wheat landrace Wangshuibai and its FHB‐susceptible mutant NAUH117, with a deleted chromosomal region carrying the Fhb1 locus (Xiao et al., 2011), have suggested that SA and JA signalling are associated with FHB resistance (Xiao et al., 2013). The Fhb1 locus seems to contribute to FHB resistance by positively influencing the expression of JA signalling [coronatine‐insensitive protein 1 (COI1), jasmonate‐zim‐domain protein (JAZ) and MYC2] and biosynthesis [allene oxide synthase (AOS), allene oxide cyclase (AOC) and 12‐oxophytodienoate reductase 3 (OPR3)] genes in Wangshuibai (Xiao et al., 2013). In contrast, Ca2+ and reactive oxygen species (ROS) signalling are associated with FHB susceptibility (Xiao et al., 2013). Finally, more recent RNA‐seq experiments have identified glucanases, nucleotide‐binding site‐leucine‐rich repeat (NBS‐LRR), WRKY transcription factors and UDP‐glucosyltransferases, differentially expressed after infection in NILs, with resistant and susceptible alleles of Fhb1 and Qfhs.ifa‐5A (Hofstad et al., 2016; Kugler et al., 2013). Fhb1 has also been implicated in harbouring a locus involved in the conversion of DON into non‐toxic DON‐3‐glucoside (Lemmens et al., 2005). More recently, the Fhb1 locus has been cloned from Sumai 3 and has been shown to encode a chimeric lectin containing two agglutinin domains involved in carbohydrate binding. This protein also contains an ETX/MTX2 domain implicated in pore forming, and thus was named PFT (pore‐forming toxin‐like) (Rawat et al., 2016). The mode of protective action of PFT is currently unknown. However, the cloning of Fhb1 has also shown that the DON‐detoxifying ability previously attributed to Fhb1 using a doubled haploid population (Lemmens et al., 2005) is not conferred by PFT, but, most likely, by a putative Uridine diphosphate (UDP)‐glucosyltransferase (UGT) also located on the chromosomal region introgressed from Sumai 3 (Rawat et al., 2016).
Qfhs.ifa‐5A
Located on chromosome 5A, this QTL also provides effective FHB resistance (20%–25% reduction). In contrast with Fhb1, Qfhs.ifa‐5A provides type 1 resistance, which makes the heads less susceptible to initial infection (Buerstmayr et al., 2003). Through comparative transcriptomic analyses of NILs, a type 1 non‐specific lipid transfer protein (LTP), constitutively expressed at higher levels in the lines carrying Qfhs.ifa‐5A, has been identified (Schweiger et al., 2013a). Several possible mechanisms of action for this LTP have been proposed based on the previously known roles of LTPs in plant disease resistance. Although, no functional data are available for this LTP, the over‐expression of TaLTP5, another LTP, in wheat confers increased FHB resistance (Zhu et al., 2012).
Fhb2
Fhb2, located on chromosome 6BS, also confers increased type 2 FHB resistance (Cuthbert et al., 2007). A comparative transcriptomic and metabolomic analysis showed enhanced levels of defence‐related metabolites, such as phenylpropanoids, lignin, glycerophospholipids, flavonoids, fatty acids and terpenoids, whereas transcriptomic analysis identified the significant induction of genes encoding receptor kinases, transcription factors, signalling and mycotoxin detoxification proteins in the recombinant inbred line carrying the Fhb2 resistant allele (Dhokane et al., 2016). Several putative defence‐associated genes [e.g. 4‐Coumarate:CoA ligase (4CL), callose synthase (CS), basic helix loop helix (bHLH041) transcription factor, glutathione S‐transferase (GST), ABC transporter‐4 (ABC4) and cinnamyl alcohol dehydrogenase (CAD)] are located in this QTL region. Therefore, it appears that the detoxification of DON and cell wall reinforcement through the action of Fhb2‐regulated genes restrict the spread of the pathogen within the spike (Dhokane et al., 2016).
In conclusion, the recent cloning of Fhb1 and 2DL (Kage et al., 2017; Rawat et al., 2016) from wheat has shown that much can be learned about the processes involved in host resistance through the cloning of genes providing quantitative FHB resistance.
Comparative Transcriptomics of FHB‐Resistant and FHB‐Susceptible NILs in other Cereals
Similar to wheat, genetic studies conducted on barley and maize have identified QTL regions conditioning resistance to F. graminearum, and NILs have been subsequently generated and subjected to transcriptomic analyses. For instance, transcriptomic analyses of barley NILs carrying contrasting alleles of three QTLs (2Hb8, 2Hb10 and 3Hb6) have shown largely different gene expression patterns. This led to the conclusion that the mode of action of each QTL may be distinct (Jia et al., 2011). More recently, to understand the molecular action of FHB resistance derived from the barley cultivar Chevron, the transcriptomes of the resistant NILs 2Hb8 and 6Hb7 and their susceptible recurrent parental genotypes, M69 and Lacey, respectively, were analysed by RNA‐seq after F. graminearum inoculation (Huang et al., 2016). These analyses suggested that 2Hb8‐mediated resistance is a result of both higher levels of basal defences and their more rapid activation during infection. Similarly, 6Hb7‐mediated resistance in barley appears to be a result of the ability of the host to mount an earlier defence response at the seedling stage. This transcriptomic study also identified hundreds of F. graminearum‐inducible long‐non‐coding RNAs (lncRNAs) with potential roles in the regulation of barley defences during F. graminearum infection (Huang et al., 2016). Transcriptomic analyses comparing lines with and without QTLs (qRfg1 and qRfg2) that provide resistance to stalk rot (Yang et al., 2010) in maize also showed that defence‐associated genes are induced more quickly and more strongly in the resistant than the susceptible NIL (Liu et al., 2016; Ye et al., 2013).
In summary, the rapid accumulation of transcriptomic data enables researchers to compare and contrast responses of different cereal species to F. graminearum. To the best of our knowledge, no QTL conferring F. graminearum resistance has yet been cloned from barley or maize. It will be interesting to test whether the transgenic expression of wheat genes underlying different QTLs in other cereals, or vice versa, would provide increased FHB resistance, as shown previously for wheat expressing a barley UDP‐glucosyltransferase involved in DON detoxification (Li et al., 2015).
Do Different Wheat Tissues Respond Differently to F. graminearum Infection?
Given the ability of F. graminearum to infect different host tissues, one intriguing question is whether different host tissues respond differently to this pathogen. In one recent study, transcriptomic analyses were conducted to identify genes expressed in the infected heads of different developmental stages (e.g. cell division, cell differentiation and grain filling) in the susceptible French wheat cultivar Recital using a wheat NimbleGen microarray that included 39 019 unigenes (Chetouhi et al., 2016). These analyses identified a number of wheat genes that shared either similar or contrasting expression profiles during development and F. graminearum infection (Chetouhi et al., 2016). Host genes involved in polyamine biosynthesis (e.g. agmatine) were up‐regulated in the second developmental stage, during which DON starts to accumulate. Polyamines are known inducers of DON biosynthesis in F. graminearum (Gardiner et al., 2009b) and the induction of these genes by F. graminearum indicates the possibility that the pathogen deliberately activates polyamine production (Gardiner et al., 2010a).
Additional transcriptomic analyses are required to determine whether different host tissues, such as head, stem and roots, respond differently to infection by F. graminearum. Such analyses have been conducted recently in Arabidopsis against a different Fusarium species (Lyons et al., 2015). It is also largely unknown whether QTLs effective against head infections might play a role in protecting other tissues from infection. These studies will potentially reveal new insights into whether similar strategies can be employed to protect different tissues from F. graminearum.
Transcriptomic Analyses of DON‐Induced Host Responses
DON is known to activate host defence gene expression, including the expression of PR genes (Desmond et al., 2008a). Numerous genes encoding ABC transporters and UGTs are activated by F. graminearum and the related DON‐producing pathogen F. pseudograminearum (Desmond et al., 2008b; Hill‐Ambroz et al., 2006; Hofstad et al., 2016; Lulin et al., 2010; Powell et al., 2017b; Steiner et al., 2009; Walter et al., 2008), suggesting that DON detoxification during FHB is an active plant defence strategy.
Transcriptomic analyses of two FHB‐resistant doubled haploid wheat lines, GS‐1‐EM0040 and GS‐1‐EM0168, which contain the Fhb1 locus, as well as their susceptible parental genotype, the Canadian wheat genotype Superb, were conducted to measure systemic responses to spike infection by DON‐producing and non‐producing F. graminearum strains and also by direct DON application (Foroud et al., 2012). TaLTP3 associated with the 5A QTL conferring type 1 FHB resistance showed a high basal expression level in the two resistant genotypes, suggesting that this LTP may be a regulator of systemic defence responses in wheat.
Another transcriptomic study analysed the heads of a toxin‐sensitive wheat cultivar following DON treatment. DON‐induced transcripts were again associated with processes such as detoxification, JA biosynthesis, carbohydrate metabolism and phenylpropanoid metabolism (Walter and Doohan, 2011). DON‐triggered transcriptional changes in barley were investigated after inoculations with either the wild‐type pathogen or tri5 mutant unable to synthesize DON. Host genes involved in DON detoxification and transport, as well as programmed cell death, gene regulation and cytochrome P450s, showed differential expression in plants infected with the wild‐type pathogen (Boddu et al., 2007). Transcriptomic studies also identified UGT‐encoding barley genes that are responsive to exogenous DON and infection by wild‐type F. graminearum, but not the tri5 mutant (Boddu et al., 2006, 2007; Gardiner et al., 2010b). A DON‐sensitive yeast strain was then used to test whether these UGTs could confer DON resistance. These analyses identified HvUGT13248 conferring DON resistance in yeast by converting DON to D3G (3‐β‐d‐glucoside) (Schweiger et al., 2010).
Fusarium graminearum isolates have the capability to produce either 15‐acetyl‐deoxynivalenol (15ADON) or 3‐acetyl‐deoxynivalenol (3ADON). 3ADON‐producing F. graminearum isolates are often more aggressive and produce higher levels of DON than 15ADON‐producing strains (Goswami and Kistler, 2004; Puri and Zhong, 2010; Ward et al., 2008). The question of whether the strains with different DON chemotypes trigger different gene expression profiles on the same wheat genotype was investigated through transcriptomic analyses (Al‐Taweel et al., 2014). Various defence genes, such as UDP‐glucosyltransferases, thaumatin‐like proteins, glucosidases, chitinases and peroxidases, were expressed more strongly in wheat in response to 15ADON‐producing isolates than to those producing 3ADON, suggesting that these latter isolates either do not trigger as strong a defence response as 15ADON‐producing isolates or have the ability to subdue the defence responses of the host (Al‐Taweel et al., 2014).
One of the major outcomes of transcriptomic studies was the identification of DON‐responsive genes, in particular ABC transporters, UGT‐glucosyltransferases potentially involved in DON detoxification. For instance, the barley UDP‐glucosyltransferase HvUGT13248, first identified as a gene inducible by F. graminearum and DON in earlier transcriptomic studies (Boddu et al., 2007; Gardiner et al., 2010b), increases DON tolerance in both yeast and Arabidopsis by converting DON to D3G (Schweiger et al., 2010; Shin et al., 2012). Transgenic expression of HvUGT13248 also provides enhanced FHB resistance in wheat (Li et al., 2015). The Brachypodium orthologue of HvUGT13248, Bradi5g03300, also confers DON tolerance and F. graminearum resistance in Brachypodium (Pasquet et al., 2016).
Microarray experiments conducted on wheat also showed that TaABCC3, an ABC transporter‐encoding gene, was activated by F. graminearum (Jia et al., 2009). Similar microarray experiments conducted on barley using the F. graminearum tri5 mutant showed that ABCC3, a putative orthologue of TaABCC3, was also expressed in a DON‐dependent manner (Boddu et al., 2007). Previously, TaABCC3 has been proposed to be the candidate gene at the Fhb1 locus that provides resistance by mediating DON detoxification (Walter et al., 2008). Although the recent cloning of the Fhb1 locus did not support this proposal, the inhibition of TaABCC3.1 expression through virus‐induced gene silencing (VIGS) increased the toxin sensitivity in treated heads (Walter et al., 2015). It is currently unknown whether TaABCC3 provides FHB resistance. However, these findings suggest that the transgenic expression of genes involved in DON detoxification could be a useful strategy to increase FHB resistance in cereals.
Combining Transcriptomics with Proteomics and Metabolomics
Transcriptomic analyses provide a snapshot of the interaction at a given time point. In addition, not all transcriptional changes observed are translated into proteins. Therefore, the complementation of transcriptomic data with those from other ‘omic’ (metabolomics, proteomics) approaches can be useful (Fig. 2). For instance, metabolomics studies conducted on NILs with and without the 2DL QTL showed that hydroxycinnamic acid amides (HCAs), such as coumaroylagmatine, associated with resistance, were differentially produced between the resistant and susceptible NILs. Subsequent genetic mapping, followed by functional analyses using VIGS and the complementation of the Arabidopsis act mutant, identified the TaACT gene encoding an agmatine coumaroyltransferase as the gene underlying the 2DL QTL (Kage et al., 2017), demonstrating the usefulness of undertaking combinatorial approaches (Fig. 2).
Another transcriptomics–metabolomics approach undertaken in barley identified HvCERK1 encoding a putative chitin elicitor receptor kinase. The silencing of HvCERK1 by VIGS increased the susceptibility to F. graminearum, suggesting that HvCERK1 is a positive regulator of defence responses effective against FHB in barley (Karre et al., 2017). In contrast, metabolomic comparisons of Fhb1 and Qfhs.ifa‐5A NILs after DON treatment did not reveal any QTL‐specific metabolites, although a faster accumulation of various defence‐related metabolites, including phenylpropanoids, amines, HCAs and auxin, was found in NILs carrying the Fhb1 locus (Warth et al., 2015).
In conclusion, the successful cloning of the 2DL QTL using a combined transcriptomic–metabolomic analysis suggests the importance of integrating data from multiple sources when attempting to understand what lies behind 2DL (Fig. 2).
How Does F. graminearum Infect its Host? Does it use Distinct or Similar Strategies when Infecting Different Host Tissues or Genotypes?
Transcriptomic analyses of F. graminearum to identify genes expressed in planta during the development of FHB have been beneficial for the understanding of infection‐related fungal processes (Fig. 2). Lysøe et al. (2011a) identified 355 F. graminearum genes expressed during its interaction with wheat. Functional groups were predicted to be involved in transport, detoxification, primary, secondary and carbohydrate metabolism, and the degradation of polysaccharide compounds. These analyses also identified several putative effector‐encoding genes expressed in wheat during the infection (Lysøe et al., 2011a).
Transcriptomic analyses have been employed to determine whether F. graminearum uses similar or different infection strategies to infect different organs and tissues (Lysøe et al., 2011a; Zhang et al., 2012). The host too shows tissue‐specific responses to F. graminearum infection. An earlier microarray study investigating the gene expression patterns in six different organs (glume, lemma, palea, anther, ovary and rachis) within the wheat spike concluded that each organ showed a distinct expression pattern when responding to F. graminearum infection (Golkari et al., 2007).
In addition to similarities, other studies have revealed both similarities and differences in fungal gene expression patterns in different tissues. For instance, in contrast with their strong expression patterns during head infection, trichothecene biosynthesis genes did not show increased in planta expression during coleoptile infection, suggesting that DON is not required for pathogen virulence during the infection of young seedlings (Zhang et al., 2012). However, DON‐non‐producing mutants were not employed to test this hypothesis. Indeed, other studies have suggested a role for DON during the colonization of wheat stems by F. graminearum and the related pathogens F. pseudograminearum and F. culmorum (Desmond et al., 2008a; Mudge et al., 2006; Powell et al., 2017b; Scherm et al., 2011). Nevertheless, of the 133 F. graminearum genes affecting pathogen virulence during head infection, only 17 showed increased in planta expression during coleoptile infection, suggesting that the infection strategies used by F. graminearum for head and seedling tissue might be somewhat different. Of the seven F. graminearum mutants identified by Zhang et al. (2012) as being required for coleoptile infection, four also showed reduced infection on heads. Together, these analyses show that F. graminearum employs mostly distinct, but also similar, colonization strategies when infecting different wheat tissues.
More recently, using RNA‐seq analyses, very few differentially expressed F. graminearum genes found in both infected spikelet and rachis samples were identified, suggesting that the strategies used by the pathogen when colonizing different tissues might be different. In addition, trichothecene biosynthesis genes found in the TRI gene cluster of F. graminearum were mostly up‐regulated in the pathogen when infecting Fhb1‐resistant NILs, suggesting that the pathogen uses somewhat different strategies when infecting resistant and susceptible genotypes (Hofstad et al., 2016).
Three distinct infection phases with distinguishable patterns of fungal gene expression were identified during the crown rot disease of wheat caused by F. graminearum (Stephens et al., 2008). Similar phasic profiles of gene expression were also observed in a coleoptile infection assay (Zhang et al., 2012). The comparison of fungal gene expression profiles of F. graminearum used during crown and head infection suggests that similar processes are involved during crown rot infection and the early stages of head infection by this pathogen (Stephens et al., 2008). Indeed, during both stem base and head infection, a period of asymptomatic disease development has been observed, suggesting that the pathogen acts as a hemibiotroph, although, in the different diseases, the relative duration of asymptomatic growth, and hence phases of gene expression, seem to be different (Brown et al., 2010).
Does F. graminearum Employ Similar Strategies when Infecting Different Host Species?
Despite their close genetic similarities, different cereal species often produce different metabolites and/or phytoalexins to defend themselves. For instance, the defence‐associated metabolite DIMBOA is produced by wheat and maize, but not by barley (Dutartre et al., 2012). Similarly, hordatines, another class of defence compounds, are produced by barley, but not wheat and maize. Therefore, one intriguing question is whether F. graminearum uses similar or different strategies when infecting different hosts that can produce different defence compounds. To answer this question, the transcriptome of the same F. graminearum strain has been comparatively analysed on wheat, barley and maize (Harris et al., 2016). These analyses revealed that some F. graminearum genes are expressed during the colonization of all three hosts, whereas others are only expressed when infecting a single host. In addition, the transcriptome of F. graminearum on wheat was more similar to that on barley than that on maize. In particular, fungal genes expressed during the colonization of maize kernels by F. graminearum were more different than those expressed during the colonization of wheat and barley heads. For instance, genes involved in transport and secondary metabolism were expressed in a host‐specific manner. In addition, the functional categories of genes induced in each host were different. Fusarium graminearum‐induced genes in wheat were enriched for processes involved in protein degradation, gene regulation and signalling, whereas, in maize, enrichment was observed for carbon metabolism, cell rescue and defence (Harris et al., 2016). These host‐specific responses to infection indicate that F. graminearum has the ability to adapt to a range of different host environments (Harris et al., 2016).
Although the transcriptomic studies reviewed here have started to reveal new clues with regard to the species specificity of the F. graminearum infection process, available pathogen mutants with altered virulence on wheat should be tested on other cereals to determine whether similar or different infection strategies are used by the pathogen when infecting different host species.
Fusarium graminearum Transcriptome during Biotrophic Versus Necrotrophic and Pathogenic Versus Saprophytic Lifestyle
As indicated above, the infection by F. graminearum remains non‐symptomatic for a period before the development of a necrotrophic phase during both head and stem infections (Brown et al., 2010, 2011; Stephens et al., 2008), suggesting that F. graminearum is a hemibiotrophic pathogen. Indeed, the genes associated with the non‐symptomatic stage are involved in primary metabolic pathways, whereas the genes involved in cell wall degradation dominate the later growth stage (Zhang et al., 2012). Nevertheless, once necrosis develops at the infected tissue, the pathogen further colonizes the dead tissue, most probably by switching to a saprophytic lifestyle. It is therefore expected that pathogen genes expressed during different phases of disease development might be different. Indeed, the comparison of the transcriptomes of F. graminearum on living and dead tissues suggests that the pathogen uses host signals to modulate the expression of many genes (Boedi et al., 2016). Similarly, some fungal genes are repressed by host signals (Boedi et al., 2016). In addition, despite significant fungal growth, the production of DON on dead tissue remains low (Boedi et al., 2016), supporting previous suggestions that the host plays a role in triggering DON biosynthesis in the pathogen (Gardiner et al., 2009b; Mudge et al., 2006). The knowledge to be gained on the lifestyle of F. graminearum potentially will aid in the design of more effective disease control options.
Transcriptomic Analysis Reveals New Insights into Don Biosynthesis in F. graminearum
The mycotoxin DON is not only an effector required for pathogen virulence on wheat, but also modulates the interaction of F. graminearum with its broader environment (Audenaert et al., 2013). Genetic analyses undertaken in F. graminearum have identified many genes involved in virulence, mainly through the analysis of knock‐out mutants. Some of these genes also affect toxin biosynthesis (reviewed by Kazan et al., 2012). Comparative analyses of the transcriptomes of such mutants and their respective wild‐type strains provide a useful tool to understand the mechanisms involved in toxin biosynthesis and pathogen virulence (Fig. 2). The transcription factors TRI6 and TRI10 regulate the expression of trichothecene biosynthesis genes in F. graminearum (Gardiner et al., 2009a; Seong et al., 2009). The transcriptomic analysis of tri6 and tri10 mutants revealed significant alterations in the expression of a large number of genes which included, in addition to known genes, many genes that have not been implicated previously in toxin production. These transcriptomic studies also facilitated the identification of cis‐acting elements enriched at the promoters of genes regulated by these transcription factors, which, in turn, helped to further define the regulons of these transcription factors (Gardiner et al., 2009a; Seong et al., 2009).
As stated earlier, various polyamine compounds, such as putrescine, arginine and cadaverine, produced by the host plant during infection, are known to induce DON biosynthesis in F. graminearum (Gardiner et al., 2009a). Interestingly, F. graminearum infection activates the genes involved in host polyamine biosynthesis, suggesting that the pathogen hijacks this response to elevate the production of DON which acts as a virulence factor (Gardiner et al., 2010a). To understand the molecular mechanisms of amine‐induced DON biosynthesis in F. graminearum, the transcriptome of F. graminearum under DON‐inducing and non‐inducing conditions (e.g. presence or absence of agmatine) was comparatively analysed. This analysis revealed large numbers of amine‐regulated pathogen genes involved in secondary metabolite biosynthesis. The expression of some of these genes was dependent on TRI6. Interestingly, knock‐out mutants generated for two of these genes showed significantly increased DON levels accompanied by increased virulence (Gardiner et al., 2009a). This unexpected finding revealed through the combined use of transcriptomic and genetic analyses suggests that F. graminearum has the potential to increase its virulence through mutation of the genes that act as negative regulators of toxin biosynthesis (Gardiner et al., 2009a).
Comparative analysis of the transcriptome of two F. graminearum strains differing in trichothecene toxin chemotype, 3ADON and 15ADON, showed that more than 1500 genes are differentially expressed by more than two‐fold between these strains, despite their otherwise close similarities (Walkowiak et al., 2015). Another RNA‐seq study conducted on strains with different DON chemotypes identified members of the C2H2 transcription factor gene family and a set of transporter‐encoding genes differentially expressed in 3ADON strains (Puri et al., 2016). The expression of TRI genes involved in DON biosynthesis also seems to be higher in 3ADON than 15ADON and nivalenol (NIV) strains (Amarasinghe and Fernando, 2016). Additional analyses are probably required to determine the relevance of these differentially expressed genes in pathogen virulence.
Transcriptomic Data and Towards a Systems‐Level Understanding of the Lifestyle of a Cosmopolitan Pathogen
As reviewed here, relatively large amounts of transcriptomic data generated from host–F. graminearum interactions over the last decade are now available. In addition, a relatively large number of F. graminearum genes affecting pathogen virulence, toxin biosynthesis and development (Blum et al., 2016; Kim et al., 2015; Lysøe et al., 2011b; Qi et al., 2006; Sikhakolli et al., 2012; Son et al., 2011; Song et al., 2014) have been functionally characterized (Kazan et al., 2012). Exploitation of these resources to build models can provide new insights, not only into the biology of this pathogen, but also for the development of new protection strategies. Liu et al. (2010) have developed a network approach to predict the pathogen genes potentially involved in virulence‐related functions based on predicted protein interactions and gene expression data available at the time. The so‐called ‘pathogenic network’ was constructed by identifying differentially expressed genes predicted to interact with at least two other genes with known roles in pathogen virulence. These analyses identified two network modules enriched for genes involved in G‐protein‐coupled receptor and mitogen‐activated protein kinase (MAPK) signalling pathways. The genes predicted to be associated with virulence were then validated by checking whether orthologous genes in other pathogenic fungi, such as Magnaporthe oryzae, Ustilago maydis and Botrytis cinerea, were also associated with virulence (Liu et al., 2010). As predicted, these analyses identified several F. graminearum orthologues associated with virulence in these pathogens, suggesting that the predictive power of this approach is reasonably strong (Liu et al., 2010).
More recently, using Bayesian network inference, the first gene regulatory network (Fig. 2) for F. graminearum has been constructed from a 166 transcriptomic dataset available at PLEXdb (Plant Expression Database) (Guo et al., 2016). The network model, which takes the correlations observed for expression levels of regulators and their potential targets into consideration, identified the following regulatory modules in F. graminearum based on the following significantly enriched physiological processes: cell cycle and development; detoxification and secondary metabolism; cytochrome P450‐mediated detoxification processes; metabolism and RNA processing; protein synthesis; transcription and cell transport; stress response and cell differentiation; carbohydrate metabolism and detoxification; and metabolism and rRNA processing. Each of these modules contains 8–21 master regulators predicted to modulate the expression of large numbers (2743–6807) of target genes in F. graminearum. Given their broad regulatory effects, these master regulators can be potential targets for genetic (e.g. host‐derived RNAi) or chemical (e.g. fungicide applications) interference. The compartmentalization of the F. graminearum genome into a conserved and slowly evolving genome and a variable and fast evolving genome has been demonstrated previously (Ma et al., 2010, 2013). The network analysis also showed that master regulators, through their target genes, are associated with either the core or variable genome, suggesting that, similar to the genome, the gene regulatory networks are also compartmentalized in F. graminearum (Guo et al., 2016).
In summary, the re‐analysis of large amounts of diverse transcriptomic data has allowed the construction of regulatory networks (Figs 1 and 2) and has revealed new insights into pathogen biology. It will be interesting to see whether the results from these analyses will be exploited to develop novel pathogen interference strategies.
Conclusions and Future Prospects
The studies reviewed in this article clearly show that transcriptomic analyses, which are summarized in Fig. 1 in terms of the general classes of genes altered in either the host or the pathogen, have been very useful in advancing our overall understanding of cereal–F. graminearum interactions.
In wheat alone, many QTLs conferring varying degrees of FHB resistance have been identified (Buerstmayr et al., 2009). So far, transcriptomic analyses have helped to better understand the potential mechanisms behind QTL‐mediated defence against F. graminearum for a few loci with relatively large effects. However, even for some of the better studied QTLs, such as Fhb1, results from different transcriptomic analyses are somewhat difficult to compare because of the use of different germplasm, methodology (inoculation, sampling time, different pathogen isolate), expression platform and data analysis methods (Nussbaumer et al., 2015). Many other QTLs have not yet been subjected to detailed transcriptomic analyses, most probably because of their relatively small effects on FHB resistance and/or the unavailability of suitable genetic resources, such as NILs. However, new approaches, such as expressed QTL (eQTL) analyses, which combine transcriptomic analyses with QTL mapping, are also being developed to identify genomic regions potentially harbouring genes affecting FHB resistance in the host. In a recent eQTL approach, loci affecting the transcript abundances of wheat genes responding to F. graminearum infection have been mapped using a double haploid wheat population segregating for FHB resistance genes (Samad‐Zamini et al., 2017) (Fig. 2). These analyses identified a novel QTL located on chromosome 6A, as well as the previously identified QTLs Fhb1 and Qfhs.ifa‐5A. Furthermore, three major regulatory hotspots that do not co‐localize with known QTLs have been identified on wheat chromosomes 2B, 4A and 5A (Samad‐Zamini et al., 2017). The cloning of genes underlying these new eQTLs, as well as those identified previously, is necessary to better understand their possible modes of action.
Tetraploid (durum) wheats are more susceptible to FHB than hexaploid wheats. However, only limited transcriptomic analyses have been undertaken on durum wheats (Soresi et al., 2015). Interestingly, in addition to having its unique resistance, durum and bread wheats are known to share many QTLs conferring FHB resistance (reviewed by Prat et al., 2014). Fhb1 has recently been successfully introgressed from hexaploid to durum wheat (Prat et al., 2017). Conducting transcriptomic analyses on Fhb1‐containing durum wheat would reveal additional clues with regard to the molecular mode of action of this QTL in the durum background. In addition, most transcriptomic analyses have focused on what makes the host plant resistant to F. graminearum. An alternative line of inquiry would be what makes the host plant susceptible to this pathogen (Garvin et al., 2015). Host susceptibility genes can be disabled through mutational (Fitzgerald et al., 2015b) or Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9)‐mediated (Wang et al., 2014) approaches. This obviously requires a better understanding of both host defence and virulence‐associated processes in the pathogen. The data available from F. graminearum transcriptomic analyses have started to provide the basic information needed for the construction of a systems‐level understanding of the biology of this pathogen (Guo et al., 2016). The master regulators controlling virulence‐associated processes in the pathogen can be useful targets for interference through genetic and chemical approaches. For instance, the feasibility of increased disease resistance obtained through host‐induced silencing of F. graminearum genes has been demonstrated recently (Cheng et al., 2015; Koch et al., 2013).
As stated earlier, a three‐stage root infection process, resembling the process used by root pathogens, has been described for F. graminearum (Wang et al., 2015). However, no transcriptomic analyses have been conducted to dissect the genes expressed in the roots of the host plant and in the pathogen during this infection process.
As reviewed here, the interaction between cereals and F. graminearum is one of the most widely studied transcriptomic analyses of all plant–microbe interactions. Although many candidate host genes have been identified through transcriptomic analyses, only a few such genes have so far been functionally characterized through over‐expression and silencing in host plants. Genetic transformation methods for cereals are beyond the reach of many laboratories. VIGS seems to be a valuable tool for gene function analysis (Lee et al., 2012; Perochon et al., 2015). Cereal mutants with disabled susceptibility gene products can also be exploited to generate resistant germplasm to be incorporated into breeding programmes. However, as a result of the polyploid nature of the wheat genome, not all single subgenome (A, B or D) mutants show detectable phenotypes (Fitzgerald et al., 2015b). Another complexity associated with the polyploid wheat genome is that most transcriptomic studies are unable to distinguish whether the differential gene expression observed affects all three homoeologues (homologues of the same gene on different subgenomes) equally. Nevertheless, recent studies that were able to differentiate a substantial percentage of homoeologues indicated that the D genome of hexaploid wheat preferentially contributes to gene expression when responding to F. graminearum and F. pseudograminearum challenge (Nussbaumer et al., 2015; Powell et al., 2017a).
The large and complex genomes of wheat and other cereals seem to be another factor that has impeded genomic studies (Brewer and Hammond‐Kosack, 2015). For this reason, in the past, the interaction between F. graminearum and the model dicot host Arabidopsis has been used to understand host resistance to F. graminearum (Brewer and Hammond‐Kosack, 2015). The genetic and genomic resources available in the model monocot plant Brachypodium distachyon, which is known to be susceptible to many cereal pathogens, including F. graminearum (Fitzgerald et al., 2015a; Pasquet et al., 2014, 2016; Peraldi et al., 2011), can also be exploited to overcome potential difficulties encountered as a result of the genetic complexity of cereal genomes, and to identify host genes regulating disease/toxin resistance or susceptibility. This knowledge can then be applied to cereal crops (Fitzgerald et al., 2015a; Schweiger et al., 2013b). It is also worthwhile to stress that not all genes involved in disease resistance are responsive to pathogen infection. In these cases, transcriptomic analyses offer little value. For instance, a recent report has shown that genes encoding ABA receptors, despite their low or no responsiveness to pathogen infection, can make wheat and Brachypodium more susceptible to F. graminearum (Gordon et al., 2016).
In conclusion, as reviewed here, transcriptomic analyses have significantly advanced our understanding of this economically important plant–microbe interaction. Figure 2 illustrates the approaches discussed in this article that have employed transcriptomic analyses. Briefly, from the host side, QTLs conferring partial resistance to F. graminearum can be integrated into adapted backgrounds through marker‐assisted selection. The identification of QTLs can also make the construction of NILs that differ by FHB resistance possible. Transcriptomic analyses comparing gene expression patterns of resistant and susceptible NILs reveal candidate genes that can be modified through traditional or molecular approaches. Similarly, transcriptomic analyses conducted on the pathogen reveal new insights into the potential virulence mechanisms employed. Together, this knowledge makes significant contributions towards the development of effective plant protection strategies against this pathogen.
Acknowledgements
We apologize to colleagues whose relevant work could not be reviewed because of space restrictions. We thank Jason Carere and Friday Obanor for providing the photographs of diseased plants used in Fig. 2, and anonymous reviewers for useful comments on the manuscript.
References
- Al‐Taweel, K. , Fernando, W.G. and Brûlé‐Babel, A.L. (2014) Transcriptome profiling of wheat differentially expressed genes exposed to different chemotypes of Fusarium graminearum . Theor. Appl. Genet. 127, 1703–1718. [DOI] [PubMed] [Google Scholar]
- Amarasinghe, C.C. and Fernando, W.G. (2016) Comparative analysis of deoxynivalenol biosynthesis related gene expression among different chemotypes of Fusarium graminearum in spring wheat. Front Microbiol. 7, 1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audenaert, K. , Vanheule, A. , Höfte, M. and Haesaert, G. (2013) Deoxynivalenol: a major player in the multifaceted response of Fusarium to its environment. Toxins, 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, G. and Shaner, G. (2004) Management and resistance in wheat and barley to fusarium head blight. Annu. Rev. Phytopathol. 42, 135–161. [DOI] [PubMed] [Google Scholar]
- Bernardo, A. , Bai, G. , Guo, P. , Xiao, K. , Guenzi, A.C. and Ayoubi, P. (2007) Fusarium graminearum‐induced changes in gene expression between Fusarium head blight‐resistant and susceptible wheat cultivars. Funct. Integr. Genomics, 7, 69–77. [DOI] [PubMed] [Google Scholar]
- Blum, A. , Benfield, A.H. , Stiller, J. , Kazan, K. , Batley, J. and Gardiner, D.M. (2016) High‐throughput FACS‐based mutant screen identifies a gain‐of‐function allele of the Fusarium graminearum adenylyl cyclase causing deoxynivalenol over‐production. Fungal Genet. Biol. 90, 1–11. [DOI] [PubMed] [Google Scholar]
- Boddu, J. , Cho, S. , Kruger, W.M. and Muehlbauer, G.J. (2006) Transcriptome analysis of the barley–Fusarium graminearum interaction. Mol. Plant–Microbe Interact. 19, 407–417. [DOI] [PubMed] [Google Scholar]
- Boddu, J. , Cho, S. and Muehlbauer, G.J. (2007) Transcriptome analysis of trichothecene‐induced gene expression in barley. Mol. Plant–Microbe Interact. 20, 1364–1375. [DOI] [PubMed] [Google Scholar]
- Boedi, S. , Berger, H. , Sieber, C. , Münsterkötter, M. , Maloku, I. , Warth, B. , Sulyok, M. , Lemmens, M. , Schuhmacher, R. , Güldener, U. and Strauss, J. (2016) Comparison of Fusarium graminearum transcriptomes on living or dead wheat differentiates substrate‐responsive and defence‐responsive genes. Front Microbiol. 7, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, H.C. and Hammond‐Kosack, K.E. (2015) Host to a stranger: Arabidopsis and Fusarium ear blight. Trends Plant Sci. 20, 651–663. [DOI] [PubMed] [Google Scholar]
- Brown, N.A. , Urban, M. , van de Meene, A.M.L. and Hammond‐Kosack, K.E. (2010) The infection biology of Fusarium graminearum: defining the pathways of spikelet to spikelet colonisation in wheat ears. Fungal Biol. 114, 555–571. [DOI] [PubMed] [Google Scholar]
- Brown, N.A. , Bass, C. , Baldwin, T.K. , Chen, H. , Massot, F. , Carion, P.W.C. , Urban, M. , van de Meene, A.M.L. and Hammond‐Kosack, K.E. (2011) Characterisation of the Fusarium graminearum wheat floral interaction. J. Pathog. 2011, e626345, 9 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerstmayr, H. , Steiner, B. , Hartl, L. , Griesser, M. , Angerer, N. , Lengauer, D. , Miedaner, T. , Schneider, B. and Lemmens, M. (2003) Molecular mapping of QTLs for Fusarium head blight resistance in spring wheat. II. Resistance to fungal penetration and spread. Theor. Appl. Genet. 107, 503–508. [DOI] [PubMed] [Google Scholar]
- Buerstmayr, H. , Ban, T. and Anderson, J.A. (2009) QTL mapping and marker‐assisted selection for Fusarium head blight resistance in wheat: a review. Plant Breed. 128, 1–26. [Google Scholar]
- Buhrow, L.M. , Cram, D. , Tulpan, D. , Foroud, N.A. and Loewen, M.C. (2016) Exogenous abscisic acid and gibberellic acid elicit opposing effects on Fusarium graminearum infection in wheat. Phytopathology, 106, 986–996. [DOI] [PubMed] [Google Scholar]
- Chen, X. , Steed, A. , Travella, S. , Keller, B. and Nicholson, P. (2009) Fusarium graminearum exploits ethylene signalling to colonize dicotyledonous and monocotyledonous plants. New Phytol. 182, 975–983. [DOI] [PubMed] [Google Scholar]
- Cheng, W. , Song, X.S. , Li, H.P. , Cao, L.H. , Sun, K. , Qiu, X.L. , Xu, Y.B. , Yang, P. , Huang, T. , Zhang, J.B. , Qu, B. and Liao, Y.C. (2015) Host‐induced gene silencing of an essential chitin synthase gene confers durable resistance to Fusarium head blight and seedling blight in wheat. Plant Biotechnol J. 13, 1335–1345. [DOI] [PubMed] [Google Scholar]
- Chetouhi, C. , Bonhomme, L. , Lasserre‐Zuber, P. , Cambon, F. , Pelletier, S. , Renou, J.P. and Langin, T. (2016) Transcriptome dynamics of a susceptible wheat upon Fusarium head blight reveals that molecular responses to Fusarium graminearum infection fit over the grain development processes. Funct. Integr. Genomics, 16, 183–201. [DOI] [PubMed] [Google Scholar]
- Close, T.J. , Wanamaker, S.I. , Caldo, R.A. , Turner, S.M. , Ashlock, D.A. , Dickerson, J.A. , Wing, R.A. , Muehlbauer, G.J. , Kleinhofs, A. and Wise, R.P. (2004) A new resource for cereal genomics: 22K barley GeneChip comes of age. Plant Physiol. 134, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo, C.A. , Güldener, U. , Xu, J.R. , Trail, F. , Turgeon, B.G. , Di Pietro, A. , Walton, J.D. , Ma, L.J. , Baker, S.E. , Rep, M. , Adam, G. , Antoniw, J. , Baldwin, T. , Calvo, S. , Chang, Y.L. , Decaprio, D. , Gale, L.R. , Gnerre, S. , Goswami, R.S. , Hammond‐Kosack, K. , Harris, L.J. , Hilburn, K. , Kennell, J.C. , Kroken, S. , Magnuson, J.K. , Mannhaupt, G. , Mauceli, E. , Mewes, H.W. , Mitterbauer, R. , Muehlbauer, G. , Münsterkötter, M. , Nelson, D. , O'Donnell, K. , Ouellet, T. , Qi, W. , Quesneville, H. , Roncero, M.I. , Seong, K.Y. , Tetko, I.V. , Urban, M. , Waalwijk, C. , Ward, T.J. , Yao, J. , Birren, B.W. and Kistler, H.C. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science, 317, 1400–1402. [DOI] [PubMed] [Google Scholar]
- Cuthbert, P.A. , Somers, D.J. , Thomas, J. , Cloutier, S. and Brulé‐Babel, A. (2006) Fine mapping Fhb1, a major gene controlling fusarium head blight resistance in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 112, 1465–1472. [DOI] [PubMed] [Google Scholar]
- Cuthbert, P.A. , Somers, D.J. and Brulé‐Babel, A. (2007) Mapping of Fhb2 on chromosome 6BS: a gene controlling Fusarium head blight field resistance in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 114, 429–437. [DOI] [PubMed] [Google Scholar]
- Dean, R. , Van Kan, J.A. , Pretorius, Z.A. , Hammond‐Kosack, K.E. , Di Pietro, A. , Spanu, P.D. , Rudd, J.J. , Dickman, M. , Kahmann, R. , Ellis, J. and Foster, G.D. (2012) The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond, O.J. , Manners, J.M. , Stephens, A.E. , MacLean, D.J. , Schenk, P.M. , Gardiner, D.M. , Munn, A.L. and Kazan, K. (2008a) The Fusarium mycotoxin deoxynivalenol elicits hydrogen peroxide production, programmed cell death and defence responses in wheat. Mol. Plant. Pathol. 9, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond, O.J. , Manners, J.M. , Schenk, P.M. , Maclean, D.J. and Kazan, K. (2008b) Gene expression analysis of the wheat response to infection by Fusarium pseudograminearum . Physiol. Mol. Plant Pathol. 73, 40–47. [Google Scholar]
- Dhokane, D. , Karre, S. , Kushalappa, A.C. and McCartney, C. (2016) Integrated metabolo‐transcriptomics reveals Fusarium Head Blight candidate resistance genes in wheat QTL‐Fhb2. PLoS One, 11, e0155851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, L. , Xu, H. , Yi, H. , Yang, L. , Kong, Z. , Zhang, L. , Xue, S. , Jia, H. and Ma, Z. (2011) Resistance to hemi‐biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS One, 6, e19008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutartre, L. , Hilliou, F. and Feyereisen, R. (2012) Phylogenomics of the benzoxazinoid biosynthetic pathway of Poaceae: gene duplications and origin of the Bx cluster. BMC Evol. Biol. 12, 64–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CONTAM Panel (EFSA Panel on Contaminants in the Food Chain) (2013) Statement on the risks for public health related to a possible increase of the maximum level of deoxynivalenol for certain semi‐processed cereal products. EFSA J. 11, 3490. [Google Scholar]
- Erayman, M. , Turktas, M. , Akdogan, G. , Gurkok, T. , Inal, B. , Ishakoglu, E. , Ilhan, E. and Unver, T. (2015) Transcriptome analysis of wheat inoculated with Fusarium graminearum . Front. Plant Sci. 6, 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, T.L. , Powell, J.J. , Schneebeli, K. , Hsia, M.M. , Gardiner, D.M. , Bragg, J.N. , McIntyre, C.L. , Manners, J.M. , Ayliffe, M. , Watt, M. , Vogel, J.P. , Henry, R.J. and Kazan, K. (2015a) Brachypodium as an emerging model for cereal–pathogen interactions. Ann. Bot. 115, 717–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, T.L. , Powell, J.J. , Stiller, J. , Weese, T.L. , Abe, T. , Zhao, G. , Jia, J. , McIntyre, C.L. , Li, Z. , Manners, J.M. and Kazan, K. (2015b) An assessment of heavy ion irradiation mutagenesis for reverse genetics in wheat (Triticum aestivum L.). PLoS One, 10, e0117369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroud, N.A. , Ouellet, T.A. , Laroche, A. , Oosterveen, B. , Jordan, M.C. , Ellis, B.E. and Eudes, F. (2012) Differential transcriptome analyses of three wheat genotypes reveal different host response pathways associated with Fusarium head blight and trichothecene resistance. Plant Pathol. 61, 296–314. [Google Scholar]
- Gardiner, D.M. , Kazan, K. and Manners, J.M. (2009a) Novel genes of Fusarium graminearum that negatively regulate deoxynivalenol production and virulence. Mol. Plant–Microbe Interact. 22, 1588–1600. [DOI] [PubMed] [Google Scholar]
- Gardiner, D.M. , Kazan, K. and Manners, J.M. (2009b) Nutrient profiling reveals potent inducers of trichothecene biosynthesis in Fusarium graminearum . Fungal Genet. Biol. 46, 604–613. [DOI] [PubMed] [Google Scholar]
- Gardiner, D.M. , Kazan, K. , Praud, S. , Torney, F.J. , Rusu, A. and Manners, J.M. (2010a) Early activation of wheat polyamine biosynthesis during Fusarium head blight implicates putrescine as an inducer of trichothecene mycotoxin production. BMC Plant Biol. 10, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner, S.A. , Boddu, J. , Berthiller, F. , Hametner, C. , Stupar, R.M. , Adam, G. and Muehlbauer, G.J. (2010b) Transcriptome analysis of the barley–deoxynivalenol interaction: evidence for a role of glutathione in deoxynivalenol detoxification. Mol. Plant–Microbe Interact. 23, 962–976. [DOI] [PubMed] [Google Scholar]
- Garvin, D.F. , Porter, H. , Blankenheim, Z.J. , Chao, S. and Dill‐Macky, R. (2015) A spontaneous segmental deletion from chromosome arm 3DL enhances Fusarium head blight resistance in wheat. Genome, 58, 479–488. [DOI] [PubMed] [Google Scholar]
- Glenn, A.E. , Hinton, D.M. , Yates, I.E. and Bacon, C.W. (2001) Detoxification of corn antimicrobial compounds as the basis for isolating Fusarium verticillioides and some other Fusarium species from corn. Appl. Environ. Microbiol. 67, 2973–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkari, S. , Gilbert, J. , Prashar, S. and Procunier, J.D. (2007) Microarray analysis of Fusarium graminearum‐induced wheat genes: identification of organ‐specific and differentially expressed genes. Plant Biotechnol. J. 5, 38–49. [DOI] [PubMed] [Google Scholar]
- Golkari, S. , Gilbert, J. , Ban, T. and Procunier, J.D. (2009) QTL‐specific microarray gene expression analysis of wheat resistance to Fusarium head blight in Sumai‐3 and two susceptible NILs. Genome, 52, 409–418. [DOI] [PubMed] [Google Scholar]
- Gordon, C.S. , Rajagopalan, N. , Risseeuw, E.P. , Surpin, M. , Ball, F.J. , Barber, C.J. , Buhrow, L.M. , Clark, S.M. , Page, J.E. , Todd, C.D. , Abrams, S.R. and Loewen, M.C. (2016) Characterization of Triticum aestivum abscisic acid receptors and a possible role for these in mediating Fusarium head blight susceptibility in wheat. PLoS One, 11, e0164996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami, R.S. and Kistler, H.C. (2004) Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525. [DOI] [PubMed] [Google Scholar]
- Gottwald, S. , Samans, B. , Lück, S. and Friedt, W. (2012) Jasmonate and ethylene dependent defence gene expression and suppression of fungal virulence factors: two essential mechanisms of Fusarium head blight resistance in wheat?. BMC Genomics, 13, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guangsheng, Y. , Zhiming, Z. , Kui, Z. , Maojun, Z. , Yaou, S. and Guangtang, P. (2012) Large‐scale identification of differentially expressed genes in maize inbreds susceptible and resistant to Fusarium ear rot. Plant Omics J. 5, 471–475. [Google Scholar]
- Güldener, U. , Seong, K.Y. , Boddu, J. , Cho, S. , Trail, F. , Xu, J.R. , Adam, G. , Mewes, H.W. , Muehlbauer, G.J. and Kistler, H.C. (2006) Development of a Fusarium graminearum Affymetrix GeneChip for profiling fungal gene expression in vitro and in planta. Fungal Genet. Biol. 43, 316–325. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Zhao, G. , Xu, J.R. , Kistler, H.C. , Gao, L. and Ma, L.J. (2016) Compartmentalized gene regulatory network of the pathogenic fungus Fusarium graminearum . New Phytol. 211, 527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzehzarghani, H. , Paranidharan, V. , Abu‐Nada, Y. , Kushalappa, A.C. , Mamer, O. and Somers, D. (2008) Metabolic profiling to discriminate wheat near isogenic lines, with quantitative trait loci at chromosome 2DL, varying in resistance to fusarium head blight. Can. J. Plant Sci. 88, 789–797. [Google Scholar]
- Harris, L.J. , Balcerzak, M. , Johnston, A. , Schneiderman, D. and Ouellet, T. (2016) Host‐preferential Fusarium graminearum gene expression during infection of wheat, barley, and maize. Fungal Biol. 120, 111–123. [DOI] [PubMed] [Google Scholar]
- Hill‐Ambroz, K. , Webb, C.A. , Matthews, A.R. , Li, W.L. , Gill, B.S. and Fellers, J.P. (2006) Expression analysis and physical mapping of a cDNA library of Fusarium head blight infected wheat spikes. Crop Sci. 46, S15–S26. [Google Scholar]
- Hofstad, A.N. , Nussbaumer, T. , Akhunov, E. , Shin, S. , Kugler, K.G. , Kistler, H.C. , Mayer, K.F. and Muehlbauer, G.J. (2016) Examining the transcriptional response in wheat Fhb1 near‐isogenic lines to infection and deoxynivalenol treatment. Plant Genome, 9, doi: 10.3835/plantgenome2015.05.0032. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Li, L. , Smith, K.P. and Muehlbauer, G.J. (2016) Differential transcriptomic responses to Fusarium graminearum infection in two barley quantitative trait loci associated with Fusarium head blight resistance. BMC Genomics, 17, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, H. , Cho, S. and Muehlbauer, G.J. (2009) Transcriptome analysis of a wheat near‐isogenic line pair carrying Fusarium head blight‐resistant and ‐susceptible alleles. Mol. Plant–Microbe Interact. 22, 1366–1378. [DOI] [PubMed] [Google Scholar]
- Jia, H. , Millett, B.P. , Cho, S. , Bilgic, H. , Xu, W.W. , Smith, K.P. and Muehlbauer, G.J. (2011) Quantitative trait loci conferring resistance to Fusarium head blight in barley respond differentially to Fusarium graminearum infection. Funct. Integr. Genomics, 11, 95–102. [DOI] [PubMed] [Google Scholar]
- Kage, U. , Karre, S. , Kushalappa, A.C. and McCartney, C. (2017) Identification and characterization of a fusarium head blight resistance gene TaACT in wheat QTL‐2DL. Plant Biotechnol J. 15, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karre, S. , Kumar, A. , Dhokane, D. and Kushalappa, A.C. (2017) Metabolo‐transcriptome profiling of barley reveals induction of chitin elicitor receptor kinase gene (HvCERK1) conferring resistance against Fusarium graminearum . Plant Mol. Biol. 93, 247–267. [DOI] [PubMed] [Google Scholar]
- Kazan, K. and Lyons, R. (2014) Intervention of phytohormone pathways by pathogen effectors. Plant Cell, 26, 2285–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazan, K. , Gardiner, D.M. and Manners, J.M. (2012) On the trail of a cereal killer: recent advances in Fusarium graminearum pathogenomics and host resistance. Mol. Plant Pathol. 13, 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle, A.J. , Carere, J. , Batley, J. , Benfield, A.H. , Manners, J.M. , Kazan, K. and Gardiner, D.M. (2015) A γ‐lactamase from cereal infecting Fusarium spp. catalyses the first step in the degradation of the benzoxazolinone class of phytoalexins. Fungal Genet. Biol. 83, 1–9. [DOI] [PubMed] [Google Scholar]
- Kim, H.K. , Jo, S.M. , Kim, G.Y. , Kim, D.W. , Kim, Y.K. and Yun, S.H. (2015) A large‐scale functional analysis of putative target genes of mating‐type loci provides insight into the regulation of sexual development of the cereal pathogen Fusarium graminearum . PLoS Genet. 11, e1005486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, A. , Kumar, N. , Weber, L. , Keller, H. , Imani, J. and Kogel, K.H. (2013) Host‐induced gene silencing of cytochrome P450 lanosterol C14α‐demethylase‐encoding genes confers strong resistance to Fusarium species. Proc . Natl. Acad. Sci. USA, 110, 19 324–19 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, L. , Ohm, H.W. and Anderson, J.M. (2007) Expression analysis of defense‐related genes in wheat in response to infection by Fusarium graminearum . Genome, 50, 1038–1048. [DOI] [PubMed] [Google Scholar]
- Kosaka, A. , Manickavelu, A. , Kajihara, D. , Nakagawa, H. and Ban, T. (2015) Altered gene expression profiles of wheat genotypes against Fusarium head blight. Toxins, 7, 604–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugler, K.G. , Siegwart, G. , Nussbaumer, T. , Ametz, C. , Spannagl, M. , Steiner, B. , Lemmens, M. , Mayer, K.F. , Buerstmayr, H. and Schweiger, W. (2013) Quantitative trait loci‐dependent analysis of a gene co‐expression network associated with Fusarium head blight resistance in bread wheat (Triticum aestivum L.). BMC Genomics, 14, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, W.S. , Hammond‐Kosack, K.E. and Kanyuka, K. (2012) Barley stripe mosaic virus‐mediated tools for investigating gene function in cereal plants and their pathogens: virus‐induced gene silencing, host‐mediated gene silencing, and virus‐mediated overexpression of heterologous protein. Plant . Physiol. 160, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens, M. , Scholz, U. , Berthiller, F. , Dall'asta, C. , Koutnik, A. , Schuhmacher, R. , Adam, G. , Buerstmayr, H. , Mesterházy, A. , Krska, R. and Ruckenbauer, P. (2005) The ability to detoxify the mycotoxin deoxynivalenol colocalizes with a major quantitative trait locus for Fusarium head blight resistance in wheat. Mol. Plant–Microbe Interact. 18, 1318–1324. [DOI] [PubMed] [Google Scholar]
- Li, G.L. and Yen, Y. (2008) Jasmonate and ethylene signaling pathway may mediate Fusarium head blight resistance in wheat. Crop Sci. 48, 1888–1896. [Google Scholar]
- Li, X. , Shin, S. , Heinen, S. , Dill‐Macky, R. , Berthiller, F. , Nersesian, N. , Clemente, T. , McCormick, S. and Muehlbauer, G.J. (2015) Transgenic wheat expressing a barley UDP‐glucosyltransferase detoxifies deoxynivalenol and provides high levels of resistance to Fusarium graminearum . Mol. Plant–Microbe Interact. 28, 1237–1246. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Tang, W.H. , Zhao, X.M. and Chen, L. (2010) A network approach to predict pathogenic genes for Fusarium graminearum . PLoS One, 5, e13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Guo, Y. , Ma, C. , Zhang, D. , Wang, C. , Yang, Q. and Xu, M. (2016) Transcriptome analysis of maize resistance to Fusarium graminearum . BMC Genomics, 17, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, X.Y. , Balcerzak, M. , Gulden, S. , Cao, W. , Fedak, G. , Wei, Y.‐M. , Zheng, Y.‐L. , Somers, D. and Ouellet, T. (2015) Expression profiling identifies differentially expressed genes associated with the fusarium head blight resistance QTL 2DL from the wheat variety Wuhan‐1. Physiol. Mol. Plant Pathol. 90, 1–11. [Google Scholar]
- Lulin, M. , Yi, S. , Aizhong, C. , Zengjun, Q. , Liping, X. , Peidu, C. , Dajun, L. and Xiu‐E, W. (2010) Molecular cloning and characterization of an up‐regulated UDP‐glucosyltransferase gene induced by DON from Triticum aestivum L. cv. Wangshuibai. Mol. Biol. Rep. 37, 785–795. [DOI] [PubMed] [Google Scholar]
- Lyons, R. , Stiller, J. , Powell, J. , Rusu, A. , Manners, J.M. and Kazan, K. (2015) Fusarium oxysporum triggers tissue‐specific transcriptional reprogramming in Arabidopsis thaliana . PLoS One, 10, e0121902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysøe, E. , Seong, K.Y. and Kistler, H.C. (2011a) The transcriptome of Fusarium graminearum during the infection of wheat. Mol. Plant–Microbe Interact. 24, 995–1000. [DOI] [PubMed] [Google Scholar]
- Lysøe, E. , Pasquali, M. , Breakspear, A. and Kistler, H.C. (2011b) The transcription factor FgStuAp influences spore development, pathogenicity, and secondary metabolism in Fusarium graminearum . Mol. Plant–Microbe Interact. 24, 54–67. [DOI] [PubMed] [Google Scholar]
- Ma, L.J. , van der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.B. , Woloshuk, C. , Xie, X. , Xu, J.R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L.J. , Geiser, D.M. , Proctor, R.H. , Rooney, A.P. , O'Donnell, K. , Trail, F. , Gardiner, D.M. , Manners, J.M. and Kazan, K. (2013) Fusarium pathogenomics. Annu. Rev. Microbiol. 67, 399–416. [DOI] [PubMed] [Google Scholar]
- Mackintosh, C.A. , Lewis, J. , Radmer, L.E. , Shin, S. , Heinen, S.J. , Smith, L.A. , Wyckoff, M.N. , Dill‐Macky, R. , Evans, C.K. , Kravchenko, S. , Baldridge, G.D. , Zeyen, R.J. and Muehlbauer, G.J. (2007) Overexpression of defence response genes in transgenic wheat enhances resistance to Fusarium head blight. Plant Cell Rep. 26, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesterhazy, A. (1995) Types and components of resistance to Fusarium head blight of wheat. Plant Breed. 114, 377–386. [Google Scholar]
- Mudge, A.M. , Dill‐Macky, R. , Dong, Y. , Gardiner, D.M. , White, R.G. and Manners, J.M. (2006) A role for the mycotoxin deoxynivalenol in stem colonisation during crown rot disease of wheat caused by Fusarium graminearum and Fusarium pseudograminearum . Physiol. Mol. Plant Pathol. 69, 73–85. [Google Scholar]
- Muhovski, Y. , Batoko, H. and Jacquemin, J.M. (2012) Identification, characterization and mapping of differentially expressed genes in a winter wheat cultivar (Centenaire) resistant to Fusarium graminearum infection. Mol. Biol. Rep. 39, 9583–9600. [DOI] [PubMed] [Google Scholar]
- Nussbaumer, T. , Warth, B. , Sharma, S. , Ametz, C. , Bueschl, C. , Parich, A. , Pfeifer, M. , Siegwart, G. , Steiner, B. , Lemmens, M. , Schuhmacher, R. , Buerstmayr, H. , Mayer, K.F. , Kugler, K.G. and Schweiger, W. (2015) Joint transcriptomic and metabolomic analyses reveal changes in the primary metabolism and imbalances in the subgenome orchestration in the bread wheat molecular response to Fusarium graminearum . G3 (Bethesda), 5, 2579–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquet, J.C. , Chaouch, S. , Macadré, C. , Balzergue, S. , Huguet, S. , Martin‐Magniette, M.L. , Bellvert, F. , Deguercy, X. , Thareau, V. , Heintz, D. , Saindrenan, P. and Dufresne, M. (2014) Differential gene expression and metabolomic analyses of Brachypodium distachyon infected by deoxynivalenol producing and non‐producing strains of Fusarium graminearum . BMC Genomics, 15, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquet, J.C. , Changenet, V. , Macadré, C. , Boex‐Fontvieille, E. , Soulhat, C. , Bouchabké‐Coussa, O. , Dalmais, M. , Atanasova‐Pénichon, V. , Bendahmane, A. , Saindrenan, P. and Dufresne, M. (2016) A Brachypodium UDP‐glycosyltransferase confers root tolerance to deoxynivalenol and resistance to Fusarium infection. Plant Physiol. 172, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peraldi, A. , Beccari, G. , Steed, A. and Nicholson, P. (2011) Brachypodium distachyon: a new pathosystem to study Fusarium head blight and other Fusarium diseases of wheat. BMC Plant Biol. 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perochon, A. , Jianguang, J. , Kahla, A. , Arunachalam, C. , Scofield, S.R. , Bowden, S. , Wallington, E. and Doohan, F.M. (2015) TaFROG encodes a Pooideae orphan protein that interacts with SnRK1 and enhances resistance to the mycotoxigenic fungus Fusarium graminearum . Plant Physiol. 169, 2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J.J. , Fitzgerald, T.L. , Stiller, J. , Berkman, P.J. , Gardiner, D.M. , Manners, J.M. , Henry, R.J. and Kazan, K. (2017a) The defence‐associated transcriptome of hexaploid wheat displays homoeolog expression and induction bias. Plant Biotechnol. J. 15, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J.J. , Carere, J. , Fitzgerald, T.L. , Stiller, J. , Covarelli, L. , Xu, Q. , Gubler, F. , Colgrave, M.L. , Gardiner, D.L. , Manners, J.M. , Henry, R.J. and Kazan, K. (2017b) The Fusarium crown rot pathogen Fusarium pseudograminearum triggers a suite of transcriptional and metabolic changes in bread wheat (Triticum aestivum L). Ann. Bot. 119, 853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat, N. , Buerstmayr, M. , Steiner, B. , Robert, O. and Buerstmayr, H. (2014) Current knowledge on resistance to Fusarium head blight in tetraploid wheat. Mol. Breed. 34, 1689–1699. [Google Scholar]
- Prat, N. , Guilbert, C. , Prah, U. , Wachter, E. , Steiner, B. , Langin, T. , Robert, O. and Buerstmayr, H. (2017) QTL mapping of Fusarium head blight resistance in three related durum wheat populations. Theor. Appl. Genet. 130, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, K.D. and Zhong, S. (2010) The 3ADON population of Fusarium graminearum found in North Dakota is more aggressive and produces a higher level of DON than the prevalent 15ADON population in spring wheat. Phytopathology, 100, 1007–1014. [DOI] [PubMed] [Google Scholar]
- Puri, K.D. , Yan, C. , Leng, Y. and Zhong, S. (2016) RNA‐Seq revealed differences in transcriptomes between 3ADON and 15ADON populations of Fusarium graminearum in vitro and in planta. PLoS One, 11, e0163803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, W. , Kwon, C. and Trail, F. (2006) Microarray analysis of transcript accumulation during perithecium development in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Mol. Genet. Genomics, 276, 87–100. [DOI] [PubMed] [Google Scholar]
- Ravensdale, M. , Rocheleau, H. , Wang, L. , Nasmith, C. , Ouellet, T. and Subramaniam, R. (2014) Components of priming‐induced resistance to Fusarium head blight in wheat revealed by two distinct mutants of Fusarium graminearum . Mol. Plant Pathol. 15, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat, N. , Pumphrey, M.O. , Liu, S. , Zhang, X. , Tiwari, V.K. , Ando, K. , Trick, H.N. , Bockus, W.W. , Akhunov, E. , Anderson, J.A. and Gill, B.S. (2016) Wheat Fhb1 encodes a chimeric lectin with agglutinin domains and a pore‐forming toxin‐like domain conferring resistance to Fusarium head blight. Nat. Genet. 48, 1576–1580. [DOI] [PubMed] [Google Scholar]
- Samad‐Zamini, M. , Schweiger, W. , Nussbaumer, T. , Mayer, K.F. and Buerstmayr, H. (2017) Time‐course expression QTL atlas of the global transcriptional response of wheat to Fusarium graminearum . Plant Biotechnol. J. in press. doi: 10.1111/pbi.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, P.M. , Kazan, K. , Wilson, I. , Anderson, J.P. , Richmond, T. , Somerville, S.C. and Manners, J.M. (2000) Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc. Natl. Acad. Sci. USA, 97, 11 655–11 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherm, B. , Orru, M. , Balmas, V. , Spanu, F. , Azara, E. , Delogu, G. , Hammond, T.M. , Keller, N.P. and Migheli, Q. (2011) Altered trichothecene biosynthesis in TRI6‐silenced transformants of Fusarium culmorum influences the severity of crown and foot rot on durum wheat seedlings. Mol. Plant Pathol. 12, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger, W. , Boddu, J. , Shin, S. , Poppenberger, B. , Berthiller, F. , Lemmens, M. , Muehlbauer, G.J. and Adam, G. (2010) Validation of a candidate deoxynivalenol‐inactivating UDP‐glucosyltransferase from barley by heterologous expression in yeast. Mol. Plant–Microbe Interact. 23, 977–986. [DOI] [PubMed] [Google Scholar]
- Schweiger, W. , Steiner, B. , Ametz, C. , Siegwart, G. , Wiesenberger, G. , Berthiller, F. , Lemmens, M. , Jia, H. , Adam, G. , Muehlbauer, G.J. , Kreil, D.P. and Buerstmayr, H. (2013a) Transcriptomic characterization of two major Fusarium resistance quantitative trait loci (QTLs), Fhb1 and Qfhs.ifa‐5A, identifies novel candidate genes. Mol. Plant Pathol. 14, 772–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger, W. , Pasquet, J.C. , Nussbaumer, T. , Paris, M.P. , Wiesenberger, G. , Macadré, C. , Ametz, C. , Berthiller, F. , Lemmens, M. , Saindrenan, P. , Mewes, H.W. , Mayer, K.F. , Dufresne, M. and Adam, G. (2013b) Functional characterization of two clusters of Brachypodium distachyon UDP‐glycosyltransferases encoding putative deoxynivalenol detoxification genes. Mol. Plant–Microbe Interact. 26, 781–792. [DOI] [PubMed] [Google Scholar]
- Schweiger, W. , Steiner, B. , Vautrin, S. , Nussbaumer, T. , Siegwart, G. , Zamini, M. , Jungreithmeier, F. , Gratl, V. , Lemmens, M. , Mayer, K.F. , Bérgès, H. , Adam, G. and Buerstmayr, H. (2016) Suppressed recombination and unique candidate genes in the divergent haplotype encoding Fhb1, a major Fusarium head blight resistance locus in wheat. Theor. Appl. Genet. 129, 1607–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong, K.Y. , Pasquali, M. , Zhou, X. , Song, J. , Hilburn, K. , McCormick, S. , Dong, Y. , Xu, J.R. and Kistler, H.C. (2009) Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 72, 354–367. [DOI] [PubMed] [Google Scholar]
- Shin, S. , Torres‐Acosta, J.A. , Heinen, S.J. , McCormick, S. , Lemmens, M. , Paris, M.P. , Berthiller, F. , Adam, G. and Muehlbauer, G.J. (2012) Transgenic Arabidopsis thaliana expressing a barley UDP‐glucosyltransferase exhibit resistance to the mycotoxin deoxynivalenol. J. Exp. Bot. 63, 4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikhakolli, U.R. , López‐Giráldez, F. , Li, N. , Common, R. , Townsend, J.P. and Trail, F. (2012) Transcriptome analyses during fruiting body formation in Fusarium graminearum and Fusarium verticillioides reflect species life history and ecology. Fungal Genet. Biol. 49, 663–673. [DOI] [PubMed] [Google Scholar]
- Sobrova, P. , Adam, V. , Vasatkova, A. , Beklova, M. , Zeman, L. and Kizek, R. (2010) Deoxynivalenol and its toxicity. Interdiscip. Toxicol. 3, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son, H. , Seo, Y.S. , Min, K. , Park, A.R. , Lee, J. , Jin, J.M. , Lin, Y. , Cao, P. , Hong, S.Y. , Kim, E.K. , Lee, S.H. , Cho, A. , Lee, S. , Kim, M.G. , Kim, Y. , Kim, J.E. , Kim, J.C. , Choi, G.J. , Yun, S.H. , Lim, J.Y. , Kim, M. , Lee, Y.H. , Choi, Y.D. and Lee, Y.W. (2011) A phenome‐based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum . PLoS Pathog. 7, e1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X.S. , Li, H.P. , Zhang, J.B. , Song, B. , Huang, T. , Du, X.M. , Gong, A.D. , Liu, Y.K. , Feng, Y.N. , Agboola, R.S. and Liao, Y.C. (2014) Trehalose 6‐phosphate phosphatase is required for development, virulence and mycotoxin biosynthesis apart from trehalose biosynthesis in Fusarium graminearum . Fungal Genet. Biol. 63, 24–41. [DOI] [PubMed] [Google Scholar]
- Soresi, D. , Carrera, A.D. , Echenique, V. and Garbus, I. (2015) Identification of genes induced by Fusarium graminearum inoculation in the resistant durum wheat line Langdon (Dic‐3A)10 and the susceptible parental line Langdon. Microbiol. Res. 177, 53–66. [DOI] [PubMed] [Google Scholar]
- Steiner, B. , Kurz, H. , Lemmens, M. and Buerstmayr, H. (2009) Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor. Appl. Genet. 118, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, A.E. , Gardiner, D.M. , White, R.G. , Munn, A.L. and Manners, J.M. (2008) Phases of infection and gene expression of Fusarium graminearum during crown rot disease of wheat. Mol. Plant–Microbe Interact. 21, 1571–1581. [DOI] [PubMed] [Google Scholar]
- Sutton, J.C. (1982) Epidemiology of wheat head blight and maize ear rot caused by Fusarium graminearum . Can. J. Plant Pathol. 4, 195–209. [Google Scholar]
- Trail, F. (2009) For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 149, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkowiak, S. , Bonner, C.T. , Wang, L. , Blackwell, B. , Rowland, O. and Subramaniam, R. (2015) Intraspecies interaction of Fusarium graminearum contributes to reduced toxin production and virulence. Mol. Plant–Microbe Interact. 28, 1256–1267. [DOI] [PubMed] [Google Scholar]
- Walter, S. and Doohan, F. (2011) Transcript profiling of the phytotoxic response of wheat to the Fusarium mycotoxin deoxynivalenol. Mycotoxin Res. 27, 221–230. [DOI] [PubMed] [Google Scholar]
- Walter, S. , Brennan, J.M. , Arunachalam, C. , Ansari, K.I. , Hu, X. , Khan, M.R. , Trognitz, F. , Trognitz, B. , Leonard, G. , Egan, D. and Doohan, F.M. (2008) Components of the gene network associated with genotype‐dependent response of wheat to the Fusarium mycotoxin deoxynivalenol. Funct. Integr. Genomics, 8, 421–427. [DOI] [PubMed] [Google Scholar]
- Walter, S. , Kahla, A. , Arunachalam, C. , Perochon, A. , Khan, M.R. , Scofield, S.R. and Doohan, F.M. (2015) A wheat ABC transporter contributes to both grain formation and mycotoxin tolerance. J. Exp. Bot. 66, 2583–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Vera‐Buxa, S. , Furch, A. , Friedt, W. and Gottwald, S. (2015) Insights into Triticum aestivum seedling root rot caused by Fusarium graminearum . Mol. Plant–Microbe Interact. 28, 1288–1303. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Cheng, X. , Shan, Q. , Zhang, Y. , Liu, J. , Gao, C. and Qiu, J.L. (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 32, 947–951. [DOI] [PubMed] [Google Scholar]
- Ward, T.J. , Clear, R.M. , Rooney, A.P. , O'Donnell, K. , Gaba, D. , Patrick, S. , Starkey, D.E. , Gilbert, J. , Geiser, D.M. and Nowicki, T.W. (2008) An adaptive evolutionary shift in Fusarium head blight pathogen populations is driving the rapid spread of more toxigenic Fusarium graminearum in North America. Fungal Genet. Biol. 45, 473–484. [DOI] [PubMed] [Google Scholar]
- Warth, B. , Parich, A. , Bueschl, C. , Schoefbeck, D. , Neumann, N.K. , Kluger, B. , Schuster, K. , Krska, R. , Adam, G. , Lemmens, M. and Schuhmacher, R. (2015) GC‐MS based targeted metabolic profiling identifies changes in the wheat metabolome following deoxynivalenol treatment. Metabolomics, 11, 722–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegulo, S.N. (2012) Factors influencing deoxynivalenol accumulation in small grain cereals. Toxins, 4, 1157–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, J. , Jia, X. , Wang, H. , Zhao, R. , Fang, Y. , Gao, R. , Wu, Z. , Cao, A. , Wang, J. , Xue, Z. , Zhao, W. , Kang, J. , Chen, Q. , Chen, P. and Wang, X. (2011) A fast‐neutron induced chromosome fragment deletion of 3BS in wheat landrace Wangshuibai increased its susceptibility to Fusarium head blight. Chromosome Res. 19, 225–234. [DOI] [PubMed] [Google Scholar]
- Xiao, J. , Jin, X. , Jia, X. , Wang, H. , Cao, A. , Zhao, W. , Pei, H. , Xue, Z. , He, L. , Chen, Q. and Wang, X. (2013) Transcriptome‐based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics, 14, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q. , Yin, G. , Guo, Y. , Zhang, D. , Chen, S. and Xu, M. (2010) A major QTL for resistance to Gibberella stalk rot in maize. Theor. Appl. Genet. 121, 673–687. [DOI] [PubMed] [Google Scholar]
- Ye, J. , Guo, Y. , Zhang, D. , Zhang, N. , Wang, C. and Xu, M. (2013) Cytological and molecular characterization of quantitative trait locus qRfg1, which confers resistance to Gibberella stalk rot in maize. Mol . Plant–Microbe Interact. 26, 1417–1428. [DOI] [PubMed] [Google Scholar]
- Yuen, G.Y. and Schoneweis, S.D. (2007) Strategies for managing Fusarium head blight and deoxynivalenol accumulation in wheat. Int. J. Food Microbiol. 119, 126–130. [DOI] [PubMed] [Google Scholar]
- Zhang, X.W. , Jia, L.J. , Zhang, Y. , Jiang, G. , Li, X. , Zhang, D. and Tang, W.H. (2012) In planta stage‐specific fungal gene profiling elucidates the molecular strategies of Fusarium graminearum growing inside wheat coleoptiles. Plant Cell, 24, 5159–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Li, Z. , Xu, H. , Zhou, M. , Du, L. and Zhang, Z. (2012) Overexpression of wheat lipid transfer protein gene TaLTP5 increases resistances to Cochliobolus sativus and Fusarium graminearum in transgenic wheat. Funct. Integr. Genomics, 12, 481–488. [DOI] [PubMed] [Google Scholar]
- Zhuang, Y. , Gala, A. and Yen, Y. (2013) Identification of functional genic components of major fusarium head blight resistance quantitative trait loci in wheat cultivar Sumai 3. Mol. Plant–Microbe Interact. 26, 442–450. [DOI] [PubMed] [Google Scholar]