

Summary
By integrating next‐generation sequencing (NGS), bioinformatics, electron microscopy and conventional molecular biology tools, a new virus infecting kiwifruit vines has been identified and characterized. Being associated with double‐membrane‐bound bodies in infected tissues and having a genome composed of RNA segments, each one containing a single open reading frame in negative polarity, this virus shows the typical features of members of the genus Emaravirus. Five genomic RNA segments were identified. Additional molecular signatures in the viral RNAs and in the proteins they encode, together with data from phylogenetic analyses, support the proposal of creating a new species in the genus Emaravirus to classify the novel virus, which is tentatively named Actinidia chlorotic ringspot‐associated virus (AcCRaV). Bioassays showed that AcCRaV is mechanically transmissible to Nicotiana benthamiana plants which, in turn, may develop chlorotic spots and ringspots. Field surveys disclosed the presence of AcCRaV in four different species of kiwifruit vines in five different provinces of central and western China, and support the association of the novel virus with symptoms of leaf chlorotic ringspots in Actinidia. Data on the molecular features of small RNAs of 21–24 nucleotides, derived from AcCRaV RNAs targeted by host RNA silencing mechanisms, are also reported, and possible molecular pathways involved in their biogenesis are discussed.
Keywords: AcCRaV, Actinidia, chlorotic ringspot, family Bunyaviridae, genus Emaravirus
Introduction
The recently established genus Emaravirus (Mühlbach and Mielke‐Ehret, 2012) includes European mountain ash ringspot‐associated virus (EMARaV) as the type species (Mielke‐Ehret and Mühlbach, 2007) plus four additional definitive species: Fig mosaic virus (FMV) (Elbeaino et al., 2009a, 2009b), Rose rosette virus (RRV) (Laney et al., 2011), Raspberry leaf blotch virus (RLBV) (McGavin et al., 2012) and Pigeonpea sterility mosaic virus (PPSMV‐1) (Elbeaino et al., 2014; Kumar et al., 2003) (http://www.ictvonline.org/virusTaxonomy.asp). Other tentative emaraviruses are redbud yellow ringspot‐associated virus (RYRSaV) (Laney et al., 2010), pigeonpea sterility mosaic virus 2 (PPSMV‐2) (Elbeaino et al., 2015), woolly burdock yellow vein virus (WBYVV) (Bi et al., 2012), maize red stripe virus (MRStV) (Skare et al., 2006) and wheat mosaic virus (WMoV) (Tatineni et al., 2014). The last two viruses have been proposed to be joined in a single species that is waiting for International Committee on Taxonomy of Viruses (ICTV) ratification (https://talk.ictvonline.org/search?q=High%20plains). All of these viruses are characterized by a multipartite negative‐sense RNA genome composed of four to eight genomic components contained within spherical virions enveloped by a double membrane (Mielke‐Ehret and Mühlbach, 2012). Open reading frames (ORFs) encoding the polymerase, putative glycoprotein, nucleocapsid and movement proteins (MPs), and other proteins with unknown functions, are separately contained in the genomic segments of emaraviruses. Most of these viruses are vectored in nature by eriophyid mites (Flock and Wallace, 1955; Kulkarni et al., 2002; Mielke‐Ehret and Mühlbach, 2012).
China is the site of origin of Actinidia spp. (Ferguson and Bollard, 1990; Ferguson and Huang, 2007; Huang, 2010) and is also the largest commercial kiwifruit producer in the world, Actinidia chinensis and A. delicious being the most widely cultivated species. Virus‐like diseases of kiwifruit have been reported previously in southern China (Lin and Gao, 1995), but their aetiology has not been determined. Recently, a strain of the capillovirus Apple stem grooving virus (ASGV) in A. chinensis (Clover et al., 2003), two vitiviruses, Actinidia virus A (AcVA) and Actinidia virus B (AcVB) (Blouin et al., 2012; Zheng et al., 2014), and a citrivirus, Citrus leaf blotch virus (CLBV) (Zhu et al., 2016), infecting Actinidia spp. from China, have been reported. More recently, Tomato zonate spot virus (TZSV), a putative member of the genus Tospovirus, has been detected in kiwifruit plants (Y. Zheng et al., unpublished data).
In 2013, during a field survey in the province of Hubei (central China), virus‐like symptoms on leaves of kiwifruit vines were observed. To verify whether a virus was associated with these symptoms, next‐generation sequencing (NGS) of small RNAs (sRNAs) accumulating in symptomatic leaves was carried out. As reported here, a hitherto undescribed virus was identified, with molecular, structural and biological properties characterizing species of the genus Emaravirus.
Results
Virus identification by high‐throughput sequencing of sRNA library
In 2013, during the course of field surveys in actinidia plantings of Hubei Province (China), several vines were found showing chlorotic spots and ringspots on their leaves (Fig. 1A–F), sometimes accompanied by vein yellowing (Fig. 1B,C,F).
Figure 1.

Symptoms on leaves of Actinidia chinensis and Nicotiana benthamiana infected by Actinidia chlorotic ringspot‐associated virus (AcCRaV). Ringspots (A), vein yellowing (B) and chlorotic spots (C) on leaves of the kiwifruit plant used for deep sequencing of a cDNA library of small RNAs. Ringspots (D), vein yellowing (E) and chlorotic spots (F) in leaves of kiwifruit vines infected by AcCRaV. (G) Necrotic spots on a mechanically inoculated leaf of N. benthamiana. (H, I) Chlorotic spots and rings, respectively, on leaves of N. benthamiana systemically infected by AcCRaV.
Sequencing by Illumina technology of a cDNA library of sRNAs from symptomatic leaves (Fig. 1A–C) of a kiwifruit plant (HN‐6), and computational processing to remove the 3′ adaptors, the low‐quality tags and sequences with a length shorter than 18 nucleotides (nt), gave rise to 8 711 573 high‐quality reads (Table S1, see Supporting Information), which generated a total of 13 764 contigs after de novo assembly. A blastx search in the National Center for Biotechnology Information (NCBI) database identified 35 contigs with significant amino acid (aa) sequence similarity to several proteins encoded by members of the genus Emaravirus, including RYRSaV, which frequently showed the best matches. The identified contigs were further aligned along the RNA components of the RYRSaV genome (Fig. S1, see Supporting Information), generating a preliminary scaffold of at least four RNA genomic components of a potential novel emaravirus infecting kiwifruit, which was tentatively named Actinidia chlorotic ringspot‐associated virus (AcCRaV). In addition, blastx analysis identified additional five contigs with sequences almost identical to genes encoded by AcVB. Reverse transcription‐polymerase chain reaction (RT‐PCR) using specific primers (Table S2, see Supporting Information) and sequencing of the amplified products confirmed the presence of AcVB in HN‐6 (data not shown). However, only sequences relevant to the possible novel emaravirus were analysed in the present study.
Determination of the genome sequence of AcCRaV
To obtain the complete sequence of AcCRaV genomic components, specific primers (Table S3, see Supporting Information) were designed considering the relative positions of contigs mapped in the RYRSaV RNAs (Fig. S1). Overlapping sequences of the amplicons generated by RT‐PCR and 5′ and 3′ rapid amplification of cDNA ends (RACE) allowed the determination of the complete sequences of four RNAs (RNA 1–RNA 4) of 7061, 2267, 1678 and 1664 nt in length (Fig. 2A) (GenBank accession numbers KT861481–KT861484). The first 13 nucleotides at both 5′ and 3′ termini of all RNA segments were almost complementary to each other, and conserved in the four viral genomic RNA components. These terminal sequences are also almost completely conserved in the genomic RNAs of all emaraviruses reported so far (Elbeaino et al., 2009b, 2014; Laney et al., 2011; Mielke‐Ehret and Mühlbach, 2007).
Figure 2.
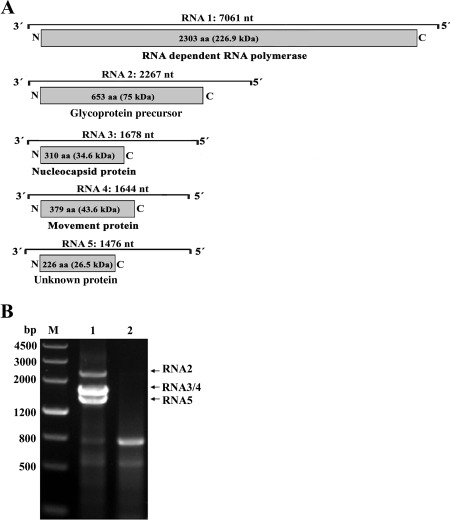
(A) Genomic organization of Actinidia chlorotic ringspot‐associated virus (AcCRaV). The nucleotide (nt) length of each RNA is represented as a black line. Black boxes at the end of each line indicate the 13 nt conserved at the RNA termini. The expression product of each RNA is shown as a grey box, with the amino acid (aa) length, estimated molecular weight (kDa) and function of the putative protein reported. (B) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis of full‐length RNA genomic segments of AcCRaV. Lane 1, symptomatic leaves of the kiwifruit vine HN6 infected by AcCRaV; lane 2, asymptomatic leaves of a kiwifruit vine not infected by this virus; M, DNA molecular weight marker with sizes (bp) indicated on the left. Cloning and sequencing of the amplification products specific to the symptomatic sample showed that they corresponded to AcCRaV genomic RNAs 2, 3, 4 and 5 as indicated on the right.
Based on this feature, the primer set AcCRaV 5H/3C (Table S3) was designed in the conserved stretch sequences at the 5′ and 3′ ends of the four AcCRaV viral RNAs known at this stage, and used to amplify full‐length cDNAs of all genomic components of the emaravirus by RT‐PCR. Three amplicons, not observed in the healthy negative control, were obtained from the vine HN‐6. According to gel migration (Fig. 2B), the largest amplicon (2300 bp) has the size expected for the full‐length cDNA of AcCRaV RNA 2 (2267 nt). Considering that RNAs 3 and 4 have similar sizes (1678 and 1664 nt), the respective full‐length amplicons are likely to co‐migrate in the gel as a single band of approximately 1700 bp. Finally, the unexpected smaller amplicon of about 1500 bp was tentatively identified as an additional genomic component of AcCRaV. Conventional sequencing of these cloned cDNAs confirmed that the full‐length sequences of AcCRaV RNAs 2, 3 and 4, determined previously by overlapping partial genomic cDNA fragments, were correct. Moreover, the full‐length sequence of the potential additional AcCRaV genomic component, tentatively designated as RNA 5, was determined to be 1474 nt in size (KT861485). No band with a size expected for RNA 1 (about 7000 bp) was obtained using this specific set of primers, probably because of the low processivity of the polymerases and/or lower stability of this long RNA.
In an attempt to identify sequences deriving from additional AcCRaV genomic RNAs, the deep‐sequenced sRNA library from HN‐6 and the in silico‐generated contigs were further screened. In particular, using the 13‐nt stretches conserved in the 5′ and 3′ termini of RNAs 1–5 as a hallmark of the viral genomic RNAs, contigs and sRNAs (18–26 nt) containing these terminal sequences were specifically searched for. All the retrieved contigs had sequences coincident with the 3′ or 5′ terminal ends of AcCRaV RNAs 2–5, and all the retrieved sRNAs mapped at the 5′ or 3′ termini of the AcCRaV genomic RNAs 1–5 (Table S5, see Supporting Information), whereas no sRNA containing the terminal 13‐nt stretches and possibly deriving from other RNA(s) was identified.
It is worth noting that RNAs 1–5 were also closely associated with each other in other AcCRaV‐infected vines grown far from Hubei Province, the geographical area in which the potential new emaravirus was first identified. Indeed, three symptomatic kiwifruit vines (AH‐3, AH‐20 and AH‐21) grown in Anhui Province, which were shown to contain RNA 1 by sequencing RT‐PCR products obtained using the primer pair AcCRaV 1AF/1CR (Tables 1 and S7, see Supporting Information), also contained AcCRaV RNAs 2–5, as shown by RT‐PCR using the primer pair AcCRaV 5H/3C. The cloning and sequencing of some of the amplification products confirmed that RNAs 1 and 3 from the vine AH‐21, RNA 4 from the vines AH‐21 and AH‐3, and RNA‐5 from all three vines were almost identical (nt sequence identity 93%–97%) to the corresponding viral genomic RNAs previously found in the HN‐6 vine (Table S4, see Supporting Information), thus demonstrating that RNAs 1–5 are genomic components of a single virus.
Table 1.
Primers used for the detection of Actinidia chlorotic ringspot‐associated virus (AcCRaV) by reverse transcription‐polymerase chain reaction (RT‐PCR).
| Virus | Target gene | Primer | Primer sequence (5′–3′) | Position (nt) | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|---|---|
| Emaraviruses* | RdRp | 1AF | GATGCATCDAAATGGTCWGC | 3239–3256 | 387 | 53 |
| 1CR | ATCATCWGARTGHACCAT | 3606–3625 | ||||
| AcCRaV | CP † | CP‐F | CGAGCTCAGTGGAAGAACCACAATATT | 649–667 | 945 | 54 |
| CP‐R | CGGGATCCATGCCAAAGCCTATGCAAGG | 1559–1578 | ||||
| CP | 3F | ATCCAAGAATTCCTTAACAGCA | 683–704 | 477 | 55 | |
| 3R | TGTGCAATCATGGCTTATCAGA | 1138–1159 |
*Primer set 1AF/1CR, generated for the detection of most emaraviruses, was designed by Elbeaino et al. (2013). ‘W’ is A or T; ‘R’ is A or G; ‘D’ is G or A or T; and ‘H’ is A or C or T.
†Nucleotides added for enzyme restriction and cloning are shown in italic type.
Genomic organization of AcCRaV
The overall genomic organization of AcCRaV is similar to that of other emaraviruses, with each RNA containing a single ORF in negative polarity (Fig. 2A). RNA 1 is 7061 nt in length and contains an ORF (ORF1, nt positions 7023 to 112) that encodes a putative protein (P1) of 2303 aa with a predicted molecular mass of 226.9 kDa. Pairwise comparisons of the deduced aa sequence showed similarities between AcCRaV P1 and the RNA‐dependent RNA polymerase (RdRp) proteins of other emaraviruses, ranging from 33.4% (WMoV) to 64.6% (RYRSaV) (Table S6, see Supporting Information). In the region delimited by aa 1135 to 1322 of the viral RdRp (Fig. S2A, see Supporting Information), there are the five motifs conserved in members of the genus Emaravirus and family Bunyaviridae, which correspond to the core polymerase module of the RdRp active site (Elbeaino et al., 2015; Laney et al., 2011) (Fig. S2A). In addition, the N‐terminus of AcCRaV P1 contains the endonuclease domains (Fig. S2A) probably involved in cap‐snatching, a genome expression strategy to cap viral mRNAs proposed for members of the family Bunyaviridae and genus Tenuivirus (Duijsings et al., 2001; Reguera et al., 2010).
RNA 2 is 2267 nt in length and contains ORF2, encoding a glycoprotein precursor (P2: GP) of 653 aa with a predicted molecular mass of 75 kDa. It shows 24.3%–48.3% identity at the aa level with the GP precursors of the other emaraviruses (Table S6), with which AcCRaV P2 shares several conserved motifs (Fig. S2B), including two predicted cleavage sites (VNT23↓K24V and VKA196↓E197D) that may generate two larger glycoproteins, Gn (19.8 kDa) and Gc (53.1 kDa), and a smaller glycoprotein (Gs), consisting of 23 amino acids (2.6 kDa) (Fig. S2B).
RNA 3 is 1678 nt in length and contains ORF3 (nt positions 1033 to 101), encoding a protein of 310 aa (34.6 kDa) predicted to be the nucleocapsid (P3: CP). It shares significant aa identity with CP of RYRSaV (55.6%) and other emaraviruses (Table S6), and contains three conserved aa stretches (NVLSFNK134–140, NRLA183–186 and GYEF204–207) (Elbeaino et al., 2009b). According to BindN prediction (Wang and Brown, 2006), P3 contains stretches of positively charged amino acids (BindN specificity, 80%) that are probably involved in RNA binding, as also predicted for RRV CP (Laney et al., 2011).
RNA 4 is 1664 nt in length and contains ORF4 (nt positions 1238 to 99). It encodes a protein (P4) of 379 aa (43.6 kDa) that shares the highest identity (51.9%) with the protein encoded by RNA4 of RYRSaV (Table S6). Interestingly, AcCRaV P4 protein shares several structural features with other emaravirus MPs (Yu et al., 2013). A signal peptide (positions 1–29, Fig. S3, see Supporting Information) was identified by Signal P (Emanuelsson et al., 2007). In addition, multiple protein alignment using the software PROMALS (Pei and Grishin, 2007) showed that the central part of AcCRaV P4 contains structural elements similar to the consensus secondary structure of the 30K superfamily of plant virus MPs (Melcher, 2000) (Fig. S3). According to the COILS program (Lupas et al., 1991), a coiled‐coil motif was found in AcCRaV P4 downstream of the 30K central conserved domain. Similar structural elements have been reported previously in P4 of RRV, RYRSaV, FMV and RLBV (Yu et al., 2013). Interestingly, in the case of FMV and RLBV, experimental data based on the transient expression of P4 proteins fused to a fluorescent reporter protein support a role of P4 in virus trafficking (Ishikawa et al., 2012; Yu et al., 2013). Altogether, these data suggest that P4 is the MP of AcCRaV.
RNA 5 (1476 nt) contains a single ORF5 (nt positions 777 to 95) encoding a protein (P5) consisting of 226 aa with a molecular mass of 26.5 kDa. Sequence similarity between P5 and proteins encoded by other emaraviruses is very low (Table S6). Moreover, no protein or conserved domain homologous to P5 was found in the databases, and thus the function of this viral protein remains unknown.
AcCRaV RNAs have untranslated regions (UTRs) at their 5′ and 3′ termini. 3′ UTRs, ranging in size from 38 to 100 nt (Fig. 2A), correspond to 0.5% (in RNA 1) up to 6.5% (in RNA 4) of the respective full‐length RNA sequences. The sizes of 5′ UTRs of the AcCRaV RNAs are more heterogeneous, with those of RNAs 3, 4 and 5 being particularly long and covering 38%, 26% and 47%, respectively, of the respective full‐length RNA sequences.
Phylogenetic analysis
To investigate the phylogenetic relationships of AcCRaV with closely related viruses, amino acid sequences of putative RdRp (P1), GP (P2), CP (P3) and MP (P4) of AcCRaV and all known emaraviruses were aligned with the homologous proteins from representative members of most genera in the family Bunyaviridae and genus Tenuivirus, and phylogenetic trees were generated. Regardless of the considered protein, AcCRaV always clustered together with emaraviruses with high bootstrap values (>99), and was clearly separated from other viruses in the family Bunyaviridae, thus confirming a close phylogenetic relationship of AcCRaV with members in the genus Emaravirus (Fig. 3). In the case of RdRp, GP and CP proteins, three subclades were identified in the emaravirus clade, one of which contained AcCRaV, RYRSaV and EMARaV (Fig. 3). However, when P4 was considered, AcCRaV clustered only with RYRSaV, which is in agreement with the low similarity between the P4 of EMARaV and the MP of the other emaraviruses.
Figure 3.
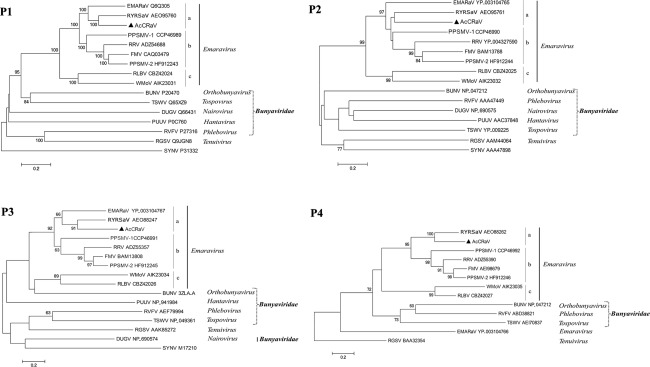
Phylogenetic trees generated using the amino acid sequences of P1–P4 of emaraviruses and the corresponding proteins of representative members in the family Bunyaviridae and genus Tenuivirus. The bar represents the number of amino acid replacements per site. The GenBank accession numbers of the proteins used for phylogenetic analyses are reported. Sequences of Sonchus yellow net virus (SYNV), a member of the genus Nucleorabdovirus, were used as outgroup in the analyses of P1, P2 and P3. In the case of P4, the protein P4 of rice grassy stunt virus genome (RGSV) in the genus Tenuivirus was used as an outgroup. BUNV, Bunyamwera virus; DUGV, Dugbe virus; PUUV, Puumala virus; RVFV, Rift valley fever virus; TSWV, Tomato spotted wilt virus. The names of the other viruses are reported in the main text.
Identification of double‐membrane‐bound bodies (DMBs) in AcCRaV‐infected kiwifruit leaves
Thin‐sectioned tissues of plants infected by several emaraviruses have been shown to contain DMBs, which are assumed to be virions (Mielke‐Ehret and Mühlbach, 2012). DMBs are found frequently near the endoplasmic reticulum (ER) and Golgi cisternae (Ahn et al., 1998; Kumar et al., 2002; Martelli et al., 1993; Silvestro and Chapman, 2004), suggesting that their surrounding membranes could originate from these intracellular host membranes. Transmission electron microscopy (TEM) observations of ultrathin sections of AcCRaV‐infected kiwifruit leaves showed the presence of typical DMBs in the cytoplasm of mesophyll cells (Fig. 4A–D). The size of these DMBs, ranging from 95 to 110 nm (average size, 100 nm) in diameter, is in line with the DMB sizes reported for other emaraviruses. Interestingly, DMBs were frequently located near ER membranes in the cytoplasm (Fig. 4C,D) and never occurred in clusters. Similar structures were never observed in the healthy controls, thus supporting a close association of DMBs with the presence of AcCRaV.
Figure 4.
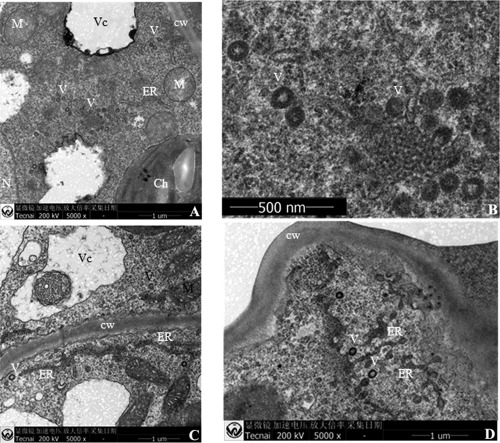
Ultrastructural analyses by transmission electron microscopy of kiwifruit leaves infected by Actinidia chlorotic ringspot‐associated virus (AcCRaV). (A) Virions with surrounding double membranes in the cytoplasm of a mesophyll cell. (B) Close up of (A). (C, D) Virions near endoplasmic reticulum in the cytoplasm of AcCRaV‐infected cells. Ch, chloroplast, cw, cell wall; ER, endoplasmic reticulum; M, mitochondria; N, nucleus; V, virion; Vc, vacuole.
Characterization of AcCRaV‐derived sRNAs
When AcCRaV sRNAs were grouped according to size, a profile with prevalent peaks corresponding to sizes of 21 and 22 nt was observed for all viral RNAs (Fig. S4, see Supporting Information). Similar proportions of AcCRaV sRNAs were derived from the genomic and anti‐genomic RNA strands, regardless of the size classes and the RNAs considered, with the only exception of AcCRaV RNA 5, in which 21‐nt anti‐genomic sRNAs largely prevailed (Fig. S4). Sequences of non‐redundant AcCRaV sRNAs cover 33%, 99%, 96%, 99% and 95% of RNAs 1 to 5, respectively. Moreover, when all (redundant) sRNAs deriving from genomic and anti‐genomic AcCRaV RNAs are considered, they generate hotspots unevenly distributed along each viral RNA (Fig. 5), thus resembling the distribution profiles of virus‐derived sRNAs (v‐sRNAs) reported for plus‐sense RNA and DNA viruses (Donaire et al., 2009). The hotspots of AcCRaV sRNAs mapped prevalently within the ORFs, with much lower hotspot sRNAs located in the UTRs. This uneven distribution was particularly evident in the case of RNAs 3, 4 and 5, which are characterized by long 5′ UTRs with a lower GC content than in the respective ORFs (Fig. 5). In contrast, a more uniform sRNA distribution was observed between the 5′ UTR and ORF2 in AcCRaV RNA 2. Interestingly, in this case, the GC content was higher in the 5′ UTR than in ORF2, thus supporting a correlation between GC content and density of the mapped sRNAs (Fig. 5). Finally, analysis of the 5′‐terminal nucleotide in the AcCRaV sRNAs revealed the prevalence of U and A in sRNAs derived from all the viral RNAs, regardless of their polarity (Fig. S5, see Supporting Information).
Figure 5.
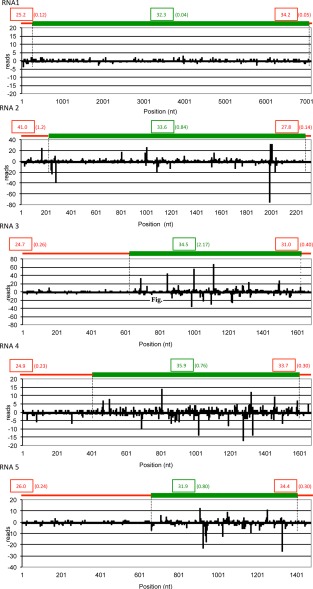
Distribution of Actinidia chlorotic ringspot‐associated virus (AcCRaV) sRNAs along the viral genomic RNAs. The locations and frequencies in each viral genomic RNA (RNAs 1–5 in 5′–3′ orientation) of the 5′ termini of the genomic (positive values) and anti‐genomic (negative values) AcCRaV sRNA reads are shown. A schematic representation of untranslated regions (UTRs) (red lines) and open reading frames (ORFs) (green lines) of each viral RNA is shown above the histogram. Numbers in boxes and in parentheses indicate the GC content (%) and the sRNA density (number of reads per nucleotide position), respectively, in the UTRs (in red) and ORFs (in green) of each genomic RNA.
Transmission of AcCRaV to herbaceous hosts
Fourteen Nicotiana benthamiana plants were mechanically inoculated with sap from a symptomatic vine infected by AcCRaV. At 15 days post‐inoculation, newly developed leaves from all inoculated plants were tested by RT‐PCR using the specific primer set CP‐F/CP‐R, designed to amplify the ORF3 sequence coding for the putative CP, and the less specific primer pair 1AF/1CR, designed to detect RNA 1 of most emaraviruses (Elbeaino et al., 2013) (Table 1). Amplicons of the expected size were obtained from seven inoculated plants using both primer pairs. Sequencing of the amplification products generated by both sets of primers (two clones from each infected plant) showed that AcCRaV RNAs 1 and 3 from infected N. benthamiana plants were almost identical to the corresponding genomic AcCRaV RNAs from actinidia (sequence identity 99% and 98% at the nt and aa level, respectively), thus confirming the mechanical transmission of the virus to the experimental host. RT‐PCR assays showed that the inoculated N. benthamiana plants were not infected by AcVA, AcVB, CLBV and ASGV, viruses previously reported in actinidia (data not shown). In this bioassay, only one of the infected plants showed necrotic spots on inoculated leaves, followed by extensive chlorotic spots on systemic leaves (Fig. 1G–I). However, serial inoculations of N. benthamiana, followed by RT‐PCR assays (Fig. S6, see Supporting Information) and sequencing of amplification products, confirmed that AcCRaV is mechanically transmissible to N. benthamiana, eliciting the symptoms reported above.
Development and validation of an AcCRaV‐specific detection method and field survey
To develop a specific method for AcCRaV diagnosis in actinidia, the primer set AcCRaV 3F/3R was designed in the sequence of AcCRaV ORF3 (Table 1), whose efficiency was compared with that of the degenerate primer set 1AF/1CR (Elbeaino et al., 2013). As primers 1AF/1CR may also detect the tospovirus TZSV in kiwifruit vines (Y. Zheng et al., unpublished data), a TZSV‐infected sample was used as an additional control. The efficiency and specificity of RT‐PCRs performed with the two primer pairs were tested using RNA preparations from 10 kiwifruit vines, seven of which (YN‐11, HN‐13, HN‐14, HN‐15, HN‐16, HN‐17 and G‐20) were known to be infected by AcCRaV, and one (YN‐4) by TZSV. In addition, two healthy seedlings of A. chinensis were used as negative controls. These analyses showed that the universal primer pair 1AF/1CR detected efficiently both TZSV and AcCRaV, generating the expected product of about 360 bp from all the plants infected by one of the two viruses (Fig. 6A). In contrast, the primer pair AcCRaV 3R/3F detected selectively only AcCRaV (Fig. 6B). Interestingly, coupling RT‐PCR assays performed with generic (1AF/1CR) and specific (AcCRaV 3F/3R) primers, it is relatively easy to discriminate AcCRaV infection from that of another emaravirus and/or TSZV.
Figure 6.
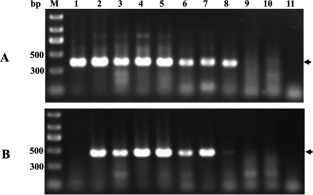
Reverse transcription‐polymerase chain reaction (RT‐PCR) assay of Actinidia chlorotic ringspot‐associated virus (AcCRaV) in kiwifruit plants using primer pairs 1AF/1CR (A) and 3F/3R (B). Agarose gels were stained with ethidium bromide. Arrows on the right indicate the expected products amplified with each primer pair from the AcCRaV‐infected samples or, only for the pair 1AF/1CR, from the Tomato zonate spot virus (TZSV)‐infected vine. 1, YN‐4 infected by TZSV; 2–8, YN‐11, HN‐13, HN‐14, HN‐15, HN‐16, HN‐17 and G‐20 infected by AcCRaV; 9 and 10, seedlings of A. chinensis not infected by TZSV or AcCRaV; 11, water control.
Therefore, both primer pairs, 1AF/1CR and 3F/3R, were chosen to undertake a field survey in different accessions of Actinidia spp. grown in China and coincident results were obtained, thus disclosing the presence of AcCRaV in several actinidia species and geographical areas and excluding infections by TSZV. In particular, 38 of 146 samples tested positive to AcCRaV, with a higher incidence in A. chinensis than in A. delicious (Table 2). The virus was also detected in one A. kolomikta and two A. eriantha plants and in some other unidentified Actinidia spp. (Table 2).
Table 2.
Preliminary survey, based on reverse transcription‐polymerase chain reaction (RT‐PCR) detection, of Actinidia chlorotic ringspot‐associated virus (AcCRaV) in several Actinidia species.
| Actinidia species | Tested samples | Positive samples | Frequency (%) |
|---|---|---|---|
| A. chinensis | 84 | 22 | 26.1 |
| A. delicious | 42 | 4 | 9.5 |
| A. arguta | 3 | 0 | – |
| A. kolomikta | 1 | 1 | – |
| A. polygama | 1 | 0 | – |
| A. eriantha | 3 | 2 | – |
| Others | 12 | 9 | 75 |
| Total | 146 | 38 | 26.0 |
–, frequency not calculated because of the limited number of tested samples.
The AcCRaV‐positive samples were also tested for the presence of AcVA, AcVB, ASGV and CBLV, already reported in Actinidia spp. (Blouin et al., 2013). Thus, in total, 15 of the 36 AcCRaV‐infected vines tested positive only to AcCRaV, whereas one or more additional viruses infected the remaining plants (Table S7). Although most of the infected actinidia vines showed symptoms of leaf chlorosis and/or malformation, the chlorotic ringspots were observed exclusively on the leaves of nine plants that were infected by AcCRaV alone (four samples) or in combination (five samples) with other viruses.
To further exclude concurrent infection by other emaraviruses, one to three clones of the cDNAs amplified using the primer set 1AF/1CR from 17 samples representative of four different actinidia species (A. chinensis, A. delicious, A. kolomikta, A. eriantha) and five Chinese geographical areas (provinces of Anhui, Hubei, Zhejiang, Shanxi and Yunnan) were sequenced, thus generating data also useful for a preliminary view on the sequence variability of AcCRaV recovered. The sequence identity of these amplified fragments with the corresponding RNA 1 sequence from the AcCRaV reference variant (AN: KT861481) was 88%–92% at the nt level and 96%–98% at the aa level (Table S8, see Supporting Information), thus conclusively excluding infection by another emaravirus and/or TZSV.
Discussion
NGS, coupled with metagenomic analysis, has been effectively used for the identification of known and previously unreported plant viruses in several host species (Adams et al., 2009; Al Rwahnih et al., 2009; Hagen et al., 2011; Kreuze et al., 2009; Wu et al., 2015). In the case of emaraviruses, RRV, WMoV, PPSMV‐1 and PPSMV‐2 have been discovered and/or sequenced by Illumina technology (Bi et al., 2012; Elbeaino et al., 2014, 2015; Laney et al., 2011). In this article, we used NGS based on Illumina technology to identify a novel virus, AcCRaV, in an actinidia vine showing symptoms of chlorotic ringspot, mottle and vein yellowing of the leaves. Integration of NGS and conventional sequencing technology allowed the determination of the full‐length sequences of five genomic components of AcCRaV, whose molecular features are consistent with those of all members in the genus Emaravirus. The five genomic components were completely sequenced and found in combination with each other in at least four AcCRaV‐infected kiwifruit vines from different geographical regions, thus strongly indicating that they are the genomic components of a single novel emaravirus. Although attempts to identify additional AcCRaV genomic fragment(s) by RT‐PCR and deep analyses of AcCRaV‐derived sRNAs and contigs failed, we cannot exclude the existence of one or more additional genomic AcCRaV RNAs. Indeed, the recent identification in the emaraviruses RRV and RLBV of genomic RNAs (Di Bello et al., 2015; Lu et al., 2015), which remained unnoticed in the first characterization of these viruses (Laney et al., 2011; McGavin et al., 2012), poses the question of whether additional genomic RNAs could also exist in other emaraviruses, including AcCRaV.
AcCRaV has a multipartite negative‐sense RNA genome with a single ORF encoded by each genomic segment and with a stretch of almost complementary sequences at the 3′ and 5′ termini, which probably hybridize to each other, forming a panhandle structure considered as a hallmark of viruses with a negative‐sense multipartite ssRNA genome (Kormelink et al., 2011). Moreover, despite the overall low sequence identity with other emaraviruses, the presence of domains and structural motifs conserved in homologous proteins in other emaraviruses and/or in members of family Bunyaviridae and genus Tenuivirus strongly indicate that the P1–P4 proteins encoded by AcCRaV correspond to the viral RdRp, GP, CP and MP, respectively.
With respect to the putative AcCRaV RdRp, the presence of motifs potentially involved in cap‐snatching, a viral capping strategy proposed for FMV (Walia and Falk, 2012) and RRV (Laney et al., 2011), indicates that this mechanism could also operate in AcCRaV genome expression. AcCRaV‐encoded GPs should contribute to the formation of DMBs, as reported previously in the case of other emaraviruses (Mielke‐Ehret and Mühlbach, 2012), tospoviruses and members of the family Bunyaviridae (Kormelink et al., 2011). In this study, the association of AcCRaV with DMBs has been shown by TEM observations, which also suggests that the surrounding membrane of DMBs could derive from the ER, in line with the data reported for some tospoviruses (Ie, 1964) and other emaraviruses (Kumar et al., 2002; Martelli et al., 1993).
In contrast with P1–P4, the possible functional role(s) of AcCRaV P5 remains unknown. Proteins with a similar molecular weight (c. 27 kDa), but with no sequence homology to each other, and a role still to be determined, have also been reported in other emaraviruses (Di Bello et al., 2015; Elbeaino et al., 2014, 2015; Ishikawa et al., 2012; Lu et al., 2015; McGavin et al., 2012; Mielke‐Ehret and Mühlbach, 2007; Tatineni et al., 2014).
Contrary to the notion that the mechanical transmission of emaraviruses is difficult (Mielke‐Ehret and Mühlbach, 2012), AcCRaV was readily mechanically transmitted to N. benthamiana plants, which developed leaf chlorotic spot and ringspots. As for other emaraviruses, however, mechanical transmission probably has no epidemiological relevance in the case of AcCRaV. A natural vector, still to be identified, is probably involved in the spread of this virus in the field. It remains to be investigated whether, similar to other emaraviruses (Flock and Wallace, 1955; Kulkarni et al., 2002; Kumar et al., 2003), eriophyid mites could be involved in the natural transmission of AcCRaV.
The use of a specific RT‐PCR detection method for a preliminary survey of AcCRaV in China showed that this virus: (i) occurs in several Actinidia species and in several Chinese provinces; (ii) is frequently accompanied by co‐infecting viruses; and (iii) is associated with the presence of chlorotic ringspots in the leaves of infected kiwifruit plants. However, the involvement of AcCRaV in the symptomatology shown by naturally infected kiwifruit plants requires fulfilment of Koch's postulates.
Finally, the analyses of sRNAs from AcCRaV‐infected kiwifruit vine revealed that AcCRaV sRNAs of both polarity strands accumulated in infected tissues. Interestingly, the distribution profiles of AcCRaV sRNAs along the viral genomic RNAs revealed hotspots mapping prevalently within ORFs. Several possible reasons can be advocated for this distribution profile, including possible protection of AcCRaV UTRs by interacting proteins, as proposed previously in the case of the intergenic regions (IRGs) of M and S RNAs of tomato spotted wilt virus and tomato yellow ring virus, two negative/ambisense viruses infecting tomato, in which the almost complete absence of v‐sRNAs in non‐coding regions was also observed (Hedil et al., 2014; Mitter et al., 2013). Alternatively, considering the correlation between sRNA density and GC content observed in several AcCRaV RNAs (Fig. 5), the uneven distribution of sRNAs could also be a result of the preferential targeting of GC‐rich RNA regions by the Dicer‐like proteins involved in their generation (Ho et al., 2007). However, it cannot be excluded that the genome expression strategy of these viruses, which is based on the transcription of viral mRNAs using, as template, the negative strand of the genomic viral RNAs, could also be implicated in this differential distribution of v‐sRNAs in AcCRaV and in other negative‐sense RNA viruses.
Experimental Procedures
Plant material
In early June 2013, leaf samples exhibiting chlorotic ringspots and vein yellowing were collected from a symptomatic kiwifruit plant (A. chinensis, HN‐6) grown in a field in Hubei Province (central China), and used as source for the generation of a cDNA library of sRNAs. During two consecutive springs (2013–2014), leaf samples from a total of 146 kiwifruit plants, most of which showed symptoms of virus‐like diseases (mainly chlorosis and leaf malformation), were collected from six provinces (60 samples from Hubei Province, 49 from Anhui Province, 18 from Yunnan Province, 16 from Shanxi Province, one from Zhejiang Province and two from Jiangxi Province).
RNA isolation and sRNA sequencing
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and sRNAs (16–30 nt) were purified and processed as reported previously (Di Serio et al., 2010) to generate a cDNA library of sRNAs that was sequenced on an Illumina Genome Analyzer (Biomarker Biology Technology Ltd Company, Beijing, China). The raw reads were filtered for quality, reduced to unique reads and de novo assembled into larger contigs using Velvet Software1.2.08 (Zerbino and Birney, 2008) with a k‐mer of 15–17. Following assembly, contigs were screened for sequence homologies using blastx and blastn (http://www.ncbi.nlm.nih.gov/) programs. Coverage and distribution of viral‐specific siRNAs were determined using the program SearchSmallRNA (de Andrade and Vaslin, 2014) and the genomic RNA sequences of AcCRaV RNAs as reference viral sequences.
Determination of complete genome sequences
Once assembled, the viral RNAs were completely sequenced through several RT‐PCR amplifications using specific primers (Table S3) designed on the basis of the sequences of the contigs. The PCR products were then cloned, sequenced and used to assemble and validate the complete sequences of viral genomic RNA components. The terminal genomic regions were determined using the RACE strategy employing a commercial kit (GeneRacer kit, Invitrogen, Carlsbad, CA, USA) and specific primers (Table S3), according to the manufacturer's instructions. RT‐PCR solutions and conditions were similar to those reported previously (Qu et al., 2014), except that 2 mm of each deoxynucleoside triphosphate (dNTP) was used in a 25‐µL PCR volume, with annealing for 45 s at 50–54°C (depending on the primer set used in each reaction) and extension for 1–3 min (depending on the size of the expected PCR product) at 72°C. PCR products were gel purified and ligated into the pMD18‐T vector (Takara, Dalian, China), following the manufacturer's instructions. At least three positive clones of each PCR product were sequenced at Shanghai Sangon Biological Engineering & Technology and Service Co. Ltd, Shanghai, China. The obtained sequences were assembled into contiguous sequences by overlapping common regions (generally about 100 bp) of the amplicons. Full‐length cDNA amplicons of RNA 2 to RNA 5 were generated by reverse transcription using the specific primer 3C, amplified by PCR using the primer pair AcCRaV 5H/3C (Table S3) and finally cloned and sequenced as reported above.
Sequence analyses
ORF Finder at NCBI was used to search for potential ORFs in the virus genomic and anti‐genomic RNAs. Sequence comparisons of proteins were performed using ClustalW in the software DNASTAR_Lasergene.v7.1 (Thompson et al., 1997). For phylogenetic tree construction, multiple alignments of amino acid sequences were conducted using MAFFT version 6.85, as implemented at http://www.ebi.ac.uk/Tools/msa/mafft/202 with default settings; the resulting data were imported into the program MEGA 5.1 (Tamura et al., 2011) and phylogenetic trees were inferred by the neighbour‐joining method with the p‐distance matrix and pairwise, and with 1000 bootstrap replicates.
Protein analyses were performed using the software for the prediction of glycosylation sites (NetNGlyc 1.0 Server: http://www.cbs.dtu.dk/services/NetNGlyc/), transmembrane helices (TMHMM 2.0 vers.: http://www.ch.embnet.org/software/TMPRED_form.html), signal peptides (SignalP 4.1: http://www.cbs.dtu.dk/services/SignalP/), potential DNA‐ or RNA‐binding residues (BindN: http://bioinfo.ggc.org/bindn/) and coiled coils (COILS: http://embnet.vital-it.ch/software/COILS_form.html). Alignments for multiple protein sequences and/or structures were performed using PROMALS (http://prodata.swmed.edu/promals/promals.php).
Bioassays
Sap extracted from leaves of A. chinensis vines infected by the novel emaravirus was used for mechanical transmission tests. Leaf tissue (about 1 g) was homogenized in 5 mL of 0.02 m phosphate buffer, pH 7.4, containing 0.15% 2‐mercaptoethanol and 0.45% diethyldithiocarbamic acid (DIECA). The homogenate was mixed with 400‐mesh carborundum powder and mechanically inoculated onto the leaves of N. benthamiana. Plants inoculated only with the buffer reported above were used as negative controls. All plants were maintained in an insect‐proof growth room at about 24°C for up to 5 weeks, observed for symptoms and tested for virus infection by RT‐PCR assays and sequencing. For serial inoculations, AcCRaV‐infected N. benthamiana plants were used as the source of inoculum. The inoculated plants were tested for AcCRaV infection by RT‐PCR using the primer pair CP‐F/CP‐R and the degenerated primer pair 1AF/1CR (Table 1), following the protocols reported in the next section, with the exception that the annealing temperature was 54°C for primer pair CP‐F/CP‐R.
Virus detection in field survey
In the field survey, AcCRaV infecting actinidia was detected by RT‐PCR using RNA preparations obtained from leaves (only in one case from dormant twigs), as reported by Li and Mock (2008), and the primers AcCRaV 3F/3R, designed in the sequence of ORF3 of AcCRaV (Table 1), or the degenerate primer set 1AF/1CR, designed previously by Elbeaino et al. (2013) in the conserved domains A and C of RdRP of most emaraviruses. Reverse transcription was performed with Moloney murine leukaemia virus (M‐MLV) reverse transcriptase (Promega, Madison, WI, USA). PCR was performed in a 25‐µL volume of reaction mixture containing 2.5 µL of 10 × PCR buffer, 0.5 mm dNTPs, 0.5 mm of each primer, one unit of Taq DNA polymerase (TaKaRa, Dalian, China) and 2 µL of cDNA, using an iCycler Thermocycler (Bio‐Rad, Hercules, CA, USA). The PCR profile consisted of an initial denaturation at 95°C for 3 min, followed by 35 cycles of 95°C for 30 s, 53°C (for primer set 1AF/1CR) or 55°C (for primer set 3F/3R) for 30 s, 72°C for 1 min and a final extension for 10 min at 72°C. The PCR products were separated by electrophoresis on a 1.2% agarose gel, stained with ethidium bromide and visualized under UV light.
Electron microscopy
For thin sectioning, tissue fragments were excised from young leaves of a kiwifruit plant (A. chinensis) infected with the novel emaravirus and processed according to standard procedures (Martelli and Russo, 1984). Ultrathin sections (60–80 nm), collected on formvar‐coated, carbon‐stabilized copper grids (75 mesh), were stained with 2% uranyl acetate for 15 min, followed by 2% lead acetate for 1.5 min, and then examined with an electron microscope (Tecncai G2 20 TEM, FEI, Oregon, USA). The diameter of the particles was determined by thirty measurements statistically analysed by Kaleidagraph 4.1 (Sinergy Software: http://www.synergy.com/wordpress_650164087/kaleidagraph/).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1 Summary of lllumina deep sequencing results.
Table S2 Primers used for the reverse transcription‐polymerase chain reaction (RT‐PCR) detection of Apple stem grooving virus (ASGV), Actinidia virus A (AcVA), Actinidia virus B (AcVB) and Citrus leaf blotch virus (CLBV).
Table S3 Primers designed for polymerase chain reaction (PCR) amplification of partial‐length and full‐length cDNAs of Actinidia chlorotic ringspot‐associated virus (AcCRaV) RNAs.
Table S4 Comparison of full‐length sequences of the genomic RNAs 3, 4 and 5 of the Actinidia chlorotic ringspot‐associated virus (AcCRaV) HN‐6 isolate (from a vine grown in Hubei Province) with the corresponding RNAs of three AcCRaV isolates from Anhui Province.
Table S5 sRNAs from the HN‐6 sequenced library that contain the conserved termini of Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic RNAs.
Table S6 Nucleotide and amino acid sequence identities (%) between Actinidia chlorotic ringspot‐associated virus (AcCRaV) and other emaraviruses.
Table S7 Reverse transcription‐polymerase chain reaction (RT‐PCR) detection of five viruses in symptomatic kiwifruit vines of different species grown in five Chinese provinces.
Table S8 Nucleotide (below diagonal) and amino acid (above diagonal) sequence identities (%) of the 387‐bp fragment amplified from 18 kiwifruit samples using the primer set 1AF/1CR.
Fig. S1 (a) Preliminary scaffold generated by aligning the contigs (red boxes) with significant amino acid sequence similarity to emaravirus‐encoded proteins along the redbud yellow ringspot‐associated virus (RYRSaV) genomic RNAs (blue boxes). Primers designed for the amplification of Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic cDNAs based on this scaffold are indicated by arrows. The sequences of the primers are reported in Table S3. (b) Length and coverage of the contigs shown in (a).
Fig. S2 Conserved motifs in Actinidia chlorotic ringspot‐associated virus (AcCRaV) proteins P1 (A) and P2 (B). (A) The five motifs (A, DASKWS1135–1140; B, QGNLNMTSS1220–1228; C, SDD1261–1263; D, NK1282–1283; E, EFLST1318–1322) corresponding to the core polymerase module of the RNA‐dependent RNA polymerase (RdRp) active site (A–E) and the N‐terminal endonuclease domains [H103‐PD133–134‐(D/E)xK156–158] characteristic of the nuclease superfamily are indicated by grey boxes with the conserved amino acids and their positions reported above. (B) Conserved motifs present in the glycoprotein encoded by AcCRaV and other emaraviruses: the N‐terminal signal sequence and three transmembrane (TM) domains are denoted by black boxes; five putative glycosylation sites and the conserved motif of the glycoprotein precursor of phleboviruses are indicated by arrowheads with the corresponding aa sequences above; the scissors indicate the two predicted cleavage sites, with the corresponding amino acid sequence and the cleavage site reported above. The predicted glycoproteins generated by cleavages at the reported sites are shown by grey boxes, with the respective estimated molecular weights in parentheses.
Fig. S3 Multiple alignment of the P4 protein of Actinidia chlorotic ringspot‐associated virus (AcCRaV) with P4 of the emaraviruses Raspberry leaf blotch virus (RLBV), Rose rosette virus (RRV), Fig mosaic virus (FMV) and redbud yellow ringspot‐associated virus (RYRSaV). Consensus secondary structure elements are shown below the sequences according to Yu et al. (2013). The central part of P4, similar to 30K MPs, is boxed according to the report of Yu et al. (2013). The position of the coiled‐coil domain (positions 241–269) in the AcCRaV P4 protein is indicated by the horizontal dotted line.
Fig. S4 Size distribution of 18–26‐nt sRNAs derived from the Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic and anti‐genomic RNA strands.
Fig. S5 Relative frequency of 5′‐terminal nucleotides of 18–26‐nt Actinidia chlorotic ringspot‐associated virus (AcCRaV) sRNAs.
Fig. S6 Detection of Actinidia chlorotic ringspot‐associated virus (AcCRaV) in Nicotiana benthamiana plants. Reverse transcription‐polymerase chain reaction (RT‐PCR) performed using the primer sets 1AF/1CR (top panel) and CP‐F/CP‐R (bottom panel). Lanes 1–11, plants of N. benthamiana mechanically inoculated with sap from a plant testing positive for AcCRaV; lanes ck– and ck+, virus‐free seedling and AcCRaV‐infected kiwifruit sample, respectively. Samples 2, 5 and 6 showed systemic symptoms of chlorotic leaf spots and ringspots.
Acknowledgements
This work was funded partly by the agricultural projects (nyhyzx‐201203076‐03 and nyhyzx‐200903004) administered by the Chinese Ministry of Agriculture. Work in the FDS and BN laboratories was partly supported by a dedicated grant (CISIA) from the Ministero dell'Economia e Finanze Italiano to the CNR (Legge 191/2009). We thank Dr Angelo De Stradis Istituto per la Protezione Sostenibile delle Piante, National Research Council (IPSP‐CNR, Italy) and Professor Jian Hong (Zhejiang University, Hangzhou, China) for help with the interpretation and presentation of transmission electron microscopy (TEM) results. We are indebted to Professor Giovanni Paolo Martelli and Dr Luisa Rubino for suggestions and critical reading of the manuscript.
Contributor Information
Francesco Di Serio, Email: whni@mail.hzau.edu.cn.
Ni Hong, Email: francesco.diserio@ipsp.cnr.it.
References
- Adams, I.P. , Glover, R.H. , Monger, W.A. , Mumford, R. , Jackeviciene, E. , Navalinskiene, M. , Samuitiene, M. and Boonham, N. (2009) Next‐generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol. Plant Pathol. 10, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, K. , Kim, K. , Gergerich, R. and Jensen, S. (1998) High plains disease of corn and wheat: ultrastructural and serological aspects. J. Submicrosc. Cytol. Pathol. 30, 563–571. [PubMed] [Google Scholar]
- Al Rwahnih, M. , Daubert, S. , Golino, D. and Rowhani, A. (2009) Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology, 387, 395–401. [DOI] [PubMed] [Google Scholar]
- de Andrade, R.R. and Vaslin, M.F. (2014) SearchSmallRNA: a graphical interface tool for the assemblage of viral genomes using small RNA libraries data. Virol. J. 11, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, Y. , Tugume, A.K. and Valkonen, J.P. (2012) Small‐RNA deep sequencing reveals Arctium tomentosum as a natural host of Alstroemeria virus X and a new putative Emaravirus. PLoS One, 7, e42758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin, A.G. , Chavan, R.R. , Pearson, M.N. , MacDiarmid, R.M. and Cohen, D. (2012) Detection and characterisation of two novel vitiviruses infecting Actinidia. Arch. Virol. 157, 713–722. [DOI] [PubMed] [Google Scholar]
- Blouin, A.G. , Pearson, M. , Chavan, R. , Woo, E. , Lebas, B. , Veerakone, S. , Ratti, C. , Biccheri, R. , MacDiarmid, R. and Cohen, D. (2013) Viruses of kiwifruit (Actinidia species). J. Plant Pathol. 95, 221–235. [Google Scholar]
- Clover, G.R.G. , Pearson, M.N. , Elliott, D.R. , Tang, Z. , Smales, T.E. and Alexander, B.J.R. (2003) Characterization of a strain of Apple stem grooving virus in Actinidia chinensis from China. Plant Pathol. 52, 371–378. [Google Scholar]
- Di Bello, P.L. , Ho, T. and Tzanetakis, I.E. (2015) The evolution of emaraviruses is becoming more complex: seven segments identified in the causal agent of Rose rosette disease. Virus Res. 210, 241–244. [DOI] [PubMed] [Google Scholar]
- Di Serio, F. , de Alba, A.E.M. , Navarro, B. , Gisel, A. and Flores, R. (2010) RNA‐dependent RNA polymerase 6 delays accumulation and precludes meristem invasion of a viroid that replicates in the nucleus. J. Virol. 84, 2477–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaire, L. , Wang, Y. , Gonzalez‐Ibeas, D. , Mayer, K.F. , Aranda, M.A. and Llave, C. (2009) Deep‐sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology, 392, 203–214. [DOI] [PubMed] [Google Scholar]
- Duijsings, D. , Kormelink, R. and Goldbach, R. (2001) In vivo analysis of the TSWV cap‐snatching mechanism: single base complementarity and primer length requirements. EMBO J. 20, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbeaino, T. , Digiaro, M. , Alabdullah, A. , De Stradis, A. , Minafra, A. , Mielke, N. , Castellano, M.A. and Martelli, G.P. (2009a) A multipartite single‐stranded negative‐sense RNA virus is the putative agent of fig mosaic disease. J. Gen. Virol. 90, 1281–1288. [DOI] [PubMed] [Google Scholar]
- Elbeaino, T. , Digiaro, M. and Martelli, G.P. (2009b) Complete nucleotide sequence of four RNA segments of fig mosaic virus. Arch. Virol. 154, 1719–1727. [DOI] [PubMed] [Google Scholar]
- Elbeaino, T. , Whitfield, A. , Sharma, M. and Digiaro, M. (2013) Emaravirus‐specific degenerate PCR primers allowed the identification of partial RNA‐dependent RNA polymerase sequences of Maize red stripe virus and Pigeonpea sterility mosaic virus. J. Virol. Methods, 188, 37–40. [DOI] [PubMed] [Google Scholar]
- Elbeaino, T. , Digiaro, M. , Uppala, M. and Sudini, H. (2014) Deep sequencing of Pigeonpea sterility mosaic virus discloses five RNA segments related to emaraviruses. Virus Res. 188, 27–31. [DOI] [PubMed] [Google Scholar]
- Elbeaino, T. , Digiaro, M. , Uppala, M. and Sudini, H. (2015) Deep sequencing of dsRNAs recovered from mosaic‐diseased pigeonpea reveals the presence of a novel emaravirus: pigeonpea sterility mosaic virus 2. Arch. Virol. 160, 2019–2029. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O. , Brunak, S. , von Heijne, G. and Nielsen, H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971. [DOI] [PubMed] [Google Scholar]
- Ferguson, A.R. and Bollard, G.E. (1990) Domestication of the kiwifruit In: Kiwifruit: Science and Management, (Warrington I.J., Weston, G.C. eds.) pp. 165–246. Auckland: Ray Richards Publisher and NZ Society for Horticultural Science. [Google Scholar]
- Ferguson, A.R. and Huang, H. (2007) Genetic resources of kiwifruit: domestication and breeding. Hortic. Rev. 33, 1–121. [Google Scholar]
- Flock, R. and Wallace, J. (1955) Transmission of fig mosaic by the eriophyid mite Aceria ficus . Phytopathology, 45, 52–54. [Google Scholar]
- Hagen, C. , Frizzi, A. , Kao, J. , Jia, L. , Huang, M. , Zhang, Y. and Huang, S. (2011) Using small RNA sequences to diagnose, sequence, and investigate the infectivity characteristics of vegetable‐infecting viruses. Arch. Virol. 156, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Hedil, M. , Hassani‐Mehraban, A. , Lohuis, D. and Kormelink, R. (2014) Analysis of the AU rich hairpin from the intergenic region of tospovirus S RNA as target and inducer of RNA silencing. PLoS One, 9, e106027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, T. , Wang, H. , Pallett, D. and Dalmay, T. (2007) Evidence for targeting common siRNA hotspots and GC preference by plant Dicer‐like proteins. FEBS Lett. 581, 3267–3272. [DOI] [PubMed] [Google Scholar]
- Huang, H. ed. (2010) Advances in Actinidia Research, pp. 3–18. Beijing: China Science Press. [Google Scholar]
- Ie, T. (1964) An electron microscope study of tomato spotted wilt virus in the plant cell. Neth. J. Plant Pathol. 70, 114–115. [Google Scholar]
- Ishikawa, K. , Maejima, K. , Komatsu, K. , Kitazawa, Y. , Hashimoto, M. , Takata, D. , Yamaji, Y. and Namba, S. (2012) Identification and characterization of two novel genomic RNA segments of fig mosaic virus, RNA5 and RNA6. J. Gen. Virol. 93, 1612–1619. [DOI] [PubMed] [Google Scholar]
- Kormelink, R. , Garcia, M.L. , Goodin, M. , Sasaya, T. and Haenni, A.L. (2011) Negative‐strand RNA viruses: the plant‐infecting counterparts. Virus Res. 162, 184–202. [DOI] [PubMed] [Google Scholar]
- Kreuze, J.F. , Perez, A. , Untiveros, M. , Quispe, D. , Fuentes, S. , Barker, I. and Simon, R. (2009) Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology, 388, 1–7. [DOI] [PubMed] [Google Scholar]
- Kulkarni, N. , Kumar, P.L. , Muniyappa, V. , Jones, A.T. and Reddy, D. (2002) Transmission of Pigeon pea sterility mosaic virus by the eriophyid mite, Aceria cajani (Acari: Arthropoda). Plant Dis. 86, 1297–1302. [DOI] [PubMed] [Google Scholar]
- Kumar, P.L. , Duncan, G. , Roberts, I. , Teifion Jones, A. and Reddy, D. (2002) Cytopathology of Pigeonpea sterility mosaic virus in pigeonpea and Nicotiana benthamiana: similarities with those of eriophyid mite‐borne agents of undefined aetiology. Ann. Appl. Biol. 140, 87–96. [Google Scholar]
- Kumar, P.L. , Jones, A.T. and Reddy, D. (2003) A novel mite‐transmitted virus with a divided RNA genome closely associated with pigeonpea sterility mosaic disease. Phytopathology, 93, 71–81. [DOI] [PubMed] [Google Scholar]
- Laney, A.G. , Gergerich, R. , Keller, K. , Martin, R. and Tzanetakis, I. (2010) Rose rosette and redbud yellow ringspot are caused by two new emaraviruses. Phytopathology, 100, S67. [Google Scholar]
- Laney, A.G. , Keller, K.E. , Martin, R.R. and Tzanetakis, I.E. (2011) A discovery 70 years in the making: characterization of the Rose rosette virus. J. Gen. Virol. 92, 1727–1732. [DOI] [PubMed] [Google Scholar]
- Li, R. and Mock, R. (2008) Characterization of a flowering cherry strain of Cherry necrotic rusty mottle virus. Arch. Virol. 153, 973–978. [DOI] [PubMed] [Google Scholar]
- Lin, Y. and Gao, R. (1995) Survey and identification of Actinidia spp. diseases in Fujian, China. J. Fujian Agric. Univ. 24, 49–53. [Google Scholar]
- Lu, Y. , McGavin, W. , Cock, P.J. , Schnettler, E. , Yan, F. , Chen, J. and MacFarlane, S. (2015) Newly identified RNAs of raspberry leaf blotch virus encoding a related group of proteins. J. Gen. Virol. 96, 3432–3439. [DOI] [PubMed] [Google Scholar]
- Lupas, A. , Van Dyke, M. and Stock, J. (1991) Predicting coiled coils from protein sequences. Science, 252, 1162–1164. [DOI] [PubMed] [Google Scholar]
- Martelli, G.P. and Russo, M. (1984) Use of thin sectioning for visualization and identification of plant viruses. Methods Virol. 8, 143–224. [Google Scholar]
- Martelli, G.P. , Castellano, M.A. and Lafortezza, R. (1993) An ultrastructural study of fig mosaic. Phytopathol. Mediterr. 32, 33–43. [Google Scholar]
- McGavin, W.J. , Mitchell, C. , Cock, P.J. , Wright, K.M. and MacFarlane, S.A. (2012) Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J. Gen. Virol. 93, 430–437. [DOI] [PubMed] [Google Scholar]
- Melcher, U. (2000) The ‘30K'superfamily of viral movement proteins. J. Gen. Virol. 81, 257–266. [DOI] [PubMed] [Google Scholar]
- Mielke‐Ehret, N. and Mühlbach, H.P. (2007) A novel, multipartite, negative‐strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J. Gen. Virol. 88, 1337–1346. [DOI] [PubMed] [Google Scholar]
- Mielke‐Ehret, N. and Mühlbach, H.P. (2012) Emaravirus: a novel genus of multipartite, negative strand RNA plant viruses. Viruses, 4, 1515–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitter, N. , Koundal, V. , Williams, S. and Pappu, H. (2013) Differential expression of Tomato spotted wilt virus‐derived viral small RNAs in infected commercial and experimental host plants. PLoS One, 8, e76276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlbach, H.P. and Mielke‐Ehret, N. (2012) Emaravirus In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses (King A.M.Q., Adams M.J., Carstens E.B. and Lefkowitz E.J., eds.), pp. 767–769. London: Elsevier‐Academic Press. [Google Scholar]
- Pei, J. and Grishin, N.V. (2007) PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics, 23, 802–808. [DOI] [PubMed] [Google Scholar]
- Qu, L.N. , Cui, H.G. , Wu, G.W. , Zhou, J.F. , Su, J.M. , Wang, G.P. and Hong, N. (2014) Genetic diversity and molecular evolution of Plum bark necrosis stem pitting‐associated virus from China. PLoS One, 9, e105443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera, J. , Weber, F. and Cusack, S. (2010) Bunyaviridae RNA polymerases (L‐protein) have an N‐terminal, influenza‐like endonuclease domain, essential for viral cap‐dependent transcription. PLoS Pathol. 6, 8871–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestro, S.R. and Chapman, G.B. (2004) A transmission electron microscope study of “New Dawn” climber rose (Rosa wichuraiana × safrano) exhibiting rose rosette disease. Plant Cell Rep. 23, 345–351. [DOI] [PubMed] [Google Scholar]
- Skare, J. , Wijkamp, I. , Denham, I. , Rezende, J. , Kitajima, E. , Park, J. , Desvoyes, B. , Rush, C. , Michels, G. , Scholthof, K. and Scholthof, H. (2006) A new eriophyid mite‐borne membrane‐enveloped virus‐like complex isolated from plants. Virology, 347, 343–353. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. and Kumar, S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol . Biol. Evol. 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatineni, S. , McMechan, A.J. , Wosula, E.N. , Wegulo, S.N. , Graybosch, R.A. , French, R. and Hein, G.L. (2014) An eriophyid mite‐transmitted plant virus contains eight genomic RNA segments with unusual heterogeneity in the nucleocapsid protein. J Virol. 88, 11 834–11 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D. , Gibson, T.J. , Plewniak, F. , Jeanmougin, F. and Higgins, D.G. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia, J.J. and Falk, B.W. (2012) Fig mosaic virus mRNAs show generation by cap‐snatching. Virology, 426, 162–166. [DOI] [PubMed] [Google Scholar]
- Wang, L. and Brown, S.J. (2006) BindN: a web‐based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 34, W243–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Q. , Ding, S.W. , Zhang, Y. and Zhu, S. (2015) Identification of viruses and viroids by next‐generation sequencing and homology‐dependent and homology‐independent algorithms. Annu. Rev. Phytopathol. 53, 425–444. [DOI] [PubMed] [Google Scholar]
- Yu, C. , Karlin, D.G. , Lu, Y. , Wright, K. , Chen, J. and MacFarlane, S. (2013) Experimental and bioinformatic evidence that raspberry leaf blotch Emaravirus P4 is a movement protein of the 30K superfamily. J. Gen. Virol. 94, 2117–2128. [DOI] [PubMed] [Google Scholar]
- Zerbino, D.R. and Birney, E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Y. , Wang, G. , Hong, N. , Zhou, J. , Yang, Z. and Hong, N. (2014) First report of Actinidia virus A and Actinidia virus B on kiwifruit in China. Plant Dis. 98, 1590–1590. [DOI] [PubMed] [Google Scholar]
- Zhu, C. , Wang, G. , Zheng, Y. , Yang, Z. , Wang, L. , Xu, W. and Hong, N. (2016) RT‐PCR detection and sequence analysis of coat protein gene of Citrus leaf blotch virus infecting kiwifruit trees. Acta Phytopathol. Sin. 46, 11–16. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1 Summary of lllumina deep sequencing results.
Table S2 Primers used for the reverse transcription‐polymerase chain reaction (RT‐PCR) detection of Apple stem grooving virus (ASGV), Actinidia virus A (AcVA), Actinidia virus B (AcVB) and Citrus leaf blotch virus (CLBV).
Table S3 Primers designed for polymerase chain reaction (PCR) amplification of partial‐length and full‐length cDNAs of Actinidia chlorotic ringspot‐associated virus (AcCRaV) RNAs.
Table S4 Comparison of full‐length sequences of the genomic RNAs 3, 4 and 5 of the Actinidia chlorotic ringspot‐associated virus (AcCRaV) HN‐6 isolate (from a vine grown in Hubei Province) with the corresponding RNAs of three AcCRaV isolates from Anhui Province.
Table S5 sRNAs from the HN‐6 sequenced library that contain the conserved termini of Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic RNAs.
Table S6 Nucleotide and amino acid sequence identities (%) between Actinidia chlorotic ringspot‐associated virus (AcCRaV) and other emaraviruses.
Table S7 Reverse transcription‐polymerase chain reaction (RT‐PCR) detection of five viruses in symptomatic kiwifruit vines of different species grown in five Chinese provinces.
Table S8 Nucleotide (below diagonal) and amino acid (above diagonal) sequence identities (%) of the 387‐bp fragment amplified from 18 kiwifruit samples using the primer set 1AF/1CR.
Fig. S1 (a) Preliminary scaffold generated by aligning the contigs (red boxes) with significant amino acid sequence similarity to emaravirus‐encoded proteins along the redbud yellow ringspot‐associated virus (RYRSaV) genomic RNAs (blue boxes). Primers designed for the amplification of Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic cDNAs based on this scaffold are indicated by arrows. The sequences of the primers are reported in Table S3. (b) Length and coverage of the contigs shown in (a).
Fig. S2 Conserved motifs in Actinidia chlorotic ringspot‐associated virus (AcCRaV) proteins P1 (A) and P2 (B). (A) The five motifs (A, DASKWS1135–1140; B, QGNLNMTSS1220–1228; C, SDD1261–1263; D, NK1282–1283; E, EFLST1318–1322) corresponding to the core polymerase module of the RNA‐dependent RNA polymerase (RdRp) active site (A–E) and the N‐terminal endonuclease domains [H103‐PD133–134‐(D/E)xK156–158] characteristic of the nuclease superfamily are indicated by grey boxes with the conserved amino acids and their positions reported above. (B) Conserved motifs present in the glycoprotein encoded by AcCRaV and other emaraviruses: the N‐terminal signal sequence and three transmembrane (TM) domains are denoted by black boxes; five putative glycosylation sites and the conserved motif of the glycoprotein precursor of phleboviruses are indicated by arrowheads with the corresponding aa sequences above; the scissors indicate the two predicted cleavage sites, with the corresponding amino acid sequence and the cleavage site reported above. The predicted glycoproteins generated by cleavages at the reported sites are shown by grey boxes, with the respective estimated molecular weights in parentheses.
Fig. S3 Multiple alignment of the P4 protein of Actinidia chlorotic ringspot‐associated virus (AcCRaV) with P4 of the emaraviruses Raspberry leaf blotch virus (RLBV), Rose rosette virus (RRV), Fig mosaic virus (FMV) and redbud yellow ringspot‐associated virus (RYRSaV). Consensus secondary structure elements are shown below the sequences according to Yu et al. (2013). The central part of P4, similar to 30K MPs, is boxed according to the report of Yu et al. (2013). The position of the coiled‐coil domain (positions 241–269) in the AcCRaV P4 protein is indicated by the horizontal dotted line.
Fig. S4 Size distribution of 18–26‐nt sRNAs derived from the Actinidia chlorotic ringspot‐associated virus (AcCRaV) genomic and anti‐genomic RNA strands.
Fig. S5 Relative frequency of 5′‐terminal nucleotides of 18–26‐nt Actinidia chlorotic ringspot‐associated virus (AcCRaV) sRNAs.
Fig. S6 Detection of Actinidia chlorotic ringspot‐associated virus (AcCRaV) in Nicotiana benthamiana plants. Reverse transcription‐polymerase chain reaction (RT‐PCR) performed using the primer sets 1AF/1CR (top panel) and CP‐F/CP‐R (bottom panel). Lanes 1–11, plants of N. benthamiana mechanically inoculated with sap from a plant testing positive for AcCRaV; lanes ck– and ck+, virus‐free seedling and AcCRaV‐infected kiwifruit sample, respectively. Samples 2, 5 and 6 showed systemic symptoms of chlorotic leaf spots and ringspots.