

Summary
A conserved mitogen‐activated protein kinase (MAPK) cascade homologous to the yeast Fus3/Kss1 mating/filamentation pathway is involved in the regulation of vegetative development and pathogenicity in Fusarium graminearum. However, little is known about the downstream transcription factors of this pathway. In Saccharomyces cerevisiae, the homeodomain protein Ste12 is a key transcription factor activated by Fus3/Kss1. In this study, we characterized a Ste12 orthologue FgSte12 in F. graminearum. The FgSTE12 deletion mutant (ΔFgSte12) was impaired in virulence and in the secretion of cellulase and protease, although it did not show recognizable phenotype changes in hyphal growth, conidiation or deoxynivalenol (DON) biosynthesis. In addition, ΔFgSte12 and the FgGPMK1 (a FUS3/KSS1‐related MAPK gene) mutant shared several phenotypic traits. Furthermore, we found that FgGpmk1 controls the nuclear localization of FgSte12. Yeast two‐hybrid and affinity capture assays indicated that FgSte12 interacts with the FgSte11–Ste7–Gpmk1 complex. Taken together, these results indicate that FgSte12 is a downstream target of FgSte11–Ste7–Gpmk1 and plays an important role in pathogenicity in F. graminearum.
Keywords: Fusarium graminearum, FgGpmk1, pathogenicity, transcription factor FgSte12
Introduction
Fusarium graminearum (teleomorph Gibberella zeae) causes Fusarium head blight (FHB), which is a devastating disease of cereal crops worldwide. Infection of cereal crops with F. graminearum may lead to huge yield losses in severe epidemic years (Goswami and Kistler, 2004). More importantly, trichothecene mycotoxins, including deoxynivalenol (DON), acetyldeoxynivalenol (3‐ADON or 15‐ADON), nivalenol (NIV) and acetylnivalenol (4‐ANIV), produced by F. graminearum in infested grains, pose a serious threat to human and animal health (McMullen et al., 1997; Pestka and Smolinski, 2005). Despite the serious damage caused by FHB, efficient strategies for the management of FHB are not available to date (Steiner et al., 2009). Understanding the fundamental biology of this fungal pathogen can provide the basis for sustainable, long‐term disease management.
In several plant‐pathogenic fungi, the mitogen‐activated protein kinase (MAPK), homologous to the Saccharomyces cerevisiae mating/filamentation MAPKs Fus3/Kss1, has been found to be involved in pathogenicity (Qi and Elion, 2005). The essential role of this MAPK (named Pmk1) during infection was first reported in the rice blast fungus Magnaporthe grisea. Fungal mutants lacking the PMK1 gene were defective in appressorium formation and were unable to infect host plants. Subsequently, Fus3/Kss1 orthologues were also found to be essential for pathogenicity in other plant pathogens, including Colletotrichum lagenarium and Botrytis cinerea (Park et al., 2006; Takano et al., 2000; Zhao et al., 2005, 2007; Zheng et al., 2000). In F. graminearum, FgGpmk1 (the orthologue of Fus3/Kss1) is involved in vegetative differentiation and virulence (Jenczmionka et al., 2003; Jenczmionka and Schafer, 2005). FgGpmk1 is known to control a variety of virulence‐related functions (Zhao et al., 2007); however, questions remain unanswered on how this MAPK regulates the pathogenicity of F. graminearum.
The transcription factor Ste12, which functions downstream of the MAPKs Fus3/Kss1, binds to pheromone and filamentation response elements in the promoters of target genes involved in invasive growth in S. cerevisiae (Madhani et al., 1997). Ste12 orthologues have been identified and characterized in several plant‐pathogenic fungal species. In M. oryzae, the appressoria formed by MST12 mutants have normal appressorium turgor, but fail to develop penetration pegs (Park et al., 2002, 2004). The orthologue of Ste12 in C. lagenarium is Cst1. Unlike Mst12 of M. oryzae, Cst1 is essential for penetration and infectious growth, but dispensable for appressorium formation (Tsuji et al., 2003). Similarly, disruption of the Ste12 orthologue in Cryphonectria parasitica results in dramatically reduced virulence, suggesting an essential role of CpSte12 during the infection process (Wong Sak Hoi et al., 2007).
A whole‐genome search revealed that F. graminearum contains an orthologue of Ste12 (hereafter named FgSte12). Based on the previous studies, we hypothesized that FgSte12 might play an important role in the pathogenicity of F. graminearum. To address this hypothesis, the main objective of this study was to analyse the biological, genetic and biochemical functions of FgSte12. Our study revealed that the FgSTE12 deletion mutants did not show recognizable phenotype changes in vegetative growth, conidiation or DON biosynthesis, but were impaired in pathogenicity on host plants. FgSte12 may function downstream of the FgGpmk1 in F. graminearum.
Results
In silico analysis of FgSte12 in F. graminearum
The Ste12 orthologue of F. graminearum was retrieved by blastp search of the Fusarium genome database (http://www.broadinstitute.org/annotation/genome/fusarium_group/MultiHome.html) with the S. cerevisiae Ste12 protein as a query. FgSte12 was predicted to encode a 695‐amino‐acid protein, which shares 60% similarity with the S. cerevisiae Ste12. In addition, protein domain analysis by SMART (http://smart.embl‐heidelberg.de/) revealed that FgSte12 contains a STE domain and two C2H2 zinc finger domains (Fig. 1a), which are highly homologous to those of other Ste12 orthologues (Fig. 1a–c).
Figure 1.

Sequence alignment of Ste12 orthologues from Fusarium graminearum and other fungi. (a) Each Ste12 orthologue contains two functional protein domains, STE and C2H2 domains, which were identified by SMART (http://smart.embl‐heidelberg.de/). (b) Alignments of amino acid sequences of STE domain of the Ste12 orthologue of F. graminearum with those of Saccharomyces cerevisiae, Magnaporthe grisea, Candida albicans and Neurospora crassa. The Boxshade program was used to highlight identical (black shading) or similar (grey shading) amino acids. (c) Alignments of amino acid sequences of two C2H2 domains of FgSte12 with those of M. grisea and N. crassa. The Boxshade program was used to highlight identical (black shading) or similar (grey shading) amino acids.
Deletion and complementation of FgSTE12 in F. graminearum
For a detailed functional analysis of FgSte12, we generated gene deletion mutants using a homology recombination strategy (Fig. 2a). Among 50 hygromycin‐resistant transformants, 24 FgSTE12 deletion mutants were identified by PCR analysis with the primer pair A5 + A6 (Table S1, see Supporting Information). The primer pair amplified 1865‐bp and 1548‐bp fragments from the deletion mutants and the wild‐type progenitor HN9‐1, respectively. Reverse transcription‐polymerase chain reaction (RT‐PCR) with the primer pair A7 + A8 amplified an expected 407‐bp fragment from the wild‐type progenitor HN9‐1 and complemented strain ΔFgSte12‐C, but not from the deletion mutant ΔFgSte12 (Fig. 2b). When probed with a 668‐bp DNA fragment of FgSTE12, ΔFgSte12 had an anticipated 4076‐bp band, but lacked a 2446‐bp band, which was present in the wild‐type progenitor (Fig. 2c). The Southern hybridization pattern confirmed that ΔFgSte12 is a null mutant resulting from the homologous recombination events at the FgSTE12 locus. The complemented strain ΔFgSte12‐C contains a single copy of the wild‐type FgSTE12, which was inserted into the genome of ΔFgSte12 (Fig. 2c).
Figure 2.

Generation and identification of the FgSTE12 deletion mutant and complemented strain. (a) Gene deletion strategy for FgSTE12. The hygromycin resistance cassette (HPH) is denoted by the large black arrow. Primer binding sites are indicated by arrows (see Table S1 for the primer sequences). A 668‐bp downstream fragment of FgSTE12 was used as a probe in Southern blot hybridization analysis. (b) Reverse transcription‐polymerase chain reaction (RT‐PCR) analysis for the transcription of FgSTE12 in the wild‐type HN9‐1, the FgSTE12 deletion mutant (ΔFgSte12) and the complemented strain (ΔFgSte12‐C). NC is a negative control without cDNA template. (c) Southern blot hybridization analysis of FgSTE12 locus in HN9‐1, ΔFgSTE12 and ΔFgSte12‐C. The genomic DNA preparation of each strain was digested with XhoI.
FgSte12 is dispensable for hyphal growth, conidiation and conidial germination
The FgSTE12 mutant (ΔFgSte12) did not show obvious phenotypic changes when compared with the wild‐type progenitor in hyphal growth and colony morphology on potato dextrose agar (PDA) or minimal medium (MM) (Fig. 3). In addition, to determine the effects of FgSTE12 deletion on conidiation, 10 mycelial plugs of each strain were inoculated in mung bean liquid (MBL) medium. After 4 days of incubation in a shaker, we found that ΔFgSte12 did not exhibit any defects in conidiation (Fig. S1a, see Supporting Information). Furthermore, the conidia of ΔFgSte12 germinated normally in 2% (w/v) sucrose solution (Fig. S1b).
Figure 3.
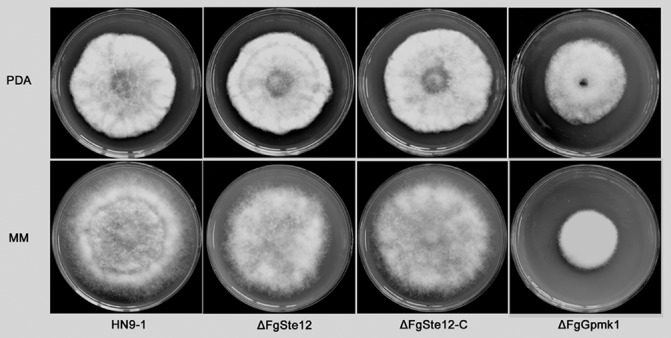
Effects of FgSTE12 and FgGPMK1 deletion on colony morphology. The wild‐type HN9‐1, ΔFgSte12, ΔFgSte12‐C and FgGPMK1 deletion mutant (ΔFgGpmk1) were grown on potato dextrose agar (PDA) and minimal medium (MM) at 25 °C for 3 days.
FgSte12 is required for pathogenicity of F. graminearum
Pathogenicity of ΔFgSte12 was evaluated by point inoculation of conidial suspensions on flowering wheat heads. Fifteen days after inoculation, scab symptoms failed to develop, even in the spikelet inoculated with conidia of ΔFgSte12. Under the same conditions, however, the wild‐type HN9‐1 and the complemented strain caused serious scab symptoms on wheat heads (Fig. 4a). In addition, each strain was incubated on wheat‐head medium. As revealed in Fig. 4b, the mutant showed no detectable vegetative defects when compared with the wild‐type HN9‐1, indicating that, although a growth medium made with wheat heads provides the necessary nutrients for growth, the mutant is still unable to cause disease on wheat heads.
Figure 4.

Virulence of FgSTE12 and FgGPMK1 deletion mutants. (a) Wheat heads were point inoculated with conidial suspensions of the wild‐type HN9‐1, ΔFgSte12, ΔFgSte12‐C and ΔFgGpmk1. Infected wheat heads were photographed 15 days after inoculation. The negative control (NC) was only injected with water. (b) Each strain was inoculated on wheat‐head medium and incubated at 25 °C for 3 days. (c) Tomatoes were inoculated with a conidial suspension of each strain and infected fruits were photographed 3 days after inoculation. The negative control (NC) was only injected with water (without fungal conidia).
DON is an important virulence factor in F. graminearum (Proctor et al., 1995; Seong et al., 2009; Starkey et al., 2007). As ΔFgSte12 is impaired in the infection of flowering wheat heads, we were also interested in analysing DON production for the mutant. After incubation on wheat kernels for 20 days, ΔFgSte12 produced similar amounts of DON as the wild‐type HN9‐1 (Fig. S2, see Supporting Information), suggesting that FgSte12 is not involved in the regulation of DON biosynthesis in F. graminearum.
To examine the ability of ΔFgSte12 to colonize tomato, a 10‐μL aliquot of conidial suspension of each strain was injected into wounded tomato after surface sterilization. As shown in Fig. 4c, the disease lesion caused by ΔFgSte12 on wounded tomato was dramatically smaller than that caused by the wild‐type HN9‐1. To test whether the reduction in virulence is associated with a penetration defect of the mutant, we performed a cellophane penetration assay. As shown in Fig. 5a, ΔFgSte12 was unable to, but the wild‐type and complemented strains were able to, penetrate cellophane sheet. Furthermore, the addition of NaNO3 or NH4NO3 had no positive effects on the penetration of ΔFgSte12 into the cellophane sheet (Fig. S3, see Supporting Information). In addition, electron scanning microscopy examination showed that the hyphae of the wild‐type progenitor formed penetration structures on wheat spikelets, but such penetration structures were not observed for ΔFgSte12 (Fig. 5b). These results suggest that FgSte12 is required for penetration and extension during infection of F. graminearum on host plants.
Figure 5.

Penetration of FgSTE12 and FgGPMK1 deletion mutants into cellophane membrane and wheat glumes. (a) Fungal colonies were grown for 2 days at 25 °C on top of cellophane membranes placed on minimal medium (Before). The cellophane membranes with the fungal colonies were removed, and the plates were incubated for an additional day to examine the presence of mycelial growth on the plate, indicating penetration of the cellophane (After). (b) Infection assay on dissected wheat glumes. Wheat head spikelets were inoculated with conidia of each strain. Samples were taken 8 days after inoculation, and examined with a scanning electron microscope. Infection structures (FS, foot structure; LA, lobate appressorium indicated by the arrows) were observed in the wild‐type (HN9‐1), but not in the mutant ΔFgSte12. Bar, 5 μm.
FgSte12 regulates sexual development
As a homothallic fungus, sexual reproduction plays a significant role in the infection cycle of F. graminearum (Min et al., 2012; Trail, 2009). In order to characterize the function of FgSte12 in sexual development, we determined the ability of the mutant to form perithecia on carrot agar. Consistent with the finding from a previous report (Jenczmionka et al., 2003), we found that the FgGPMK1 mutant was sexually sterile. The FgSTE12 deletion mutant produced significantly (P < 0.05) less perithecia than the wild‐type HN9‐1 (Fig. 6).
Figure 6.

Effects of the deletion of FgSTE12 and FgGPMK1 on sexual development of Fusarium graminearum. The wild‐type HN9‐1, ΔFgSte12 and ΔFgGpmk1 were incubated on carrot agar for 2 weeks to induce the formation of perithecia. Line bars denote standard errors of three repeated experiments. Values on the open bars followed by the same letter are not significantly different according to Fisher's least significant difference (LSD) test at P = 0.05.
FgSte12 regulates the secretion of cell wall‐degrading enzymes
Cell wall‐degrading enzymes secreted by F. graminearum have been found to be important pathogenicity factors. A previous study has indicated that FgGpmk1 regulates the activities of extracellular endoglucanase, and xylanolytic and proteolytic enzymes (Jenczmionka and Schafer, 2005). Furthermore, this MAPK was also responsible for the induction of secreted lipolytic activities (Jenczmionka and Schafer, 2005). As ΔFgSte12 was unable to penetrate a cellophane sheet, we were also interested in analysing the secreted proteolytic and endoglucanase activity for the mutant. As shown in Fig. 7a,b, ΔFgSte12 exhibited reduced proteolytic and cellulase activities, which could partially explain the reduced virulence of the mutant on the host plant. However, ΔFgSte12 did not show a detectable alteration in lipolytic activity compared with the wild‐type HN9‐1 (Fig. S4, see Supporting Information).
Figure 7.

Comparison of the activities of cell wall‐degrading enzymes secreted by the wild‐type HN9‐1, ΔFgSte12 and ΔFgGpmk1. (a) Cellulase activity was measured spectrophotometrically using carboxymethylcellulose as substrate. One unit of enzymatic activity is defined as 1 nmol/min reducing sugars released from the substrate. Line bars denote the standard errors of three repeated experiments. (b) Proteolytic activity. One unit of enzymatic activity is defined as 1 nmol/min aromatic amino acids released from the substrate. Line bars denote standard errors of three repeated experiments.
In S. cerevisiae, the cis‐acting sequence of TGAACA was recognized by Ste12 (Yuan and Fields, 1991). We also identified four cellulase genes which contain the sequence of TGAACA in their promoters. Quantitative real‐time PCR assays showed that the expression levels of these four genes were decreased significantly in ΔFgSte12 (Fig. 8). In contrast, the transcription levels were similar to the wild‐type for four other cellulase genes without the sequence of TGAACA in their promoters (Fig. S5, see Supporting Information); this suggests that FgSte12 might normally bind to the cis‐acting sequence in F. graminearum.
Figure 8.

Effects of FgSTE12 deletion on the transcription of cellulase genes. The relative transcription level of each cellulase gene in ΔFgSte12 is the relative amount of mRNA of each gene in the wild type HN9‐1. These four genes contain the cis‐element TGAAACA in their promoters. Line bars in each column denote standard errors of three repeated experiments. Values on the grey bars followed by the same letter are not significantly different according to Fisher's least significant difference (LSD) test at P = 0.05.
Identification of FgSte12‐interacting proteins
To identify FgSte12‐interacting proteins, we conducted an affinity capture experiment. Briefly, FgSte12 was tagged with green fluorescent protein (GFP), and proteins co‐purified with FgSte12‐GFP were analysed by mass spectrometry. Interestingly, FgSte7 (encoded by the FGSG_09897 gene) was co‐purified with FgSte12‐GFP (Table 1). blastp analysis revealed that FgSte7 is homologous to S. cerevisiae ScSte7, which functions as a kinase upstream of FgGpmk1. In F. graminearum, the interaction of FgGpmk1 with FgSte7, but not with FgSte12, was demonstrated by yeast two‐hybrid assay (Fig. 9).
Table 1.
Identification of FgSte12 interacting proteins by affinity capture assays
| Proteins | Putative encoding protein |
|---|---|
| FGSG_09897 | Ste7 |
| FGSG_10042 | Signal transduction histidine kinase |
| FGSG_07397 | Predicted unusual protein kinase |
| FGSG_04382 | Serine/threonine protein kinase nrc‐2 |
| FGSG_06957 | Serine/threonine protein kinase Cla4 |
| FGSG_09903 | Serine/threonine protein kinase carbon catabolite derepressing protein kinase |
| FGSG_05912 | Mevalonate kinase |
| FGSG_11938 | ATP phosphoribosyltransferase |
| FGSG_10198 | Aconitase A |
| FGSG_09908 | Regulatory subunit of cAMP‐dependent protein kinases |
| FGSG_09892 | Uncharacterized conserved protein, contains RING Zn‐finger |
| FGSG_09616 | Cullin, a subunit of E3 ubiquitin ligase |
| FGSG_09572 | 5,10‐Methylenetetrahydrofolate reductase |
| FGSG_09555 | Electron transfer flavoprotein, β subunit |
| FGSG_09020 | Cation transport ATPase |
| FGSG_08529 | Phosphoserine phosphatase |
| FGSG_05683 | Predicted flavoprotein involved in K+ transport |
| FGSG_05447 | Rho5 |
| FGSG_05293 | Predicted divalent heavy‐metal cation transporter |
| FGSG_05132 | Predicted phosphatase |
| FGSG_04948 | Uncharacterized conserved protein, contains RING Zn‐finger |
| FGSG_04555 | Uncharacterized conserved protein |
| FGSG_03412 | Amino acid transporters |
| FGSG_02570 | Translation elongation factors (GTPases) |
| FGSG_02460 | Cation transport ATPase |
| FGSG_01947 | Sulphite oxidase and related enzymes |
| FGSG_01866 | Predicted GTPases (dynamin‐related) |
| FGSG_01329 | Adenylylsulphate kinase and related kinases |
| FGSG_01006 | Sulphate permease and related transporters |
| FGSG_00852 | Diadenosine tetraphosphatase and related serine/threonine protein phosphatases |
Figure 9.

Yeast two‐hybrid analyses of interactions between FgSte7, FgGpmk1 and FgSte12. Serial decimal dilutions of yeast cells (cells/mL) transferred with the bait and prey constructs indicated in the figure were assayed for growth on SD‐Leu‐Trp‐His [synthetic medium lacking leucine (Leu), tryptophan (Trp) and histidine (His)] plates. The transformant bearing the pair of plasmids pGBKT7‐53 and pGADT7 served as a positive control. The transformants bearing the pairs of plasmids pGBKT7‐Lam and pGADT7, pGBKT7 and pGADT7‐FgGpmk1, pGBKT7‐FgSte7 and pGADT7, and pGBKT7‐FgSte12 and pGADT7 were used as negative controls.
To further investigate the relationship between FgGpmk1 and FgSte12, we generated FgGPMK1 mutants. Phenotypic analyses showed that, similar to FgSTE12 mutants, the FgGPMK1 mutant exhibited defects in pathogenicity, penetration of cellophane sheet, secretion of protease and cellulose, and was unable to form perithecia (Figs 4, 5, 6, 7). In addition, the transcription level of FgSTE12 was increased three‐fold in the FgGPMK1 mutant (Fig. S6a, see Supporting Information), indicating that FgGpmk1 positively regulates the transcription of FgSTE12. Western blotting assays showed that the phosphorylation of FgGpmk1 in ΔFgSte12 did not change dramatically when compared with that in the wild‐type progenitor (Fig. S7, see Supporting Information). To further understand how FgGpmk1 regulates FgSte12, we analysed the subcellular localization of FgSte12 in ΔFgGpmk1. Briefly, the entire FgSte12 protein coding sequence was fused in‐frame to GFP under the strong constitutive promoter RP27, and introduced into ΔFgGpmk1 and ΔFgSte12, respectively. As shown in Fig. 10, FgSte12‐GFP localized to the nucleus in mycelia in the ΔFgSte12 background strain. However, FgSte12‐GFP localized to both the nucleus and cytoplasm in the ΔFgGpmk1 background strain, indicating that FgGpmk1 regulates the subcellular localization of FgSte12. Taken together, these results indicate that FgSte12 may function downstream of FgGpmk1 in F. graminearum.
Figure 10.

Subcellular localization of FgSte12 in Fusarium graminearum. The entire FgSte12 protein coding sequence was fused to green fluorescent protein (GFP) with the constitutive promoter RP27, and introduced into ΔFgGpmk1 and ΔFgSte12. FgSte12‐GFP was localized in the nucleus in mycelia of the ΔFgSte12 background strain (left panel), and into both cytoplasm and nucleus in the ΔFgGpmk1 background strain (right panel). Bar, 5 μm. DIC, differential interference contrast. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI).
Discussion
The Ste12 and Ste12‐like proteins are transcription factors found exclusively in the fungal kingdom. Ste12 was originally reported to be involved in the regulation of mating and invasive growth as a target of the Fus3 and Kss1 MAPK cascade in S. cerevisiae (Errede and Ammerer, 1989; Gavrias et al., 1996; Madhani and Fink, 1997). In this study, we identified a Ste12 orthologue from F. graminearum. Similar to other Ste12 orthologues, FgSte12 contains a characteristic STE homeodomain in the N‐terminal region of the protein (Fig. 1a). Although the fungal STE homeodomain is highly divergent, a sequence of KQKVFFWFSVA in this domain is considerably conserved (Yuan and Fields, 1991). In S. cerevisiae, a pheromone response element (PRE) containing a sequence of TGAAACA is able to interact with the Ste12 protein (Yuan and Fields, 1991). The STE domain has also been found to be required for DNA binding in Cryptococcus neoformans (Chang et al., 2004). A recent study has shown that each base pair from the cis‐element sequence of TGAAACA recognized by Ste12 is important for binding, except for the last A, in Colletotrichum lindemuthianum (Wong Sak Hoi et al., 2007). In F. graminearum, we found that four cellulase genes (FGSG_10922, FGSG_04678, FGSG_03795 and FGSG_11487) contain this cis‐element in their promoter regions. Quantitative real‐time PCR assays showed that the transcription levels of these genes were decreased dramatically, but the transcription levels of other cellulase genes without the cis‐element in their promoters did not change significantly, in ΔFgSte12. These results indicate that FgSte12 may regulate the transcription of target genes by binding to this cis‐element in F. graminearum.
Ste12 proteins have been shown to be required for infection in several pathogenic fungi, including Alternaria brassicicola, M. grisea, Fusarium oxysporum, C. lagenarium and C. lindemuthianum (Asuncion Garcia‐Sanchez et al., 2010; Park et al., 2002; Schamber et al., 2010). In M. grisea, the MST12 mutant showed a defect in microtubule reorganization during penetration peg formation, which possibly leads to penetration defects (Park et al., 2004). In C. lindemuthianum, the STE12 mutant showed decreased pectinase activity (Wong Sak Hoi et al., 2007). However, Ste12 is not required for the activation of pectinase genes in F. oxysporum (Asuncion Garcia‐Sanchez et al., 2010). In contrast with pectinase activity, the activities of extracellular amylase and cellulase were positively regulated by Ste12 in F. oxysporum (Asuncion Garcia‐Sanchez et al., 2010). In our study, we observed that FgSte12 is required for F. graminearum penetration of the cellophane sheet and host plant tissue. The defects in penetration may partially result from the low levels of secretion of the cell wall‐degrading enzymes in ΔFgSte12. In A. brassicicola, the STE12 mutant failed to penetrate host plants to cause disease symptoms. The mutants produced multiple hyphae at several locations of immature conidia and were unable to form appressoria (Cho et al., 2009). These results indicate that, although Ste12 proteins are required for infection in various plant‐pathogenic fungi, the mechanisms for the involvement of Ste12 in pathogenicity vary dramatically in different fungal species.
To test whether FgSte12 functions downstream of the MAPK, FgGpmk1, we also generated FgGPMK1 mutants and compared the phenotypes of ΔFgSte12 and ΔFgGpmk1 systematically. ΔFgSte12 and ΔFgGpmk1 share several phenotypic traits, including defects of penetration into cellophane, induction of cell wall‐degrading enzymes, sexual development and pathogenicity. In addition, a high transcript level of FgSTE12 was detected in the FgGPMK1 mutant. Furthermore, FgGpmk1 regulates the nuclear localization of FgSte12. These results indicate that FgSte12 may be associated with the FgGpmk1 pathway. In the yeast two‐hybrid assay, we did not observe the direct interaction between FgSte12 and the MAPK FgGpmk1 (Fig. 9). However, we found that FgSte7 was co‐purified with FgSte12 in the affinity capture assay (Table 1). As reported recently, FgGpmk1 and FgSte11–FgSte7 form a complex (Wang et al., 2011). In our yeast two‐hybrid assays, we observed the physical interaction between FgSte7 and FgGpmk1 (Fig. 9). Thus, we propose that FgSte12 interacts with the FgSte11–FgSte7–FgGpmk1 complex in F. graminearum. Furthermore, Western blotting assays showed that FgGpmk1 phosphorylation was not detectable in the Fgste11 and Fgste7 mutants (Ramamoorthy et al., 2007), indicating that FgSte11 and FgSte7 function upstream of FgGpmk1. However, phosphorylation of FgGpmk1 was not affected by the deletion of FgSTE12 (Fig. S7), indicating that FgSte12 may be a downstream target of the FgSte11–Ste7–Gpmk1 complex.
Although ΔFgSte12 and ΔFgGpmk1 shared several characteristic phenotypes, we also identified some defects of ΔFgGpmk that were not observed for ΔFgSte12. The FgGPMK1 mutant exhibited decreased hyphal growth and conidiation. A delay in conidial germination was observed in ΔFgGpmk1 (Jenczmionka et al., 2003). By contrast, deletion of FgSTE12 had no detectable effects on hyphal growth, conidiation or conidial germination (Fig. 3, Fig. S1a,b), indicating that, although FgSte12 may be one of the outputs of FgGmpk1, other downstream transcription factor(s) might be involved in the regulation of vegetation differentiation in F. graminearum.
In S. cerevisiae, the phosphorylation of Ste12 by either Fus3 or Kss1 was established as a key step for the induction of the activity of Ste12 (Wong Sak Hoi and Dumas, 2010). Phosphorylated Ste12, dissociated from its regulatory proteins (Dig1 and Dig2), is input into the nucleus and activates its target genes (Bardwell et al., 1998; Cook et al., 1996). In the current study, we found that deletion of FgGpmk1only had a partial effect on the nuclear localization of FgSte12 (Fig. 10), indicating that other unknown upstream component(s) may also act on FgSte12 (Fig. 11). In our affinity capture assays, we found that several kinases (including FGSG_10042, FGSG_07397, FGSG_06957 and FGSG_04382) interact with FgSte12 (Table 1). These kinases might also play important roles in the phosphorylation of the FgSte12 protein. Thus, further characterization of these FgSte12‐interacting proteins will be helpful for a full understanding of the functions of Ste12 in filamentous fungi.
Figure 11.

Proposed functions of FgSte12 in Fusarium graminearum. FgSte12 regulates penetration, sexual development and activities of lytic enzymes downstream of the FgSte11–Ste7–Gpmk1 complex, whereas the remaining FgGpmk1‐controlled functions may be mediated by other unidentified factors. In addition, other unknown kinase(s) may also regulate FgSte12.
Experimental Procedures
Fungal strains, growth assays and sporulation tests
Fusarium graminearum strain HN9‐1 was used as the wild‐type progenitor for the construction of various gene deletion mutants. The wild‐type strain, resultant mutants and complemented strains were cultured at 25 °C on PDA (200 g potato, 20 g dextrose, 20 g agar and 1 L water), MM (10 mm K2HPO4, 10 mm KH2PO4, 4 mm (NH4)2SO4, 2.5 mm NaCl, 2 mm MgSO4, 0.45 mm CaCl2, 9 mm FeSO4, 10 mm glucose and 1 L water, pH 6.9) or wheat‐head medium (200 g ground fresh wheat heads and 20 g agar in 1 L double‐distilled H2O) for mycelial growth tests.
For conidiation assays, 10 mycelial plugs (5 mm in diameter) of each strain, taken from the periphery of a 3‐day‐old colony, were inoculated in a 50‐mL flask containing 20 mL of MBL (10 g mung beans boiled in 1 L water for 20 min and filtered through cheesecloth) (Mirocha et al., 1989). The flasks were incubated at 25 °C for 4 days in a shaker (180 rpm). The number of conidia was determined for each strain using a haemocytometer. The experiment was repeated three times independently.
Constructed gene deletion and complemented strains
The gene deletion vectors and transformation of F. graminearum were carried out using the protocols described previously (Jiang et al., 2011a). The primers used to amplify the flanking sequences for each gene are listed in Table S1. Putative gene deletion mutants were identified by PCR assays with the relevant primers (Table S1), and were further analysed by the Southern blotting assay (Fig. 2c). All of the mutants generated in this study were preserved in 15% glycerol at −80 °C.
Microscopic examinations of hyphal and conidial morphology
The hyphal morphology of each mutant was examined with a Leica TCS SP5 imaging system (Wetzlar, Hesse‐Darmstadt, Germany) using fresh mycelia harvested from a 3‐day‐old colony of each strain growing on PDA plates. For the observation of GFP signals (GFP fused with FgSte12), fresh mycelia were examined with a Zeiss LSM780 confocal microscope (Gottingen, Niedersachsen, Germany). To observe nuclei, fresh mycelia were washed with sterilized water and stained with 10 μg/mL of 4′,6‐diamidino‐2‐phenylindole (DAPI) (Sigma, St. Louis, MO, USA).
Assays for sexual development
Aerial hyphae of a 3‐day‐old culture of each strain on carrot agar were pressed down with 300 μL of sterilized 0.1% Tween 60. Perithecium formation was examined after 1 week of incubation at 25 °C, as described previously (Jenczmionka et al., 2003). The experiment was repeated three times.
Pathogenicity assays
The pathogenicity of each strain on flowering wheat heads was assessed as described previously (Jiang et al., 2011a). Briefly, each strain was cultured in MBL for 5 days. The resultant conidia were collected by filtration through three layers of cheese cloth and subsequently resuspended in sterilized water to a concentration of 106 conidia/mL. A 10‐μL aliquot of conidial suspension was injected into a floret in the central section spikelet of a single flowering wheat head of susceptible cultivar Zimai22. The control heads were inoculated with 10 μL of sterilized water. There were 20 replicates for each strain. After inoculation, the plants were kept under 100% humidity at 22 ± 2 °C for 2 days, and then maintained in a glasshouse. Fifteen days after inoculation, the infected spikelets in each inoculated wheat head were recorded. The experiment was repeated four times independently. To analyse the virulence defect of the mutants in detail, infected wheat spikelets were sampled 8 days after inoculation. Hyphal structures of each strain growing in the spikelet were examined with a Hitachi TM‐1000 tabletop microscope (Hitachi, Tokyo, Japan).
To examine the ability to colonize tomato, a 10‐μL aliquot of a conidial suspension was injected into wounded tomato after surface sterilization. There were five replicates for each strain. Inoculated tomatoes were incubated at 25 °C and 100% humidity with 12 h of daylight, and were photographed 3 days after inoculation. The experiment was repeated three times.
To analyse the virulence defect of the mutants in detail, the penetration behaviour of each strain was examined on cellophane membranes, as described previously (Lopez‐Berges et al., 2010). Briefly, each strain was grown on MM covered with a cellophane membrane. After 2 days of incubation, the cellophane membrane with the colony was removed from each plate. After the plates had been incubated for an additional day, mycelial growth on each plate was examined. The presence of mycelial growth on the plate indicated penetration of the cellophane membrane. The experiment was repeated three times.
Determination of DON production
A 50‐g aliquot of healthy wheat kernels was sterilized and inoculated with five mycelial plugs of each strain. A negative control was prepared with wheat kernels inoculated with five agar plugs. After incubation at 25 °C for 20 days, DON was extracted using a previously described protocol (Mirocha et al., 1998). The DON extracts were purified with a PuriToxSR DON column TC‐T200 (Trilogy Analytical Laboratory, Washington, MO, USA), and the amount of DON (per milligram of fungal DNA) in each sample was determined using a Waters 1525 high‐performance liquid chromatography (HPLC) system (Shimadzu Corporation, Kyoto, Japan) (Jiang et al., 2011b). The amount of F. graminearum DNA in each sample was determined using a quantitative real‐time PCR method (Yin et al., 2009). The experiment was repeated three times, and the data were analysed using analysis of variance (SAS version 8.0; SAS Institute, Cary, NC, USA).
RNA extraction and quantitative real‐time PCR
To extract total RNA, mycelia of each strain were inoculated in potato dextrose broth (PDB) and cultured for 2 days at 25 °C in the dark. Mycelia were harvested by filtration over two layers of Miracloth and washed with sterilized water. Harvested mycelia were then lyophilized and ground in liquid nitrogen. Total RNA was extracted from mycelia of each sample using the TaKaRa RNAiso Reagent (TaKaRa Biotechnology Co., Dalian, China), and 10 mg of each RNA sample was used for reverse transcription with a RevertAid H Minus First Strand cDNA Synthesis Kit employing the oligo(dT)18 primer (Fermentas Life Sciences, Burlington, ON, Canada). The expression levels of FgSTE12 and cellulase genes were determined by quantitative real‐time PCR with the primers listed in Table S1. For each sample, PCR amplification with the primer pair actin‐F + actin‐R (Table S1) for the quantification of the expression of the actin gene was performed as a reference. The experiment was repeated three times independently.
Cell wall‐degrading enzyme activity assays
Total proteolytic activity was assayed by measuring the release of aromatic amino acids from the substrate using a previously described protocol (Jenczmionka and Schafer, 2005). The activity of cellulase was assayed by measuring the reducing sugars set free from the substrates using a previously published protocol (Jenczmionka and Schafer, 2005). For the determination of extracellular lipolytic activity, fresh mycelia of each strain were incubated in 50 mL of water supplemented with 2% (v/v) wheat germ oil (a lipase inducer) for different times, as indicated in the legend to Fig. S4. Lipolytic activity was determined using a previously published protocol (Voigt et al., 2005). The experiment was repeated three times, and data were analysed using analysis of variance (SAS version 8.0; SAS Institute).
Western blotting assay
Six mycelial plugs were inoculated into 150 mL PDB and incubated at 25 °C in a shaker (200 rpm) for 36 h. Mycelia were then harvested, washed with deionized water and ground in liquid nitrogen. Approximately 200 mg of finely ground mycelia were resuspended in 1 mL of extraction buffer [50 mm Tris‐HCl, pH 7.5, 100 mm NaCl, 5 mm ethylenediaminetetraacetic acid (EDTA), 1% Triton X‐100, 2 mm phenylmethylsulphonylfluoride (PMSF)] and 10 μL of protease inhibitor cocktail (Sangon, Shanghai, China). After homogenization with a vortex shaker, the lysate was centrifuged at 14 000 g in a microcentrifuge for 20 min at 4 °C. The resulting proteins were separated on 10% denaturing polyacrylamide gel (sodium dodecylsulphate‐polyacrylamide gel electrophoresis, SDS‐PAGE) and transferred to Immobilon‐P transfer membrane (Millipore, Billerica, MA, USA) with a Bio‐Rad electroblotting apparatus (Hercules, CA, USA). The total and phosphorylated FgGpmk1 were detected with a PhosphoPlus p44/42 MAPK antibody kit (Cell Signaling Technology Inc., Danvers, MA, USA). The total FgSte12‐GFP was detected with anti‐GFP antibody (Abcam, Cambridge, MA, USA). The input protein samples were also detected with anti‐histone H3 antibody (Abcam) as a reference. Incubation with a secondary antibody and chemiluminescent detection were performed as described previously (Yang et al., 2012). The experiment was conducted three times independently.
Yeast two‐hybrid assays
To construct plasmids for yeast two‐hybrid analyses, the coding sequence of each tested gene was amplified from cDNA of HN9‐1 with the primer pairs indicated in Table S1. The cDNA fragment was inserted into the yeast GAL4 binding domain vector pGBKT7 and GAL4 activation domain vector pGADT7 (Clontech, Mountain View, CA, USA). The pairs of yeast two‐hybrid plasmids were co‐transformed into S. cerevisiae strain AH109 following the LiAc/SS‐DNA/PEG transformation protocol (Schiestl and Gietz, 1989). In addition, a pair of plasmids, pGBKT7‐53 and pGADT7, served as a positive control. The following pairs of plasmids, pGBKT7‐Lam and pGADT7, pGBKT7 and pGADT7‐FgGpmk1, pGADT7 and pGBKT7‐FgSte7, pGADT7 and pGBKT7‐FgSte12, were used as negative controls. Transformants were grown at 30 °C for 3 days on synthetic medium lacking leucine (Leu) and tryptophan (Trp), and were then transferred to medium lacking histidine (His), Leu and Trp, and containing 5 mm 3‐aminotriazole (3‐AT), to assess binding activity. The experiment was repeated twice independently.
Affinity capture assays
FgSte12 was tagged with GFP and transferred to the FgSTE12 deletion mutant. The resultant transformant was used for protein extraction as described above. After centrifugation at 14 000 g in a microcentrifuge for 20 min at 4 °C, supernatant (800 μL) was transferred into a sterilized tube. About 50 μL of anti‐GFP agarose (Abmart, Shanghai, China) was added to capture FgSte12‐GFP‐interacting proteins, following the manufacturer's instructions. After incubation at 4 °C overnight, the agarose was washed three times with 500 μL of TBS (20 mm Tris‐HCl, 500 mm NaCl, pH 7.5). Proteins binding to the beads were boiled with 60 μL of TBS supplemented with 10 μL of 10% SDS. After centrifugation at 14 000 g in a microcentrifuge for 5 min at 4 °C, the supernatant was digested with trypsin as described previously (Tao et al., 2005; Zhou et al., 2007). Tryptic peptides were analysed by mass spectrometry by BGI Co. (Shenzheng, China), as described previously (Ding et al., 2010).
Supporting information
Fig. S1 Effect of FgSTE12 deletion on conidiation and conidial germination of Fusarium graminearum. (a) Conidia were counted after incubation of the wild‐type HN9‐1, the mutant ΔFgSte12 and the complemented strain ΔFgSte12‐C in mung bean broth for 4 days in a shaker. Line bars in each column denote the standard errors of three experiments. (b) Conidia of HN9‐1, ΔFgSte12 and ΔFg12‐C were suspended in 2% (w/v) sucrose solution. After incubation for 6 h, conidial germination of 150 conidia was examined. Line bars in each column denote standard errors of three repeated experiments. Values on the black bars followed by the same letter are not significantly different at P = 0.05.
Fig. S2 Effect of FgSTE12 deletion on deoxynivalenol (DON) production in Fusarium graminearum. The amount of DON (per milligram of fungal ergosterol) produced by each strain in infected wheat kernels was determined after 20 days of inoculation. Line bars in each column denote the standard errors of three replicated experiments. Values on the black bars followed by the same letter are not significantly different at P = 0.05.
Fig. S3 Penetration of FgSTE12 and FgGPMK1 deletion mutants into cellophane membrane (CM) on minimal medium (MM) supplied with different nitrogen sources. Fungal colonies were grown for 2 days at 25 °C on top of CM placed on MM containing 50 mm NaNO3 or NH4NO3 (Before). CMs with the fungal colonies were then removed, and the plates were incubated for an additional day to examine the presence of mycelial growth on the plate, indicating the penetration of the CM (After). All the strains were unable to penetrate CM on MM supplied with 50 mm HN4NO3. The wild‐type HN9‐1 and complemented strain ΔFgSte12‐C, but not the mutants ΔFgSte12 and ΔFgGpmk1, were able to penetrate CM on MM amended with 50 mm NaNO3.
Fig. S4 Extracellular lipolytic activity of the wild‐type HN9‐1 and the mutants ΔFgSte12 and ΔFgGpmk1. One unit of lipolytic activity was defined as 1 nmol/min p‐nitrophenol released from the substrate. Bars denote the standard errors of three repeated experiments.
Fig. S5 Effects of FgSTE12 deletion on the expression of cellulase genes. Transcription levels of cellulase genes in the wild‐type HN9‐1 and ΔFgSte12 were determined by quantitative real‐time PCR. The relative transcription level of each cellulase gene in ΔFgSte12 is the relative amount of mRNA of each gene in the wild type HN9‐1. These four genes do not contain the cis‐element TGAAACA in their promoters. PCR amplifications with the primer pair actin‐F + actin‐R for the quantification of expression of the actin gene were performed as a reference. Line bars in each column denote the standard errors of three repeated experiments. Values on the open bars followed by the same letter are not significantly different at P = 0.05.
Fig. S6 FgSTE12 was up‐regulated in the FgGPMK1 deletion mutant. (a) The relative transcription level of FgSTE12 in ΔFgGpmk1 is the relative amount of mRNA of FgSTE12 in the wild type HN9‐1. The line bar in the column denotes the standard errors of three repeated experiments. Values on the grey bars followed by the same letter are not significantly different at P = 0.05. (b) The protein levels of FgSte12 in wild‐type and ΔFgGpmk1. The entire FgSte12 protein coding sequence was fused to green fluorescent protein (GFP) under the native promoter, and introduced into ΔFgGpmk1 and ΔFgSte12. The anti‐GFP antibody was used to detect the protein levels of FgSte12‐GFP.
Fig. S7 The deletion of FgSTE12 did not affect FgGpmk1 phosphorylation dramatically. The phosphor‐p44/42 mitogen‐activated protein kinase (MAPK) antibody and p44/42 MAPK antibodies were used to detect phosphorylated FgGpmk1 and total FgGpmk1, respectively.
Table S1 Oligonucleotide primers used in this study.
Acknowledgements
This research was supported by the National Key Basic Research and Development Program (2013CB127802), Doctoral Fund of Ministry of Education of China (20120101110071), Special Fund for Agro‐scientific Research in the Public Interest (No. 201303023) and China Agriculture Research System (CARS‐3‐1‐15).
References
- Asuncion Garcia‐Sanchez, M. , Martin‐Rodrigues, N. , Ramos, B. , de Vega‐Bartol, J.J. , Perlin, M.H. and Diaz‐Minguez, J.M. (2010) FoSt12, the Fusarium oxysporum homolog of the transcription factor Ste12, is upregulated during plant infection and required for virulence. Fungal Genet. Biol. 47, 216–225. [DOI] [PubMed] [Google Scholar]
- Bardwell, L. , Cook, J.G. , Voora, D. , Baggott, D.M. , Martinez, A.R. and Thorner, J. (1998) Repression of yeast Ste12 transcription factor by direct binding of unphosphorylated Kss1 MAPK and its regulation by the Ste7 MEK. Genes Dev. 12, 2887–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y.C. , Wright, L.C. , Tscharke, R.L. , Sorrell, T.C. , Wilson, C.F. and Kwon‐Chung, K.J. (2004) Regulatory roles for the homeodomain and C2H2 zinc finger regions of Cryptococcus neoformans Ste12alphap. Mol. Microbiol. 53, 1385–1396. [DOI] [PubMed] [Google Scholar]
- Cho, Y. , Kim, K.H. , La Rota, M. , Scott, D. , Santopietro, G. , Callihan, M. , Mitchell, T.K. and Lawrence, C.B. (2009) Identification of novel virulence factors associated with signal transduction pathways in Alternaria brassicicola . Mol. Microbiol. 72, 1316–1333. [DOI] [PubMed] [Google Scholar]
- Cook, J.G. , Bardwell, L. , Kron, S.J. and Thorner, J. (1996) Two novel targets of the MAP kinase Kss1 are negative regulators of invasive growth in the yeast Saccharomyces cerevisiae . Genes Dev. 10, 2831–2848. [DOI] [PubMed] [Google Scholar]
- Ding, S.L. , Liu, W.D. , Iliuk, A. , Ribot, C. , Vallet, J. , Tao, A. , Wang, Y. , Lebrun, M.H. and Xu, J.R. (2010) The Tig1 histone deacetylase complex regulates infectious growth in the Rice Blast Fungus Magnaporthe oryzae . Plant Cell, 22, 2495–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede, B. and Ammerer, G. (1989) STE12, a protein involved in cell‐type‐specific transcription and signal transduction in yeast, is part of protein–DNA complexes. Genes Dev. 3, 1349–1361. [DOI] [PubMed] [Google Scholar]
- Gavrias, V. , Andrianopoulos, A. , Gimeno, C.J. and Timberlake, W.E. (1996) Saccharomyces cerevisiae TEC1 is required for pseudohyphal growth. Mol. Microbiol. 19, 1255–1263. [DOI] [PubMed] [Google Scholar]
- Goswami, R.S. and Kistler, H.C. (2004) Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525. [DOI] [PubMed] [Google Scholar]
- Jenczmionka, N.J. and Schafer, W. (2005) The Gpmk1 MAP kinase of Fusarium graminearum regulates the induction of specific secreted enzymes. Curr. Genet. 47, 29–36. [DOI] [PubMed] [Google Scholar]
- Jenczmionka, N.J. , Maier, F.J. , Losch, A.P. and Schafer, W. (2003) Mating, conidiation and pathogenicity of Fusarium graminearum, the main causal agent of the head‐blight disease of wheat, are regulated by the MAP kinase gpmk1. Curr. Genet. 43, 87–95. [DOI] [PubMed] [Google Scholar]
- Jiang, J. , Yun, Y. , Yang, Q. , Shim, W.B. , Wang, Z. and Ma, Z.H. (2011b) A type 2C protein phosphatase FgPtc3 is involved in cell wall integrity, lipid metabolism, and virulence in Fusarium graminearum . PLoS ONE, 6, e25311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J.H. , Liu, X. , Yin, Y.N. and Ma, Z.H. (2011a) Involvement of a velvet protein FgVeA in the regulation of asexual development, lipid and secondary metabolisms and virulence in Fusarium graminearum . PLoS ONE, 6, e28291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Berges, M.S. , Rispail, N. , Prados‐Rosales, R.C. and Di Pietro, A. (2010) A nitrogen response pathway regulates virulence functions in Fusarium oxysporum via the protein kinase TOR and the bZIP protein MeaB. Plant Cell, 22, 2459–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani, H.D. and Fink, G.R. (1997) Combinatorial control required for the specificity of yeast MAPK signaling. Science, 275, 1314–1317. [DOI] [PubMed] [Google Scholar]
- Madhani, H.D. , Styles, C.A. and Fink, G.R. (1997) MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell, 91, 673–684. [DOI] [PubMed] [Google Scholar]
- McMullen, M. , Jones, R. and Gallenberg, D. (1997) Scab of wheat and barley: a re‐emerging disease of devastating impact. Plant Dis. 81, 1340–1348. [DOI] [PubMed] [Google Scholar]
- Min, K. , Shin, Y. , Son, H. , Lee, J. , Kim, J.C. , Choi, G.J. and Lee, Y.W. (2012) Functional analyses of the nitrogen regulatory gene areA in Gibberella zeae . FEMS Microbiol. Lett. 334, 66–73. [DOI] [PubMed] [Google Scholar]
- Mirocha, C.J. , Abbas, H.K. , Windels, C.E. and Xie, W. (1989) Variation in deoxynivalenol, 15‐acetyldeoxynivalenol, 3‐acetyldeoxynivalenol, and zearalenone production by Fusarium graminearum isolates. Appl. Environ. Microbiol. 55, 1315–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirocha, C.J. , Kolaczkowski, E. , Xie, W. , Yu, H. and Jelen, H. (1998) Analysis of deoxynivalenol and its derivatives (Batch and Single Kernel) using gas chromatography/mass spectrometry. J. Agric. Food Chem. 46, 1414–1418. [Google Scholar]
- Park, G. , Xue, C. , Zheng, L. , Lam, S. and Xu, J.R. (2002) MST12 regulates infectious growth but not appressorium formation in the rice blast fungus Magnaporthe grisea . Mol. Plant–Microbe Interact. 15, 183–192. [DOI] [PubMed] [Google Scholar]
- Park, G. , Bruno, K.S. , Staiger, C.J. , Talbot, N.J. and Xu, J.R. (2004) Independent genetic mechanisms mediate turgor generation and penetration peg formation during plant infection in the rice blast fungus. Mol. Microbiol. 53, 1695–1707. [DOI] [PubMed] [Google Scholar]
- Park, G. , Xue, C. , Zhao, X. , Kim, Y. , Orbach, M. and Xu, J.R. (2006) Multiple upstream signals converge on the adaptor protein Mst50 in Magnaporthe grisea . Plant Cell, 18, 2822–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka, J.J. and Smolinski, A.T. (2005) Deoxynivalenol: toxicology and potential effects on humans. J. Toxicol. Environ. Health B Crit. Rev. 8, 39–69. [DOI] [PubMed] [Google Scholar]
- Proctor, R.H. , Hohn, T.M. , McCormick, S.P. and Desjardins, A.E. (1995) Tri6 encodes an unusual zinc finger protein involved in regulation of trichothecene biosynthesis in Fusarium sporotrichioides . Appl. Environ. Microbiol. 61, 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, M. and Elion, E.A. (2005) MAP kinase pathways. J. Cell Sci. 118, 3569–3572. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy, V. , Zhao, X. , Snyder, A.K. , Xu, J.R. and Shah, D.M. (2007) Two mitogen‐activated protein kinase signalling cascades mediate basal resistance to antifungal plant defensins in Fusarium graminearum . Cell. Microbiol. 9, 1491–1506. [DOI] [PubMed] [Google Scholar]
- Schamber, A. , Leroch, M. , Diwo, J. , Mendgen, K. and Hahn, M. (2010) The role of mitogen‐activated protein (MAP) kinase signalling components and the Ste12 transcription factor in germination and pathogenicity of Botrytis cinerea . Mol. Plant Pathol. 11, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl, R.H. and Gietz, R.D. (1989) High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 16, 339–346. [DOI] [PubMed] [Google Scholar]
- Seong, K.Y. , Pasquali, M. , Zhou, X. , Song, J. , Hilburn, K. , McCormick, S. , Dong, Y.H. , Xu, J.R. and Kistler, H.C. (2009) Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 72, 354–367. [DOI] [PubMed] [Google Scholar]
- Starkey, D.E. , Ward, T.J. , Aoki, T. , Gale, L.R. , Kistler, H.C. , Geiser, D.M. , Suga, H. , Toth, B. , Varga, J. and O'Donnell, K. (2007) Global molecular surveillance reveals novel Fusarium head blight species and trichothecene toxin diversity. Fungal Genet. Biol. 44, 1191–1204. [DOI] [PubMed] [Google Scholar]
- Steiner, B. , Kurz, H. , Lemmens, M. and Buerstmayr, H. (2009) Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor. Appl. Genet. 118, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, Y. , Kikuchi, T. , Kubo, Y. , Hamer, J.E. , Mise, K. and Furusawa, I. (2000) The Colletotrichum lagenarium MAP kinase gene CMK1 regulates diverse aspects of fungal pathogenesis. Mol. Plant–Microbe Interact. 13, 374–383. [DOI] [PubMed] [Google Scholar]
- Tao, W.A. , Wollscheid, B. , O'Brien, R. , Eng, J.K. , Li, X.J. , Bodenmiller, B. , Watts, J.D. , Hood, L. and Aebersold, R. (2005) Quantitative phosphoproteome analysis using a dendrimer conjugation chemistry and tandem mass spectrometry. Nat. Methods, 2, 591–598. [DOI] [PubMed] [Google Scholar]
- Trail, F. (2009) For blighted waves of grain: Fusarium graminearum in the postgenomics era. Plant Physiol. 149, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, G. , Fujii, S. , Tsuge, S. , Shiraishi, T. and Kubo, Y. (2003) The Colletotrichum lagenariu Ste12‐like gene CST1 is essential for appressorium penetration. Mol. Plant–Microbe Interact. 16, 315–325. [DOI] [PubMed] [Google Scholar]
- Voigt, C.A. , Schafer, W. and Salomon, S. (2005) A secreted lipase of Fusarium graminearum is a virulence factor required for infection of cereals. Plant J. 42, 364–375. [DOI] [PubMed] [Google Scholar]
- Wang, C. , Zhang, S. , Hou, R. , Zhao, Z. , Zheng, Q. , Xu, Q. , Zheng, D.W. , Wang, G.H. , Liu, H.Q. , Gao, X.L. , Ma, J.W. , Kistler, H.C. , Kang, Z.S. and Xu, J.R. (2011) Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum . Plos Pathog. 7, e1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Sak Hoi, J. and Dumas, B. (2010) Ste12 and Ste12‐like proteins, fungal transcription factors regulating development and pathogenicity. Eukaryot. Cell, 9, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Sak Hoi, J. , Herbert, C. , Bacha, N. , O'Connell, R. , Lafitte, C. , Borderies, G. , Rossignol, M. , Rouge, P. and Dumas, B. (2007) Regulation and role of a STE12‐like transcription factor from the plant pathogen Colletotrichum lindemuthianum . Mol. Microbiol. 64, 68–82. [DOI] [PubMed] [Google Scholar]
- Yang, Q. , Yan, L. , Gu, Q. and Ma, Z. (2012) The mitogen‐activated protein kinase kinase kinase BcOs4 is required for vegetative differentiation and pathogenicity in Botrytis cinerea . Appl. Microbiol. Biotechnol. 96, 481–492. [DOI] [PubMed] [Google Scholar]
- Yin, Y. , Liu, X. and Ma, Z. (2009) Simultaneous detection of Fusarium asiaticum and Fusarium graminearum in wheat seeds using a real‐time PCR method. Lett. Appl. Microbiol. 48, 680–686. [DOI] [PubMed] [Google Scholar]
- Yuan, Y.L. and Fields, S. (1991) Properties of the DNA‐binding domain of the Saccharomyces cerevisiae STE12 protein. Mol. Cell. Biol. 11, 5910–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Kim, Y. , Park, G. and Xu, J.R. (2005) A mitogen‐activated protein kinase cascade regulating infection‐related morphogenesis in Magnaporthe grisea . Plant Cell, 17, 1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Mehrabi, R. and Xu, J.R. (2007) Mitogen‐activated protein kinase pathways and fungal pathogenesis. Eukaryot. Cell, 6, 1701–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, L. , Campbell, M. , Murphy, J. , Lam, S. and Xu, J.R. (2000) The BMP1 gene is essential for pathogenicity in the gray mold fungus Botrytis cinerea . Mol. Plant–Microbe Interact. 13, 724–732. [DOI] [PubMed] [Google Scholar]
- Zhou, F. , Galan, J. , Geahlen, R.L. and Tao, W.A. (2007) A novel quantitative proteomics strategy to study phosphorylation‐dependent peptide–protein interactions. J. Proteome Res. 6, 133–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Effect of FgSTE12 deletion on conidiation and conidial germination of Fusarium graminearum. (a) Conidia were counted after incubation of the wild‐type HN9‐1, the mutant ΔFgSte12 and the complemented strain ΔFgSte12‐C in mung bean broth for 4 days in a shaker. Line bars in each column denote the standard errors of three experiments. (b) Conidia of HN9‐1, ΔFgSte12 and ΔFg12‐C were suspended in 2% (w/v) sucrose solution. After incubation for 6 h, conidial germination of 150 conidia was examined. Line bars in each column denote standard errors of three repeated experiments. Values on the black bars followed by the same letter are not significantly different at P = 0.05.
Fig. S2 Effect of FgSTE12 deletion on deoxynivalenol (DON) production in Fusarium graminearum. The amount of DON (per milligram of fungal ergosterol) produced by each strain in infected wheat kernels was determined after 20 days of inoculation. Line bars in each column denote the standard errors of three replicated experiments. Values on the black bars followed by the same letter are not significantly different at P = 0.05.
Fig. S3 Penetration of FgSTE12 and FgGPMK1 deletion mutants into cellophane membrane (CM) on minimal medium (MM) supplied with different nitrogen sources. Fungal colonies were grown for 2 days at 25 °C on top of CM placed on MM containing 50 mm NaNO3 or NH4NO3 (Before). CMs with the fungal colonies were then removed, and the plates were incubated for an additional day to examine the presence of mycelial growth on the plate, indicating the penetration of the CM (After). All the strains were unable to penetrate CM on MM supplied with 50 mm HN4NO3. The wild‐type HN9‐1 and complemented strain ΔFgSte12‐C, but not the mutants ΔFgSte12 and ΔFgGpmk1, were able to penetrate CM on MM amended with 50 mm NaNO3.
Fig. S4 Extracellular lipolytic activity of the wild‐type HN9‐1 and the mutants ΔFgSte12 and ΔFgGpmk1. One unit of lipolytic activity was defined as 1 nmol/min p‐nitrophenol released from the substrate. Bars denote the standard errors of three repeated experiments.
Fig. S5 Effects of FgSTE12 deletion on the expression of cellulase genes. Transcription levels of cellulase genes in the wild‐type HN9‐1 and ΔFgSte12 were determined by quantitative real‐time PCR. The relative transcription level of each cellulase gene in ΔFgSte12 is the relative amount of mRNA of each gene in the wild type HN9‐1. These four genes do not contain the cis‐element TGAAACA in their promoters. PCR amplifications with the primer pair actin‐F + actin‐R for the quantification of expression of the actin gene were performed as a reference. Line bars in each column denote the standard errors of three repeated experiments. Values on the open bars followed by the same letter are not significantly different at P = 0.05.
Fig. S6 FgSTE12 was up‐regulated in the FgGPMK1 deletion mutant. (a) The relative transcription level of FgSTE12 in ΔFgGpmk1 is the relative amount of mRNA of FgSTE12 in the wild type HN9‐1. The line bar in the column denotes the standard errors of three repeated experiments. Values on the grey bars followed by the same letter are not significantly different at P = 0.05. (b) The protein levels of FgSte12 in wild‐type and ΔFgGpmk1. The entire FgSte12 protein coding sequence was fused to green fluorescent protein (GFP) under the native promoter, and introduced into ΔFgGpmk1 and ΔFgSte12. The anti‐GFP antibody was used to detect the protein levels of FgSte12‐GFP.
Fig. S7 The deletion of FgSTE12 did not affect FgGpmk1 phosphorylation dramatically. The phosphor‐p44/42 mitogen‐activated protein kinase (MAPK) antibody and p44/42 MAPK antibodies were used to detect phosphorylated FgGpmk1 and total FgGpmk1, respectively.
Table S1 Oligonucleotide primers used in this study.