

Summary
Genetic and genomics tools to characterize host–pathogen interactions are disproportionately directed to the host because of the focus on resistance. However, understanding the genetics of pathogen virulence is equally important and has been limited by the high cost of de novo genotyping of species with limited marker data. Non‐resource‐prohibitive methods that overcome the limitation of genotyping are now available through genotype‐by‐sequencing (GBS). The use of a two‐enzyme restriction‐associated DNA (RAD)‐GBS method adapted for Ion Torrent sequencing technology provided robust and reproducible high‐density genotyping of several fungal species. A total of 5783 and 2373 unique loci, ‘sequence tags’, containing 16 441 and 9992 single nucleotide polymorphisms (SNPs) were identified and characterized from natural populations of Pyrenophora teres f. maculata and Sphaerulina musiva, respectively. The data generated from the P. teres f. maculata natural population were used in association mapping analysis to map the mating‐type gene to high resolution. To further validate the methodology, a biparental population of P. teres f. teres, previously used to develop a genetic map utilizing simple sequence repeat (SSR) and amplified fragment length polymorphism (AFLP) markers, was re‐analysed using the SNP markers generated from this protocol. A robust genetic map containing 1393 SNPs on 997 sequence tags spread across 15 linkage groups with anchored reference markers was generated from the P. teres f. teres biparental population. The robust high‐density markers generated using this protocol will allow positional cloning in biparental fungal populations, association mapping of natural fungal populations and population genetics studies.
Keywords: fungi, genotype‐by‐sequencing, Pyrenophora teres, Sphaerulina musiva
Introduction
The virulence of plant‐pathogenic fungi can be difficult to characterize genetically because of a combination of factors including: (i) the complexity of inheritance; (ii) the inability to conduct controlled crosses among different pathogen genotypes; (iii) the large amount of genetic diversity present in natural populations; (iv) limited genomic information on many of these organisms; and (v) the high cost of de novo genotyping. The generation of low‐cost, high‐density genotype‐by‐sequencing (GBS) data can overcome these roadblocks by combining next‐generation sequencing (NGS) technology with sequence restriction‐associated DNA (RAD) tagging. Adaptations of this procedure have been used to generate high‐density genotype datasets for a fungal genome, and several plant and animal genomes, typically utilizing Illumina NGS technology (Baird et al., 2008; Baxter et al., 2011; Chutimanitsakun et al., 2011; Elshire et al., 2011; Ma et al., 2012; Pfender et al., 2011; Poland et al., 2012). A recently reported GBS protocol optimized the system by utilizing two enzymes (PstI and MspI) and Y‐adaptors to reduce genome complexity and enrich for target DNA in a sequencing library preparation (Poland et al., 2012). The efficiency of RAD‐GBS for specific species can be optimized by using combinations of restriction enzymes that effectively reduce the complexity of the genome, whilst maintaining adequate numbers of evenly distributed sequence tags. An enzyme combination that adjusts the number of sequence tags produced to achieve adequate sequence reads for each tag is important when balancing data quality and cost. This optimization allows for accurate determination of the number of individuals that can be sequenced in parallel in each reaction, thus reducing the cost, whilst maintaining an acceptable level of missing data points. Draft genome sequences can be used to help select optimal enzyme combinations based on: (i) genome size (used to determine the use of rare or frequent enzyme recognition sequences); (ii) GC content and/or the percentage of AT‐rich repetitive elements; and (iii) gene density, where methylation‐sensitive enzymes can be utilized to select against methylated non‐transcribed or gene‐poor repetitive regions of the fungal genome.
Poland et al. (2012) optimized the two‐enzyme GBS method for the large repeat‐rich grass genomes and successfully generated high‐density single nucleotide polymorphism (SNP) data for the ∼5.5‐ and 16‐Gb (Arumuganathan and Earle, 1991) genomes of barley and wheat, respectively. They also used the Y‐adaptor procedure for the development of pooled barcoded libraries to be sequenced on the Illumina platform. The Y‐adaptors allowed for polymerase chain reaction (PCR)‐mediated amplification of the libraries, whilst enriching for DNA fragments containing a high proportion of heterogeneous adaptors ligated to the termini, universal adaptor at one terminus and the unique barcoded adaptor at the other terminus. They determined that this efficient and reproducible two‐enzyme system could be adopted to generate high‐density SNP markers in species lacking reference genomes, which would include many important plant‐pathogenic fungi (Poland et al., 2012).
These methods were originally developed for use with the Illumina sequencing platform, which is very low cost per base of sequence, because of the large datasets generated per run, yet with a potential prohibitively high cost per sequencing run. Mascher et al. (2013), comparing the Illumina and Ion Torrent semiconductor sequencing technology platforms, concluded that the bench top Ion Torrent personal genomics machine (PGM) did not produce sufficient reads for GBS in large complex plant genomes, such as barley. Although it is likely that this technology would be sufficient for smaller fungal genomes, typically 10–60 Mb in size (Gregory et al., 2007), it has remained largely untested to date. To this end, a RAD‐GBS protocol was optimized and tested using the PGM platform on two natural populations and a biparental mapping population of two different pathogenic fungi. This GBS procedure was adapted from those described above by: (i) adding a size selection step to reduce the sequence tag complexity; and (ii) utilizing the ion sphere enrichment to eliminate the use of Y‐adaptors. The method described here typically resulted in ∼12 000–20 000 sequence tags depending on the species studied. The GBS protocol produced robust SNP markers, which were validated by identifying marker trait associations (MTAs) by association mapping (AM) in a small natural population, as well as by producing a genetic map of a biparental population containing anchored simple sequence repeat (SSR) and amplified fragment length polymorphism (AFLP) markers. The procedure was also shown to be reliable and repeatable with DNA isolations and library preparations performed across three laboratories and sequencing performed on two separate PGMs located at North Dakota State University and Washington State University. Although this protocol is adapted to the PGM, it could easily be applied to the Illumina platform.
Results
The two‐enzyme RAD‐GBS protocol adapted for Ion Torrent sequencing technology (Fig. 1) was used to genotype two natural populations of phylogenetically diverse species of necrotrophic plant‐pathogenic fungi in the phylum Ascomycota belonging to the Dothideomycetidae: Pyrenophora teres Drechs f. maculata Smedeg and Sphaerulina musiva (Peck) Verkeley, Quaedvlieg, & Crous (syn. Septoria musiva Peck). This protocol was also used to genotype a biparental cross between two isolates of P. teres Drechs f. teres Smedeg, 15A and 0–1, and a robust genetic map was generated with SNP markers validated using anchored SSRs and AFLPs.
Figure 1.
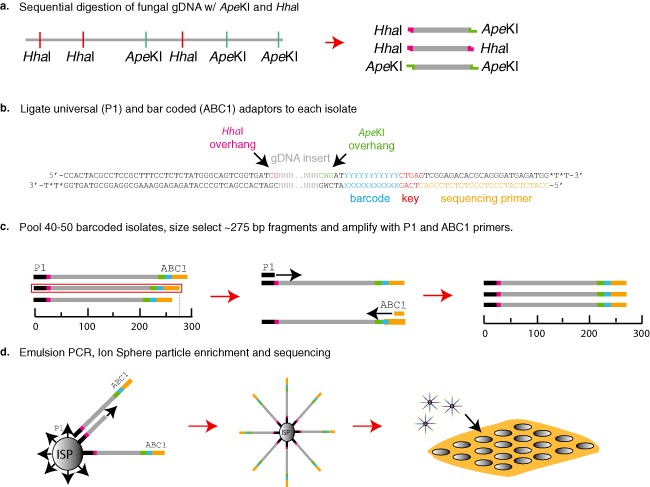
Genotype‐by‐sequencing (GBS) protocol for fungal genomes. (a) Fungal genomic DNA was sequentially digested with the restriction enzymes HhaI then ApeKI, producing three different combinations of restriction termini. (b) The ABC1 adaptor containing the sequencing primer site (orange), Ion Torrent key site (red), barcodes (blue) and ApeKI sticky ends (green), and the P1 adaptor containing the Ion Sphere Particle attachment nucleotides (black) and HhaI sticky ends (pink), were ligated to the restriction‐digested genomic DNA fragment. (c) The barcoded fragments were pooled, unligated adaptors were removed and fragments were size selected for ∼275‐bp fragments. The 275‐bp fraction from the pooled libraries was amplified using the Ion Torrent sequencing primers and Ion Sphere Particle (ISP) primer, without sphere particles attached. (d) The polymerase chain reaction (PCR) products were quantified and emulsion PCR was performed to add monoclonal DNA templates to a single ISP. The ISPs with DNA templates were enriched on the Ion One Touch 2 bead enrichment station and loaded into single wells on the 318 chip and sequenced on the Ion Torrent PGM™.
The GBS library preparation and sequencing produced 2.5, 5.4 and 5.9 million reads for the S. musiva, P. teres f. teres and P. teres f. maculata genomes, respectively. The value given for P. teres f. teres is the average of three separate sequencing runs needed to genotype the population of 118 isolates. The number of reads per isolate that were barcoded and sequenced in parallel ranged from 34 316 to 432 627, with a mean value of 148 643 reads per isolate. Utilizing the analysis of sequencing reads per isolate, determined by the barcode sequestration, it was shown that approximately 80 000 reads are required from an individual isolate to have <15% missing data (>85% data present; Fig. 2). Averaging all six of the sequencing runs reported here, <15% missing data was achieved utilizing our bioinformatics pipeline.
Figure 2.

Graph showing the distribution of sequence tags and percentage of data points present for each isolate. Bars and dots above correspond to a single isolate, and red indicates Pyrenophora teres f. maculata (Ptm) sequences, orange P. teres f. teres (Ptt) and yellow Sphaerulina musiva (Sm). The left axis scale is the number of reads corresponding to the bar chart, ranging from 34 316 to 432 627 sequence reads. The right axis scale is the percentage of data present corresponding to the dots. The asterisks next to the four dots indicate P. teres f. teres isolates that were originally isolated from the natural P. teres f. maculata collections in North Dakota. The line indicates the mean change in percentage data present with increased sequence reads per isolate.
The GBS analysis of 38 isolates of P. teres collected in North Dakota yielded 7672 sequence tags containing 18 957 quality SNPs with an average of 2.47 SNPs per tag. The subsequent phenotypic analysis of the 38 putative P. teres f. maculata isolates, performed at the seedling stage on a barley differential set in a controlled growth chamber environment, confirmed that 34 of the isolates yielded the spot‐form phenotype, consistent with P. teres f. maculata, and the four outlier isolates (Fig. 2; data points denoted with asterisks) gave rise to net‐form net blotch lesions, consistent with those caused by P. teres f. teres. These isolates were subsequently removed from the analysis, which resulted in 16 441 informative SNPs at 5783 unique loci for the remaining 34 P. teres f. maculata isolates. Based on the predicted genome size of P. teres f. maculata, this would place a sequence tag with at least one SNP every 5.7 kb on average across the genome.
Utilizing the 16 441 SNPs located on 5783 sequence tags, markers were identified in the North Dakota P. teres f. maculata natural population; two highly significant MTAs were identified using the mating type data collected from the 34 isolates analysed. The two GBS‐SNP markers SNP‐06979_81 and SNP‐17724_48 were identified with highly significant association to mating type using both logistic regression (data not shown) and the general linearized model (Fig. 3a). These two SNP markers, which had a −log10(p) value of 32, were perfectly associated with the mating type gene (Fig. 3b) and were located 6.2 and 7.5 kb from the MAT2 locus, respectively (Fig. 3c), based on alignment to the previously published genome of the closely related P. teres f. teres (Ellwood et al., 2010). A third SNP marker, SNP‐14232_7, had a −log10(p) value of 3.1 and was located 11.2 kb from the MAT2 gene (Fig. 3c)
Figure 3.
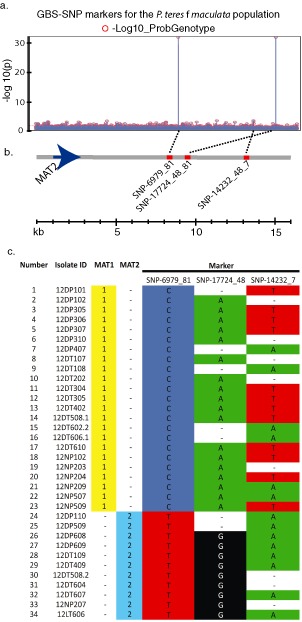
Data validating the polymerase chain reaction‐genotype‐by‐sequencing (PCR‐GBS) data for association mapping of the mating type gene in the Pyrenophora teres f. maculata natural population. (a) Manhattan plot showing significant marker trait associations (MTAs) between the 16 441 GBS‐single nucleotide polymorphisms (GBS‐SNPs) and the mating type trait using a general linearized model approach in JMP Genomics. (b) The physical sequence at the mating type locus showing significant MTAs in relation to the mating type 2 gene (MAT2). The grey horizontal line represents the sequence from the mating type locus of P. teres f. teres with the blue arrow depicting the MAT2 gene and the red bars showing the relative position of the significant SNP markers. The broken black lines show where the MTAs for the specific markers are located on the Manhattan plot above. (c) A graph showing the correlation of the genotypes of all 24 isolates for mating type and significant markers. Mating type and SNP markers are perfectly correlated for markers SNP‐06979_81 and SNP‐17724_48.
Three independent GBS libraries were generated to sequence 118 progeny isolates and parental lines from the biparental population developed by crossing isolates 15A and 0–1, as described in Ellwood et al. (2010). The population utilized in this GBS analysis was expanded from the original 78 individuals to 118 progeny isolates. Including the parents, DNA from the 120 isolates was used to construct three GBS libraries with 40 barcoded isolates per library. Each library was sequenced in parallel on three separate Ion Torrent 318 chips. The GBS data generated 16 206 and 9554 sequence tags for the parental lines 15A and 0–1, respectively. The parental sequence tags containing at least four reads per isolate were aligned using dnastar software to identify quality SNPs. In this initial SNP analysis stage, more quality SNPs would have been identified; however, only 50 118 reads were generated for the 0–1 parental genotype, which reduced our quality sequence tags by ∼40% compared with the number generated for the parental isolate 15A, in which we obtained 100 625 sequence reads. The sequence tags generated from the 15A parental line were then utilized as templates for the alignment of the progeny isolates and to call SNPs utilized in the development of genetic linkage maps. Analysis of the SNP calls in which we had parental genotype data for both 15A and 0–1 showed that the calls based solely on the 15A parental genotype were 100% accurate, and thus the polymorphic SNPs from the progeny isolates that deviated from the 15A sequence were called as the 0–1 genotype. A further analysis of the 0–1 sequence tags determined that 27% of the 0–1 tags generated did not match with the 15A tag sequences. Utilizing these additional 2406 0–1 sequence tags for further SNP analyses identified 41 quality SNPs. Additional informative SNPs may also have been identified if the analyses had been performed on all progeny in aggregate and parental genotypes imputed after linkage analyses. However, for the stringent validation of the markers reported here, we wanted to rely on markers with quality data calls for at least one of the parental genotypes without relying on data imputation. MapDisto software was used to generate a map, which contained 1393 SNPs on 997 sequence tags spread across 15 linkage groups (Fig. 4 shows linkage group 1). The map and GBS‐SNP markers placed on the genetic map were validated by analysis with the anchored SSR and AFLP markers previously utilized to develop a skeletal genetic map of the population (Ellwood et al., 2010). The GBS‐SNP markers corresponded with the anchored markers, but had higher marker saturation, giving better definition of the regions of recombination within the progeny to a much higher resolution than reported previously (Fig. 4). The sequence tags that were used to develop the genetic map were aligned to the P. teres f. teres genome scaffolds (Ellwood et al., 2010) utilizing the dnastar SeqMan NGen genome assembly program set to the templated assembly. The sequence tags containing the GBS‐SNP markers aligned or physically mapped to common P. teres f. teres isolate 0–1 scaffolds in nearly perfect agreement with the genetic data (data not shown).
Figure 4.
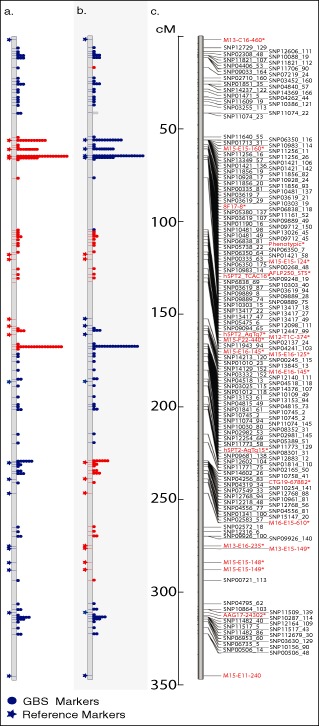
Linkage group 1 from the genetic map developed using the biparental population created from the cross between Pyrenophora teres f. teres isolates 15A and 0–1. (a, b) Individual progeny lines with genotype indicated by colour: red is 15A and blue is 0–1. The stars to the left of the vertical grey bar indicate the genetic position and genotype of anchored simple sequence repeat (SSR) and amplified fragment length polymorphism (AFLP) markers. The dots to the right of the vertical bars indicate the position and number of restriction‐associated DNA‐genotype‐by‐sequencing (RAD‐GBS) single nucleotide polymorphism (SNP) markers positioned from the data generated with the protocol described here. A transition in colour indicates the region delimiting a recombination event in the specific progeny line. (c) Genetic map containing the GBS SNP markers with SNP markers in black and anchored SSR and AFLP markers in red. The scale corresponding to all three maps is in centimorgans (cM).
The alignment of the sequences generated from the RAD fragments using the dnastar software reduced the complexity of the approximately 30‐Mb S. musiva genome to 10 481 contiguous assemblies or sequence tags. After SNP analysis and the removal of sequence tags that did not contain informative SNPs, described above, 9992 SNPs were localized on 2373 sequence tags. This analysis of a small subset of isolates (n = 18) of a natural population of S. musiva identified approximately 4.21 SNPs per sequence tag. The SNP data generated from this Ion Torrent PGM analysis predicted the placement of an SNP marker approximately every 12.6 kb, based on the ∼30 Mb of the sequenced S. musiva genome (http://genome.jgi‐psf.org/Sepmu1/Sepmu1.info.html) (Fig. 5). Alignment of the unique sequence tags to the S. musiva genome available on the JGI database indicated that 2314 of the 2373 SNP loci aligned to the annotated genome sequence at unique loci, showing that the analysis and stringency utilized to identify SNP markers were robust. As predicted, these alignments placed the sequence tags at unique loci, which were relatively evenly distributed across the fungal genome at approximately 12.6‐kb intervals (Fig. 5).
Figure 5.
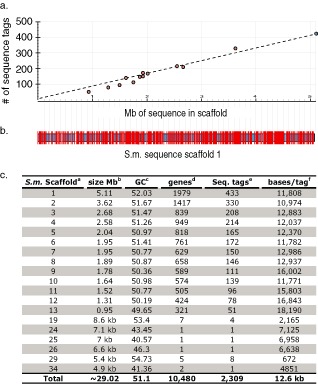
Alignment of 434 genotype‐by‐sequencing (GBS) single nucleotide polymorphism (SNP) markers to the JGI Septoria musiva (Scaffold 1; ∼5.1 Mb) sequence assembly. (a) The graph shows the distribution of all 2309 sequence tags containing informative SNPs across the 13 largest S. musiva scaffolds ranging from 0.95 to 5.1 Mb. The x‐axis is the Mb of sequence in the sequence scaffold and the y‐axis is the number of sequence tags containing informative SNPs. The dots represent the 13 major scaffolds in the S. musiva genome assembly. The blue dot represents the largest (5.1 Mb) scaffold used for analysis in Fig. 1b. (b) The horizontal blue/black bordered bar represents the 5.1‐Mb S. musiva genome Scaffold 1 sequence from the JGI database (http://genome.jgi‐psf.org/Sepmu1/Sepmu1.info.html). The vertical white lines show the position of missing sequence between contigs within the scaffold. The red vertical lines show the positions of the 433 GBS ‘sequence tags’ containing informative SNPs. (c) Table showing the alignment of GBS sequence tags containing SNPs to the S. musiva genome sequence. aScaffold numbers from the JGI website (http://genome.jgi‐psf.org/Sepmu1/Sepmu1.info.html). bSize of sequence scaffolds given in megabases unless designated by kb (kilobases). c GC content of designated sequence scaffolds. dNumber of predicted genes present on sequence scaffold. eNumber of sequence tags aligning to scaffold. fNumber of nucleotide bases per sequence tag marker.
Utilizing the partially methylation‐sensitive, GC‐rich, five‐base recognition enzyme ApeKI [recognition site; GC(A/T)CG] and a second methylation‐sensitive GC‐rich enzyme HhaI (recognition site; GCGC), we would expect the sequence tags to be concentrated in gene‐rich regions of the genome. The blastx analysis of the identified sequence tags containing SNPs determined that ∼95% of the S. musiva sequence tags aligned to predicted genes.
Discussion
GBS data have been underutilized for the characterization of fungal pathogens because of the need for protocols and sequencing technologies that are less expensive per run and generate sequencing datasets proportional to the smaller size of fungal genomes. There are limited examples of GBS protocols being utilized for fungal genomes and the limited reports have used a shotgun sequencing strategy on the Illumina NGS platform, where hundreds of millions of reads were generated and aligned to hundreds of thousands of different sequence tags (Dalman et al., 2013). For example, Dalman et al. (2013) used the shotgun approach on the Illumina platform to sequence 24 Heterobasidion annosum s.s isolates in parallel, generating approximately 163 million sequences with a range of 2.6–12.6 × mean coverage of each genome. Utilizing this strategy, hundreds of millions of reads were generated for the small number of isolates sequenced in parallel to achieve minimal coverage of hundreds of thousands of sequence tags. This method does not effectively reduce the complexity of the fungal genome; thus, even with the large number of sequence reads, only the most minimal amount of coverage (two sequences for each tag per isolate) was achieved for many regions of the genome and used to call SNPs. This coverage spread throughout the genome results in a higher number of SNP markers, 33 018 SNPs in this case (Dalman et al., 2013). Adequate marker density is necessary for AM in natural populations, where linkage decay can be very rapid because of the large amount of ancient recombination. Dalman et al. (2013) were able to use this small population and saturated marker density to perform AM for virulence. Our protocol does not provide this marker density; however, we show that it still produces a sufficient number of markers for AM analyses in a small population of P. teres f maculata at a much lower cost per isolate.
A robust method of reducing the genome complexity is RAD‐GBS; however, most of the data generated utilizing RAD sequencing to characterize host–pathogen interactions have focused on large host genomes, including maize, barley, wheat and ryegrass (Baird et al., 2008; Baxter et al., 2011; Chutimanitsakun et al., 2011; Elshire et al., 2011; Ma et al., 2012; Pfender et al., 2011; Poland et al., 2012). To generate GBS data for biparental populations of these large grass genomes, with acceptable levels of missing data, hundreds of millions of reads are necessary and a large percentage of missing data still remains. The relatively small size of fungal genomes, 10–60 Mb (Gregory et al., 2007), requires less sequence coverage to achieve adequate sequencing depth to minimize the amount of missing data (reported above). This coverage can be achieved for a single biparental or natural fungal population of 100–200 individuals with tens of millions of reads. The reduced coverage requirements of small fungal genomes lend themselves to GBS conducted on small bench top NGS platforms, such as the Ion Torrent PGM. This platform is capable of generating four to six million 200–400‐bp reads per sequencing run. The relatively low number of reads generated by this NGS platform, and the barcode sequestration from the Ion Torrent server, simplifies the bioinformatics pipeline required to analyse the data, generating genomic resources that were previously underdeveloped for many non‐characterized fungal pathogen populations.
Reducing the complexity of the genome to achieve adequate sequence coverage alleviates the problem of missing data points, which is currently the major disadvantage of GBS. This protocol and analysis produced GBS data with, on average, fewer than 15% missing values (range, 1%–30% missing data; Fig. 2). It is interesting to note that the four outliers from the P. teres f. maculata natural population dataset with a high percentage of missing data per number of sequences were determined to be from a very closely related, yet different form, of the pathogen P. teres f. teres. We hypothesize that the loss of data points per read number for these four isolates, representing different forms of the fungus, is related to divergence of restriction sites across the two forms. However, the robustness of the sequence tag development by fragment size selection is apparent by the fact that >65% of the data points were still conserved across the two forms of P. teres.
The number and genome coverage of GBS‐SNP markers are more than adequate for the development of genetic maps of biparental populations or for use in AM studies. It would also be possible to improve the quality of the data by correcting the missing data by imputation (Scheet and Stephens, 2006), but data imputation is more accurate if a priori genome sequence or genetic maps with anchored markers are available. However, imputation of missing SNP data for unordered markers can be accomplished utilizing several imputation methods (Rutkoski et al., 2013). In order to characterize quantitatively inherited traits, such as virulence loci in necrotrophic pathogens (Friesen and Faris, 2010), it is not only important to have a minimal number of missing data points, but also molecular markers evenly distributed across the genome. The distribution of sequence tags containing informative SNPs was even across the physical S. musiva sequence at approximately 12.6‐kb intervals, as predicted by the number of sequence tags and genome size (Fig. 4). Considering that ∼90% of the S. musiva genome is made up of unique non‐repetitive regions, it was expected that a large proportion of the tags would hit transcribed genes. This was achieved for S. musiva using this protocol, where ∼95% of the sequence tags aligned to predicted gene space. The question still remains as to whether or not DNA methylation is important in fungi, but the limited research to address this question suggests that methylation within fungal genomes is variable from trace to up to 4.34% (Antequera et al., 1984; Binz et al., 1998). DNA sequence analysis determined that methylated DNA in fungal genomes is mostly repetitive or transposon sequences. A study of Neurospora crassa determined that the methylated component of the genome is almost exclusively an artefact of transposons that underwent repeat‐induced point mutation (Selker et al., 2003), the mechanism in fungi that defends against genome expansion of repetitive elements by rapidly mutating duplicate sequences (Selker, 2002). Thus, although the use of methylation‐sensitive enzymes to avoid non‐transcribed regions of the genome may not be as important in fungi as it is in higher plants, where as much as 25% of the cytosines are methylated (Paszkowski and Whitham, 2001), it still allows for some enrichment of gene encoding regions of the genome which is beneficial.
Excellent marker coverage was achieved in all three populations utilized in this study, despite the fact that DNA samples were isolated with different procedures in three independent laboratories, and library preparations and sequencing reactions were performed in two independent laboratories. The P. teres GBS libraries were sequenced at the North Dakota State University Barley/Fungal genomics laboratory, Fargo, ND, USA, and the S. musiva sequencing was performed at the Molecular Biology Core, Washington State University, Pullman, WA, USA. These successful results across several laboratories show that the protocol is repeatable and robust.
This GBS‐RAD protocol requires a limited amount of fungal genomic DNA (gDNA) and has recently been successfully conducted with one‐tenth of the fungal gDNA described above (data not shown). This allows researchers to genotype isolates from a small number of spores or mass of hyphae, a benefit when genotyping fungi that are difficult or impossible to culture. The authors have also demonstrated that it is possible to multiplex up to 46 isolates on a single PGM 318 chip and that this number could potentially be increased to 50 isolates per chip with balanced libraries. However, we recommend sequencing 40 isolates in parallel to achieve <15% missing data. From the 96 isolates utilized here, it was shown that ∼80 000 reads per isolate were needed to achieve <15% missing data (Fig. 2). With the ∼30‐Mb S. musiva genome (http://genome.jgi‐psf.org/Sepmu1/Sepmu1.info.html), which is relatively smaller in size compared with the ∼42‐Mb P. teres f. teres genome (Ellwood et al., 2010), the data did not follow the average trend (Fig. 2). For S. musiva, where 10 481 sequence tags were identified, compared with the 16 206 for P. teres f. teres, fewer missing data were achieved with lower numbers of reads per isolate, suggesting that, as the fungal genome size decreases, more isolates could be sequenced on a single 318 chip.
Reducing the complexity of the fungal genomes using the ApeKI and HhaI enzyme combination was shown to give the best results of the three combinations of enzymes tested (ApeKI/HhaI, ApeKI/PhoI, ApeKI/HindIII). The PhoI blunt end adaptor resulted in ligation efficiency problems during library preparation, and the HindIII digestion with the six‐base recognition enzyme resulted in inadequate numbers of sequence tags in the size‐selected fraction (data not shown). The ApeKI/HhaI combination has given consistent results of 10 000–20 000 sequence tags in six different fungi, including S. musiva, P. teres f. maculata, P. teres f. teres, Sclerotinia sclerotiorum Lib. de Bary, Parastagonospora nodorum Berkley Quaedvlieg, Verkeley, & Crous and Puccinia helianthi Schwein. This protocol consistently size selects sequence tags using the Pippin Prep system across sequencing libraries and fungal species. A critical factor is the quality of the library preparations, with normalized DNA concentrations identified as the major factor contributing to balanced read numbers across isolates and reduced missing data points. It was determined with the fungal DNA preparations that the NanoDrop system (Thermo Scientific, Waltham, MA, USA) did not accurately quantify the DNA concentrations as determined by gel imaging and fluorescent quantification (data not shown). With the fungal DNA preparations, the Qubit fluorometer (Life Technologies, Carlsbad, CA, USA) was shown to more accurately and consistently quantify the DNA.
This procedure is currently being used for the genetic characterization of natural and biparental fungal populations to identify virulence genes. To validate the robustness of the SNP markers generated by this protocol, we used the method on a characterized biparental population of P. teres f. teres. The analysis and comparison with known anchored SSR and AFLP markers demonstrated an almost perfect correlation between the previous recombination data and the higher resolution map generated here (Fig. 4). The future and current work to characterize biparental and natural fungal populations will utilize the GBS data to produce robust genetic maps and perform AM studies, allowing for the identification of candidate effectors involved in virulence from these populations. This technology is also a powerful tool for phylogenetic and evolutionary studies of closely related fungi. The GBS data generated for the P. teres population described above allowed the authors to accurately differentiate isolates of P. teres f. teres from P. teres f. maculata.
Here, we report the use of a two‐enzyme RAD‐GBS system adapted for use on the smaller genomes of plant‐pathogenic fungi. The protocol allows for the high‐density SNP marker saturation of a diversity of fungal genomes using the small platform Ion Torrent PGM. The low cost of the microprocessor chip sequencing technology and quick sequencing allow for high‐throughput and high‐density genotyping of fungal populations at a low cost that could be adapted to other sequencing platforms. This method has allowed the discovery and characterization of the genetic diversity present in natural fungal populations, the performance of population genetics studies and AM where robust virulence phenotyping data can be generated. This relatively inexpensive GBS protocol can serve as a template for a range of fungal genome characterization projects and bring genomic resources to a wider range of host–pathogen interactions than has been realized previously.
Experimental Procedures
Biological material
Pyrenophora teres f. maculata natural population
Thirty‐eight isolates of P. teres were collected from barley leaves with spot‐type lesions during the 2012 growing season from three geographically distinct locations in North Dakota: 26 isolates originated from Dickinson (Southwest ND), 11 from the Nesson Valley Township (Northwest ND) and one from Langdon (Northeast ND). Monoconidial isolates were obtained from symptomatic leaf tissue that was incubated in the dark on water agar for 2–7 days to induce sporulation, and single spores consistent with P. teres were selected and transferred to V8PDA [150 mL V8 juice, 10 g Difco (Difco Laboratories Inc, Franklin Lakes, NJ, USA) potato dextrose agar (PDA), 3 g calcium carbonate, 10 g agar]. Isolates were single spored a second time to ensure purity and grown on V8PDA for 7–10 days before being air dried as 4‐mm plugs and stored at −80 °C in 2‐mL screw‐cap microcentrifuge tubes.
Pyrenophora teres f. teres biparental population
The isolates 15A and 0–1 that were used to develop the biparental P. teres f. teres population utilized in this study were collected from California, USA and Ontario, Canada, respectively (Lai et al., 2007). These two parental isolates were shown to have differential virulence patterns on barley. A population of 118 progeny from the 15A and 0–1 cross was created as described by Lai et al. (2007). The original SSR and AFLP map was described in Ellwood et al. (2010); however, an additional 40 progeny were included in this GBS analysis and biparental map construction.
Sphaerulina musiva natural population
Septoria cankers were collected from dormant branches of 18 genotypes of hybrid poplar at the University of Minnesota Duluth Belle River Nursery in Garfield, MN, USA. Isolations were conducted as described previously in the literature (LeBoldus et al., 2009). Pure cultures of the fungus were stored as mycelial plugs, suspended in a 50% glycerol solution, in 2‐mL cryotubes at −80 °C.
Fungal growth and DNA isolation
Pyrenophora teres f. maculata natural population
The 38 isolates of P. teres f. maculata were grown at room temperature on V8PDA for 7–10 days in the dark. Hyphae were scraped from the agar surface using clean glass slide cover slips, placed in 1.5‐mL microcentrifuge tubes and lyophilized. Total gDNA was extracted from the P. teres f. teres isolates using a modified cetyltrimethylammonium bromide (CTAB) method. Approximately 100 mg of lyophilized fungal tissue was macerated in 2‐mL screw‐cap microcentrifuge tubes with one‐third volume of 1‐mm glass beads. The ground tissue was mixed with 500 μL DNA extraction buffer [0.140 m sorbitol, 220 mm Tris‐HCl, 22 mm ethylenediaminetetraacetic acid (EDTA), 0.8 m NaCl, 0.8% CTAB and 1.0% sarcosine] and incubated at 65 °C for 30–45 min. The resulting slurry was homogenized gently with 300 μL of chloroform–isoamyl alcohol (24:1) and centrifuged for 15 min at 13 000 g. The aqueous layer was collected and mixed well with an equal volume of isopropanol and centrifuged at 13 000 g for 25 min. Isopropanol was removed and pellets were washed with 70% ethanol, dissolved in ∼200 μL of TE buffer (pH 8.0) containing 10 μg/mL RNase, incubated overnight at 4 °C, vortexed and stored at −20 °C.
Pyrenophora teres f. teres biparental population
The DNA isolation for the 118 15A and 0–1 progeny and parental isolates was conducted as described in Shjerve et al. (2014).
Sphaerulina musiva natural population
The 18 isolates of S. musiva were grown in Erlenmeyer flasks with 250 mL of liquid V‐8 juice amended with 0.5 g of CaCO3. Mycelial mats were removed from the flasks after 7 days of growth at room temperature, placed in a Büchner funnel with P5 filter paper (Fisher brand), vacuum filtered and rinsed three times with sterile distilled water. Total gDNA was extracted from 1 g of each of the 18 isolates of S. musiva using the Plant mini kit (Qiagen, Valencia, CA, USA) following the manufacturer's protocol.
Library construction and sequencing
The basic protocol for GBS library construction is shown in Fig. 1. Approximately 600 ng of RNA‐free gDNA were digested with 5 units of HhaI enzyme (NEB, Ipswich, MA, USA). After 2.5 h of digestion at 37 °C, 1 unit of ApeKI enzyme (NEB) was added to the reaction and placed at 65 °C for an additional 2.5 h (Fig. 1a). The digestion reactions were extracted using 1 vol of chloroform–isoamyl alcohol (24:1) and DNA precipitated with 0.3 m sodium acetate (NaOAc) and 2.5 vol of 95% ethanol. The pelleted gDNA was washed once with 75% ethanol and air dried for 15 min at 37 °C. The gDNA of each isolate was resuspended in the ligation reaction components (total reaction volume, 15 μL) with 1 × DNA ligase buffer, 1 unit of T4 DNA ligase (Promega, Madison, WI, USA), 100 μm P1‐HhaI adaptor and 100 μm of each A1–A40 ApeKI specific barcoded adaptor (see adaptor sequences below). Ligation reactions were allowed to proceed for 16 h at 4 °C (Fig. 1b). The T4 ligase was heat inactivated at 65 °C for 20 min, the reactions were then combined and unligated adaptors were removed using a cycle pure DNA purification spin column (Omega Bio‐Tek, Norcross, GA, USA). Two different adaptors were used in this protocol and were modified from the Ion Torrent sequencing adaptors (Life Technologies, Grand Island, NY, USA) to contain two different restriction site‐compatible sticky ends. The unique barcoded adaptors contained 48 different 10‐bp barcodes at the 3′ terminus (Table S1, see Supporting Information), a four‐base key sequence, the priming site that initiates sequencing and a three‐base overhang on the 5′ end of the bottom strand (CWG). This three‐base overhang is complimentary to the ‘sticky’ ends generated by ApeKI digestion of gDNA. The sphere particle or ‘common’ adaptor contains a sequence that allows for the loading of gDNA templates onto the ion sphere particles via emulsion PCR utilizing the Ion One Touch 2 instrument (Life Technologies). The sphere particle adaptor contains a two‐base overhang on the 3′ end of the top strand (CG) that is complimentary to the ‘sticky’ ends generated by the HhaI digestion of gDNA (Fig. 1). The sequences of the oligonucleotides that are annealed together to generate the barcoded (ABC1‐48) adaptors are 5′‐CCATCTCATCCCTGCGTGTCTCCGACTCAGxxxxxxxxxxGAT‐3′ and 5′‐CWGATCyyyyyyyyyyCTGAGTCGGAGACACGCAGGGATGAGATGG*T*T‐3′, and the sequence oligos of the sphere particle adaptor (P1) are 5′‐CCACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGATCG‐3′ and 5′‐ATCACCGACTGCCCATAGAGAGGAAAGCGGAGGCGTAGTGG*T*T‐3′.
The ‘x’ and ‘y’ in the sequence denote the barcode and barcode complement sequences, respectively (Table S1). The asterisks indicate phosphorothioated S‐oligo linkages with an (*) between the two bases at which the link occurs. This linkage inhibits endonuclease activity, minimizing non‐specific blunt end ligations occurring as a result of single‐strand ‘chew‐back’. The oligos were denatured at 95 °C for 2 min in 100 mL of water, in a 500‐mL beaker, on a hot plate set on high. After boiling for 2 min, the oligos were allowed to anneal by cooling to room temperature slowly, approximately 1 h, to produce double‐stranded adaptors. The adaptor‐ligated gDNA fragments were size selected for approximately 275‐bp fragments using the Pippin Prep (Sage Science, Beverly, MA, USA) size selection system on 2% agarose cassettes. Subsequently, 2 μL of the 30 μL of gDNA collected from the Pippin Prep size selection cartridge were used as the template for library amplification. Amplification was performed in a Mastercycler pro (Eppendorf, Hauppauge, NY, USA) thermocycler using the following parameters: 95 °C for 1 min, 6–10 cycles of 95 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s, followed by 72 °C for 5 min. Library sequencing for the P. teres f. teres biparental and P. teres f. maculata natural populations was performed at the Barley Pathology/Fungal Molecular Genetics Laboratory at North Dakota State University, Fargo, ND, USA. The S. musiva sequencing was performed at the Molecular Biology Core, Washington State University, Pullman, WA, USA. The sequencing was performed on Ion Torrent PGM™ Sequencers (Life Technologies) using Ion 318™ Microprocessor Chips, and current Ion Torrent PGM standard sequencing protocols.
Sequence analysis and SNP marker calling analysis
Sequencing data were provided from the Ion Torrent server with isolate sequences sequestered by specific barcoded adaptor sequences (Table S1). These sequences were aligned using dnastar software (dnastar, Madison, WI, USA) and the SNPs were called using the dnastar software standard parameters. The alignments of the Ion Torrent data were conducted using dnastar, a CLCbio Genomics Workbench (Qiagen, Venlo, the Netherlands), a Burrows‐Wheeler Alignment Tool BWA‐SW (Li, 2012) and BWA‐MEM. All software utilized did not produce alignment problems as a result of the Ion Torrent sequence technology difficulties with homopolymer regions that introduce indel sequencing errors. The SNP data files produced in dnastar were exported as BAM files, opened using SAMTools and read column by column utilizing the following criteria. The SNP call for any nucleotide within a sequence tag from a specific isolate was defined as a single copy locus if the alignments were nearly identical and the called SNPs per sequence tag were unique to the individual isolate. We utilized a >75% cut‐off for this analysis, yet the majority of the called SNPs were within the 95%–100% identity range, well within the confidence interval for calling a homozygous individual from a haploid genome. The called SNPs from the isolate and parental alignments also had to occur in >75% of the reads within the alignment of each sequence tag. Sequence tag alignments that did not meet these criteria were discarded and considered as missing data points. The data files were then exported as Excel files for downstream data analyses.
Biparental genetic map construction
Linkage groups were created using MapDisto 1.7.6.5.2.2 (Lorieux, 2012) as described in Shjerve et al. (2014). In short, the RAD‐GBS SNP markers and anchored AFLP and SSR markers were used to create groups using ‘find groups’ with a limit of detection (LOD) threshold setting of 7.0 and an r‐max threshold setting of 0.3 with the Kosambi mapping function. The best order of markers was determined using the ‘check inversions’ and ‘ripple order’ functions. Markers that were potentially problematic and/or expanding an interval by more than 3 cM were dropped one at a time to generate high‐quality linkage groups using the ‘drop locus’ command. After assembling the linkage groups, maps were generated.
Genome alignment and analysis
The sequence tags generated from the P. teres f. teres and S. musiva GBS were aligned to the P. teres f. teres genome scaffolds (Ellwood et al., 2010) and 72 sequence scaffolds of the Septoria musiva SO2202 genome assembly release version 1.0 (http://genome.jgi‐psf.org/Sepmu1/Sepmu1.info.html), respectively. The scientific name of Septoria musiva was recently changed to Sphaerulina musiva; the JGI genome assembly still uses the Septoria musiva nomenclature. The alignments were performed using the dnastar SeqMan NGen genome assembly program set to templated assembly. The analysis to determine whether sequence tags coded for predicted proteins was performed by submitting multiple query sequences in single blastx searches at the National Center for Biotechnology Information (NCBI) (http://blast.ncbi.nlm.nih.gov).
Marker trait (mating type) association analysis
For the 34 P. teres f. maculata isolates, AM analysis was used to determine MTA between the GBS‐SNP markers and mating type using JMP Genomics version 6.1 (SAS Institute Inc., Cary, NC, USA). Two mating types occur in P. teres f. maculata, MAT1‐1 and MAT1‐2, and they do not occur simultaneously in the same individual; thus, the mating type trait can be considered as binary and was determined using PCR‐specific genetic markers described by Lu et al. (2010). Logistic regression and a general linear model were used for the MTA analysis. For both approaches, a randomly mating population was assumed. The significant sequence tags containing the MTAs were aligned to the closely related P. teres f. teres genome assembly (Ellwood et al., 2010; Genome Biology 11:R109; http://genomebiology.com/2010/11/11/R109) to determine the proximity of the SNP markers identified as MTAs to the previously characterized MAT1‐2 gene using the dnastar program.
Supporting information
Table S1 Consensus barcoding adaptor (ABC) and barcode sequences used for genotype‐by‐sequencing (GBS). aThe consensus adaptor sequence with ‘x’ designating the position at which the different 10‐bp barcode sequences are located. bThe 46 10‐bp barcodes utilized in this study. Barcode #30 was omitted because it did not work.
Acknowledgements
We received valuable technical help and expertise from Danielle Holmes at the US Department of Agriculture‐Agricultural Research Service (USDA‐ARS), Cereal Crops Research Unit, Fargo, ND, USA. This project was supported by the National Research Initiative Competitive Grants CAP project 2011‐68002‐30029 from the USDA‐National Institute of Food and Agriculture (USDA‐NIFA) and USDA‐NIFA‐Regional Integrated Pest Management (USDA‐NIFA‐RIPM) 2012‐34103‐19771, and the National Science Foundation CAREER project Award Number 1253987.
References
- Antequera, F. , Tamame, M. , Villanueva, J.R. and Santos, T. (1984) DNA methylation in the fungi. J. Biol. Chem. 259, 8033–8036. [PubMed] [Google Scholar]
- Arumuganathan, K. and Earle, E. (1991) Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218. [Google Scholar]
- Baird, N.A. , Etter, P.D. , Atwood, T.S. , Currey, M.C. , Shiver, A.L. , Lewis, Z.A. , Selker, E.U. , Cresko, W.A. and Johnson, E.A. (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE, 3, e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter, S.W. , Davey, J.W. , Johnston, J.S. , Shelton, A.M. , Heckel, D.G. , Jiggins, C.D. and Blaxter, M.L. (2011) Linkage mapping and comparative genomics using next‐generation RAD sequencing of a non‐model organism. PLoS ONE, 6, e19315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binz, T. , D'Mello, N. and Horgen, P.A. (1998) A comparison of DNA methylation levels in selected isolates of higher fungi. Mycologia, 90, 785–790. [Google Scholar]
- Chutimanitsakun, Y. , Nipper, R.W. , Cuesta‐Marcos, A. , Cistue, L. , Filichkina, T. , Johnson, E.A. and Hayes, P.M. (2011) Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genomics, 12, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman, K. , Himmelstrand, K. , Olson, A. , Lind, M. , Brandstrom‐Durling, M. and Stenlid, J. (2013) Genome‐wide association study identifies genomic regions for virulence in the non‐model organism Heterobasidion annosum s.s. PLoS One, 8, e53525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwood, S.R. , Liu, Z. , Syme, R.B. , Lai, Z. , Hane, J.K. , Keiper, F. , Moffat, C.S. , Oliver, R.P. and Friesen, T.L. (2010) A first genome assembly of the barley fungal pathogen Pyrenophora teres f. teres . Genome Biology, 11, R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire, R.J. , Glaubitz, J.C. , Sun, Q. , Poland, J.A. , Kawamoto, K. , Buckler, E.S. and Mitchell, S.E. (2011) A robust, simple Genotyping‐by‐Sequencing (GBS) approach for high diversity species. PLoS ONE, 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen, T.L. and Faris, J.D. (2010) Characterization of the wheat–Stagonospora nodorum disease system: what is the molecular basis of this quantitative necrotrophic disease interaction. Can. J. Plant Pathol. 32, 20–28. [Google Scholar]
- Gregory, T.R. , Nicol, J.A. , Tamm, H. , Kullman, B. , Kullman, K. , Leitch, I.J. , Murray, B.G. , Kapraun, D.F. , Greilhuber, J. and Bennett, M. (2007) Eukaryotic genome size database. Nucleic Acids Res. 35, D332–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, Z. , Faris, J.D. , Weiland, J.J. , Steffenson, B.J. and Friesen, T.L. (2007) Genetic mapping of Pyrenophora teres f. teres genes conferring avirulence on barley. Fungal Genet. Biol. 44, 323–329. [DOI] [PubMed] [Google Scholar]
- LeBoldus, J.M. , Blenis, P.V. , Thomas, B.R. , Feau, N. and Bernier, L. (2009) Susceptibility of Populus balsamifera to Septoria musiva: a field study and greenhouse experiment. Plant Dis. 93, 1146–1150. [DOI] [PubMed] [Google Scholar]
- Li, H. (2012) Exploring single‐sample SNP and INDEL calling with whole‐genome de novo assembly. Bioinformatics, 28, 1838–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorieux, M. (2012) MapDisto: fast and efficient computation of genetic linkage maps. Mol. Breed. 30, 1231–1235. [Google Scholar]
- Lu, S. , Platz, G.J. , Edwards, M.C. and Friesen, T.L. (2010) Mating type (MAT) locus‐specific PCR markers for differentiation of Pyrenophora teres f. teres and P. teres f. maculata, the causal agents of barley net blotch. Phytopathology 100: 1298–1306. [DOI] [PubMed] [Google Scholar]
- Ma, X.‐F. , Jensen, E. , Alexandrov, N. , Troukham, M. , Zhang, L. , Thomas‐Jones, S. , Farrar, K. , Clifton‐Brown, J. , Donnison, I. , Swaller, T. and Flavell, R. (2012) High resolution genetic mapping by genome sequencing reveals genome duplication and tetraploid genetic structure of the diploid Miscanthus sinensis . PLoS ONE, 7, e33821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Wu, S. , St. Amand, P. , Stein, N. and Poland, J. (2013) Application of genotyping‐by‐sequencing on semiconductor sequencing platforms: a comparison of genetic and reference‐based marker ordering in barley. PLoS One, 8, e76925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkowski, J. and Whitham, S.A. (2001) Gene silencing and DNA methylation processes. Curr. Opin. Plant Biol. 4, 123–129. [DOI] [PubMed] [Google Scholar]
- Pfender, W.F. , Saha, M.C. , Johnson, E.A. and Slabaugh, M.B. (2011) Mapping with RAD (restriction‐site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne . Theor. Appl. Genet. 122, 1467–1480. [DOI] [PubMed] [Google Scholar]
- Poland, J.A. , Brown, P.J. , Sorrells, M.E. and Jannink, J.‐L. (2012) Development of a high‐density genetic map for barley and wheat using a novel two‐enzyme genotype‐by‐sequencing approach. PLoS ONE, 7, e32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkoski, J.E. , Poland, J. , Jannink, J.L. and Sorrells, M.E. (2013) Imputation of unordered markers and the impact on genomic selection accuracy. G3: Genes Genomes Genetics, 3, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheet, P. and Stephens, M. (2006) A fast and flexible statistical model for large‐scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selker, E. (2002) Repeat‐induced gene silencing in fungi. Adv. Genet. 46, 439–450. [DOI] [PubMed] [Google Scholar]
- Selker, E.U. , Tountas, N.A. , Cross, S.H. , Margolin, B.S. , Murphy, J.G. , Bird, A.P. and Freitag, M. (2003) The methylated component of the Neurospora crassa genome. Nature, 422, 893–897. [DOI] [PubMed] [Google Scholar]
- Shjerve, R.A. , Faris, J.D. , Brueggeman, R.S. , Yan, C. , Zhu, Y. , Koladia, V. and Friesen, T.L. (2014) Evaluation of a Pyrenophora teres f. teres mapping population reveals multiple independent interactions with the barley 6H chromosome region. Fungal Genet. Biol. 70, 104–112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Consensus barcoding adaptor (ABC) and barcode sequences used for genotype‐by‐sequencing (GBS). aThe consensus adaptor sequence with ‘x’ designating the position at which the different 10‐bp barcode sequences are located. bThe 46 10‐bp barcodes utilized in this study. Barcode #30 was omitted because it did not work.