

Summary
Among the Eukaryotes, Fungi have relatively small genomes (average of 44.2 Mbp across 1850 species). The order Pucciniales (Basidiomycota) has the largest average genome size among fungi (305 Mbp), and includes the two largest fungal genomes reported so far (Puccinia chrysanthemi and Gymnosporangium confusum, with 806.5 and 893.2 Mbp, respectively). In this work, flow cytometry was employed to determine the genome size of the Bidens pilosa rust pathogen, Uromyces bidentis. The results obtained revealed that U. bidentis presents a surprisingly large haploid genome size of 2489 Mbp. This value is almost three times larger than the previous largest fungal genome reported and over 50 times larger than the average fungal genome size. Microscopic examination of U. bidentis nuclei also showed that they are not as different in size from the B. pilosa nuclei when compared with the differences between other rusts and their host plants. This result further reinforces the position of the Pucciniales as the fungal group with the largest genomes, prompting studies addressing the role of repetitive elements and polyploidy in the evolution, pathological specialization and diversity of fungal species.
Keywords: Bidens pilosa, flow cytometry, genome size, Uromyces bidentis
Genome size in Eukaryotes varies greatly, as a result of polyploidization and/or structural chromosomal rearrangements. Genome size and its dynamics have direct implications on the evolutionary fitness, reproduction and non‐sexual mechanisms for diversity creation (D'Hondt et al., 2011). Among Fungi, previous studies have revealed that genomes vary from <1 to 893 Mbp (Kullman et al., 2005; Tavares et al., 2014). Recently, rust fungi (Basidiomycota, Pucciniales) have been recognized as the order with the largest average genome size (305 Mbp) (Tavares et al., 2014), with species with genomes as large as 806.5 and 893.2 Mbp (Puccinia chrysanthemi Roze and Gymnosporangium confusum Dietel, respectively; Tavares et al., 2014). Genome sequences available for a few rust fungi have shown that these genomes are populated with transposable elements and other non‐coding repetitive sequences (Cantu et al., 2011; Duplessis et al., 2011; Nemri et al., 2014; Tan et al., 2014). As most of the genome sequences available for rusts represent relatively small genomes, it is expected that larger rust genomes could comprise even higher proportions of such repetitive elements, which is corroborated by preliminary data obtained from Hemileia vastatrix Berk. & Broome (Cristancho et al., 2014) and Uromyces fabae de Bary ex Cooke (Link et al., 2014). Polyploidy could also account for these large genomes, but no evidence supporting its occurrence has been gathered.
The objective of this work was to employ flow cytometry to determine the genome size of the Bidens pilosa L. rust fungus Uromyces bidentis Lagerheim, showing that this organism has the largest fungal genome ever reported.
Bidens pilosa L. (black‐jack, or Spanish needle, among other common names) is an Asteraceae species originating from the Americas, being widely disseminated in tropical and temperate regions of the world. The genome size of this species has been estimated as 2C = 3325 Mbp (Bennett and Leitch, 1997). Plants exhibiting rust symptoms were identified and collected at Tapada da Ajuda, Lisbon, Portugal. Cross‐sections of infected leaves (20–25 μm) were stained with cotton blue for the observation of fungal structures, or with 4′,6‐diamidino‐2‐phenylindole (DAPI; Sigma‐Aldrich, St. Louis, MO, USA) and diethanol (Uvitex 2B, Advanced Technology & Industrial Co., Hong Kong, China) for the visualization of nuclei and fungal cell walls (respectively), as described previously (Tavares et al., 2014). Observations were made with a Leica DM‐2500 microscope (Leica Microsystems GmbH, Wetzlar, Germany), under bright field or ultraviolet (UV) illumination (excitation filter BP 340–380; barrier filter LP 430). For comparison purposes, nuclei from Coffea arabica L. leaves infected with H. vastatrix and of Avena sterilis L. infected with Puccinia coronata Corda were also analysed using the same procedure.
The genome size of the rust and host species was estimated by flow cytometry using a Partec CyFlow Space flow cytometer (Partec GmbH, Görlitz, Germany). For this, nuclei from rust and host species, together with those of Solanum lycopersicum L. ‘Stupické’ (internal reference standard; 2C = 1.96 pg or 1917 Mbp; Doležel et al., 1992), were isolated by simultaneously chopping infected leaves of B. pilosa with leaves of the reference standard, as described previously (Loureiro et al., 2007; Tavares et al., 2014). The nuclear suspension was then filtered through a 30‐μm nylon filter to remove plant and fungal debris, and 50 μg/mL of propidium iodide (Fluka, Buchs, Switzerland) and 50 μg/mL of RNase (Fluka) were added to stain DNA only. Data were acquired using Partec FloMax software v2.4d (Partec GmbH), as described previously (Tavares et al., 2014). Fluorescence peaks of fungal nuclei were identified by comparing fluorescence histograms of rust‐infected leaves and of healthy B. pilosa leaves.
Rust symptoms were observed on leaves of flowering B. pilosa plants between May 2014 and January 2015 (Fig. 1a,b). The symptoms began with the appearance of brownish spots on both sides of B. pilosa leaves. On the abaxial surface of the leaves, the spots gradually became raised, and distinct circular light‐brown uredinia, first subepidermal then erumpent, appeared on the spots. Premature leaf chlorosis, senescence and defoliation were common on leaves where sori were abundant. Microscopic examination of urediniospores and sori (Fig. 1c,d) indicated that the pathogen is Uromyces bidentis Lagerheim, Bull. Soc. Myc. Fr. 11, 213, 1895 (=U. bidenticola Arth., Mycologia, 9, 71, 1917) (Baker, 1955; Sydow, 1910). A herbarium specimen was deposited in the ‘João de Carvalho e Vasconcellos’ herbarium (LISI), under accession LISI‐FUNGI‐00026.
Figure 1.
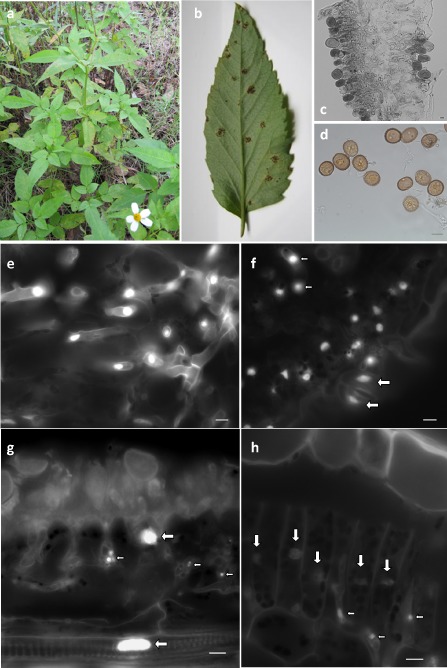
Uromyces bidentis symptoms and structures, and nuclei by comparison with nuclei of other organisms. (a) Symptoms in Bidens pilosa plants in the field (Tapada da Ajuda, Lisbon, Portugal; November 2014). (b) Uredinia on the abaxial surface of B. pilosa leaflet. (c) Cross‐section of an infected leaf showing uredinia. (d) Urediniospores. (e–h) 4′,6‐Diamidino‐2‐phenylindole (DAPI)‐stained nuclei (bar, 10 μm; small arrow, fungal nuclei; large arrow, plant nuclei), visualized under fluorescence, of U. bidentis (e), of U. bidentis and Bidens pilosa including stoma (f), of Puccinia coronata and Avena sterilis (g) and of Hemileia vastatrix and Coffea arabica (h).
At the nuclear level, it has already been shown that genome size is correlated with both nuclear (Baetcke et al., 1967) and chromosome (Bennett et al., 1983) volume. Therefore, the larger the genome size, the greater the minimum nuclear volume. Considering that the microscopic examination of infected leaf sections revealed that the fungal nuclei were not very different in size from the host plant nuclei (Fig. 1e,f), similar genome sizes were expected. To further explore the correlation between genome size and nuclear volume, and to provide further evidence that could support a genome size of U. bidentis close to that of B. pilosa, A. sterilis leaves infected with P. coronata and C. arabica leaves infected with H. vastatrix were also analysed microscopically. Although large differences in nuclear size were observed between P. coronata and A. sterilis nuclei (Fig. 1g), whose genome sizes are 244 Mbp (Tavares et al., 2014) and 26 699 Mbp (Bennett and Smith, 1976), respectively, i.e. over 100 times difference in genome size, the use of H. vastatrix and C. arabica (Fig. 1h) revealed smaller differences in nuclei size, which is in accordance with the genome sizes of both the rust and host species of 797 and 2347 Mbp, respectively (Tavares et al., 2014), i.e. less than three times difference.
The comparison between flow cytometric histograms of healthy and rust‐infected B. pilosa leaves clearly enabled the identification of U. bidentis peaks (Fig. 2b,c), and enabled us to estimate the 1C DNA content of U. bidentis as 2.54 pg, corresponding to a haploid genome of 2489 ± 43 Mbp [coefficient of variation (CV), 1.7%; n = 12]. As noted previously for other rusts (Tavares et al., 2014), 2C peaks were always observed for U. bidentis together with the 1C peak typical of fungal species. The nuclear DNA content of the host species, B. pilosa, was also estimated by comparison with the internal reference standard S. lycopersicum (Fig. 2a), and revealed that the B. pilosa genome is larger than previously thought (6.04 ± 0.15 pg/2C, i.e. 5908 Mbp vs. 3325 Mbp). This large difference in genome size is probably caused by the use of different methodologies. Although our estimation was obtained using flow cytometry, the estimation of Bennett and Leitch (1997) was obtained using Feulgen densitometry, which presents many critical procedural points (e.g. fixation, slide preparation and storage, acid hydrolysis) that determine its precision.
Figure 2.
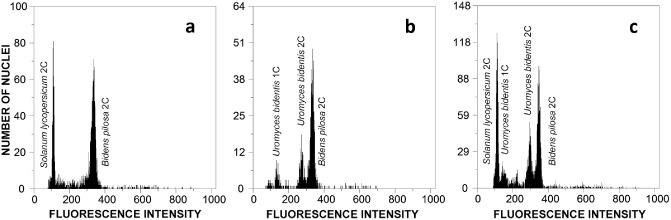
Flow cytometric histograms of relative fluorescence intensities of propidium iodide‐stained nuclei isolated from healthy Bidens pilosa leaves and the plant DNA reference standard Solanum lycopersicum (a), Uromyces bidentis‐infected B. pilosa leaves (b), and U. bidentis‐infected B. pilosa leaves and the plant DNA reference standard S. lycopersicum (c).
The genome size estimated for U. bidentis, 2489 Mbp, is within the range of that of the host plant, which is further corroborated by the similar nuclear sizes indicated by fluorescence microscopy observation (Fig. 1e,f). Although very large, it is interesting to note that the rust genome is smaller than the host plant genome, a situation that was recurrently observed in a study involving 32 rust–host species pairs (Tavares et al., 2014). Nevertheless, the U. bidentis genome is approximately three times larger than the largest fungal genome reported previously (G. confusum, 893 Mbp), and over 50 times larger than the average fungal genome size (44.2 Mbp). Indeed, adding the genome size of U. bidentis to the list of 1850 fungal genomes with known size (Kullman et al., 2005; Tavares et al., 2014) shifts the average fungal genome size by c. 1.5 Mbp to 45.9 Mbp, the average Basidiomycota genome size from 70.4 to 76.2 Mbp, and the average Pucciniales genome size from 305.5 to 350.9 Mbp.
This result further reinforces the unparalleled position of the Pucciniales among fungi as a group with large or very large genomes. It also stresses the huge variation in genome size occurring within the group, highlighted by the over 30 times difference between Cronartium quercuum f. sp. fusiforme Burds. & G.A. Snow (with 76.6 Mbp; Anderson et al., 2010) and U. bidentis (at 2489 Mbp). Interestingly, genome size information available for seven species in the Uromyces genus ranges from 276.8 Mbp for Uromyces rumicis (Schumach.) G. Winter to 2489 Mbp for U. bidentis, illustrating important fluctuations in genome size even within a genus. Such large genome size variations within fungal genera are rare, paralleled only by the genera Hymenoscyphus (9–54 Mbp), Suillus (20–94 Mbp) and Glomus (14–264 Mbp), together with the rust genus Puccinia (77–806 Mbp) (Kullman et al., 2005; Tavares et al., 2014), and could suggest that variations in genome size may be an active element of fungal evolution, speciation and host specialization in some taxonomic groups. The occurrence and evolutionary significance of genome size expansion, mostly through polyploidy, and the subsequent ecological, structural and functional consequences have been shown in diverse fungi (Albertin and Marullo, 2012). Among rusts, such fluctuations in genome size are clear across their phylogeny (Tavares et al., 2014). For example, H. vastatrix, one of the most ancestral rust lineages (Aime, 2006), has one of the largest rust genomes (797 Mbp), whereas Puccinia malvacearum Bertero ex Mont., basal to the Pucciniaceae (van der Merwe et al., 2008), has a smaller genome (178 Mbp); nevertheless, Pucciniaceae, encompassing the genera Puccinia and Uromyces, comprises the entire genome size range among rusts. Rapid adaptive evolution in many pathogenic fungi has been shown to be linked to chromosomal rearrangements and ploidy changes (as reviewed by Croll & McDonald, 2012 and Stukenbrock and Croll, 2014), including the examples arising from Fusarium (Ma et al., 2010) and Mycosphaerella graminicola (Fuckel) J. Schröt. (Goodwin et al., 2011; Stukenbrock et al., 2010). Intraspecific variability of genome size in fungi has been documented (Bourne et al., 2014), in some cases being related to host–pathogen interactions, such as in Geosmithia (Veselská and Kolařík, 2015) or Leptosphaeria maculans Ces. & De Not. (Grandaubert et al., 2014). Moreover, rust fungi with large genomes, such as H. vastatrix, Phakopsora pachyrhizi Syd. & P. Syd. and, to some extent, P. chrysanthemi, all rely on asexual reproduction (Tavares et al., 2014). Similarly, U. bidentis is a hemicyclic rust (Baker, 1955), with no known aecial host. Therefore, polyploidy and/or the activity of transposable elements could be important diversity‐creating factors in such mostly asexual organisms, being the main mechanisms driving genome expansions. Such relations between genome size variation and sexual abstinence and/or population size have been documented in several organisms, including fungi (Hu et al., 2014; Spanu, 2012), and have been the subject of theoretical analyses in evolutionary biology (e.g. Startek et al., 2013; Whitney & Garland, 2010). With diversified reproduction strategies (sexual, asexual or, rarely, sexual), host ranges (single or few species, or several species from diverse taxa) and large but variable genome sizes, we anticipate that rust fungi may also prove to be relevant for such studies.
Acknowledgements
Eng. Filomena Caetano, Ms Ana Paula Paes and Eng. Teresa Vasconcelos (Instituto Superior de Agronomia, University of Lisbon, Portugal) are acknowledged for support in the identification and mapping of host species and of rust‐infected plants. Dr Andreia Loureiro and Ms Paula Leandro are acknowledged for support in the preparation of samples for microscopy analyses. This work was supported by Fundação para a Ciência e a Tecnologia (FCT, Portugal) through project PTDC/AGR‐GPL/114949/2009 and postdoctoral grants awarded to Sílvia Tavares and Pedro Talhinhas (SFRH/BPD/65965/2009 and SFRH/BPD/88994/2012, respectively).
The authors declare no conflicts of interest.
References
- Aime, M.C. (2006) Toward resolving family‐level relationships in rust fungi (Uredinales). Mycoscience, 47, 112–122. [Google Scholar]
- Albertin, W. and Marullo, P. (2012) Polyploidy in fungi: evolution after whole‐genome duplication. Proc. R. Soc. London, B: Biol. Sci. 279, 2497–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, C.L. , Kubisiak, T.L. , Nelson, C.D. , Smith, J.A. and Davis, J.M. (2010) Genome size variation in the pine fusiform rust pathogen Cronartium quercuum f. sp. fusiforme as determined by flow cytometry. Mycologia, 102, 1295–1302. [DOI] [PubMed] [Google Scholar]
- Baetcke, K.P. , Sparrow, A.H. , Naumann, C.H. and Schwemme, S.S. (1967) The relationship of DNA content to nuclear and chromosome volumes and to radiosensitivity (LD50). Proc. Natl. Acad. Sci. USA, 58, 533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, S. (1955) Additions to the rust fungi of New Zealand, II. Trans. Proc. R. Soc. N. Z. 83, 453–463. [Google Scholar]
- Bennett, M.D. and Leitch, I.J. (1997) Nuclear DNA amounts in angiosperms—583 new estimates. Ann. Bot. 80, 169–196. [Google Scholar]
- Bennett, M.D. and Smith, J.B. (1976) Nuclear DNA amounts in angiosperms. Philos. Trans. R. Soc. B , 274, 227–274. [DOI] [PubMed] [Google Scholar]
- Bennett, M.D. , Heslop‐Harrison, J.S. , Smith, J.B. and Ward, J.P. (1983) DNA density in mitotic and meiotic metaphase chromosomes of plants and animals. J. Cell Sci. 63, 173–179. [DOI] [PubMed] [Google Scholar]
- Bourne, E.C. , Mina, D. , Gonçalves, S.C. , Loureiro, J. , Freitas, H. and Muller, L.A.H. (2014) Large and variable genome size unrelated to serpentine adaptation but supportive of cryptic sexuality in Cenococcum geophilum . Mycorrhiza, 24, 13–20. [DOI] [PubMed] [Google Scholar]
- Cantu, D. , Govindarajulu, M. , Kozik, A. , Wang, M. , Chen, X. , Kojima, K.K. , Jurka, J. , Michelmore, R.W. and Dubcovsky, J. (2011) Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS ONE, 6, e24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristancho, M.A. , Botero‐Rozo, D.O. , Giraldo, W. , Tabima, J. , Riaño‐Pachón, D.M. , Escobar, C. , Rozo, Y. , Rivera, L.F. , Durán, A. , Restrepo, S. , Eilam, T. , Anikster, Y. and Gaitán, A.L. (2014) Annotation of a hybrid partial genome of the coffee rust (Hemileia vastatrix) contributes to the gene repertoire catalog of the Pucciniales. Front. Plant Sci. 5, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll, D. and McDonald, B.A. (2012) The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 8, e1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hondt, L. , Hofte, M. , Van Bockstaele, E. and Leus, L. (2011) Applications of flow cytometry in plant pathology for genome size determination, detection and physiological status. Mol. Plant Pathol. 12, 815–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležel, J. , Sgorbati, S. and Lucretti, S. (1992) Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 85, 625–631. [Google Scholar]
- Duplessis, S. , Cuomo, C.A. , Lin, Y.C. , Aerts, A. , Tisserant, E. , Veneault‐Fourrey, C. , Joly, D.L. , Hacquard, S. , Amselem, J. , Cantarel, B.L. , Chiu, R. , Coutinho, P.M. , Feau, N. , Field, M. , Frey, P. , Gelhaye, E. , Goldberg, J. , Grabherr, M.G. , Kodira, C.D. , Kohler, A. , Kües, U. , Lindquist, E.A. , Lucas, S.M. , Mago, R. , Mauceli, E. , Morin, E. , Murat, C. , Pangilinan, J.L. , Park, R. , Pearson, M. , Quesneville, H. , Rouhier, N. , Sakthikumar, S. , Salamov, A.A. , Schmutz, J. , Selles, B. , Shapiro, H. , Tanguay, P. , Tuskan, G.A. , Henrissat, B. , Van de Peer, Y. , Rouzé, P. , Ellis, J.G. , Dodds, P.N. , Schein, J.E. , Zhong, S. , Hamelin, R.C. , Grigoriev, I.V. , Szabo, L.J. and Martin, F. (2011) Obligate biotrophy features unravelled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA, 108, 9166–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, S.B. , M'barek, S.B. , Wittenberg, A.H.J. , Crane, C.F. , Hane, J.K. , van der Lee, T.A.J. , Grimwood, J. , Aerts, A. , Antoniw, J. , Bowler, J. , van der Burgt, A. , Coutinho, P.M. , Csukai, M. , Dehal, P. , Hammond‐Kosack, K.E. , Henrissat, B. , Kilian, A. , Lindquist, E. , Mehrabi, R. , Rudd, J.J. , Salamov, A. , Schmutz, J. , Schouten, H.J. , Shapiro, H. , Stergiopoulos, I. , Torriani, S.F.F. , de Vries, R.P. , Waalwijk, C. , Ware, S.B. , Zwiers, L.‐H. , Oliver, R.P. , Grigoriev, I.V. and Kema, G.H.J. (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 7, e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandaubert, J. , Lowe, R.G.T. , Soyer, J.L. , Schoch, C.L. , van de Wouw, A.P. , Fudal, I. , Robbertse, B. , Lapalu, N. , Links, M.G. , Ollivier, B. , Linglin, J. , Barbe, V. , Mangenot, S. , Cruaud, C. , Borhan, H. , Howlett, B.J. , Balesdent, M.‐H. and Rouxel, T. (2014) Transposable element‐assisted evolution and adaptation to host plant within the Leptosphaeria maculans–Leptosphaeria biglobosa species complex of fungal pathogens. BMC Genomics, 15, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, X. , Xiao, G. , Zheng, P. , Shang, Y. , Su, Y. , Zhang, X. , Liu, X. , Zhan, S. , St. Leger, R.J. and Wang, C. (2014) Trajectory and genomic determinants of fungal‐pathogen speciation and host adaptation. Proc. Natl. Acad. Sci. USA, 111, 16 796–16 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullman, B. , Tamm, H. and Kullman, K. (2005) Fungal Genome Size Database. http://www.zbi.ee/fungal‐genomesize [accessed on March 4, 2015].
- Link, T. , Seibel, C. and Voegele, R.T. (2014) Early insights into the genome sequence of Uromyces fabae . Front. Plant Sci. 5, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro, J. , Rodriguez, E. , Doležel, J. and Santos, C. (2007) Two new nuclear isolation buffers for plant DNA flow cytometry: a test with 37 species. Ann. Bot. 100, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L.‐J. , van der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.‐J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.‐B. , Woloshuk, C. , Xie, X. , Xu, J.‐R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A.E. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G.J. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.‐H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.‐Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe, M.M. , Walker, J. , Ericson, L. and Burdon, J.J. (2008) Coevolution with higher taxonomic host groups within the Puccinia/Uromyces rust lineage obscured by host jumps. Mycol. Res. 112, 1387–1408. [DOI] [PubMed] [Google Scholar]
- Nemri, A. , Saunders, D.G. , Anderson, C. , Upadhyaya, N.M. , Win, J. , Lawrence, G.J. , Jones, D.A. , Kamoun, S. , Ellis, J.G. and Dodds, P.N. (2014) The genome sequence and effector complement of the flax rust pathogen Melampsora lini . Front. Plant Sci. 5, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanu, P.D. (2012) The genomics of obligate (and nonobligate) biotrophs. Ann. Rev. Phytopathol. 50, 91–109. [DOI] [PubMed] [Google Scholar]
- Startek, M. , Le Rouzic, A. , Capy, P. , Grzebelus, D. and Gambin, A. (2013) Genomic parasites or symbionts? Modeling the effects of environmental pressure on transposition activity in asexual populations. Theor. Pop. Biol. 90, 145–151. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. and Croll, D. (2014) The evolving fungal genome. Fungal Biol. Rev. 28, 1–12. [Google Scholar]
- Stukenbrock, E.H. , Jørgensen, F.G. , Zala, M. , Hansen, T.T. , McDonald, B.A. and Schierup, M.H. (2010) Whole‐genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola . PLoS Genet. 6, e1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow, P. and Sydow, H. (1910) Genus Uromyces. Monogr. Uredinearum , 2, 1–144. [Google Scholar]
- Tan, M.‐K. , Collins, D. , Chen, Z. , Englezou, A. and Wilkins, M.R. (2014) A brief overview of the size and composition of the myrtle rust genome and its taxonomic status. Mycology, 5, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares, S. , Ramos, A.P. , Pires, A.S. , Azinheira, H.G. , Caldeirinha, P. , Link, T. , Abranches, R. , Silva, M.C. , Voegele, R.T. , Loureiro, J. and Talhinhas, P. (2014) Genome size analyses of Pucciniales reveal the largest fungal genomes. Front. Plant Sci. 5, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselská, T. and Kolařík, M. (2015) Application of flow cytometry for exploring the evolution of Geosmithia fungi living in association with bark beetles: the role of conidial DNA content. Fungal Ecol. 13, 83–92. [Google Scholar]
- Whitney, K. and Garland, Jr., T. (2010) Did genetic drift drive increases in genome complexity? PLoS Genet. 6, e1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]